
Abstract
Coronavirus disease 2019 (COVID-19) is a multi-systemic illness that became a pandemic in March 2020. Although environmental factors and comorbidities can influence disease progression, there is a lack of prognostic markers to predict the severity of COVID-19 illness. Identifying these markers is crucial for improving patient outcomes and appropriately allocating scarce resources. Here, an RNA-sequencing study was conducted on blood samples from unvaccinated, hospitalized patients divided by disease severity; 367 moderate, 173 severe, and 199 critical. Using a bioinformatics approach, we identified differentially expressed genes (DEGs), alternative splicing (AS) and alternative polyadenylation (APA) events that were severity-dependent. In the severe group, we observed a higher expression of kappa immunoglobulins compared to the moderate group. In the critical cohort, a majority of AS events were mutually exclusive exons and APA genes mostly had longer 3’UTRs. Interestingly, multiple genes associated with cytoskeleton, TUBA4A, NRGN, BSG, and CD300A, were differentially expressed, alternatively spliced and polyadenylated in the critical group. Furthermore, several inflammation-related pathways were observed predominantly in critical vs. moderate. We demonstrate that integrating multiple downstream analyses of transcriptomics, from moderate, severe, and critical patients confers a significant advantage in identifying relevant dysregulated genes and pathways.
Similar content being viewed by others

Introduction
In December 2019, the first case of Coronavirus disease 2019 (COVID-19) emerged in Wuhan, China, caused by the severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) virus1. In March 2020, the situation evolved to a global pandemic. As of September 9th, 2024, it had affected more than 775 million individuals worldwide resulting in more than seven million fatalities according to the World Health Organization2. Originally categorized as a respiratory infection, COVID-19 is now recognized as a multi-systemic disease. Approximately 80% of unvaccinated cases are either asymptomatic or will develop mild to moderate symptoms, while 15% develop a severe disease and the remaining 5% face critical disease, which might lead to death3.
The use of transcriptomic techniques enables a comprehensive understanding of how gene expression can vary between conditions. In the context of COVID-19, multiple differentially expressed genes (DEGs) have already been identified. Transcriptomics also offers the ability to investigate post-transcriptional modifications, such as alternative splicing (AS) and alternative polyadenylation (APA), crucial for diversifying and regulating the proteome. AS involves differential exon and intron inclusion, creating diverse transcripts which can have different functions based on their domains or results in non-functioning and altered proteins. There are five subtypes of AS events: alternative 3’ splice site (A3SS), alternative 5’ splice site (A5SS), mutually exclusive exons (MXE), retained intron (RI) and skipped exon (SE). SE is the most prevalent subtype, accounting for around 40% of AS events in humans4. They are often found in protein domains and may affect the protein secondary structure leading to altered interaction with other proteins. A3SS and A5SS often lead to more subtle changes to the sequence at the beginning or end of the exon5. As for RIs, they may lead to preservation of a stop codon in the intron, that normally should have been removed or it may have a less drastic effect and change the expression of the resulting protein6. MXE is the rarest form of AS and around 65% of events are tissue-specific7,8. MXEs are also the most complex, keeping only one of two available exons by activating one and inactivating the spliced one. This leads to a change in sequence from one exon to the other depending on the resulting isoform, but not a change in structure like other AS types7,9. The study of AS in infectious disease is particularly interesting as viruses can manipulate the spliceosome to aid their replication and evade host immune responses affecting the host cell cycle10. Moreover, a previous study suggested that the degree in which the spliceosome is altered could serve as a predictor of severity in COVID-19 patients10,11. APA, a less explored regulatory mechanism, influences messenger RNA translation and stability by changing the 3’ untranslated region (UTR) length of the poly(A) tail of a gene12. APA is the last step in post-transcriptional modification and more than 70% of genes are thought to be prone to APA13. Like AS, APA also has some tissue-specific impact. For example, transcripts in the blood tend to have shorter 3’ UTR, while the brain prefers longer 3’ UTR13. The length of the 3’UTR can have an important impact on gene expression by affecting the messenger RNA stability, degradation rate or microRNA binding14,15. Most of the studies on APA were done in cancer cell lines. They found that genes with longer 3’ UTR were associated with apoptosis and cell death, while genes with shorter 3’ UTR were implicated in cell cycle, metabolic pathways, glucose transport and antigen processing and presentation13,16. Another study on cancer cells, found that shorter 3’ UTR increased gene stability and led to higher protein production in some genes17. In COVID-19, APA can up-regulate immune genes, and a global shortening of APA was observed in moderate and severe patients with SARS-CoV-2 infection compared to non-infected controls18. While AS and APA have been studied in the context of COVID, there is, to our knowledge, no study that has integrated both types of analyses in the context of COVID-19 severity. Since AS and APA can both influence gene expression, it is important to study them in combination with DEGs to gain a comprehensive understanding of the changes occurring between conditions. Those changes can also be seen by doing pathway enrichment. By using DEGs as well as genes affected by AS and APA, pathways can be identified as being dysregulated according to different COVID-19 severities. Some pathways are already of interest based on previous studies, including cytokine storm, inflammation, cell cycle and cell death19,20,21. Identifying those relevant pathways could eventually lead to effective treatment of each COVID-19 severity. Beyond transcriptional diversity, the Human Leukocyte Antigen (HLA) protein complex emerged as a potential key factor in understanding COVID-19 pathogenesis since it had been associated to viral infections in the past22,23,24. Studies have identified HLA alleles related to both susceptibility and severity of COVID-19 and found that those alleles are population dependent22.
In this study, we conducted a thorough unbiased analysis of transcriptional and post-transcriptional profiles in COVID-19 patients exhibiting diverse clinical severities. Through this investigation, we aimed to pinpoint essential post-transcriptional factors and their effect on gene expression based on hospitalized moderate, severe, and critical COVID-19 patients. We also aimed to identify pathways affected by DEGs, AS and APA affected genes, that contribute to the underlying pathophysiology of the disease within these distinct patient groups.
Results
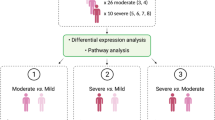
To find differences in the transcriptional profile associated with COVID-19 severity, RNA from the blood of 367 moderate, 173 severe and 199 critical hospitalized patients was extracted and sequenced by the BQC1925. The cohort was divided by disease severity; moderate; severe; critical according to defined criteria (see Methods) and cohort characteristics are resumed in Table 1. Comorbidities are included in Supplementary Table S1. Here, transcriptomic downstream analyses were integrated, including DEGs, AS, APA, HLA typing, and pathway enrichment analysis, to achieve a comprehensive atlas of COVID-19 changes in severe and critical patients compared to moderate patients.
Critical COVID-19 patients have higher gene expression changes compared to severe patients
For the DEG analyses, 29,921 genes were detected in the severe vs. moderate cohort, and 32,956 genes in the critical vs. moderate cohort. After filtering for p-value < 0.01, baseMean > 10, and an absolute value of log2FoldChange greater than one, 90 DEGs were obtained in the severe vs. moderate comparison, 84 up-regulated and 6 down-regulated (Fig. 1a, b). Similarly, the critical vs. moderate comparison yielded 674 DEGs, with 645 up-regulated and 29 down-regulated (Fig. 1c, d).

Higher number of DEGs between critical vs. moderate when compared to severe vs. moderate. (a) Volcano plot for severe vs. moderate. All the genes shown have a baseMean > 10. The red dots are up-regulated genes with p-value < 0.01 and log2FoldChange > 1. The blue dots are down-regulated genes with p-value < 0.01 and log2FoldChange < -1. The top 10 genes are shown for up-regulated genes and the top6 genes are shown for down-regulated genes. (b) Normalized gene count for a subset of genes in the top10 of DEGs in severe vs. moderate. The expression is shown for the moderate, severe, and critical groups. (c) Volcano plot for critical vs. moderate. All the genes shown have a baseMean > 10. The red dots are up-regulated genes with p-value < 0.01 and log2FoldChange > 1. The blue dots are down-regulated genes with p-value < 0.01 and log2FoldChange < -1. The top 10 genes are shown for up- and down-regulated genes. (d) Normalized gene count for a subset of genes in the top10 of DEGs in critical vs. moderate. The expression is shown for the moderate, severe, and critical groups. (e) Upset plot (UpSetR v1.4.0 in R v4.2.1) for genes in common in the critical vs. moderate (critical) and in the severe vs. moderate (severe) comparisons. The genes are separated in three categories for each comparison, up-regulated (up; pvalue < = 0.01, log2FC > 0 and baseMean > = 10), down-regulated (down; pvalue < = 0.01, log2FC < 0 and baseMean > = 10) and not differentially expressed (nonDE; pvalue > 0.01 or baseMean < 10). All figures were made with ggplot2 (v3.4.3) in R (v4.2.1). All panels were assembled with cowplot (1.1.1) in R (v4.2.1). Statistics are from a t-test. ns: padj > 0.05; *: padj < = 0.05; **: padj < = 0.01; ***: padj < = 0.001; ****: padj < = 0.0001.
In the severe vs. moderate group, four Immunoglobulin genes (IGKV4-1 (padj = 3.75E-12), IGKV1-39 (padj = 3.52E-11), IGLV9-49 (padj = 1.26E-11), and IGLV3-19 (padj = 3.75E-12)) were found in the top 10 up-regulated genes, interestingly (Fig. 1a). Among the DEGs in severe vs. moderate, only IGKV4-1, IGKV1-39 and IGLV3-19 genes showed significant (t-test; padj < 0.05) higher expression in severe cases when compared to the other groups (Fig. 1b).
Next, we focused on the DEGs from the critical vs. moderate cohort. In the top 10 up-regulated genes, two genes are implicated in the S100 Calcium Binding Protein, S100A9 (padj = 1.23E-44) and S100A12 (padj = 9.40E-46)(Fig. 1c). Among the down-regulated genes, IL7R (padj = 3.13E-30) was identified as a relevant gene. Interestingly, one gene was in the top 10 up-regulated genes for both comparison: ZDHHC19 (padj = 3.28E-11 for severe and 5.17E-54 for critical) (Fig. 1a, c). Significant DEGs between moderate and critical were also significantly changed when comparing severe and critical (Fig. 1d). The differential expression for these genes is consistent with severity. Changes in gene expression for S100A9 and ZDHHC19 were validated using Real-time quantitative PCR (RT-qPCR). S100A9 was found to be significantly higher in critical compared to moderate (padj < 0.05) and ZDHHC19 was higher in critical compared to moderate (padj < 0.001) and severe (padj < 0.01) (Supplementary Figure S1).
To compare the critical group to the severe group, the up-regulated, down-regulated, and not differentially expressed (nonDE) genes in critical vs. moderate and severe vs. moderate were compared (Fig. 1e). To do so, genes with pvalue < = 0.01, log2FC > 0 and baseMean > = 10 were considered up-regulated, genes with pvalue < = 0.01, log2FC < 0 and baseMean > = 10 were considered down-regulated and genes with pvalue > 0.01 or baseMean < 10 were nonDE. There were 3,598 genes up-regulated in common in both severe and critical, while 2,826 genes down-regulated in common in severe and critical (Fig. 1e). A total of 5,260 (3,260 up- and 2,000 down-regulated) genes differentially expressed in critical vs. moderate were nonDE in severe vs. moderate. On the other hand, only 660 (361 up- and 299 down-regulated) genes were differentially expressed in severe vs. moderate and nonDE in critical vs. moderate. We confirmed this trend by comparing critical patients to severe patients using DESeq2 (data not shown). A total of 161 DEGs were found, all were up-regulated, the most dysregulated gene was GGT5. From this list of DEGs, 142 were also significant in critical vs. moderate. This suggests that the degree of gene expression modulation is proportionate to COVID-19 severity.
Age, sex and comorbidities, can influence gene expression. To evaluate the impact of age and sex on our results, we included them as covariates in our severe vs. moderate DEG analysis using DESeq2. This showed that 75% of DEGs remained significant, including our main findings. Demonstrating that most DEGs are not influenced by age and sex in severe vs. moderate specifically. In addition, we performed Pearson correlation analysis between the gene expression of S100A9 in the severe group and multiple covariables (age, sex, obesity, diabetes, arterial hypertension and prior stroke), to assess if any could impact the DEG results. No significant correlation was observed (Supplementary Figure S2).
A majority of MXE splicing subtype affects the critical COVID-19 patients
Next, we explored the effect of AS in the severe vs. moderate group compared to the critical vs. moderate group. Events of type A3SS, A5SS, MXE, RI, and SE were identified in both cohorts with a p-value < 0.01. Positive events refer to events with more exon skipping and negative events refer to more exon inclusion in the severe or critical cohort compared to moderate. In severe vs. moderate, 3,732 events were found: 1,421 positive and 2,311 negative, and in critical vs. moderate, 10,149 events were identified: 3,749 positive and 6,400 negative. Most genes in severe vs. moderate were affected by SE while in critical vs. moderate, most events were MXE (Fig. 2a, b). This was more evident in Fig. 2c, where negative MXE events in critical vs. moderate represented 78.1% of all events. The high proportion of MXE in critical patients further supports greater transcriptional dysregulation in accordance with COVID-19 severity.

The majority of AS are MXE events. Upset plot for AS events with p-value < 0.01 in (a) severe vs. moderate and (b) critical vs. moderate, showing the intersection of genes affected by one or multiple AS types. Made with the upset (UpSetR v1.4.0) package in R (v4.2.1). (c) Table for number of AS events with p-value < 0.01 for each AS type with positive PSI (PSI > 0) and negative PSI (PSI < 0) for severe vs. moderate and critical vs. moderate groups. Made with ggtexttable (ggpubr v0.6.0) in R (v4.2.1). (d) AS events in critical vs. moderate and severe vs. moderate for both positive and negative PSI, separately with a p-value < 0.01 separated by type (A3SS, A5SS, MXE, RI and SE) that are also found in the DEGs. The grey bar represents the events that were not significant (baseMean < 10 or p-value > 0.01), the blue bar are down-regulated events (baseMean > 10, p-value < 0.01 and log2FoldChange < 0) and the red bar are AS events that are up-regulated (baseMean > 10, p-value < 0.01 and log2FoldChange > 0). Made with ggplot2 (v3.4.3) in R (v4.2.1). All panels were assembled with cowplot (1.1.1) in R (v4.2.1).
AS events, in general, have the potential to modulate gene expression. Thus, we combined AS results with DEGs to see how each event type affects gene expression (Fig. 2d). We found that in severe vs. moderate, most of the genes affected by AS events were not significantly differentially expressed. However, in the critical vs. moderate comparison, all AS types, especially the MXE subtype, lead mostly to an up-regulation of the implicated genes (Fig. 2d). In severe vs. moderate, two positive events were identified, with a threshold of PSI of 0.1: one SE event in RGS3 (PSI = 0.108; FDR = 2.06E-11) and one MXE event in PTPRC (PSI = 0.1; FDR = 6.72E-12). Visualization of the MXE event in PTPRC using the Integrated Genome Viewer (IGV) demonstrates the differential expression of two exons between severe and moderate (Supplementary Figure S1). Events with a positive PSI indicate that exons are more skipped in the severe group when compared to the moderate. These two events were not detected in critical vs. moderate. No events met the threshold of PSI <= -0.1 in severe vs. moderate. For critical vs. moderate, 20 events had a PSI higher than 0.1 and FDR < 1.0E-07. Among these events, three were also found in severe vs. moderate but were not statistically significant: ADGRE2, IKZF1, and ARAP3. Nine were also significant in severe vs. moderate but with lower scores : WSB1, WIPF1, IKBKG, AC098613.1, EPB41, AC005280.3, ARL4A, and two events in DDX3X. Finally, eight were only identified in critical vs. moderate: BPTF, DHX30, BOD1L1, SON, U2AF1L4, ITFG2, FAM160B2, and MANBA. For negative events in critical vs. moderate, four were identified in the following genes: PKIG, RPS24, CCDC142, and AOAH. Only the AOAH event was absent in the severe vs. moderate cohort. Overall, some AS events were exclusive to specific severity levels and the majority of AS events in critical vs. moderate led to a differential expression of the corresponding genes.
Longer 3’ UTR in the critical group led to a down-regulation of gene expression
To analyze the APA of different severity levels in COVID-19 patients, a comparison was made between the lengthening (positive PDUI score) and shortening (negative PDUI score) of the 3’UTR in the severe vs moderate and critical vs moderate groups. In severe vs moderate, 14 shortening events were observed in 12 unique genes, and 18 lengthening events occurred in 13 unique genes (Fig. 3a). In critical vs moderate, there were 19 shortening and 131 lengthening events, involving 16 and 94 unique genes, respectively (Fig. 3a). The length of the 3’UTR can have an important impact on gene expression by affecting the mRNA stability, degradation rate or miRNA binding. An analysis was performed to explore the influence of APA on gene expression by integrating these findings with DEGs. In severe vs moderate, no APA events led to down-regulation of gene expression, and both lengthening and shortening of APA led to around one third of up-regulation of genes. (Fig. 3b). In critical vs moderate, most APA events were also up-regulated, 76.9% for lengthening and 71.4% for shortening events. Interestingly, the lengthening of poly(A) tail in critical vs moderate also led to a diminution of expression in 1.9% of genes (Fig. 3b). The corresponding genes with longer 3’ UTR and lower expression in the critical patients are SARAF (log2FoldChange= -0.24; PDUI = 0.086; padj = 9.69E-06) and PTMA (log2FoldCahnge=-0.2; PDUI = 0.048; padj = 0.0018). This aligns with the fact that longer poly(A) tails should result in longer persistence of the mRNA.

Large proportion of genes have a lengthening of the 3’UTR in critical vs. moderate. (a) Number of unique genes with p-value < 0.01 affected by shortening (PDUI < 0; in blue) or lengthening (PDUI > 0; in red) of 3’UTR in severe vs. moderate and critical vs. moderate. (b) Pie chart for APA events with p-value < 0.01 also found in DEGs for severe vs. moderate and critical vs. moderate, in percentage. Red represents the APA events up-regulated in DEGs (baseMean > 10, p-value < 0.01 and log2FoldChange > 0), blue represents down-regulated DEGs (baseMean > 10, p-value < 0.01 and log2FoldChange < 0) and grey are not significant (baseMean < 10 or p-value > 0.01). Made with ggpie (ggpubr v0.6.0) in R (v4.2.1). (c) Bar chart of HLA genes with APA events in severe vs. moderate and critical vs. moderate. The deltaPDUI score in shown for each comparison in each HLA gene and transcript. (d) Violin plot showing the PDUI score of every sample in moderate, severe, and critical groups for HLA-E for transcript NM_005516. Statistics from t-test. a), c) and d) are made with ggplot2 (v3.4.3) in R (v4.2.1). All panels were assembled with cowplot (1.1.1) in R (v4.2.1).
In severe vs. moderate, no events respected the threshold of |PDUI| >= 0.1. In critical vs. moderate, the only lengthening that respected this filter was in CIRBP (PDUI = 0.13; padj = 5.30E-05), as supported in IGV by a higher expression of the longer 3’UTR in the critical group (Supplementary Figure S1). It was not significant in the severe vs. moderate cohort. For the shortenings in critical vs. moderate, three events were found with a PDUI <= -0.1 in the SEC14L1 (PDUI=-0.13; padj = 2.41E-06), KLF6 (PDUI=-0.11; padj 4.13E-11) and ACP1 (PDUI=-0.10; not significant after pvalue adjustment) genes. All events were not present in severe vs. moderate.
Interestingly, APA changes were observed in four HLA genes in the severe vs moderate comparisons (Fig. 3c). One APA event in HLA-C was not significant in either cohort after filtering for p-value. Events in HLA-A and HLA-B were significant in severe vs moderate only. Finally, the APA event in HLA-E is the only one that had a significant p-value in both comparisons, with a slightly longer 3’ UTR. It was also noted that the PDUI for each group in the HLA-E|NM_005516 event increased with severity, p < = 0.0001 and p < = 0.001 for critical compared to moderate and critical compared to severe, respectively (Fig. 3d). Following the findings of HLA genes affected by APA, an analysis of HLA typing was conducted on the percentage of HLA alleles in those genes, but no significant differences were found between patient groups (results shown for HLA-E; Fig. 3e).
Combining results from DEGs, AS and APA allows a global view of the transcriptome profiles
To compare genes dysregulated by different modifications, two Venn diagrams were generated, one for severe vs. moderate (Fig. 4a) and one for critical vs. moderate (Fig. 4b). In severe vs. moderate, 121 genes affected by multiple transcriptional modifications were identified; in critical vs. moderate, this number increased to 542 genes. Only one gene, EVI2B, was dysregulated in all analyses (DEGs, AS, and APA) in severe vs. moderate (Fig. 4a). It had a log2FoldChange of 0.21, a PSI of -0.001, and a PDUI of 0.047, all with a pvalue adjusted under 0.007. This gene was also among the list of genes dysregulated by all analyses in critical vs. moderate, along with ten other genes (Fig. 4b). The values for EVI2B in critical vs. moderate were 0.26, -0.002, and 0.052 for log2Foldchange, PSI, and PDUI, respectively. The adjusted pvalues were all under 9.19E-07. The additional genes dysregulated in all analyses in critical vs. moderate were: BCL6, IFITM2, TUBA4A, LYL1, TNFSF10, CCND3, NRGN, BSG, CD300A, and GNAI2 (Fig. 4b). From the dysregulated genes that are differentially expressed, spliced and polyadenylated, pathways were identified. In total, 238 KEGG pathways were identified in the severe vs. moderate comparison, including 25 affected by DEGs, AS and APA (Fig. 4c) and 264 pathways affected in critical vs. moderate, including 90 that were found using DEGs, AS and APA results (Fig. 4d). Furthermore, 23 pathways were in common between the two comparisons and affected by all the downstream analysis (Fig. 4e). Several of these pathways are related to infection, either bacterial or viral. While other pathways are related to cancer and neurodegenerative diseases. In addition, generic pathways such as apoptosis, ubiquitin mediated proteolysis, sphingolipid signaling pathway, regulation of actin cytoskeleton, rap1 signaling pathway, platelet activation, focal adhesion, phagosome and lysosome were also enriched. Moreover, the number of genes present in the pathways was bigger in the critical and the FDR of the pathways is slightly lower in the critical vs. moderate comparison (Fig. 4e). From the pathways in critical vs. moderate only, the Coronavirus disease-COVID-19 is present, as well as pathways related to cytokines and immune response, including IL-17 signaling pathway, chemokine signaling pathway, T cell receptor signaling pathway, Toll-like receptor signaling pathway, TNF signaling pathway and NOD-like receptor pathway. Overall, this suggests that the critical group may exhibit a more pronounced immune response to the SARS-CoV-2 virus.

Integrating different transcriptomic analyses demonstrates differences between COVID-19 severities. Venn diagram for genes identified as DEGs (p-value < 0.01 and baseMean > 10), AS (p-value < 0.01) and APA (p-value < 0.01) in (a) severe vs. moderate and (b) critical vs. moderate. The genes in common from all analysis (middle of Venn) are shown in a table next to the Venn diagram. Made with ggvenn (v0.1.10) and ggtexttable (ggpubr v0.6.0) in R (v4.2.1). Venn diagram of pathways affected by DEGs, AS genes and APA genes in (c) severe vs. moderate and (d) critical vs. moderate. Made with ggvenn (v0.1.10). (e) Pathways identified with DEGs, AS and APA in common in severe vs. moderate and critical vs. moderate. The dot represents the severe vs. moderate group, the triangle represents the critical vs. moderate group, the size represents the Fold Enrichment of the pathway, the color scale represents the -log10(FDR), with a higher value (lighter color) meaning a more significant pathway (lower FDR). The x axis represents the Gene ratio (number of genes dysregulated divided by total number of genes implicated in the pathway), and the y axis are the pathways. Made with ggplot2 (v3.4.3) in R (v4.2.1). All panels were assembled with cowplot (1.1.1) in R (v4.2.1).
Discussion
To our knowledge, this is the first study combining multiple transcriptional and post-transcriptional analyses across varying degrees of severity of COVID-19. RNA-seq of 367 moderate patients, 173 severe patients and 199 critical patients from the BQC19, a unique biobank from Quebec, was used to obtain information from DEGs, AS, APA, HLA typing and pathway enrichment. Interestingly, we observed more significant up-regulated DEGs in both comparisons, demonstrating an overexpression of genes in severe and critical cases following COVID-19 infection. In the splicing analysis, MXEs were the most common AS events, and in APA, the number of lengthening events in critical vs. moderate was higher compared to severe vs. moderate. Multiple pathways identified from genes with altered expression, splicing and polyadenylation were related to immune response, inflammatory response, viral life cycle, and various signaling pathways. Genes and pathways corresponding to each severity are discussed in the sections below. Overall, this study highlights the global transcriptomic dysregulation associated with COVID-19 severity.
The small number of significant events in the severe vs moderate group demonstrated that the transcriptomic profiles of severe and moderate patients are closer than in critical and moderate patients. In total, 84 DEGs were up-regulated, while only six were down-regulated (Fig. 1a). From the top 10 up-regulated genes, the kappa immunoglobulins, IGKV4-1 and IGKV1-39, and lambda immunoglobulin IGLV9-49 had higher expression in the severe group, while the other lambda immunoglobulin such as IGLV3 − 19, was not significantly changed across all severities (Fig. 1b). Immunoglobulins play a role in the immune response via antigen binding and can neutralize viruses by removing foreign material from cells26,27. Abnormal levels of kappa and lambda immunoglobulins are associated with an excessive inflammatory response28. Different kinds of immunoglobulins may have distinct effects on adaptive immunity thus, kappa immunoglobulins could be important for severe COVID-19 infection29. These results are in line with another study showing that kappa free light chain concentration in serum was the best for distinguishing severe and moderate illness30. The only two significant AS events in severe vs moderate where exons were more skipped in severe patients were in the RGS3 and PTPRC genes, both playing a role in response to chemokines and regulation of cytokine receptor signaling, respectively31,32. Both chemokines and cytokines are implicated in cytokine storms that cause an inflammatory response proportional to severity, following COVID-19 infection19,33,34. The small number of genes alternatively spliced demonstrated either minimal differences between moderate and severe patients at the spliceosome level or high heterogeneity in these groups. For APA, the number of shortening and lengthening of the 3’ UTR region was similar in severe vs moderate, with 12 and 13 genes affected, respectively (Fig. 3a). No event reached the threshold fixed for PDUI, showing that APA is even less altered than AS in severe vs moderate. Of note, for both AS and APA in severe vs moderate, the events identified mostly did not lead to a significant change in expression of the affected genes (Figs. 2d and 3b)35. Despite the lack of global expression changes, we cannot exclude the possibility that isoform-specific expression could be altered. Furthermore, AS and APA could have a functional impact without affecting gene expression by changing the protein structure, functional domains, or cellular localization. From our combined analyses in severe vs moderate, only one gene, EVI2B, was differentially expressed and had AS, and APA events (Fig. 4a), representing global changes in severe COVID-19 patients. EVI2B was also observed in critical vs moderate. The EVI2B gene plays a role in granulocyte differentiation and is involved in immune system processes. Previous studies have identified EVI2B as a DEG in COVID-19 patients; however, no emphasis on this gene in the context of COVID has been made previously36,37. In cancer, EVI2B has been identified as a prognostic biomarker associated with interferon-gamma gene signature and immune infiltration38,39. Given the global transcriptomic dysregulation of EVI2B in our study, EVI2B could be of importance to diagnose severe COVID-19 patients.
When comparing the critical and moderate groups, a higher number of DEGs, AS and APA events were identified, showing that the overall transcriptome is more altered in critical patients compared to severe patients. This is highlighted in Fig. 1e, where 9.9% of genes are differentially expressed in critical vs moderate but not in severe vs moderate. The most frequent AS type in our study was MXEs, which is known to be a rare subtype of AS (Fig. 2b)40. It represented up to 78.1% of AS events with a p-value lower than 0.01 and a negative PSI, meaning more exon inclusion, in the critical vs moderate comparison (Fig. 2c). To our knowledge, no prior studies identified such high levels of MXEs in COVID-19 patients. Other studies identified a majority of SE events, the most common AS type10,18,41. Two studies on lung tissue, found MXE to be either the second most common in moderate, severe and critical patients10 or the least common in SARS-CoV-2 infected cells 41. A third study where they used peripheral blood mononuclear cells (PBMCs) from moderate and severe patients, also found MXE events to be the least common . Since MXEs are often tissue-specific, the higher abundance of MXE events could be due to our samples coming from whole blood7. The difference between whole blood and PBMCs might be due to the fact that RNA from whole blood was found to yield more transcripts than RNA from PBMCs only42 or it might be due to the fact that some sequencing platform have a bias toward finding more MXEs43. These results require further investigation. Importantly, the effects of MXEs are less understood compared to other AS types, as they were thought to have little or no functional impact9. More recent studies however, demonstrated that amino acid changes resulting from MXE events can lead to important functional shifts by changing protein binding or structure9. Generally, AS events in critical vs moderate led mostly to an up-regulation of the affected genes (Fig. 2d). In AS, three events found only in critical vs moderate, were associated with neurological diseases, MANBA, SON and BPTF44,45,46. In line with this, is data showing that the risk of developing a neurological disorder increases with COVID-19 disease severity47; which could explain why genes associated with neurological illnesses are dysregulated in critical patients. As time progresses, it will be important to gain more information on the impact of COVID-19 on the brain, particularly in critical patients, since it was found that around 2% of patients in intensive care developed dementia, particularly Alzheimer’s disease, after infection48. A study from An et al., 202118, on APA in COVID-19, reported that there was an average shortening of 3’ UTR length following COVID-19 infection in PBMCs of moderate to severe patients, but we found no difference in average UTR length in our study. We did identify 94 genes that had longer 3’ UTRs while 16 genes had a shortening, in critical patients compared to moderate patients (Fig. 3a). One study found that longer poly(A) tail lead to increased mRNA expression49, which aligns with our results that most APA events were up-regulated in critical vs moderate (Fig. 4b). On the other hand, shortening of the 3’UTR is often observed during proliferation of cells or in response to viral infection50. This is coherent when considering that three significant APA events had shorter 3’UTR in critical vs moderate only: SEC14L1, KLF6 and ACP1. The SEC14L1 gene is known to play a role in innate antiviral signaling via the inhibition of RIG-I (retinoic acid-inducible gene-I), a molecule necessary for induction of the type 1 interferon members51,52. It was also shown that SARS-CoV-2 replication was higher after downregulation of RIG-I53. KLF6 has already been identified as a gene of interest for cytokine and inflammation activation following COVID-19 infection54. On the other hand, ACP1, from the phosphotyrosine protein phosphatase family, could impact lymphocytes, inflammatory response and cytokines and chemokines55. In the combined results of critical vs moderate, multiple significant genes and pathways were related to actin cytoskeleton (Fig. 4b, e). Indeed, multiple DEGs that were also affected by AS and APA were linked to changes in cytoskeleton: TUBA4A, NRGN, BSG, and CD300A56,57,58,59. The cytoskeleton is particularly important in neuron functions. The actin filaments that are a part of the cytoskeleton have a role in development and assembly of neurons. SARS-Cov-2 can change the cytoskeleton structure to enter the nervous system and cause brain tissue damage60. In another study, neurodegenerative biomarkers have been identified to correlate with severity of COVID-19, including levels of tau, associated with Alzheimer’s disease61,62. Thus, supporting the previous findings that critical patients are more at risk of developing neurological illnesses, particularly Alzheimer’s disease56,60. Knowledge of how SARS-CoV-2 interacts with the actin cytoskeleton is still limited but is being investigated as a potential therapy for COVID-1963,64.
To validate the DEGs identified above, an analysis of critical vs. severe was carried out. This identified 161 significant DEGs that were all up-regulated, meaning all more expressed in critical compared to severe. 88% of these DEGs were also significantly up-regulated in critical vs. moderate. This demonstrates that the transcriptomic profiles of critical and severe are close, but the dysregulation in critical patients is more pronounced. The top DEG was GGT5, a gene that is known to regulate the immune system. It is also a member of the gamma-glutamyl transferase (GGT) family, where high levels of GGT are associated with more severe forms of COVID-1965,66.
Transcriptomic profiles reveal dysregulation of multiple biological processes. Pathway analysis showed 25 pathways globally changed by all analysis in severe vs. moderate (DEGs, AS and APA) (Fig. 4c). Twenty-three of those were also found in critical vs. moderate, representing pathways involved in COVID-19 pathogenicity regardless of severity (Fig. 4e). Most of these pathways have already been linked to COVID-19, including apoptosis67, lysosome68, platelet activation69, sphingolipid signaling pathway70 and ubiquitin mediated proteolysis71. From pathways only present in critical vs. moderate, many relate to signaling pathways like MAPK, NOD-like receptor, TNF, Toll-like receptor, IL-17 and chemokine. All these members of the inflammasome are responsible for the cytokine storm often observed in critical patients33,72,73,74. This leads to tissue damage, like lung injury, often observed in patient with need for mechanical ventilation while hospitalized or impaired lung function after recovery75.
A limitation from our study is the lack of adjustment for confounding factors as covariates in the DEG, AS and APA analyses. Given that the APA analysis tool (DaPars) does not support covariate adjustment, we decided to exclude them from all analyses to ensure unbiased comparisons. However, comorbidities are provided in Supplementary Table S1. Future studies incorporating age, sex, and comorbidities as covariates will be important for strengthening the validity of our findings in severe and critical COVID-19 patient groups.
Overall, we believe that neither DEGs, AS or APA alone could act as a better predictor of severity in COVID-19. Unlike many studies that focused on single factors, our approach involved combining downstream analyses of RNA-seq to uncover potentially overlooked genes or pathways relevant to COVID-19. By examining patients across different severity levels, we gained a nuanced understanding of how the transcriptome changed with COVID-19 severity. This differentiation is crucial, especially for those requiring mechanical ventilation and facing lower chances of survival. The use of blood in this study is also an advantage since it is easily accessible and can lead to a faster diagnosis. Validation of the genes and pathways identified here is essential before considering these findings for COVID-19 therapeutic approaches. With approximately 70% of the global population vaccinated, it will be intriguing to assess its impact on moderate, severe, and critical patients separately. We also anticipate that this in-depth transcriptomics approach could be extended to other complex diseases.
Methods
Sample selection
In response to the COVID-19 pandemic, Quebec launched a Biobank program on March 26, 2020 to collect, store and share data collected during the pandemic25. The “Biobanque québécoise de la COVID-19” (BQC19) has recruited 6,272 participants, children, and adults, from 11 hospitals and research centers across Quebec, Canada. Inclusion criterion to take part in BQC19 was to undergo polymerase chain reaction testing for SARS-CoV-2, to confirm or infirm the diagnosis. Patients were recruited from emergency rooms and hospitals in the acute phase of the infection, or from outpatient clinics following the contagious period. Informed consent was obtained from all participants in the BQC19 and ethical approval for the biobank was granted by the CHUM research ethics board (REB approval number MP-02-2020-8929). Participants contributed plasma, serum, PBMCs, stool, and whole blood to the biobank. DNA and RNA were extracted from whole blood, as described previously19,25,76. The RNA samples were sequenced and made available for downstream analysis.
RNA-sequencing
For this study, we selected RNA collected between April 2020 and August 2021 from unvaccinated individuals, for a total of 1,465 RNA-sequencing (RNA-seq) samples. The sequencing was previously done with Illumina NovaSeq PE100 2500 M reads using mRNA stranded library and NEBNext dual adapters. We filtered to keep the samples that had severity and clinical data available, which left 1,279 samples from 739 unique individuals. We analyzed only the first collected sample after symptom onset to minimize sample variability. We separated samples by severity into three groups: 367 moderate samples, 173 severe samples and 199 critical samples. The moderate group were patients that did not need supplemental oxygen, the severe group required only low-flow nasal cannula for oxygen and the critical group needed either high-flow oxygen by nasal canula, non-invasive ventilation or mechanical ventilation19. The cohort characteristics are described in Table 1 and comorbidities are described in Supplementary Table S1.
Bioinformatics pipeline
The 739 RNA-seq samples went through the same bioinformatics pipeline for further analysis. The reads were trimmed using fastp (v0.20.0) to remove the adapters77. Next the reads were aligned to the hg38 reference genome using the HISAT2 (v2.2.1) aligner78. The gene expression was obtained with FeatureCounts (v2.0.1) for downstream analysis79. Once all the samples were ready, we did the following analysis for the severe group compared to the moderate group (severe vs. moderate) and the critical group compared to the moderate group (critical vs. moderate). That way we could identify differences between severe and critical patients compared to moderate patients, which are used as controls. We looked at different aspects of transcriptomics to have a global view of each profile. We looked at DEGs, AS, APA, HLA genes, and pathway enrichment. The methods are resumed in Fig. 5a, and each section is explained below in more detail.

Study flowchart. (a) Bioinformatic pipeline for downstream analysis of RNA-seq samples. From our 739 samples, we compared two sets of groups, severe vs. moderate and critical vs. moderate. For each group, we looked at DEGs, AS and APA events, using DESeq2, rMATS and DaPars, respectively. From the significant events, we looked at pathways affected by these events. Every event and pathway were compared between groups. In parallel, we compared HLA allele frequencies between moderate, severe, and critical patients. Made with draw.io from diagrams.net (v21.6.8). (b) Principal Component Analysis of expression in moderate, severe and critical. Made with ggplot2 (v3.4.3) in R (v4.2.1).
Differentially expressed genes
To obtain a list of DEGs, we used the DESeq2 (v1.38.3)80 package in R (v4.2.1). The input files were the count matrix obtained from FeatureCounts, they were merged per group (moderate, severe, and critical). The matrices for the severe vs. moderate and critical vs. moderate are available in Supplementary Tables S2 and S3, respectively. We filtered to keep only genes that had at least five counts across all samples in each comparison, severe vs. moderate, critical vs. moderate, and critical vs. severe. We normalized the counts using the median ratio from DESeq2. Next, we used the DESeq function to get the differential expression results. The Principal Component Analysis in Fig. 5b shows the variance observed between the severity groups according to the gene expression. The DEGs results were filtered to keep only genes with a p-value < 0.01, a baseMean > 10, and an absolute value of log2FoldChange higher than one. A log2FoldChange higher than zero for a gene A means that, in a severe vs. moderate comparison for example, the gene A is up-regulated in the severe group (with higher expression) compared to the moderate group. The normalized counts of certain genes were compared across groups using a t-test from the rstatix (v0.7.2) package in R (v4.2.1).
Alternative splicing
To capture AS events, we used rMATS (v3.1.0) on the bam files obtained from the alignment81. rMATS is an extension of MATS82 that allows the use of replicates. We used default parameters for paired-end and added the --readLength 101 parameter. We checked for events in critical vs. moderate and severe vs. moderate groups. We kept the JCEC output files for each AS event mentioned. The output files were further filtered to keep events with a p-value smaller than 0.01. We used the exon inclusion level or percent spliced in (PSI) as a measure of alternative splicing between the two conditions in each comparison. It is measured using “I” as the number of counts where the isoform includes the alternatively skipped exon and “S” the number of counts where the isoform skips the alternatively spliced exon. The PSI is calculated as shown in Eq. (1).
To compare each condition, the inclusion level difference is calculated as the average PSI from all the replicates in condition 1 minus the average of PSI from replicates in condition 2. For example, if we compare the severe group to the moderate group and the PSI is positive, it means that the isoform with the skipped exon is more common in the severe group. We combined all the AS event types in a single file and filtered the absolute PSI to keep events with a score higher than 0.1 to keep the most significant events.
For the APA events, we obtained results from DaPars (v0.9.1) for the two comparisons83. DaPars is a de novo tool to estimate the expression of long and short 3’ UTR usage due to APA in RNA-seq. The bam files for each sample were converted to bedgraph to use as input. For each comparison, we ran the tool with the bedgraphs from condition 1 and condition 2, we used a minimum coverage of 20, a threshold of 5 samples that passed the coverage in each group. We also set the threshold for the FDR at 0.05, distal poly(A) site usage index (PDUI) at 0.5 and fold change at 0.59, as proposed by the documentation. The PDUI is measured as presented in Eq. 2, where “L” is the expression of the long 3’ UTR and “S” is for the short 3’ UTR expression level. The mean PDUI is calculated for each condition using all the replicates and the PDUI group difference that we use for further analysis is the mean PDUI from condition 1 minus the mean PDUI from condition 2.
If the resulting delta PDUI is positive, it represents a lengthening of the 3’ UTR expression level in condition 1. Otherwise, there is a shortening of 3’ UTR if the delta PDUI is negative in condition 1. The output was further filtered to keep events with a p-value smaller than 0.01 and a delta PDUI higher than 0.1 or lower than − 0.1.
HLA typing
For HLA typing, we used the arcasHLA (v0.5.0) tool with the bam files and default options84. arcasHLA allows for detection of HLA genes to three digits. The first field is the gene and the first digit is the allele group, the second digit is the HLA protein and the third is the synonymous mutation85. arcasHLA uses Kallisto86 for pseudo-alignments and quantification of HLA genes from chromosome 6. The IMGT/HLA database was used to obtain the HLA sequences to help in the quantification87. For each HLA gene the allele abundance is calculated and the most abundant allele pair is inferred as the genotype. The tool was run on every sample separately and the results were merged into three different files for moderate, severe, and critical groups. The allele frequency was quantified by counting the number of alleles for each gene and dividing by the total number of alleles in each group.
Pathway enrichment
For pathway enrichment, we used the online tool ShinyGo (v0.8) and selected KEGG pathways88,89,90. The default parameters were used. In brief, an FDR cut-off for a pathway considered significant is less than 0.05, the minimum of genes in pathways is set at two and the maximum is set at 2000. To select the genes, we used the results from the DEGs, AS and APA analysis experiment. For the DEGs, we selected the genes with p-value < 0.01 and baseMean > 10. For the pathways affected by AS, genes with a p-value < 0.01 were selected and for APA, a p-value < 0.01 was also used to select genes.
Correlation analysis
The correlation analysis to detect correlation between age, sex, and comorbidities and gene expression was done in R (v4.2.1), using the corrplot package (v0.95) and the Pearson’s correlation coefficient to measure significance.
Real-time quantitative PCR
cDNA was prepared using superscript IV VILO (Thermo Fisher Scientific). Real-time quantitative PCR was performed using Taqman master mix (Applied Biosystems) on the QuantStudio7 (Applied Biosystems). Taqman primers (Thermo Fisher Scientific): 18 S (Hs99999901_s1), S100A9 (Hs00610058_m1), ZDHHC19 (Hs00376116_m1). A total of 20 moderate patients, 18 severe patients and 18 critical patients were used. Delta Ct was calculated by normalizing the gene expression of each target mRNA to the expression levels of the housekeeping gene 18 S that was found to be stable across experimental conditions.
Data availability
The datasets analysed during the current study are available through the “Biobanque Québécoise de la COVID-19 (BQC-19)” (Quebec COVID-19 Biobank) data repository. Access can be requested at info@bqc19.ca, or for more information, visit https://en.quebeccovidbiobank.ca. Further inquiries can be directly addressed to the corresponding author.
Abbreviations
- A3SS:
-
Alternative 3’ splice site
- A5SS:
-
Alternative 5’ splice site
- APA:
-
Alternative polyadenylation
- AS:
-
Alternative splicing
- BQC19:
-
“Biobanque québécoise de la COVID-19” (Quebec’s biobank for COVID-19)
- COVID-19:
-
Coronavirus Disease 2019
- DEGs:
-
Differentially Expressed Genes
- HLA:
-
Human Leukocyte Antigen
- IGV:
-
Integrated Genome Viewer
- MXE:
-
Mutually exclusive exons
- PBMCs:
-
Peripheral blood mononuclear cells
- PDUI:
-
Distal polyA site usage index
- PSI:
-
Percent spliced in
- RT-qPCR:
-
Real-time quantitative PCR
- RI:
-
Retained intron
- RNA-seq:
-
RNA sequencing
- SARS‑CoV‑2:
-
Severe Acute Respiratory Syndrome Coronavirus 2
- SE:
-
Skipped exon
- UTR:
-
Untranslated region
References
Wu, F. et al. A new coronavirus associated with human respiratory disease in China. Nature 579 (7798), 265–269 (2020).
Organization, W. H. Coronavirus disease (COVID-19) pandemic 2024.
Papadopoulou, G. et al. Molecular and clinical prognostic biomarkers of COVID-19 severity and persistence. Pathogens 11(3). (2022).
Lee, Y. et al. Variants affecting exon skipping contribute to complex traits. PLoS Genet. 8 (10), e1002998 (2012).
Wang, Y. et al. Mechanism of alternative splicing and its regulation (Review). Biomed. Rep. 3 (2), 152–158 (2015).
Sakabe, N. J. & de Souza, S. J. Sequence features responsible for intron retention in human. BMC Genom. 8 (1), 59 (2007).
Jin, Y., Dong, H., Shi, Y. & Bian, L. Mutually exclusive alternative splicing of pre-mRNAs. Wiley Interdiscip Rev. RNA 9 (3), e1468 (2018).
Nakanishi, T. et al. Alternative splicing in lung influences COVID-19 severity and respiratory diseases. Nat. Commun. 14 (1), 6198 (2023).
Lam, S. D., Babu, M. M., Lees, J. & Orengo, C. A. Biological impact of mutually exclusive exon switching. PLoS Comput. Biol. 17 (3), e1008708 (2021).
Wang, C. et al. Abnormal global alternative RNA splicing in COVID-19 patients. PLoS Genet. 18 (4), e1010137 (2022).
Chen, Y. et al. CASA: a comprehensive database resource for the COVID-19 alternative splicing atlas. J. Transl Med. 20 (1), 473 (2022).
de Prisco, N. et al. Alternative polyadenylation alters protein dosage by switching between intronic and 3’UTR sites. Sci. Adv. 9 (7), eade4814 (2023).
Guo, S. & Lin, S. mRNA alternative polyadenylation (APA) in regulation of gene expression and diseases. Genes Dis. 10 (1), 165–174 (2023).
Sommerkamp, P. et al. Differential alternative polyadenylation landscapes mediate hematopoietic stem cell activation and regulate glutamine metabolism. Cell. Stem Cell. 26 (5), 722–38e7 (2020).
Zhang, Y. et al. Alternative polyadenylation: methods, mechanism, function, and role in cancer. J. Exp. Clin. Cancer Res. 40 (1), 51 (2021).
Fu, Y. et al. Differential genome-wide profiling of tandem 3’ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res. 21 (5), 741–747 (2011).
Chen, W. et al. Alternative polyadenylation: methods, findings, and impacts. Genomics Proteom. Bioinf. 15 (5), 287–300 (2017).
An, S. et al. Genome-Wide profiling reveals alternative polyadenylation of innate Immune-Related mRNA in patients with COVID-19. Front. Immunol. 12, 756288 (2021).
Brunet-Ratnasingham, E. et al. Integrated Immunovirological profiling validates plasma SARS-CoV-2 RNA as an early predictor of COVID-19 mortality. Sci. Adv. 7 (48), eabj5629 (2021).
Farahani, M. et al. Molecular pathways involved in COVID-19 and potential pathway-based therapeutic targets. Biomed. Pharmacother. 145, 112420 (2022).
Cecilia, L-M. et al. Transcriptomic clustering of critically ill COVID-19 patients. Eur. Respir. J. 2200592. (2022).
Deb, P. et al. Association of HLA gene polymorphism with susceptibility, severity, and mortality of COVID-19: A systematic review. HLA 99 (4), 281–312 (2022).
Augusto, D. G. & Hollenbach, J. A. HLA variation and antigen presentation in COVID-19 and SARS-CoV-2 infection. Curr. Opin. Immunol. 76, 102178 (2022).
Augusto, D. G. et al. A common allele of HLA is associated with asymptomatic SARS-CoV-2 infection. Nature 620 (7972), 128–136 (2023).
Tremblay, K. et al. The Biobanque Québécoise de La COVID-19 (BQC19)—A cohort to prospectively study the clinical and biological determinants of COVID-19 clinical trajectories. PLOS ONE 16 (5), e0245031 (2021).
Greenspan, N. S. & Cavacini, L. A. 15 - Immunoglobulin function. In: (eds Rich, R. R., Fleisher, T. A., Shearer, W. T., Schroeder, H. W., Frew, A. J. & Weyand, C. M.) Clinical Immunology (Fifth Edition). London: Elsevier; p. (2019). 223 – 33.e1.
Khalid, Z. et al. Identification of novel therapeutic candidates against SARS-CoV-2 infections: an application of RNA sequencing toward mRNA based nanotherapeutics. Front. Microbiol. 13, 901848 (2022).
Gudowska-Sawczuk, M. & Mroczko, B. Free light chains Κ and Λ as new biomarkers of selected diseases. Int. J. Mol. Sci. 24(11). (2023).
Townsend, C. L. et al. Significant differences in physicochemical properties of human Immunoglobulin kappa and lambda CDR3 regions. Front. Immunol. 7, 388 (2016).
Małecka-Giełdowska, M., Fołta, M., Wiśniewska, A., Czyżewska, E. & Ciepiela, O. Cell population data and serum polyclonal Immunoglobulin free light chains in the assessment of COVID-19 severity. Viruses 13 (7), 1381 (2021).
Porcu, M. et al. Mutation of the receptor tyrosine phosphatase PTPRC (CD45) in T-cell acute lymphoblastic leukemia. Blood 119 (19), 4476–4479 (2012).
Ambreen K, José VP, Sadaf N. RGS3 and IL1RAPL1missense variants implicate defective neurotransmission in early-onset inherited schizophrenias.Journal of Psychiatry and Neuroscience. 2022;47(6):E379.
Coperchini, F., Chiovato, L., Croce, L., Magri, F. & Rotondi, M. The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor. Rev. 53, 25–32 (2020).
Hsu, R-J. et al. The role of cytokines and chemokines in severe acute respiratory syndrome coronavirus 2 infections. Front. Immunol. 13. (2022).
Hosseini, E., Minagar, A., Ghasemzadeh, M., Arabkhazaeli, A. & Ghasemzadeh, A. HLA-E*01:01 + HLA-E*01:01 genotype confers less susceptibility to COVID-19, while HLA-E*01:03 + HLA-E*01:03 genotype is associated with more severe disease. Hum. Immunol. 84 (4), 263–271 (2023).
Vastrad, B., Vastrad, C. & Tengli, A. Bioinformatics analyses of significant genes, related pathways, and candidate diagnostic biomarkers and molecular targets in SARS-CoV-2/COVID-19. Gene Rep. 21, 100956 (2020).
Zheng, W. et al. Host factor interaction networks identified by integrative bioinformatics analysis reveals therapeutic implications in COPD patients with COVID-19. Front. Pharmacol. 12, 718874 (2021).
Todorovic-Rakovic, N. & Whitfield, J. R. Between Immunomodulation and immunotolerance: the role of IFNgamma in SARS-CoV-2 disease. Cytokine 146, 155637 (2021).
Yonekura, S. & Ueda, K. EVI2B is a new prognostic biomarker in metastatic melanoma with IFNgamma associated immune infiltration. Cancers (Basel) 13(16). (2021).
Pohl, M., Bortfeldt, R. H., Grützmann, K. & Schuster, S. Alternative splicing of mutually exclusive exons—A review. Biosystems 114 (1), 31–38 (2013).
Sun, J. et al. Computational methods to study human transcript variants in COVID-19 infected lung cancer cells. Int. J. Mol. Sci. 22, 18 (2021).
He, D. et al. Whole blood vs PBMC: compartmental differences in gene expression profiling exemplified in asthma. Allergy Asthma Clin. Immunol. 15, 67 (2019).
Aktas Samur, A. et al. In-depth analysis of alternative splicing landscape in multiple myeloma and potential role of dysregulated splicing factors. Blood Cancer J. 12 (12), 171 (2022).
Chen, X. et al. A novel genetic variant potentially altering the expression of MANBA in the cerebellum associated with attention deficit hyperactivity disorder in Han Chinese children. World J. Biol. Psychiatry 23 (7), 548–559 (2022).
Kushary, S. T. et al. ZTTK syndrome: clinical and molecular findings of 15 cases and a review of the literature. Am. J. Med. Genet. Part. A 185 (12), 3740–3753 (2021).
Glinton, K. E. et al. Phenotypic expansion of the BPTF-related neurodevelopmental disorder with dysmorphic facies and distal limb anomalies. Am. J. Med. Genet. Part. A 185 (5), 1366–1378 (2021).
Xu, E., Xie, Y. & Al-Aly, Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 28 (11), 2406–2415 (2022).
Li, C., Liu, J., Lin, J. & Shang, H. COVID-19 and risk of neurodegenerative disorders: A Mendelian randomization study. Translational Psychiatry. 12 (1), 283 (2022).
Kiltschewskij, D. J., Harrison, P. F., Fitzsimmons, C., Beilharz Traude, H. & Cairns Murray, J. Extension of mRNA poly(A) Tails and 3′utrs during neuronal differentiation exhibits variable association with post-transcriptional dynamics. Nucleic Acids Res. 51 (15), 8181–8198 (2023).
Jia, X. et al. The role of alternative polyadenylation in the antiviral innate immune response. Nat. Commun. 8, 14605 (2017).
Li, M-T. et al. Negative regulation of RIG-I-Mediated innate antiviral signaling by SEC14L1. J. Virol. 87 (18), 10037–10046 (2013).
Matsumiya, T. & Stafforini, D. M. Function and regulation of retinoic acid-inducible gene-I. Crit. Rev. Immunol. 30 (6), 489–513 (2010).
Yamada, T. et al. RIG-I triggers a signaling-abortive anti-SARS-CoV-2 defense in human lung cells. Nat. Immunol. 22 (7), 820–828 (2021).
Wen, W. et al. Immune cell profiling of COVID-19 patients in the recovery stageby single-cell sequencing. Cell. Discovery 6 (1), 31 (2020).
Pike, K. A. & Tremblay, M. L. Protein tyrosine phosphatases: regulators of CD4 T cells in inflammatory bowel disease. Front. Immunol. 9, 2504 (2018).
Ganne, A. et al. In Silico analysis of TUBA4A mutations in amyotrophic lateral sclerosis to define mechanisms of microtubule disintegration. Sci. Rep. 13 (1), 2096 (2023).
Castrogiovanni, P. et al. Brain CHID1 expression correlates with NRGN and CALB1 in healthy subjects and AD patients. Cells [Internet] 10(4). (2021).
Muramatsu, T. Basigin (CD147), a multifunctional transmembrane glycoprotein with various binding partners. J. Biochem. 159 (5), 481–490 (2016).
Ghavampour, S., Lange, C., Bottino, C. & Gerke, V. Transcriptional profiling of human monocytes identifies the inhibitory receptor CD300a as regulator of transendothelial migration. PLOS ONE 8 (9), e73981 (2013).
Zhang, Q. & Jiu, Y. The regulation of host cytoskeleton during SARS-CoV-2 infection in the nervous system. Brain Sci. Adv. 9 (1), 43–52 (2023).
Pillai, J. A. et al. Highly elevated cerebrospinal fluid total Tau level reflects higher likelihood of Non-Amnestic subtype of Alzheimer’s disease. J. Alzheimers Dis. 70 (4), 1051–1058 (2019).
Frontera, J. A. et al. Comparison of serum neurodegenerative biomarkers among hospitalized COVID-19 patients versus non-COVID subjects with normal cognition, mild cognitive impairment, or Alzheimer’s dementia. Alzheimer’s Dement. 18 (5), 899–910 (2022).
Kloc, M., Uosef, A., Wosik, J., Kubiak, J. Z. & Ghobrial, R. M. Virus interactions with the actin cytoskeleton-what we know and do not know about SARS-CoV-2. Arch. Virol. 167 (3), 737–749 (2022).
Aminpour, M., Hameroff, S., Tuszynski, J. A. & How COVID-19 hijacks the cytoskeleton: therapeutic implications. Life (Basel) 12(6). (2022).
Wang, Y. et al. Identification of GGT5 as a novel prognostic biomarker for gastric cancer and its correlation with immune cell infiltration. Front. Genet. 13. (2022).
Liu, J. et al. The clinical implication of gamma-glutamyl transpeptidase in COVID-19. Liver Res. 5 (4), 209–216 (2021).
Yuan, C. et al. The role of cell death in SARS-CoV-2 infection. Signal. Transduct. Target. Therapy. 8 (1), 357 (2023).
Ghosh S, Dellibovi-Ragheb TA, Kerviel A, Pak E, Qiu Q, Fisher M, et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 183(6):1520-35.e14 (2020).
Jevtic, S. D. & Nazy, I. The COVID complex: A review of platelet activation and immune complexes in COVID-19. Front. Immunol. 13. (2022).
Fenizia, S., Gaggini, M. & Vassalle, C. The Sphingolipid-Signaling pathway as a modulator of infection by SARS-CoV-2. Curr. Issues Mol. Biol. 45 (10), 7956–7973 (2023).
Ciechanover, A., Orian, A. & Schwartz, A. L. Ubiquitin-mediated proteolysis: biological regulation via destruction. BioEssays 22 (5), 442–451 (2000).
Taheri, G. & Habibi, M. Comprehensive analysis of pathways in coronavirus 2019 (COVID-19) using an unsupervised machine learning method. Appl. Soft Comput. 128, 109510 (2022).
Carreto-Binaghi, L. E. et al. Reduced IL-8 secretion by NOD-like and Toll-like receptors in blood cells from COVID-19 patients. Biomedicines 11(4). (2023).
Choudhary, S., Sharma, K. & Silakari, O. The interplay between inflammatory pathways and COVID-19: A critical review on pathogenesis and therapeutic options. Microb. Pathog. 150, 104673 (2021).
Mancilla-Ceballos, R., Milne, K. M., Guenette, J. A. & Cortes-Telles, A. Inflammation associated with lung function abnormalities in COVID-19 survivors. BMC Pulm Med. 23 (1), 235 (2023).
Brunet-Ratnasingham, E. et al. Sustained IFN signaling is associated with delayed development of SARS-CoV-2-specific immunity. Nat. Commun. 15 (1), 4177 (2024).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34 (17), i884–i90 (2018).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37 (8), 907–915 (2019).
Liao, Y., Smyth, G. K. & Shi, W. FeatureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30 (7), 923–930 (2014).
Anders, S. & Huber, W. Differential expression analysis for sequence count data. Genome Biol. 11 (10), R106 (2010).
Shen, S. et al. rMATS: Robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proceedings of the National Academy of Sciences. 111(51):E5593-E601. (2014).
Shen, S. et al. MATS: a bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res. 40 (8), e61–e (2012).
Xia, Z. et al. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3′-UTR landscape across seven tumour types. Nat. Commun. 5 (1), 5274 (2014).
Orenbuch, R. et al. ArcasHLA: high-resolution HLA typing from RNAseq. Bioinformatics 36 (1), 33–40 (2020).
Kishore, A. & Petrek, M. Next-Generation sequencing based HLA typing: Deciphering Immunogenetic aspects of sarcoidosis. Front. Genet. 9. (2018).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34 (5), 525–527 (2016).
Robinson, J. et al. The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43 (D1), D423–D31 (2015).
Ge, S. X., Jung, D. & Yao, R. ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36 (8), 2628–2629 (2020).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (D1), D457–D462 (2016).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Acknowledgements
We would like to thank Eric Bareke for technical advice and Vincent Mooser for his involvement in the BQC-19 and his previous work on this data.
Funding
This project was funded by a joint grant from COVID-19 immunity task force and the Canadian Institute of Health research (CITF-CIHR) (grant VR2-173203) and American Foundation for AIDS Research (amfAR) grant 110068-68-RGCV. M.T. received a Junior 1 salary Award from “Fond de Recherche Québec – Santé (FRQS)”. A.F. was supported by a Canada Research Chair. D.E.K. was supported by a FRQS Merit Research Scholar Award. M.D. receives a clinician-researcher salary Award from FRQS. BQC19 received funding from FRQS, the Public Health Agency of Canada, Génome Québec and the Ministère de la Santé et des services sociaux du Québec.
Author information
Authors and Affiliations
Contributions
Conceptualization, A.F, D.E.K, M.T; Patient recruitment, B.R, M.D; Previous studies on BQC-19, S.R; Next-Generation Sequencing, D.A, A.M; Clinical data analysis, E.B.R.; Data Analysis, M.L.; Writing – Original Draft, M.L.; Writing – Review & Editing, E.B.R., L.K.H, D.E.K., M.T.; Funding Acquisition, A.F, D.E.K, M.T.; Supervision, D.E.K, M.T.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Labrecque, M., Brunet-Ratnasingham, E., Hamilton, L.K. et al. Transcriptomic profiling of severe and critical COVID-19 patients reveals alterations in expression, splicing and polyadenylation. Sci Rep 15, 13469 (2025). https://doi.org/10.1038/s41598-025-95905-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-025-95905-y
