
Abstract
Endoplasmic reticulum stress (ERS) is an intracellular process in which improperly folded proteins lead to a cellular stress response. How endoplasmic reticulum stress contributes to the onset and progression of laryngeal squamous cell carcinoma remains unclear. Our research aimed to find an ERS signature to forecast the prognosis of laryngeal squamous cell carcinoma and to investigate its potential biological functions. LSCC sample data obtained from The Cancer Genome Atlas (TCGA) database were co-expressed with ERS- related genes, and then a prognostic signature on the basis of endoplasmic reticulum stress- related lncRNAs (ERS-related lncRNAs) was constructed by differential analysis and Cox regression analysis. Survival analysis, TMB, consensus cluster analysis, drug sensitivity analysis, immune analysis and clinical drug prediction were carried out on the model. Finally, the function of LHX1-DT was verified by in vitro experiments. From the TCGA-LSCC cohort, 35 significantly different ERS-related lncRNAs were identified. A prognostic signature consisting of three lncRNAs (AC110611.2, LHX1-DT, and AL157373.2) was identified. Kaplan–Meier analysis demonstrated the predictive ability of the model for overall survival. Calibration curves and receiver operating characteristic curves were validated and showed high predictive accuracy. Ultimately, the experimental results verified the expression of LHX1-DT in LSCC.
Similar content being viewed by others


Introduction
Neoplasms remain the main killer worldwide1,2,3,4. Among which, laryngeal squamous cell carcinoma (LSCC) is the most common cancer in the upper respiratory tract and one of the most common malignancies of the head and neck. It is caused by squamous cell proliferation in the pharynx and larynx, which seriously harms the global human health5. Major contributors to LSCC include smoking, alcohol consumption, and HPV infection6. Although LSCC can be treated by surgery7, radiotherapy5, chemotherapy and targeted therapy, its prognosis remains bleak7. Therefore, there is a pressing need to investigate novel molecular markers and understand the mechanisms driving LSCC progression.
The endoplasmic reticulum (ER) is a cellular organelle, which is the main place for protein folding and maturation8,9,10. It is closely related to many biological processes such as protein synthesis, drug metabolism and calcium ion regulation, and is very important for the normal function of cells11. However, when the endoplasmic reticulum is subjected to abnormal stimulation, its function can be compromised, the protein folding ability is affected and destroyed, which leads to the endoplasmic reticulum stress state12. Studies have shown that ERS causes IRE1α to regulate glioma cell migration through RIDD-mediated mRNA decay13. ERS-related ATF6 upregulates the oncoprotein phosphatase 2A (CIP2A) to promote the progression of colon cancer14. ERS-induced miR-1281, whose promoter binds to p53 and targets USP39, facilitating the apoptosis of osteosarcoma cells15. ERS induces and enhances the expression of miR-27a-3p, and miR-27a-3p negatively regulates MAGI2, which leads to the up-regulation of MAGI2 through PTEN/PI1K signaling pathway, thus promoting the immune escape of breast cancer16. Therefore, ERS may be a potential target for the treatment of malignant tumors and it is essential to explore the association between ERS and LSCC progression.
LncRNAs are RNA molecules that do not code for protein, and are usually more than 200 nucleotides in length, and are vital for regulating gene expression, maintaining chromatin structure, controlling the cell cycle, cell differentiation and development17,18,19,20,21. Previous research has demonstrated the tight relationship between lncRNAs and the progression of numerous cancers: LncRNA AK023948 is upregulated in breast cancer and promotes tumor cell growth22; LncRNA RMRP affects proliferation, migration and invasion of bladder cancer by modulating miR20623; LncRNA-SNHG1 affects angiogenesis and tumorigenesis in breast cancer by altering the phosphorylation of STAT4 and changing the number of F80/206CD6 positive macrophage24; LncRNA IGKJ2-MALLP2 restricts the development of LSCC by competitively binding with miRNA-1911-3p25.
However, the connection between LSCC and ERS-related lncRNAs remains unclear, prompting our study to investigate this relationship and explore ERS mechanisms in LSCC further. In this study, we created and validated a risk signature made up of three ERS-related lncRNAs using RNA sequencing data from the TCGA database. Then, to learn more about the involvement of ERS-related lncRNAs in LSCC, we carried out GSEA and GO enrichment analysis, immune infiltration analysis, consensus clustering analysis, and chemotherapy drug sensitivity analysis. Finally, we verified the impact of two highly expressed ERS-related lncRNAs on LSCC biological functions. Our study offers insights into LSCC prognosis prediction and has the potential to advance clinical treatment approaches for LSCC.
Materials and methods
Data acquisition
The RNA transcriptome sequence data of 117 LSCC samples were downloaded from The Cancer Genome Atlas (TCGA) (https://portal.gdc.cancer.gov/) in FPKM format, with the corresponding transcriptome data and clinical features. To ensure data consistency and comparability, expression values were transformed into Transcripts Per Million (TPM) values and then log2(TPM + 1) transformed for normalization. Additionally, Z-score normalization was applied to ensure standardized data distribution across different samples. Using annotation files from Ensembl (https://asia.ensembl.org/), we distinguished between lncRNA and mRNA. For duplicate genes, we retained the one with the highest expression level across samples. To identify ERS-related gene sets, we queried "endoplasmic reticulum stress" in the Molecular Signature Database (https://gsea-msigdb.org/), refining and combining gene sets to acquire 294 genes for further analysis.
Identification of ERS-related LncRNAs
To identify ERS-related lncRNAs, we performed Pearson correlation analysis between the expression profiles of all lncRNAs and the 294 ERS-related genes using the “limma” R package; lncRNAs with an absolute Pearson correlation coefficient (|R|) > 0.4 and a significance threshold of p < 0.05 were retained for further analysis. Subsequently, differentially expressed lncRNAs between LSCC tumor tissues and adjacent normal tissues were identified using the “limma” package with the criteria set as |log₂(fold change)|> 1 and a false discovery rate (FDR)-adjusted p < 0.05. To further select lncRNAs with prognostic relevance, univariate Cox proportional hazards regression analysis was applied to these differentially expressed, ERS-related lncRNAs, and those with p < 0.05 were considered statistically significant predictors of overall survival (OS) and retained for subsequent model construction. Finally, heatmaps and volcano plots were generated using the “pheatmap” R package to visualize the expression patterns and differential expression of these ERS-related lncRNAs.
Construction and verification of the ERS-related LncRNA prognostic signature
To identify drug candidates associated with prognosis, we performed univariate Cox regression analysis by using the “Survival” and “Survminer” R packages. Subsequently, the “glmnet” R package was used for Lasso regression analysis to avoid overfitting. A prognostic signature comprising ERS-related lncRNAs was then constructed via multivariate Cox regression analysis, with the resultant risk score serving as the prognostic indicator. Patients were randomly divided into training and testing sets in a 1:1 ratio. We used the training set to build a prognostic model and plotted the associated heatmap using the pheatmap R package. The accuracy of the model was confirmed by using the testing set and all patients. Differences between clinical features among these sets was analyzed by chi-square test, with statistical significance set at p < 0.05. The risk score was computed based on the following formula:
\(Coe{f}_{i}\) in the formula represents the regression coefficient, \(Exp{r}_{i}\) indicates the expression level of lncRNA. Kaplan–Meier survival analysis was performed using ‘survival’ and ‘survminer’ R packages, while the “pheatmap” R package was used to create risk heatmaps, survival state diagrams, and risk curves. Then, combined with the risk score and the clinical information of patients’ age, gender and tumor grade, Cox regression analysis was used to carry out independent prognostic analysis. Finally, the “timeROC” R package was used to render Receiver Operating Characteristic (ROC) curves and calculate the Area Under the Curve (AUC) to evaluate the predictive capability of the signature. Nomogram was developed to estimate the survival rates of LSCC patients for 1, 3, and 5 years. The veracity was estimated by means of calibration curves.
Enrichment analysis
We conducted Gene Ontology (GO) analysis(|log2 fold change (FC)|> 1, false discovery rate (FDR) < 0.05) using the "org.Hs.eg.db" “enrichplot” “ComplexHeatmap” and “clusterProfiler” in R. This analysis aimed to elucidate the functional and biological processes associated with ERS-related lncRNAs. Visual representations, including barplot, bubble and circlize plot, were generated to illustrate the findings. To determine the biological pathways of ERS-related lncRNAs, we used GSEA (Gene Set Enrichment Analysis) to evaluate the differences between high- and low-risk patients by running the "org.hs.eg.db" “enrichplot” “ComplexHeatmap” “clusterProfiler” R packages.
Tumor mutation burden analysis
The Perl script was exploited to acquire mutation data for each sample, followed by calculating mutation frequency and mutation burden. Subsequently, samples were categorized into high TMB and low TMB groups. The “maftools” R package was used to obtain the mutation frequency of each gene, and a waterfall plot for visualization allowed us to compare mutation status between high- and low-risk groups. The R packages “ggpubr” and “reshape2” were used to perform TMB ANOVA and correlation analysis between the high and low risk groups. Kaplan–Meier curves were generated to appraise the impact of risk scores and TMB on patients’ survival. Furthermore, a combined analysis was performed to investigate survival disparities among groups with high TMB and high risk, high TMB and low risk, low TMB and high risk, and low TMB and low risk.
Evaluation of tumor immune microenvironment, immune cell infiltration and immune checkpoint
The scores of immune cells and stromal cells of each sample were computed by using “estimate” and “limma” R packages. Differences in StromalScore, ImmuneScore, and ESTIMATEScore between the two groups were analyzed using the “limma” and “ggpubr” R packages, with p < 0.05 indicating statistical significance. Then, the correlation between immune cells and risk score was studied by the usage of XCELL, MCPCOUNTER, ERIC, CIBERSORT and other algorithms, and visualized the outcomes with a bubble plot. Survival analysis of immune cell infiltration using “survival” and “survminer” R packages generated Kaplan–Meier curves and screened with P < 0.05. Next, ssGSEA (single-sample gene set enrichment analysis) analysis was used to acquire the scores of immune cells and immune-related functions, and the differences between high and low risk groups were analyzed by using “ggpubr” and “reshape2” R packages to draw box plots. In addition, to explore the variation of immune checkpoint related genes between high- and low-risk groups, we can use “Limma”, “Reshape2”, "GGPlot2 "and “GGPUBR” R packages and plot a box plot.
Consensus cluster analysis
The tumor samples were analyzed by consensus clustering with “Conensus ClusterPlus” R package and divided into different subgroups. Kaplan–Meier curves were subsequently generated using the “survival” and “survminer” R packages to assess survival differences among these clusters. We utilized the “dplyr” “ggplot2” and “ggalluvial” R packages to generate Sankey plots, aiming to uncover the relationship between sample clusters and risk. Additionally, t-distributed Stochastic Neighbor Embedding (tSNE) analysis was performed on different clusters using the “Rtsne” R package to distinguish the grouping of samples. To analyze variances in immune responses between clusters, we used the “limma” and “pheatmap” R packages and algorithms such as CIBERSOR, CIBERSORT- ABS, QUANTISE, MCPCOUNTE, XCEL and EPI. Finally, we conducted differential analysis for immune checkpoint-related genes.
Drug sensitivity prediction
After scoring drug sensitivity with the “limma” “oncoPredict” and “parallel” R packages, the differences of drug sensitivity between different clusters and between high and low risk groups were analyzed with the “ggplot2” “ggpubr” “limma” and “reshape2” R packages. p < 0.05 was considered significant.
Cell culture and transfection
Human LSCC cell lines (TU686, AMC-HN-8) and human immortalized epidermal cell line HaCaT was sourced from Procell Life Science & Technology Co., Ltd. Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA, USA) added with 10% fetal bovine serum (Bioexplore, USA) and maintained in a suitable incubator with 5%CO2 at 37 °C. SiRNA-NC and siRNA-LHX1-DT were purchased from Ribobio (Guangzhou,China) and transfected with Lipofectamine RNAiMAX reagent (Glpbio, CA, USA).
RNA extraction and quantitative real-time polymerase chain reaction(qRT-PCR)
Cells used for qRT-PCR were washed with Cold Phosphate ‐ Buffered Saline (PBS), and RNAs were extracted by RNA purification kit (RNAfast2000; Fastagen, China), reverse transcribed 1ug of total RNA with cDNA synthesis Kit (NobelabBiotech, China), and performed qRT-PCR with SYBR Green PCR Mix (NobelabBiotech, China ). Results were standardized to GAPDH, and data were calculated by 2-ΔΔCT. PCR primers were designed and composited by Guangzhou RiboBio Co., Ltd. The primer sequences can be found in Table 1.
Western blot analysis
After transfection, total proteins of AMC-HN-8 and TU686 cells were fetched by using RIPA solution (Sigma-Aldrich) including 1 × protease inhibitor and 1 × phosphatase inhibitor, and protein concentrations were determined by BCA Protein Assay Kit (Thermo Fisher). Protein samples (20 μg) were subjected to 12% SDS-PAGE gel electrophoresis and then transferred to a PVDF membrane. Subsequently, the membrane was blocked with 5% BSA Blocking Buffer for 1 h and then incubated overnight with primary antibodies (Abcam, Cambridge, MA, USA) against cleaved caspase-3 (ab2302,1:800 dilution,19 kDa) and GAPDH (ab8245, 1:2000 dilution, 36 kDa) at 4 °C. The membrane washed with PBS the next day, then treated with secondary HRP-linked goat anti-rabbit IgG antibody (ab6721, 1:5000 dilution), and signals were tested with an enhanced chemiluminescence kit (Thermo Fisher Scientific). The results were analyzed using Image J software.
Cell Counting kit-8(CCK-8) assay
AMC-HN-8 and TU686 cells were seeded into 96-well plates at a density of 8000 cells/well, then 10 μl of CCK8 reagent (Dojindo, Japan) was added at 4, 24, 48 and 72 h, followed by incubation at 37 °C for 30 min. The absorbance of each well was measured at 450 nm using an enzyme reader.
Colony formation assay
AMC-HN-8 and TU686 cells were placed in 6-well plates, and the complete medium was changed every 48 h. 2 weeks later, the cells were immobilization with 4% paraformaldehyde and stained with crystal violet and took pictures.
Transwell assays
Transwell chambers were used to assess the migratory and invasive abilities of AMC-HN-8 and TU686 cells. To assess cell migration, AMC-HN-8 and TU686 cells (5 × 104 cells/well) were seeded in the upper chamber with serum-free medium, while the complete DMEM medium was in the lower chamber. Cells that migrated to the lower chamber after a period of 24 h were fixed for 25 min with 4% paraformaldehyde and stained for 30 min with crystal violet. The detection of invasion ability is to apply a layer of matrix gel on the upper chamber, and the remaining steps were similar to the migration assay. Photos were taken with an optical microscope.
Wound healing assay
AMC-HN-8 and TU686 cells were grown in 6-well plates. When reaching 90% convergence, a scratch was made using a 1 ml sterile pipette tip. The wells were then washed with PBS to remove debris, and serum-free media was put into each well for 24 h of culture. An optical microscope was used to take images at both 0 and 24 h. Using Image J software, compute the relative migration rate.
Caspase-3 activity assay
Caspase 3/7 Activity Assay Kit was used to determine caspase3 activity. From the instructions given by the company (Procell Life Science & Technology Co., Ltd), collect and calculate the cell precipitates. Cells were handled with cell lysate and protein concentrations were measured. Samples were read on an enzyme labeler at 405 nm.
Statistical analysis
For univariate Cox regression analysis, we used the ‘Survival’ and ‘Survminer’ R packages to identify ERS-related lncRNAs associated with prognosis. Hazard ratios (HRs) and 95% confidence intervals (CIs) were calculated to assess the strength of the association. LASSO regression analysis, performed with the “glmnet” R package, was used to avoid overfitting by penalizing the coefficients of the regression model. In the process, a series of lambda values were tested, and the optimal lambda was selected based on the minimum cross-validation error. Multivariate Cox regression analysis was then carried out to construct the prognostic signature. For Kaplan–Meier survival analysis, also using the ‘survival’ and ‘survminer’ R packages, log-rank tests were applied to compare survival differences between groups. For gene enrichment analyses (GO and GSEA), false discovery rate (FDR) < 0.05 was set as the significance threshold. The experimental results were statistically and quantitatively analyzed using GraphPad Prism 8.0.2, SPSS 25, and ImageJ (v1.54 g, URL: https://imagej.net/ij/) software. Independent sample t-tests were used to compare two variables, and one-way ANOVA followed by a Tukey’s post hoc test was applied for comparisons between groups. The data presented were expressed as the mean ± standard deviation, which are derived from a minimum of three independent experiments. p values < 0.05 were considered statistically significant.
Results
Identification of ERS-related lncRNAs in LSCC patients
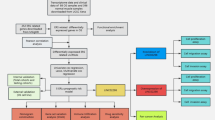
Figure 1 depicts a comprehensive flowchart detailing the data collection and analysis procedures. Initially, a total of 117 LSCC samples were extracted from the TCGA database (https://portal.gdc.cancer.gov/repository). Subsequently, co-expression analysis with 294 ERS-related genes sourced from the MsigDB database (Table S1) generated the identification of 1187 ERS-related lncRNAs (Table S2). Through differential analysis, we obtained 534 lncRNAs that were disparity expressed between normal and tumor tissues (Table S3). The heatmap in Fig. 2A visually represents the expression of these lncRNAs. Additionally, the volcano plot (Fig. 2B) highlights 506 upregulated lncRNAs and 28 downregulated lncRNAs in tumor tissues.

Flow chart of data collection and analysis.

Identification of ERS-related lncRNAs in LSCC patients. (A) The heatmap reflecting expression levels of ERS-related lncRNAs; (B) The volcano map showing the expression levels of ERS-related lncRNAs.
Construction of an ERS-related lncRNA Prognostic signature and analysis of independent prognostic potentials
Univariate Cox regression analysis was utilized to identify 35 ERs-related lncRNAs with prognostic significance (Fig. 3A). In Fig. 3B, all lncRNAs except AL121933.2 exhibited upregulated expression in tumor tissues. Next, using Lasso regression analysis to penalize the 35 ERS-related lncRNAs (Fig. 3C,D), and selected 3 optimal lncRNAs as prognostic markers for building the prognostic signature (Table S4). The risk score was computed based on the following formula:

Construction of an ERS-related lncRNA Prognostic signature and analysis of independent prognostic potentials. (A) The forest plot showing the results of univariate Cox regression analysis of 35 prognostic ERS-related lncRNAs; (B) The heatmap showing the expression levels of these 35 genes in LSCC and normal tissues; (C) Cross-validation in LASSO regression to optimize parameter selection; (D) LASSO coefficient profile; (E–H) PCA illustrates that the model lncRNA group (H) is most effective in differentiating patients in high and low risk groups than ERS-related lncRNAs (E), ERS-related genes (F) and all genes (G); (I-J) Univariate and multivariate analysis assessing relationship between risk scores and relevant clinical parameters and OS in the TCGA–LSCC cohort; (K) The Nomogram model of the risk score and clinical indicators for predicting 1-year, 3-year, and 5-year OS of LSCC patients; (L) Calibration curves of the nomogram prediction of OS of patients in TCGA-LSCC cohort.
The risk score = (0.70333 × AC110611.2) + (0.77696 × LHX1-DT) + (-0.71220 × AL157373.2).
Then, 117samples were arbitrarily assigned to the training and test groups, with clinical characteristics presented in Table S5. The risk score of each sample was calculated based on the expression level of each lncRNA and the correlation coefficients obtained from multivariate Cox regression analysis. These scores, based on the median risk score, were then used to divide the samples into high-risk and low-risk groups. Principal Component Analysis (PCA) illustrated that compared with the ERS-related lncRNAs (Fig. 3E), ERS-related genes (Fig. 3F) and all genes (Fig. 3G), the prognostic ERS-related lncRNA (Fig. 3H) signature differentiates patients in the high and low risk groups most effectively. Next, to evaluate the potential of the prognostic characteristics as independent prognostic indicators for LSCC patients, we undertook univariate and multivariate Cox regression analysis. Forest maps were generated on the basis of both the risk score and clinicopathological characteristics. Gender and prognostic signature could be used as independent prognostic indicators for LSCC patients in univariate Cox regression analysis (P < 0.001, Fig. 3I). The 3-ERS risk signature remains an independent predictor for OS rate in LSCC patients, according to multivariate Cox analysis (P < 0.001, Fig. 3J). Combining the risk score and clinicopathological features, and aggregating the values of different variables, the nomogram was used to predict OS at 1, 3 and 5 years in patients with LSCC (Fig. 3K). The calibration curves for the 1-year, 3-year, and 5-year OS models showed good agreement between the predicted and actual values (Fig. 3L).
Establishment and verification of the prognostic signature of the ERS-related lncRNA signature in the training, validation and complete sets
Kaplan–Meier survival curves showed clear differences in overall survival (OS) between the two groups in training, testing and complete groups, with lower OS rates in the high-risk group.(Figs. 4A-C). According to the three prognostic markers of lncRNA, the profile of risk scores is depicted in Figs. 4D-F. Figures 4G-I present scatter plots show the interrelation between patient survival status and risk scores, with increasing risk scores associated with increasing patient mortality. To compare the expression levels between high-risk and low-risk groups in the training, test and complete datasets, heatmaps were constructed on basis of the three prognostic ERS-related lncRNAs. (Figs. 4J-L). The heatmaps clearly show that AL157373.2 was highly enriched in the low-risk group across all datasets, whilst AC110611.2 and LHX1-DT were significantly enriched in the high-risk group. Moreover, ROC curves were constructed for the training, testing, and complete datasets over 1, 3, and 5 years, with the AUCs consistently exceeding 0.65 throughout (Figs. 4M-O). Particularly, the AUC for the risk score in the ROC curves was 0.882, 0.659, and 0.763, separately, surpassing the predictive efficiency of other clinical variables (Figs. 4P-R).

Establishment and verification of the prognostic signature of the ERS-related lncRNA signature in the training, validation and complete sets. (A-C) Kaplan–Meier survival curves of LSCC patients’ OS in the high-risk and low-risk groups in the training (A), validation (B), and complete sets (C); (D-F) Risk score distribution in the high-risk and low-risk groups in the training (D), validation (E), and complete sets (F); (G–I) Scatter plots of LSCC patient survival status distribution in the training (G), validation (H), and complete sets (I); (J–L) Risk heatmaps of the 3 ERS-related lncRNA expression in the training (J), validation (K), and complete sets (L); (M–O) ROC curves of the prognostic ERS-related lncRNAs signature at 1-, 2-, and 3-year in the training (M), validation (N) and complete sets (O); (P-R) AUC of ROC curves comparing the prognostic accuracy of the risk score and other prognostic factors in the training (P), validation (Q) and complete sets (R).
Analysis of biological functions and associated signaling pathways of ERs-related signature
To illustrate the biological functions and associated signaling pathways of ERs-related lncRNAs, we conducted GO and GSEA analysis. As displayed in Figs. 5A-C, in Biological process (BP), ERS-related lncRNAs were enriched in extracellular tissue, hormone metabolism and gastrulation. In cell component (CC), ERS-related lncRNAs were mainly enriched in Collagen-containing extracellular matrix and endoplasmic reticulum lumen. In molecular function (MF), ERS-related lncRNAs were mainly enriched in receptor ligand activity and endopeptidase activity. The results of GSEA analysis revealed that the high-risk group may display stronger immune and inflammatory responses, alterations in the extracellular matrix and cell adhesion, and abnormalities in the hematopoietic system (Fig. 5D). The low-risk group demonstrates robust drug metabolism and detoxification capabilities, a balanced carbohydrate and short-chain fatty acid metabolism, and active steroid hormone synthesis and regulation. These metabolic characteristics may contribute to reducing disease risk and maintaining normal body function and health (Fig. 5E).

Biological functional and pathway enrichment analysis of ERS-related lncRNA Signature. (A-C) The GO function enrichment analysis; (D-E) The GSEA pathway enrichment analysis in high-risk and low-risk groups.
Exploration of the association between TMB and the ERS-related lncRNA signature
TMB can indirectly reflect tumor neoantigen generation capacity and extent, as well as predict immunotherapy efficacy for a range of malignancies26,27. On the basis of the risk score, we performed an analysis of the TMB in LSCC patients with the aid of the R package “maftools”. This helped to identify the top 15 most mutated genes. The mutation frequency of high-risk group was 92.73% (Fig. 6A), while in the low-risk group was 98.18% (Fig. 6B). The waterfall plot delineates the primary mutant genes, including TP53, TTN, CSMD3, SYNE1, NSD1, LRP18, MUC16, USH2A, and PCLO, exhibiting distinct mutation rates between the high-risk and low-risk groups. We find that the TMB score of high-risk group is significantly lower than that of low-risk group, with statistical significance (p = 0.00022) (Fig. 6C). A negative correlation between TMB and risk score is shown by the correlation curve in Fig. 6D (r = -0.46, p = 5.2e-0.7). The survival curve of TMB suggested minimal influence on the survival of the sample (Fig. 6E). Irrespective of the TMB level, patients classified in the high-risk group exhibit a less favorable prognosis (Fig. 6F).

The relationship between TMB and ERS-related lncRNA signature. (A-B) The oncoplots show the top 15 gene with the highest mutation rate in the high-risk group (A) and low-risk group (B); (C)TMB differences between high-risk and low-risk groups; (D) Relationship between TMB and risk scores; (E) Kaplan–Meier survival analysis of LSCC patients between the High-TMB and Low-TMB patients; (F) Kaplan–Meier survival curves of LSCC patients across High-TMB + high risk, High-TMB + low risk, Low-TMB + high risk, and Low-TMB + low risk. TMB, tumor mutational burden.
Analysis of immunological characteristics
The tumor microenvironment (TME) is an intricate system that is formed by the interplay between tumor cells, neighboring tissues and the immune cells28. In Fig. 7A, a noticeable distinction is observed in the tumor microenvironment scores between the high-risk and low-risk groups, with significantly elevated StromalScore, ImmuneScore, and ESTIMATEScore evident in the high-risk group (p < 0.01). In Fig. 7B, most of the immune cells showed a positive correlation with the risk score, but they may also be negatively regulated in different algorithms. In addition, survival analysis indicated that patients with higher levels of B cell/M1 macrophage/M2 macrophage/CD8 + T cell infiltration have better survival outcomes (Figs.7C-H). Next, further exploration into the relationship between immune cells, immune function, and risk score is facilitated through ssGSEA enrichment analysis. As illustrated in Fig. 7I, there was a significant difference between the high and low-risk groups for several immune cell types, with higher scores in the high-risk group for all except B-cells, CD8 + T-cells, mast-cells, NK-cells, T-cells and Th2-cells. Additionally, the score of infiltrating immune cells in the high-risk group for APC co-inhibition, APC co-stimulation, chemokine receptor (CCR), checkpoint, parainflammation, T cell co-inhibition, T cell co-stimulation, type I IFN Response, and type II IFN Response were all higher than those in the low-risk group (Fig. 7J), demonstrating that these immune functions were more active in the high-risk group.

Analysis of tumor immune signature. (A) The boxplot for StromalScore, ImmuneScore, and ESTIMATEScore in the high- and low-risk groups; (B) Estimation of immune-infiltrating cells in LSCC by using different algorithms; (C-H) Kaplan–Meier survival curves of screened B cells (C), Macrophages M1 (D), Macrophages M2 (E) , plasma cells (F), B memory cells (G) and CD8 + T cells (H) in LSCC patients; (I-J) The score of the infiltrating immune cells (I) and immune-related functions (J) in the high-risk and low-risk groups.
Identification of ERs-related lncRNA Signature through consensus cluster
Consensus clustering analysis was conducted on the expression levels of ERS-related lncRNA, dividing all tumor samples into 3 clusters: cluster1 (n = 26), cluster2 (n = 65), and cluster3 (n = 25) (Figs. 8A-C). Then, Kaplan–Meier analysis was performed for survival analysis of the three clusters. The results showed that the patients in cluster1 had the best survival outcomes, while those in cluster3 exhibited the poorest survival prognosis. (Fig. 8D), with cluster 1 consisting mainly of patients at low risk and cluster 3 consisting mainly of patients at high risk (Fig. 8E). T-SNE analysis further delineated the disparate clustering patterns based on the expression profiles of model genes, facilitating the stratification of samples into different clusters and risk groups (Figs. 8F,G). Differences in immune cell infiltration between the clusters were manifested in the immune response heatmap, similar to the previous results, patients with low expression of B cells and CD8 + T cells are more frequently observed in the high-risk cluster3 (Fig. 8H). Immune checkpoint molecule expression was then compared across the three clusters. It can be observed from Fig. 8I that VTCN1, TNFSF18, TNFRSF4, CD40, and HHLA2 have the highest expression in cluster1, while CD86, TNFRSF18, CD274, CD276, PDCD1LG2, CD44, and NRP1 have the highest expression in cluster3. Through the comparative evaluation of drug sensitivity by IC50 and screening, Fig.S1A-F demonstrated significant differences in drug sensitivity between any two subgroups. It was observed that patients in cluster3, which has a higher proportion of high-risk patients, are more sensitive to BMS-536924, ERK_2440, PD0325901, Telomerase inhibitor IX, Selumetinib and VX-11e.

Identification of ERS-related lncRNA Signature through consensus cluster. (A) Consensus clustering CDF for k = 2 to 9; (B) The consensus score matrix of all samples when k = 3; (C) Length and slope of the CDF curve as the index changes from 2 to 9; (D) Kaplan–Meier survival curves of LSCC patients’OS among the three different clusters; (E) Sankey diagram of the relationship between three different subgroups and risk scores; (F) tSNE analysis between three different subgroups; (G) tSNE analysis between high- and low-risk groups; (H) Heatmap reflects differences in immune cell infiltration between the three clusters; (I) Boxplot showing the expression of immune checkpoint molecules between the three clusters.
Analysis of drug sensitivity and immunotherapy efficacy related to the ERS-related lncRNA signature
Further investigation into the correlation between drug sensitivity and risk scores was conducted by determining the IC50 values of conventionally utilized chemotherapy drugs for LSCC. Utilizing Wilcoxon analysis, the results revealed notable distinctions. For a majority of the drugs, including AZ960, AZD1332, AZD2014, AZD8055, BMS-536924, CZC4832, Dasatinib, Entospletinib, ERK_2440, JAK_8517, NU7441, Selumetinib and Staurosporine, patients in the high-risk group demonstrate greater sensitivity. Conversely, for OSI-027, Low-risk patients show heightened sensitivity (Fig. 9A). These findings may have potential implications for guiding clinical treatment in high-risk patients. Given the substantial clinical significance of immune checkpoints in tumor immunotherapy29, as depicted in Fig. 9B, we use the TIDE score for prognostic assessment of immune checkpoint inhibition therapy, it was seen that the TIDE score of low-risk group is lower, implying that its immunotherapy effect might be better.

Potential drug sensitivity analysis by IC50 and the immune checkpoint gene expression analysis between the high-risk and low-risk groups. (A) Boxplots showing the drug sensitivity analysis of commonly used chemotherapeutic agents in LSCC between high-risk and low-risk groups; (B) Violin showing expression of TIDE score between the high-risk and low-risk groups.
Elevated expression of lncRNA LHX1-DT in LSCC
To further investigate the mechanisms by which lncRNAs in the signature exert their functions, we selected three LncRNAs AC110611.2, LHX1-DT, and AL157373.2 for in-depth exploration (Figs. 10A-F). Their expression levels were observed to be elevated in LSCC, but the survival of AC110611.2 was not significant. Notably, elevated expression levels of LHX1-DT were correlated with poor prognosis (p < 0.001), whereas increased expression of AL157373.2 was associated with higher survival rates (p < 0.05). In order to select a gene with an oncogenic role in LSCC, a follow up experiment was carried out using LHX1-DT for the verification. We detected the expression of LHX1-DT in normal and LSCC tumor cells by RT-qPCR. As shown in Figs. 10G-H, the expression of LHX1-DT was upregulated in AMC-HN-8 and TU686 cells compared to HaCaT cells. The following experiments were carried out using AMC-HN-8 cells and TU686 cells.

LHX1-DT expression is up-regulated in LSCC. (A-F) Expression difference analysis and Kaplan–Meier survival analysis of AC110611.2, LHX1-DT and AL157373.2;(G-H) RT-qPCR detected the expression of LHX1-DT in normal and tumor tissues. *P < 0.05, **P < 0.01 and ***P < 0.001.
LHX1-DT regulates the malignancy of LSCC
To further explore the function of LHX1-DT, we used siRNA to knockdown its expression. The silencing efficiency of the siRNAs were shown in Fig. 10A-B.It was observed that the interference efficiency of si-LHX1-DT#002 exhibited superior interference efficiency compared to other sequences (Fig. 11A,B), and this sequence was used for subsequent experiments. In the CCK-8 assay, cell proliferation was increased after LHX1-DT knockdown in both AMC-HN-8 and TU686 cells (Fig. 11C, D). In the colony formation assay, LHX1-DT knockdown inhibited both AMC-HN-8 and TU686 colony formation (Fig. 11E). To elucidate the effects of LHX1-DT on cell metastasis, transwell assay and wound healing assay were performed. The results showed that after silencing of LHX1-DT, the ability of migration and invasion was significantly decreased (Fig. 11F,G). Western blotting was performed to detect protein levels of the apoptosis marker cleaved caspase 3 in LHX1-DT silenced AMC-HN-8 and TU686 cells, Fig. 11H revealed an augmentation of apoptosis following LHX1-DT knockdown. To determine the effect of LHX1-DT on activity of caspase-3, we measured the activity of caspase-3 by caspase 3/7 colorimetric assay and showed that caspase-3 activity was increased in AMC-HN-8 and TU686 cells with LHX1-DT knockdown (Fig. 11I). The primers for ERS-related molecules were detected with the knockdown of LHX1-DT through RT-qPCR, and it could be found that the expression of ATF6, GRP78, CHOP, and IRE1 was elevated in AMC-HN-8 and TU686 cells after the knockdown of LHX1-DT(Fig.S2A-H), and it could be suspected that LHX1-DT affects the death of LSCC by influencing the ERS.

LHX1-DT regulates the malignancy of LSCC. (A-B) Detection of relative silencing levels of LHX1-DT in AMC-HN-8 (A) and TU686 (B); (C-D) Knockdown of LHX1-DT in AMC-HN-8 (C) and TU686 (D) cells by CCK8 analysis; (E–G) Knockdown of LHX1-DT in AMC-HN-8 and TU686 cells by colony formation assay (E), transwell assay (F), Wound healing assay (G); (H) Western blotting was performed to detect protein levels of the apoptosis marker cleaved caspase 3 in knockdown of LHX1-DT in AMC-HN-8 and TU686 cells; (I) Caspase3 activity was measured in AMC-HN-8 and TU686. *P < 0.05, **P < 0.01 and ***P < 0.001.
Discussion
Laryngeal cancer is one of the most common types of malignant tumors, characterized by an escalating incidence rate and considerable mortality30. Therefore, it is crucial to find effective prognostic markers for LSCC. ERS plays important roles in tumorigenesis31. However, studies focusing on LSCC and ERS are still limited11.
Recent studies have highlighted the pivotal role of endoplasmic reticulum stress (ERS) in the development and progression of various cancers. For instance, in hepatocellular carcinoma, ER stress-related super enhancers have been shown to drive epithelial-mesenchymal transition through CREB5-mediated activation of TNC, thereby promoting tumor invasiveness and poor prognosis32. In colorectal cancer, SEC23A has been reported to enhance 5-FU resistance by regulating ER stress-mediated apoptosis, indicating its involvement in chemotherapy response33. Similarly, in non-small cell lung cancer (NSCLC), targeting the ER stress signaling pathway has emerged as a promising therapeutic strategy to counteract tumor survival and drug resistance34. These findings collectively underscore the importance of ERS not only in tumor biology but also in prognostic modeling and therapeutic targeting. Inspired by these advances, we explored ERS-related lncRNAs in LSCC to identify prognostically relevant biomarkers.
In recent years multiple studies have established prognostic signatures based on genes associated with stress responses, cell death mechanisms, or metabolic pathways to stratify patients and guide personalized treatment strategies. For example, Yao et al. developed an unfolded protein response (UPR)-related gene signature in stomach cancer that revealed significant heterogeneity in the tumor immune microenvironment and strong prognostic power35. Similarly, Zhang et al. proposed a lactylation-related gene signature in breast cancer, linking epigenetic regulation to immune infiltration and patient outcomes36. Beyond these, several recent studies have expanded this strategy to other cancers. Chen et al. identified an anoikis-related lncRNA signature predictive of immune infiltration and prognosis in gastric cancer patients, supporting the immunomodulatory role of lncRNAs37. Qin et al. constructed a disulfidptosis-associated lncRNA signature for low-grade glioma that robustly predicted patient classification, tumor microenvironment patterns, and therapy response38 Yin et al. reported a disulfidptosis-related lncRNA model in head and neck squamous carcinoma that strongly correlated with anti-tumor immune infiltration and overall survival39. These studies demonstrate the utility of gene signatures and their integration with immunogenomic features. Compared to these studies, our study distinguishes itself by focusing on ERS-associated lncRNAs in LSCC-a novel molecular angle not previously explored in this tumor type. In this study, we obtained differentially expressed ERS-related lncRNA by co-expression analysis and differential analysis of the prognosis of patients in the TCGA database and the expression profiles of ERS-related genes. Cox regression analysis and lasso analysis were used to verify, resulting in the establishment of a prognostic signature comprising 3 ERS-related lncRNAs.Our prognostic signature included LHX1-DT, AC110611.2, and AL157373.2. Of these three lncRNAs, LHX1-DT was negatively correlated with LSCC prognosis and AL157373.2 was positively correlated with prognosis. Previous investigations have elucidated that during embryonic stem cell-to-mesoderm transition, the expression of LHX1-DT is significantly upregulated, followed by a progressive reduction throughout cardiomyocyte development40. Additionally, an influential study by Yang et al. shed light on the heightened expression of LHX1-DT in breast cancer patients, linking it to an unfavorable prognosis41. However, to our knowledge, the biological role and molecular mechanisms of LHX1-DT in LSCC have not been previously reported. In this study, we provide the first evidence that LHX1-DT is significantly overexpressed in LSCC tissues and cell lines compared to normal controls. Functional experiments revealed that silencing LHX1-DT suppresses cell proliferation, migration, and invasion, while promoting apoptosis, suggesting an oncogenic role in LSCC progression. Interestingly, we also demonstrated that knockdown of LHX1-DT leads to the upregulation of several canonical ER stress markers, including ATF6, GRP78, CHOP, and IRE1 (Fig.S2A-H), indicating a mechanistic link between LHX1-DT and the modulation of endoplasmic reticulum stress. These results suggest that LHX1-DT may promote tumor progression by inhibiting ERS-induced apoptotic pathways, thereby facilitating tumor cell survival under stress conditions. Mechanistically, LHX1-DT may exert its function through interaction with ERS-related transcriptional regulators or by acting as a competing endogenous RNA (ceRNA) to modulate ERS-related signaling cascades, though this requires further elucidation. Nonetheless, our findings establish LHX1-DT as a novel ERS-associated oncogenic lncRNA in LSCC and support its potential utility as both a prognostic biomarker and a therapeutic target.
To elucidate possible interactions between the prognostic signature and the tumor immune microenvironment, we performed an extensive evaluation of immune infiltration in LSCC patients. 10 of the 16 immune cells, including DCs, neutrophils and Tregs, showed differences between high and low-risk groups. DCs, as key are antigen-presenting cells42, initiate immune responses through antigen processing and are critical for immunotherapy efficacy43. Neutrophils drive tumor progression by generating neutrophil extracellular traps (NETs), which facilitate proliferation, metastasis, and immunosuppression44. Tregs, as the primary regulator of self-tolerance, can maintain immunological homeostasis and coordinate the suppression of excessive activation of the immune system45. Within the high-risk group, the marked increase in DCs, neutrophils, Tregs, and M2 macrophages suggests a shift toward an immunosuppressive TME. Although elevated immune scores typically indicate an immune-active or “hot” tumor, in this context, the infiltrated immune components may skew toward immunosuppressive phenotypes Mechanistically, this immunosuppressive microenvironment may arise from persistent ER stress within the tumor, which has been shown to enhance the recruitment and activation of immunosuppressive cell populations via secretion of cytokines such as TGF-β, IL-10, and CCL22. These cytokines can promote Treg expansion and M2 macrophage polarization, further amplifying immune evasion. In addition, the high-risk group exhibited elevated expression of immune checkpoint molecules (e.g., PD-L1, PDCD1LG2, CD86), which can lead to functional exhaustion of cytotoxic T cells and impaired anti-tumor immunity. As a result, the immune microenvironment in these patients, despite being heavily infiltrated, is dysfunctional and unable to exert effective anti-tumor responses. This may explain the paradox of higher immune infiltration yet worse prognosis. The presence of such a microenvironment likely facilitates tumor progression by supporting immune evasion, angiogenesis, and metastatic potential. These findings underscore the importance of not only quantifying immune infiltration but also characterizing the functional state and phenotypes of immune cells. From a therapeutic perspective, this highlights the need to develop combinatorial immunotherapeutic strategies, such as immune checkpoint blockade in conjunction with agents targeting Tregs, M2 macrophages, or key ER stress pathways, to reverse immunosuppression and restore anti-tumor immunity in high-risk LSCC patients. These findings also provide important implications for optimizing immunotherapy protocols tailored to immune landscape differences between risk groups. In high-risk LSCC patients, where the tumor microenvironment is enriched with immunosuppressive cell types such as Tregs and M2 macrophages, monotherapy using PD-1/PD-L1 immune checkpoint inhibitors may be insufficient due to T cell exhaustion and active immune evasion mechanisms. Instead, combination strategies that include agents targeting immunosuppressive components may enhance treatment efficacy. For example, the use of IDO (Indoleamine 2,3-dioxygenase) inhibitors (e.g., Epacadostat) to deplete Tregs or CSF1R inhibitors to reduce M2 macrophage populations has shown promise in preclinical models and clinical trials when combined with ICIs46. Additionally, high expression of TGF-β signaling in immunosuppressive TMEs has been associated with resistance to checkpoint blockade, and combining TGF-β inhibitors (e.g., Galunisertib) with PD-L1 blockade is currently under clinical evaluation and may benefit the high-risk subgroup47,48. In contrast, the low-risk group, characterized by higher infiltration of effector immune cells (e.g., CD8 + T cells and B cells) and lower expression of immunosuppressive markers, may represent an “immune-inflamed” phenotype more amenable to PD-1/PD-L1 monotherapy. Such patients may also benefit from vaccination-based or neoantigen-targeted immunotherapies, where effective immune engagement is already present. Furthermore, our TIDE analysis showed lower TIDE scores in the low-risk group, suggesting potentially better responsiveness to immune checkpoint inhibitors. Thus, stratifying LSCC patients by our ERS-related lncRNA signature could provide a valuable reference for personalized immunotherapy planning.
In this study, we performed a firm of immune checkpoint genes differentially expressed between high- and low-risk group, notable among them being CD276, PDCD1LG2 and CD86. Extensive research has elucidated the significant involvement of CD276 across various cancers. Its heightened expression in breast cancer has been implicated in disease progression49, while in ovarian cancer, CD276 has been shown to suppress T-cell-mediated anti-tumor responses50. These findings implicate CD276 as an important immune microenvironment regulator and potential therapeutic target. PDCD1LG2 as a prognostic marker of cancer, is involved in immune infiltration. Study found that PDCD1LG2 is relevant with the prognosis of Hepatocellular Carcinoma, and its high expression had a better prognosis51. As a costimulatory molecule, CD86 is an M1 macrophage marker expressed on the surface of immune cells52. The significant upregulation of genes CD276, PDCD1LG2 and CD86 in the high-risk group indicates a significant level of immunosuppression within the tumor microenvironment. This phenomenon could represent one of the mechanisms by which tumors accomplish immune evasion via these immune checkpoint molecules, generally correlating with a less favorable prognosis. These findings not only aid in comprehending the characteristics of the tumor immune microenvironment, but also potentially present new targets and strategies for immunotherapy in LSCC.
Our study indicates that TMB alone is insufficient to stratify patient prognosis in LSCC, as observed in Fig. 6E and Fig. 6F. The lack of significant survival differences between high and low TMB groups suggests that other factors such as immune suppression and immune exclusion mechanisms contribute significantly to patient outcomes. Specifically, the high-risk group exhibited lower TMB but higher immune checkpoint expression, indicating an adaptive immune resistance mechanism. Previous studies have shown that tumors with high immune checkpoint expression may evade immune attack despite having lower TMB53. Conversely, the low-risk group had higher TMB but also higher TIDE scores, suggesting immune exclusion mechanisms may be at play, restricting immune cell infiltration and reducing immune system engagement54. These results emphasize that while TMB remains a useful biomarker in some cancer types, its role in LSCC prognosis is complex and influenced by the immune microenvironment. Future studies should consider integrating TME-related factors and functional immune profiling to refine immunotherapy response predictions.
However, our study is subject to certain limitations that warrant consideration. Firstly, the data used in this investigation were obtained from the TCGA public database, which has inherent limitations due to the relatively limited number of samples available. Consequently, validation of the clinical utility of our findings will require additional prospective studies with larger cohorts. Secondly, while our study identified LHX1-DT as a potential regulator in ERS modulation, the precise role of LHX1-DT in this context remains incompletely understood. It is conceivable that LHX1-DT may associate with other genes to regulate ERS pathways. Therefore, further exploration of the intricate interplay between LHX1-DT and other ERS-related genes is warranted to elucidate its mechanistic involvement more comprehensively.
Conclusion
This study identified and confirmed a new ERS-related lncRNA signature (LHX1-DT, AC110611.2, AL157373.2), which can divide LSCC patients into high-risk and low-risk groups, demonstrating its potential as an independent prognostic indicator and a determinant for treatment strategies in LSCC patients. Additionally, our investigation revealed that LHX1-DT plays a pivotal role in promoting the progression of LSCC, thus suggesting its potential as a promising therapeutic target for intervention.
Data availability
The study’s original contributions are included in the article/Supplementary materials; further inquiries should be directed to the corresponding author.
References
Zhang, X. et al. Research progress on the interaction between oxidative stress and platelets: Another avenue for cancer?. Pharmacol. Res. 191, 106777 (2023).
Hu, C. et al. Oleanolic Acid Induces Autophagy and Apoptosis via the AMPK-mTOR Signaling Pathway in Colon Cancer. J. Oncol. 2021, 8281718 (2021).
Li, S. et al. Exosomes: Another intercellular lipometabolic communication mediators in digestive system neoplasms?. Cytokine Growth Factor Rev. 73, 93–100 (2023).
Wu, X. et al. MicroRNA: Another Pharmacological Avenue for Colorectal Cancer?. Front. Cell Dev. Biol. 8, 812 (2020).
Zhao, R. et al. A novel tyrosine tRNA-derived fragment, tRFTyr, induces oncogenesis and lactate accumulation in LSCC by interacting with LDHA. Cell. Mol. Biol. Lett. 28, 49 (2023).
Li, D. et al. Elevated ITGA5 facilitates hyperactivated mTORC1-mediated progression of laryngeal squamous cell carcinoma via upregulation of EFNB2. Theranostics 12, 7431–7449 (2022).
Wu, Y. et al. Circular RNA circCORO1C promotes laryngeal squamous cell carcinoma progression by modulating the let-7c-5p/PBX3 axis. Mol. Cancer 19, 99 (2020).
Tian, X. et al. Salidroside attenuates myocardial ischemia/reperfusion injury via AMPK-induced suppression of endoplasmic reticulum stress and mitochondrial fission. Toxicol. Appl. Pharmacol. 448, 116093 (2022).
Liu, J. et al. Pterostilbene exerts an anti-inflammatory effect via regulating endoplasmic reticulum stress in endothelial cells. Cytokine 77, 88–97 (2016).
Li, T. et al. Snapshots: Endoplasmic Reticulum Stress in Lipid Metabolism and Cardiovascular Disease. Curr. Issues Mol. Biol. 28, 14–28 (2018).
Oakes, S. A. & Papa, F. R. The role of endoplasmic reticulum stress in human pathology. Annu. Rev. Pathol. 10, 173–194 (2015).
Chen, X. & Cubillos-Ruiz, J. R. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat. Rev. Cancer 21, 71–88 (2021).
Oakes, S. A. Endoplasmic Reticulum Stress Signaling in Cancer Cells. Am. J. Pathol. 190, 934–946 (2020).
Liu, C.-Y. et al. ER stress-related ATF6 upregulates CIP2A and contributes to poor prognosis of colon cancer. Mol. Oncol. 12, 1706–1717 (2018).
Jiang, J. et al. MiR-1281, a p53-responsive microRNA, impairs the survival of human osteosarcoma cells upon ER stress via targeting USP39. Am. J. Cancer Res. 8, 1764–1774 (2018).
Yao, X. et al. Endoplasmic reticulum stress-induced exosomal miR-27a-3p promotes immune escape in breast cancer via regulating PD-L1 expression in macrophages. J. Cell. Mol. Med. 24, 9560–9573 (2020).
Bhan, A., Soleimani, M. & Mandal, S. S. Long Noncoding RNA and Cancer: A New Paradigm. Cancer Res. 77, 3965–3981 (2017).
Wang, Y. et al. LncRNA THUMPD3-AS1 enhances the proliferation and inflammatory response of chondrocytes in osteoarthritis. Int. Immunopharmacol. 100, 108138 (2021).
Guo, Q. et al. The Regulatory Network and Potential Role of LINC00973-miRNA-mRNA ceRNA in the Progression of Non-Small-Cell Lung Cancer. Front. Immunol. 12, 684807 (2021).
Chen, C. et al. Pharmacological roles of lncRNAs in diabetic retinopathy with a focus on oxidative stress and inflammation. Biochem. Pharmacol. 214, 115643 (2023).
Sun, M. et al. A nine-lncRNA signature predicts distant relapse-free survival of HER2-negative breast cancer patients receiving taxane and anthracycline-based neoadjuvant chemotherapy. Biochem. Pharmacol. 189, 114285 (2021).
Koirala, P. et al. LncRNA AK023948 is a positive regulator of AKT. Nat. Commun. 8, 14422 (2017).
Cao, H.-L., Liu, Z.-J., Huang, P.-L., Yue, Y.-L. & Xi, J.-N. lncRNA-RMRP promotes proliferation, migration and invasion of bladder cancer via miR-206. Eur. Rev. Med. Pharmacol. Sci. 23, 1012–1021 (2019).
Zong, S., Dai, W., Guo, X. & Wang, K. LncRNA-SNHG1 promotes macrophage M2-like polarization and contributes to breast cancer growth and metastasis. Aging (Albany NY) 13, 23169–23181 (2021).
Cao, J. et al. lncRNA IGKJ2-MALLP2 suppresses LSCC proliferation, migration, invasion, and angiogenesis by sponging miR-1911-3p/p21. Cancer Sci. 111, 3245–3257 (2020).
McNamara, M. G. et al. Impact of high tumor mutational burden in solid tumors and challenges for biomarker application. Cancer Treat Rev. 89, 102084 (2020).
Li, R. et al. Choosing tumor mutational burden wisely for immunotherapy: A hard road to explore. Biochim. Biophys. Acta Rev. Cancer 1874, 188420 (2020).
Xiao, Y. & Yu, D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol. Ther. 221, 107753 (2021).
Li, B., Chan, H. L. & Chen, P. Immune Checkpoint Inhibitors: Basics and Challenges. Curr. Med. Chem. 26, 3009–3025 (2019).
Zhao, J., Li, X.-D., Wang, M., Song, L.-N. & Zhao, M.-J. Circular RNA ABCB10 contributes to laryngeal squamous cell carcinoma (LSCC) progression by modulating the miR-588/CXCR4 axis. Aging (Albany NY) 13, 14078–14087 (2021).
Mohamed, E., Cao, Y. & Rodriguez, P. C. Endoplasmic reticulum stress regulates tumor growth and anti-tumor immunity: A promising opportunity for cancer immunotherapy. Cancer Immunol. Immunother. 66, 1069–1078 (2017).
Wang, A. et al. Endoplasmic reticulum stress-related super enhancer promotes epithelial-mesenchymal transformation in hepatocellular carcinoma through CREB5 mediated activation of TNC. Cell Death Dis. 16, 73 (2025).
Su, Z., Liu, M., Krohn, M., Schwarz, S. & Linnebacher, M. The impact of SEC23A on 5-FU chemotherapy sensitivity and its involvement in endoplasmic reticulum stress-induced apoptosis in colorectal cancer. Apoptosis https://doi.org/10.1007/s10495-025-02084-2 (2025).
Jadhav, A., Menon, A., Gupta, K. & Singh, N. Molecular and therapeutic insight into ER stress signalling in NSCLC. J. Drug Target https://doi.org/10.1080/1061186X.2025.2461105 (2025).
Ouyang, W. et al. Unraveling the unfolded protein response signature: implications for tumor immune microenvironment heterogeneity and clinical prognosis in stomach cancer. Aging (Albany NY) 16, 7818–7844 (2024).
Jiao, Y., Ji, F., Hou, L., Lv, Y. & Zhang, J. Lactylation-related gene signature for prognostic prediction and immune infiltration analysis in breast cancer. Heliyon 10, e24777 (2024).
Chen, T., Jiang, Q., Wang, Z., Wang, F. & Fu, Z. LncRNA AF117829.1 is correlated with prognosis and immune infiltration and facilitates tumor progression by targeting OR7C1 in colorectal cancer. Transl. Cancer Res. 13, 5347–5364 (2024).
Qin, X., Chen, Z., Wu, L. & Ding, R. A disulfidptosis-associated long noncoding RNA signature to predict low-grade glioma classification, prognosis, tumor microenvironment, and therapy regimens: Observational study. Medicine (Baltimore) 103, e39316 (2024).
Yin, Z. et al. Identification and Verification of a Novel Disulfidptosis-Related lncRNAs Prognostic Signature to Predict the Prognosis and Immune Activity of Head and Neck Squamous Carcinoma. Iran J. Public Health 53, 2328–2340 (2024).
Yu, Q. et al. Long non-coding RNA LHX1-DT regulates cardiomyocyte differentiation through H2A.Z-mediated LHX1 transcriptional activation. iScience 26, 108051 (2023).
Yang, X. et al. Individualized Prediction of Survival by a 10-Long Non-coding RNA-Based Prognostic Model for Patients With Breast Cancer. Front. Oncol. 10, 515421 (2020).
Wculek, S. K. et al. Dendritic cells in cancer immunology and immunotherapy. Nat. Rev. Immunol. 20, 7–24 (2020).
Macri, C., Pang, E. S., Patton, T. & O’Keeffe, M. Dendritic cell subsets. Semin. Cell Dev. Biol. 84, 11–21 (2018).
Tecchio, C., Scapini, P., Pizzolo, G. & Cassatella, M. A. On the cytokines produced by human neutrophils in tumors. Semin. Cancer Biol. 23, 159–170 (2013).
Huppert, L. A. et al. Tissue-specific Tregs in cancer metastasis: opportunities for precision immunotherapy. Cell. Mol. Immunol. 19, 33–45 (2022).
Pyonteck, S. M. et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 19, 1264–1272 (2013).
Mariathasan, S. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548 (2018).
David, C. J. & Massagué, J. Contextual determinants of TGFβ action in development, immunity and cancer. Nat. Rev. Mol. Cell Biol. 19, 419–435 (2018).
Cong, F., Yu, H. & Gao, X. Expression of CD24 and B7–H3 in breast cancer and the clinical significance. Oncol. Lett. 14, 7185–7190 (2017).
Zang, X. et al. Tumor associated endothelial expression of B7–H3 predicts survival in ovarian carcinomas. Mod. Pathol. 23, 1104–1112 (2010).
Li, W. et al. Identification of PDCD1 and PDCD1LG2 as Prognostic Biomarkers and Associated with Immune Infiltration in Hepatocellular Carcinoma. Int. J. Gen. Med. 15, 437–449 (2022).
Chen, R. et al. Overexpression of CD86 enhances the ability of THP-1 macrophages to defend against Talaromyces marneffei. Immun. Inflamm. Dis. 10, e740 (2022).
Yarchoan, M. et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. JCI Insight 4, e126908 (2019).
Wang, S., He, Z., Wang, X., Li, H. & Liu, X.-S. Antigen presentation and tumor immunogenicity in cancer immunotherapy response prediction. Elife 8, e49020 (2019).
Acknowledgements
We thanked Hung Yoo Editorial Team for language editing service.
Funding
This study was funded by the Major Science and Technology Plan Project of Hainan Province (ZDKJ202005), the Science and Technology Plan Project of Hainan Province (LCYX202105, LCYX202202), and also received support from the Clinical Medical Center of Hainan Province.
Author information
Authors and Affiliations
Contributions
Xiazhi Pan: Conceptualization, Data curation, Validation, Formal analysis, Software, Visualization, Writing-original draft, Writing-review & editing. Qin Che: Writing-review & editing,Supervision.Duanshali Liu:Writing-review & editing, Writing-original draft, Software, Data curation. Yingli Xie: Software, Data curation. Beicheng Li: Software, Data curation.Shanshan Zhang: Software, Data curation.Tian Li:Writing-review & editing,Supervision.Gege Li: Supervision. Xiaohan Li: Supervision. Qiuchen Zheng: Supervision. Kai Zhao: Supervision, Writing – review & editing. Mingbo Liu: Funding acquisition, Conceptualization, Supervision. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Pan, X., Che, Q., Liu, D. et al. Development and validation of a novel endoplasmic reticulum stress-related lncRNA signature in laryngeal squamous cell carcinoma. Sci Rep 15, 12497 (2025). https://doi.org/10.1038/s41598-025-96576-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96576-5
