
Abstract
A new series of glycosyl heterocyclic scaffolds, 5a-10b with N-glycosidic linkage, were synthesized, starting with 2-acetyl-1H-benzimidazole as a precursor of the propargyl-derived substrates (2) and (3), which were then converted to the target 1,2,3-triazole glycosides (8a-10b) bearing unprotected hydroxyl groups. The new chemical entities have been assessed for their cytotoxic properties on diverse human cancer cell lines, namely HepG-2 (human liver cancer), HCT-116 (human colorectal), and MCF-7 (human breast cancer), in addition to a human normal cell line (BJ-1), following the LDH assay and with erlotinib and doxorubicin as the standard references. Most of the tested compounds demonstrated potent activity, particularly the triazole glycosides 6b, 7b, 8b, 9a, 9b, 10a, and 10b. Compound 9a was the best against all targeted cell lines, particularly HepG-2 and HCT-116, by IC50 values of 1.64 ± 0.11 and 5.00 ± 0.51 µM, superior to that of erlotinib, IC50 = 2.07 ± 0.07 and 5.14 ± 0.33 µM, respectively. Furthermore, it showed a safe profile against the tested normal cell line BJ-1. The triazole glycosides 8a-10b were investigated to assess their capability to inhibit EGFR. Remarkably, 9a and 9b exhibited noteworthy inhibitory activity against EGFR (IC50 = 0.069 ± 0.003 and 0.075 ± 0.003 µM, respectively) in comparison with erlotinib, the reference drug (0.048 ± 0.002 µM). Molecular docking confirmed these findings, suggesting that the incorporation of the α,β-unsaturated ketone function enhances compounds’ stability within the EGFR active site. Thus, these results indicate that compounds 9a and 9b disclosed potential anti-cancer agents targeting EGFR kinase.
Similar content being viewed by others
Introduction
Despite the sturdy and weariless endeavors to conflict with cancer, the implementation for treating certain types of tumors has exhibited minimal advancement owing to their highly aggressive nature and the intricate mechanisms of malignant cell metastasis. Moreover, the administration of anticancer agents manifests a toxic effect not only on the cancerous cells but also on the non-neoplastic cells, thereby lacking specificity toward the intended target and curtailing the scope of the treatment1.
The resistance by certain ailments to the prevalent medications and the potential for severe toxicity forces and urges researchers to intensify research work and represent a pressing demand to develop and provide alternative, more efficient therapeutic strategies and molecular targets2. Targeted drug therapy is among strategies that are now largely being employed, which were not common a few years ago. In this regard, designing and synthesizing new potent molecules with the lowest toxicities to normal cells is a major objective for cancer medicine research. Imidazole, 1,2,3-triazole, and pyrazole scaffolds have achieved immense significance in medicinal chemistry exerting a broad spectrum of activities. This is primarily due to their association with a diverse range of pharmaceutical properties, including but not limited to, anticancer, anti-inflammatory, antidiabetic, antimicrobial and antihypertensive effects3,4.
The presence of abundant oligosaccharide antigens on the surfaces of cancer cells is a well-established fact that endows them with the potential to serve as markers for both active and passive cancer types. Within this framework, two notable derivatives have been identified for their potentiality to inhibit cell growth in HEp-2 and NCI-H292 strains. HEp-2 cells are associated with laryngeal carcinoma, while NCI-H292 cells are linked to lung carcinoma. These compounds demonstrate significant potential in targeting and managing these specific types of cancer5,6.
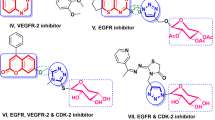
Tyrosine kinase is an important receptor of the ErbB and refers to the EGFR (epidermal growth factor receptor). It is essential for controlling numerous important biological routes, for example metastasis, apoptosis, angiogenesis, cell cycle regulation, adhesion, and cell motility7,8,9,10,11. EGFR is frequently altered in solid cancers, including breast (MCF-7), colorectal (HCT116) and Human liver carcinoma (HepG-2)12,13,14. These alterations can occur via kinase-activating mutations or over-expression of EGFR. Such changes are associated with unfavorable clinical results, including early recurrence, improved risk of metastasis, and decreased survival15. As a consequence, EGFR is a useful candidate target for drug discovery in research for breast cancer (BC) drug development. Clinically, there are several small molecules available as EGFR inhibitors, including reversible inhibitors such as lapatinib, erlotinib, and gefitinib as well as irreversible inhibitors like Osimertinib, dacomitinib, gefitinib, and afatinib16,17 (Fig. 1).

Examples of some cytotoxic agents comprising imidazole, 1,2,3-triazole, glycosyl and pyrazole scaffold.
Click chemistry has emerged as a fundamental tool in organic or bioorganic chemistry, primarily synthesizing diverse macromolecules18. To achieve this, many techniques have been devised, encompassing an array of reaction types and methods that employ a range of catalysts and solvent systems19,20,21.
1,2,3-Triazoles core possessing compounds have acquired a significant interest in organic and medicinal chemistry attributable to their revealed biological activities and easy formation over click chemistry technique Cu(I)-catalyzed azide-alkyne cycloaddition22. These compounds demonstrate diverse pharmacological effects, for instance, anti-fungal, anti-viral, anti-bacterial, anti-tubercular, α-glucosidase inhibition, anti-protozoal, antioxidant, anti-inflammatory, and anti-proliferative properties23,24. There are two synthesis pathways for 1,2,3-triazoles: The 1,4-disubstituted-1,2,3-triazoles produced utilizing a catalyzed pathway; on the other hand, a 1,5- and 1,4-disubstituted-1,2,3-triazoles mixture was afforded when the non-catalyzed pathway was applicable. The regioselectivity of the reaction is influenced by factors like substituents on the acetylenic and azido compounds and the reaction conditions25,26.
Recently, a multitude of pharmacophores incorporating sugar moieties linked by glycosidic linkages in glycoside fragments have been synthesized and revealed to exert immense potential as antitumor agents27,28,29, particularly through the pathway of EGFR inhibition30,31,32. On the other hand, benzimidazole derivatives serve as highly valuable intermediates/subunits for the development of molecules that hold significance in the domains of pharmaceuticals or biology33. Substituted benzimidazole derivatives have been employed in various therapeutic areas34, especially as potent anticancer agents3,35,36.
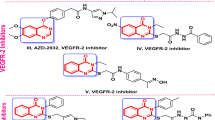
The design strategies of EGFR-targeting anticancer compounds have leveraged both 1,2,3-triazole and benzimidazole scaffolds as key pharmacophoric elements. For triazoles, click chemistry has been utilized to incorporate the moiety either as a linker or hydrogen bond acceptor within the ATP-binding pocket, with lipophilic substituents at positions 1 and 4 enhancing binding affinity37,38,39,40. Similarly, benzimidazoles exploit their rigid, planar structure with nitrogen atoms forming crucial hydrogen bonds in the same binding region. While triazole designs often feature quinazoline or quinoline cores serving as metabolically stable bioisosteric replacements for amide linkages, benzimidazole scaffolds are typically functionalized at positions 1, 2, and 5, with halogen or alkoxy substitutions at position 5 enhancing activity41,42,43. Both structural classes have been strategically modified to overcome resistance in mutant EGFR variants, particularly the challenging T790M mutation, through careful optimization of solubilizing groups and linker regions to improve pharmacokinetic properties while maintaining potent inhibitory activity44. Interestingly, benzimidazole/1,2,3-triazole hybrids VIII and IX (Fig. 1) were revealed as EGFR inhibitors with potency more than erlotinib45.
The molecular hybridization strategy for designing and synthesizing novel scaffolds of potential pharmaceutical impact acquired attention, leading to new anticancer candidates. In such a context, the pharmacological importance of several imidazole-triazole, pyrazolyl-imidazole, and benzimidazole-pyrazole-triazole hybrids has gained considerable interest (Fig. 1)46,47,48,49. Such hybrid molecules, with their reported potent anticancer and enzyme inhibition activity, comprise various linkage types of the heterocyclic core to the sugar moiety, which may be significant in enzymatic interactions, leading to promising anticancer effects, particularly on breast cancer cell lines.
Thus, our design depends on the fact that the conjugation of these potent pharmacophores forming new hybrids comprising such multiple scaffolds might result in a new structure that will have great promise for the enhancement of innovative therapeutics for the cancer treatment, Fig. 2.

Designed targets Pyrazolyl-Benzoimidazolyl-triazolyl-glycosyl hybrids.
Results and discussion
Chemistry
The synthetic route toward the formation of structurally related glycosyl heterocyclic scaffolds 5a-10b comprising the targeted diverse glycosidic linkages was depicted in Fig. 3 providing the synthesis of benzimidazole-C2 based hybrid substituted glycoside-1,2,3-triazoles. 2-Acetyl-1H-benzimidazole was alkylated with propargyl bromide and the derived terminal acetylenic product was formed. The latter was used even either as a substrate for the targeted click reaction or as a precursor for the functionalized propargyl nucleobases incorporating 4-fluorophenyl chalcone 2 and pyrazoline-linked terminal alkynes 3. The coupling arrangements for the alkynyl function for compound 1 were perceived as a triplet signal at δH = 3.34 ppm (J = 2.5 Hz) representing ≡ CH whereas they appeared at δH = 5.60 ppm as a doublet for –CH2C≡ (J = 2.5 Hz) in 1H NMR spectra. 13C NMR chart also confirmed the structure of monoalkylated nucleobases with the evidence of signals at δC = 34.98 ppm (–CH2C≡), δC = 73.32 ppm (≡ CH) and at δC = 77.53 ppm (–CH2C≡), which agree with the literature50,51.
Applying the click conditions for the reaction between the terminal acetylenic analogs 1–3 and the acetylated glycosyl azides; 2,3,4,6-tetra-O-acetyl-d-galactopyranosyl (4a) otherwise 2,3,4-tri-O-acetyl-d-xylopyranosyl azide (4b), eventually afforded targeted 1,2,3-triazole glycosides 5a-7b, respectively. Thus, sodium ascorbate and copper sulfate were used for generating the required Cu1 reactive species, in a basic medium following the previously documented procedure42. The occurrence of the click reaction affording the glycosyl-1,2,3-triazole products was verified by 1H NMR spectra by the disappearance of terminal alkyne proton signals (δH = 3.34–3.40 ppm) and the appearance of the signals representing the olefinic proton of triazole in the range δH = 7.8–7.9 ppm. The glycosyl moiety’s anomeric hydrogens were recorded as a doublet at δH = 5.20–5.55 ppm, demonstrating the β-type linkage of the sugar attached with the 1,2,3-triazole motif via their coupling constants. Furthermore, the singlet signals that were downfielded in the range δH = 1.70–2.89 ppm signified the acetyl groups’ protons besides the 1,2,3-triazolyl-H5 that was also observed at 7.75–7.89 ppm. The signals of those acetyl carbons resonated at δC = 19.88–28.14 in 13C NMR spectra.
The 1,2,3-triazole glycosides with free hydroxyl groups (8a-10b) were successfully obtained through the deprotection of acetylated 1,2,3-triazole glycosides (5a-7b) by overnight stirring at room temperature in dry methanol saturated with gaseous ammonia. The IR spectra of these triazole glycosides, which incorporate free hydroxyl groups (8a-10b), showed the disappearance of the characteristic acetyl C = O bands. Instead, strong stretching absorption bands correspond to the sugar moiety’s hydroxyl groups. The 1H NMR spectral data exposed the existence of signals representing the protons of the (OH) groups and the absence of signals that represent the acetyl protons of the protected precursors. The disappearance of the signals corresponding acetyl carbons in the 13C NMR spectra of 8a-10b validated that the chemical structure of series 8a-10b aligns with the structures of free hydroxyl glycosides based on such assignments.

The synthetic route toward the targeted conjugates 2-10b.
Biological activity
In vitro antiproliferative activity
All synthesized analogs 2-10b were assessed via in vitro screening to evaluate their activity against three diverse human cancer cell lines MCF-7 (human breast cancer), HepG-2 (human liver cancer), and HCT-116 (human colorectal cancer), along with a human normal cell line (BJ-1). This screening used the LDH assay methodology with erlotinib and doxorubicin as the standard references. The obtained findings revealed that all tested compounds inhibited the growth of the three cancer cell lines (HCT-116, HepG-2, and MCF-7) in a dose-dependent manner (Figs. S1-S3). Notably, all compounds exhibited potent activity with IC50 values surpassing those of erlotinib on the HepG-2 cell line, however derivative 8b was the best with IC50 = 1.55 ± 0.15 µM compared with IC50 = 2.07 ± 0.07 µM for erlotinib, whereas, against MCF7, the cytotoxic activities of all tested compounds showed a moderate response, though, derivatives 9a and 9b were the most active with IC50 values 1.99 ± 0.11, 2.06 ± 0.13 µM, respectively, comparable to 1.05 ± 0.03 µM for the reference used, Table 1.
Alternatively, except for compounds 6b, 7b and 10a that showed moderate activities on the HCT-116 cell line, the rest analogs demonstrated brilliant potency with IC50 values exceeding that recorded for erlotinib, nevertheless, derivatives 8b, 9a, 9b and 10b exposed the best IC50 values 5.7 ± 0.7, 5.3 ± 0.7, 5.1 ± 0.5 and 5.6 ± 0.3 µM, respectively, in comparison with that of erlotinib 5.14 ± 0.33 µM. It is worth mentioning that compound 9a revealed brilliant activity against HCT-116 with IC50 = 5.00 ± 0.51µM, superior to that recorded by erlotinib, the utilized reference drug (IC50 = 5.14 ± 0.33 µM). On the other hand, when the obtained cytotoxicity results against the tested cancer cell lines compared with that of the normal cell line BJ-1, it may be then concluded that all the tested derivatives displayed a safety profile in comparison to erlotinib, even though derivative 10a was the best, compounds 2-5a, 6b, 8b and 10b showed potential safety profile with IC50 in the range 14.67 ± 2.32–96.3 ± 5.9 µM superior to erlotinib 84 ± 1.97 µM, Fig. S4, Table 1. The results reflected that the cancer cell line most suspected by all tested compounds with observed potency is the HepG-2 cancer cell line. Trying to correlate the structural features with the activity results of the observed LDH assay revealed that the benzimidazolyl-triazole glycosides incorporating the δ,β-unsaturated ketone system attached to the benzimidazole core displayed the best potency on both HCT-116 and MCF-7 cancer cells. While towards the latter cancer cells, the activity was reduced in the same iso-structurally related derivatives. The effect of the included sugar moiety in the 1,2,3-triazole glycosides was also observed since the glycosyl-1,2,3-triazole possessing the galactopyranosyl sugar motif disclosed relatively developed activity than its xylopyranosyl analogue.
EGFR PK Inhibition assay of derivatives (8a, 8b, 9a, 9b, 10a and 10b )
The results obtained from the cytotoxic effects of the novel benzimidazole-glycoside conjugates on the three distinct human cancer cell lines (HepG-2, MCF-7, and HCT-116) revealed that compounds 8a, 8b, 9a, 9b, 10a and 10b exhibited remarkable cytotoxic effects, prompting further investigation. Importantly, these compounds demonstrated a high safety profile when tested on the normal cell line BJ-1, Table 2. Encouraged by these findings, our focus shifted to studying the in vitro inhibition of EGFR kinase by these six compounds, as their exceptional cytotoxic activities on cancer cell lines warrant exploration of their potential anti-cancer effects through EGFR inhibition assays. In light of the extraordinary cytotoxic activities displayed by compounds 8a, 8b, 9a, 9b, 10a and 10b on various cancer cell lines, we aimed to delve deeper into their mechanisms of action. Given their promising performance in cytotoxicity assays and their favorable safety profiles on normal cell lines, we directed our attention toward investigating their potential as inhibitors of EGFR kinase in vitro. By focusing on these six compounds, we seek to elucidate their ability to inhibit EGFR kinase activity, thus shedding light on their potential utility as anti-cancer agents. This strategic approach aims to capitalize on the compounds’ potent cytotoxic effects and explore their therapeutic potential through targeted inhibition of EGFR kinase.
The analysis of the EGFR inhibition results revealed promising outcomes for the six compounds tested, exhibiting significant inhibitory effects within the micromolar range. Notably, compounds 9a and 9b demonstrated the most pronounced inhibitory properties (IC50 = 0.069 ± 0.003 and 0.075 ± 0.003 µM, respectively) compared with the reference standard used erlotinib (IC50: 0.048 ± 0.002 µM). Interestingly, the smaller compounds 8a and 8b exhibited superior inhibitory activity (> 2-fold) compared to the larger molecules 10a and 10b, featuring the pyrazole ring. This observation suggests that the elongated lipophilic α,ß-unsaturated ketone located in 9a and 9b may enhance EGFR inhibition relative to compounds 8a and 8b. However, the further ring closure of α,ß-unsaturated ketones into the pyrazole derivatives 10a and 10b resulted in a substantial decrease in EGFR inhibition by approximately 10-fold, indicating poor tolerance of these larger compounds by the EGFR kinase active site.
Docking studies on EGFR
The binding interactions of the recently developed EGFR inhibitors (8–10) within the EGFR receptor’s binding pocket underwent analysis through molecular docking, alongside comparison to the renowned EGFR inhibitor, erlotinib. The latter operates by blocking EGFR activity, leading to the suppression of cell growth and division. This targeted therapy is commonly administered for cancers propelled by mutations in the EGFR gene, which foster tumor growth52. Erlotinib was selected as the reference compound for evaluating the efficacy of our novel EGFR inhibitors, given its well-established potency as an EGFR inhibitor53.
The EGFR receptor bound to erlotinib (N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)-quinazolin-4-amine) was achieved from the Protein Data Bank (PDB code: 4HJO)54, for docking simulations using Biovia Discovery Studio, water molecules as well as extraneous chains were removed during preparation, except for water molecule 1104, which participates in a bridging interaction between the N1-atom of the quinazoline moiety and the THR766 residue55. Polar hydrogens were incorporated, and partial charges were modified to maintain the accuracy of the receptor. Next, Autodock 4 software was utilized to predict the binding interactions among the newly synthesized EGFR and EGFR inhibitors (8–10)56. This allowed for the calculation of binding energies and offered insights into the positioning of the ligand inside the binding site of the enzyme, thereby improving our understanding of the binding mechanisms.
To verify the precision of the docking procedure, the co-crystallized instinctive ligand was re-docked into the EGFR active sites. The resulting pose closely mirrored the original ligand conformations, demonstrating binding affinities of -13.55 kcal/mol (Table 3). Additionally, the RMSD (root mean square deviation) between the re-docked poses and the original ligands was found to be 0.58, further confirming the reliability of the docking method. This alignment between the docked and native ligands indicates a successful overlap, confirming the validity of the docking protocol. The quinazoline core of erlotinib is responsible for the important interactions that stabilize the molecule into the EGFR kinase active pocket, Fig. 4. The N1- of quinazoline ring exhibited H-bonding with MET769 residue. Whereas, the N3- of quinazoline is engaged with water H-bonding with water molecule HOH1104 which bridges interactions with THR766 residue. Moreover, the 2-methoxy ethoxy chain includes one H-bond with CYS773 residue. The 2-methoxyethoxy chains in erlotinib are very flexible, therefore we noticed some different orientations between the native pose and the docked pose (Fig. 4). The instinctive ligand engages with multiple hydrophobic interactions (van der Waals, alkyl and carbon-hydrogen bonding) by crucial amino acids: LYS721, LEU764, ALA719, THR766, GLN767 and ASP776.

(A) Overlay of the native co-crystallized ligand erlotinib and the docked one into the binding site of EGFR kinase (PDB ID: 4HJO). The carbon atoms of the native ligand are colored in violet, while for the docked one in orange; (B) 2D interactions of docked erlotinib with EGFR kinase.
Compounds 9a and 9b exhibited the highest inhibition of EGFR kinase in this series (IC50 = 0.069 and 0.075 µM) in that order, quite similar to the native ligand erlotinib (IC50 of 0.048 µM). Through analyzing the docking interactions of these two compounds (Table 3), the carbonyl group of the α,ß-unsaturated ketones are involved with H-bonding with THR766 residue. Also, the benzimidazole moiety’s phenyl ring involves Pi-sulfur interaction with CYS773. Interestingly, the key amino acid MET769, which exhibited H-bonding with the quinazoline ring of erlotinib, showed a similar interaction with the triazole moiety of compound 9a. Additionally, MET769 is involved with H-bonding with the glycose moiety of compounds 9a and 9b. The glycoside moiety’s hydroxy groups are engaged in H-bonding with VAL821. Notably, the water residue HOH1104 exhibited a strong H-bond with the triazole moiety of compound 9a, similar to the interaction with the quinazoline moiety of erlotinib. These interactions would emphasize the strong inhibition of compound 9a to EGFR kinase compared with the native ligand, Fig. 5.

The proposed binding interactions (in both 3D and 2D) of compound 9a within the EGFR kinase active site are illustrated. The carbon atoms of compound 9a are depicted in orange.
Compound 9b showed additional interactions that would contribute to its high binding within the EGFR kinase active site. The terminal p-fluorophenyl function is involved with halogen bonding with PHE832 residue. This interaction was not observed with compound 9a, which took a slightly different pose that may have a longer distance from PHE832 residue, thus, it could not form a similar interaction. Additionally, the terminal phenyl ring, in addition to the benzimidazole moiety’s phenyl ring, is involved with Pi-sulfur interactions with MET742 and CSY773, respectively. Additional two H-bonds were also observed between THR830 and ASP831 residues and the N-atom of the benzimidazole moiety, Fig. 6. Compounds 9a and 9b are involved in plentiful hydrophobic interactions for instance van der Waals forces, pi-cation interactions, and carbon-hydrogen bonding through crucial amino acids including LYS721, LEU764, LEU820, ALA719, THR766, GLN767 and ASP776 (Figs. 5 and 6).

2D and 3D proposed binding interactions of compound 9b within the EGFR kinase active site. The carbon atoms of compound 9b are depicted in orange.
Considering the binding poses of compounds 8a,b compared to 9a,b, we propose that compounds 9a,b, which feature a longer aromatic moiety, engage in multiple hydrophobic interactions within the active site. Furthermore, the terminal halide in these series, particularly compound 9b, as depicted in Fig. 6, may participate in halogen bonding interactions with PHE832. These interactions are likely attributable to the heightened inhibitory activity of compounds 9a,b against EGFR kinase. Additionally, compounds 9a,b have higher flexibility than compounds 10a, b, which could contribute to a higher inhibitory effect.
in-silico pharmacokinetic analysis
The physicochemical possessions of the newly synthesized analogs 8a, 8b, 9a, 9b, 10a, and 10b as well as the drug-likeness were assessed using the SwissADME server. This tool provides insights into various in silico parameters, including aqueous solubility, distribution, metabolism, and excretion in humans (Table 4; Fig. 7). Overall, many of these compounds confirmed auspicious drug-like characteristics and adhered well to Lipinski’s Rule of Five, which is a guideline for assessing the compounds’ drug-likeness. However, some compounds exhibited slight deviations from these rules. For instance, the larger compounds 10a and 10b showed a molecular weight slightly exceeding 500 daltons and a marginally higher number of hydrogen bond acceptors. These deviations suggest that while the compounds are generally promising, they may require further optimization to fully comply with drug-likeness criteria.

Physicochemical space for oral availability of compound 8a,b, 9a,b, 10a,b, erlotinib and Doxorubicin. LIPO (Lipophilicity), FLEX (Flexibility), INSOLU (Insolubility), INSATU (Unsaturation), and POLAR (Polarity) are key physicochemical properties. The pink-colored zone represents the optimal physicochemical space for oral bioavailability.
Interestingly, all synthesized compounds exhibited a promising topological polar surface area (TPSA) that surpassed that of the reference compounds erlotinib and Doxorubicin. This high TPSA indicates significant polarity and corresponds to a solubility class ranging from moderately soluble to very soluble. Such properties suggest favorable gastrointestinal absorption for most of the compounds. Moreover, a distinction was observed between the compounds based on their interaction with the CYP450 2A6 isoform (Table 3). The smaller molecules (8a, 8b, 9a, and 9b) were not found to be inhibitors of CYP450 2A6. In contrast, the larger compounds (10a and 10b), which feature a pyrazole core, could potentially inhibit CYP450 2A6, similar to erlotinib and Doxorubicin. Additionally, the compounds generally demonstrated high gastrointestinal (GI) absorption. Most of them are not likely to be significantly effluxed by P-glycoprotein (P-gp), which supports their potential to exert therapeutic effects effectively.
Experimental
Chemistry
All apparatuses data used to elucidate the chemical structure of new compounds is attached in the supplementary file, The azido sugars were prepared as earlier reported57,58. Representative NMR charts are attached in the Supplementary material file (Figures S5-S31).
Synthesis of (E)-3-(4-fluorophenyl)-1-(1-(prop-2-yn-1-yl)-1 H-benzo[d]imidazol-2-yl)prop-2-en-1-one (2)33
The procedure is attached in the supplementary file.
Yield: 78%; m.p. 156–157 °C; IR (KBr) cm− 1, ν: 3342 (C-H alkyne), 2119 (C≡C alkyne), 1749 (C = O), 1597 (C = C); 1H NMR (500 MHz, CDCl3) δ/ppm: 2.35 (t, 1H, J = 2.5 Hz, CH), 5.61 (d, 2 H, CH2) 7.11 (t, 2 H, J = 8.6 Hz, Ar-H), 7.40–7.43 (m, 1H, Ar-H), 7.47–7.51 (m, 1H, Ar-H), 7.60 (d, 1H, J = 8.2 Hz, CH = CH), 7.73 (dd, 2 H, J = 8.6, 5.5 Hz, Ar-H), 7.88–7.95 (m, 2 H, Ar-H), 8.17 (d, 1H, CH = CH); 13C NMR (125 MHz, CDCl3) δ/ppm: 34.98, 73.32, 77.53, 111.09, F-Cortho: 116.25 (d, J = 21.8 Hz); 116.402 and 116.149, 122.22, 122.62, 124.34, 126.57, F-Cmeta: 131.14 (d, J = 8.6 Hz); 131.275 and 130.939, 136.21, 142.00, 143.84, 146.02, F-C: 164.47 (d, J = 252.6 Hz); 165.638 and 163.377, 182.79; Analysis calcd. for C19H13FN2O (304.32): C, 74.99; H, 4.31; N, 9.21. Found: C, 75.07; H, 4.25; N, 9.28%.
Synthesis of 1-(3-(4-fluorophenyl)-5-(1-(prop-2-yn-1-yl)-1 H-benzo[d]imidazol-2-yl)-4,5-dihydro-1 H-pyrazol-1-yl)ethan-1-one (3)
The procedure was attached in the supplementary file.
Yield: 88%; m.p. 171–173 °C; IR (KBr) cm− 1, ν: 3329 (C-H alkyne), 2122 (C≡C alkyne), 1665 (C = O); 1H NMR (500 MHz, CDCl3) δ/ppm: 2.38 (s, 3 H, CH3CO), 3.40 (t, 1H, J = 2.3 Hz, CH), 4.03 (dd, 1H, J = 6.8, 8.2 Hz, pyrazoly-H4a), 4.06 (dd, 1H, J = 6.2, 8.2 Hz, pyrazoly-H4b), 5.54–5.66 (m, 3 H, pyrazolyl-H5, CH2), 7.16 (t, 2 H, J = 8.9 Hz, Ar-H), 7.33–7.39 (m, 4 H, Ar-H), 7.42 (t, 1H, J = 7.7 Hz, Ar-H), 7.75 (dd, 1H, J = 9.8, 8.2 Hz, Ar-H); 13C NMR (125 MHz, DMSO) δ/ppm: 21.68, 34.80, 43.39, 58.29, 74.96, 78.61, 110.79, F-Cortho: 115.40 (d, J = 21.6 Hz); 115.948 and 115.014, 119.91, 122.99, 124.53, F-Cmeta: 127.76 (d, J = 8.2 Hz); 127.956 and 127.493, 135.67, 138.02, 142.17, 143.49, 146.98, F-C: 161.37 (d, J = 243.1 Hz); 162.507 and 160.194, 168.06; Analysis calcd. for C21H17FN4O (360.39): C, 69.99; H, 4.75; N, 15.55. Found: C, 70.04; H, 4.85; N, 15.47%.
General procedure for the synthesis of compounds 5a-7b.
The procedure was attached in the supplementary file.
1-(1-((1-[2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)ethan-1-one (5a)
Yield: 65%; m.p. 140–142 °C; IR (KBr) cm− 1, ν: 3142 (C-H), 2120 (N = N = N), 1751 (C = O), 1687 (C = O); 1H NMR (500 MHz, CDCl3) δ/ppm: 1.73, 2.02, 2.08, 2.22, 2.89 (5s, 15 H, CH3CO), 4.09, 4.12 (dd, 1H, J = 3.2, 11.2 Hz, H-6′′), 4.15, 4.19 (dd, 1H, J = 2.8, 11.2 Hz, H-6’), 4.23–4.27 (m, 1H, H-5′), 5.01–5.07 (m, 1H, H-4′), 5.17, 5.19 (dd, 1H, J = 8.6, 10.2 Hz, H-2′), 5.51 (s, 2 H, CH2), 5.75 (t, 1H, J = 8.8 Hz, H-3′), 5.90 (d, 1H, J = 10.2 Hz, H-1′), 7.42 (d, 1H, J = 8.4 Hz, Ar-H), 7.44 (m, 2 H, Ar-H), 7.81 (d, 1H, J = 8.6 Ar-H), 7.88 (s, 1H, triazole-H); 13C NMR (125 MHz, CDCl3) δ/ppm: δ 20.09, 20.64, 28.14, 40.55, 61.22, 66.90, 67.84, 70.76, 74.25, 76.91, 77.16, 77.42, 86.38, 111.72, 121.89, 124.22, 126.57, 134.00, 137.03, 140.62, 142.38, 143.87, 145.17, 169.13, 170.03, 170.34, 194.22; Analysis calcd. for C26H29N5O10 (571.54): C, 54.64; H, 5.11; N, 12.25. Found: C, 54.58; H, 4.99; N, 12.35%.
1-(1-((1-[2,3,4-tri-O-acetyl-β-D-xylopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)ethan-1-one (5b)
Yield: 61%; m.p. 177–178 °C; IR (KBr) cm− 1, ν: 3140 (C-H), 2119 (N = N = N), 1752 (C = O), 1686 (C = O); 1H NMR (500 MHz, CDCl3) δ/ppm: 1.70, 1.99, 2.02, 2.86 (4s, 12 H, CH3CO), 4.20, 4.22 (dd, 1H, J = 3.4, 10.8 Hz, H-5′′), 5.07–5.11 (m, 1H, H-5′), 5.29–5.33 (m, 1H, H-4′), 5.35 (s, 2 H, CH2), 5.65, 5.68 (dd, 1H, J = 8.6, 9.8 Hz, Hz, H-2′), 5.82 (t, 1H, J = 8.8 Hz, H-3′), 5.87 (d, 1H, J = 9.8 Hz, H-1′), 7.33–7.39 (m, 4 H, Ar-H), 7.87 (s, 1H, triazole-H); 13C NMR (125 MHz, CDCl3) δ/ppm: 19.93, 20.52, 27.96, 40.48, 65.47, 68.33, 70.36, 71.93, 86.31, 111.57, 121.80, 124.00, 126.44, 131.93, 137.83, 141.52, 149.50, 168.70, 169.62, 169.76, 193.55; Analysis calcd. for C23H25N5O8 (499.48): C, 55.31; H, 5.05; N, 14.02. Found: C, 55.40; H, 4.92; N, 13.97%.
(E)-1-(1-((1-[2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)prop-2-en-1-one (6a)
Yield: 71%; m.p. 178–180 °C; IR (KBr) cm− 1, ν: 2962 (C-H), 2118 (N = N = N), 1754 (C = O), 1657 (C = O), 1596 (C = C); 1H NMR (500 MHz, CDCl3) δ/ppm: 1.73, 1.97, 1.99, 2.20 (4s, 12 H, CH3CO), 4.13–4.16 (m, 3 H, H-6′′, H-6’, H-5′), 5.03 (d, 1H, H-4′), 5.19 (d, 1H, H-2′), 5.50 (s, 2 H, CH2), 5.76 (d, 1H, J = 8.7 Hz, H-3′), 5.95 (d, 1H, H-1′), 6.09 (d, 1H, J = 7.0 Hz, CH = CH), 7.09–7.14 (m, 4 H, Ar-H), 7.43 (dd, 4 H, Ar-H), 7.75 (s, 1H, triazole-H), 7.94 (d, 1H,, J = 7.0 Hz, CH = CH); 13C NMR (125 MHz, CDCl3) δ/ppm: 20.07, 20.46, 20.61, 20.69, 26.95, 40.75, 61.20, 61.29, 66.89, 67.83, 68.17, 70.80, 74.21, 86.39, 111.77, F-Cortho: 116.21 (d, J = 21.8 Hz); 116.513 and 116.086, 121.87, 122.80, 124.23, 126.55, F-Cmeta: 131.05 (d, J = 8.5 Hz); 131.369 and 130.801, 135.45, 137.95, 141.91, 143.60, F-C: 164.51 (d, J = 252.8 Hz); 165.891 and 163.523, 169.78, 170.06, 170.31, 182.43; Analysis calcd. for C33H32FN5O10 (677.64): C, 58.49; H, 4.76; N, 10.34. Found: C, 58.41; H, 4.80; N, 10.23%.
(E)-1-(1-((1-[2,3,4-tri-O-acetyl-β-D-xylopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)prop-2-en-1-one (6b)
Yield: 70%; m.p. 210–212 °C; IR (KBr) cm− 1, ν: 2956 (C-H), 2115 (N = N = N), 1750 (C = O), 1663 (C = O), 1597 (C = C); 1H NMR (500 MHz, CDCl3) δ/ppm: 1.73, 2.01, 2.04 (3s, 9 H, CH3CO), 3.53 (dd, 1H, J = 3.7, 10.2 Hz, H-5′′), 3.73–3.75 (m, 1H, H-5′), 4.23 (d, 1H, J = 7.0 Hz, H-4′), 5.10 (dd, 1H, J = 7.9, 9.8 Hz, H-2′), 5.38 (s, 2 H, CH2), 5.70 (t, 1H, J = 9.2 Hz, H-3′), 6.06 (d, 1H, J = 9.8 Hz, H-1′), 7.12–7.25 (m, 5 H, Ar-H, CH = CH), 7.27–7.47 (dd, 4 H, Ar-H), 7.87–7.89 (m, 2 H, triazole-H, CH = CH); 13C NMR (125 MHz, CDCl3) δ/ppm: 20.12, 20.63, 20.67, 41.02, 65.66, 68.45, 70.54, 72.06, 86.55, 111.57, F-Cortho: 116.30 (d, J = 22.0 Hz); 116.689 and 116.082, 121.39, 122.23, 124.67, 126.96, 130.99, F-Cmeta: 131.37 (d, J = 9.3 Hz); 131.714 and 131.177, 144.43, F-C: 164.58 (d, J = 252.7 Hz); 165.772 and 163.499, 168.84, 169.76, 169.91, 182.78; Analysis calcd. for C30H28FN5O8 (605.58): C, 59.50; H, 4.66; N, 11.56. Found: C, 59.42; H, 4.56; N, 11.48%.
1-(5-(1-((1-[2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)-4,5-dihydro-1 H-pyrazol-1-yl)ethan-1-one (7a)
Yield: 80%; m.p. 138–140 °C; IR (KBr) cm− 1, ν: 3095 (C-H), 2119 (N = N = N), 1754 (C = O), 1665 (C = O), 1511 (C = C); 1H NMR (500 MHz, CDCl3) δ/ppm: 1.77, 1.99, 2.00, 2.10, 2.37 (5s, 15 H, CH3CO), 4.06–4.19 (m, 4 H, CH2-diazole, H-6′′, H-6’), 5.22 (s, 1H, CH-diazole), 5.31–5.39 (m, 1H, H-5′), 5.50 (s, 2 H, CH2), 5.77 (d, 1H, J = 9.4 Hz, H-4′), 5.92 (m, 1H, H-2′), 5.94 (t, 1H, J = 16.0 Hz, H-3′), 6.17 (d, 1H, J = 10.2 Hz, H-1′), 7.01 (d, 2 H, J = 8.4 Hz, Ar-H), 7.38–7.44 (m, 4 H, Ar-H), 7.53–7.61 (m, 2 H, Ar-H), 7.81 (s, 1H, triazole-H); 13C NMR (125 MHz, CDCl3) δ/ppm: 20.10, 20.60, 22.11, 41.15, 43.61, 58.91, 61.13, 66.79, 67.99, 70.57, 74.27, 86.42, 110.43, F-Cortho: 115.83 (d, J = 21.7 Hz); 116.431 and 115.367, 120.29, 120.57, 123.62, 125.29, F-Cmeta: 127.72 (d, J = 8.0 Hz); 128.850 and 126.567, 136.57, 137.99, 142.69, 144.11, 145.53, 146.61, 151.66, F-C: 163.11 (d, J = 246.0 Hz); 163.997 and 160.916, 168.97, 169.97, 170.31; Analysis calcd. for C35H36FN7O10 (733.71): C, 57.30; H, 4.95; N, 13.36. Found: C, 57.26; H, 5.01; N, 13.28%.
1-(5-(1-((1-[2,3,4-tri-O-acetyl-β-D-xylopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)-4,5-dihydro-1 H-pyrazol-1-yl)ethan-1-one (7b)
Yield: 79%; m.p. 160–162 °C; IR (KBr) cm− 1, ν: 3079 (C-H), 2120 (N = N = N), 1756 (C = O), 1672 (C = O), 1513 (C = C); 1H NMR (500 MHz, CDCl3) δ/ppm: 1.78, 2.06, 2.37 (4s, 12 H, CH3CO), 3.57 (s, 2 H, CH2-diazole), 4.23 (dd, 2 H, J = 3.5, 10.4 Hz, H-5′′, H-5′), 5.06–5.12 (d, 1H, J = 7.1 Hz, H-4′), 5.17–5.26 (dd, 1H, J = 8.2, 9.5 Hz, H-2′), 5.41–5.75 (m, 4 H, pyrazolyl-H, H-3′, CH2), 6.09 (d, 1H, J = 9.8 Hz, H-1′), 7.03-7-90 (m, 2 H, Ar-H), 7.26–7.41 (m, 4 H, Ar-H), 7.54 (m, 2 H, Ar-H), 7.84 (s, 1H, triazole-H); 13C NMR (125 MHz, CDCl3) δ/ppm: 19.88, 20.42, 20.47, 22.11, 41.08, 43.41, 58.74, 65.43, 68.23, 70.48, 71.72, 86.33, 110.24, F-Cortho: 115.65 (d, J = 21.7 Hz); 115.902 and 115.228, 120.46, 123.42, 125.09, F-Cmeta: 127.62 (d, J = 8.0 Hz); 127.950 and 127.343, 136.08, 136.97, 142.71, 143.49, 147.71, F-C: 162.14 (d, J = 246.0 Hz); 163.339 and 161.093, 168.68, 168.91, 169.57, 169.65; Analysis calcd. for C32H32FN7O8 (661.65): C, 58.09; H, 4.88; N, 14.82. Found: C, 57.99; H, 4.81; N, 14.75%.
General procedure for the synthesis of compounds 8a-10b.
Procedure was attached in the supplementary file.
1-(1-((1-[β-D-galactopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)ethan-1-one (8a)
Yield: 55%; m.p. 162–164 °C; IR (KBr) cm− 1, ν: 3431 (OH), 2918 (C-H), 2122 (N = N = N), 1679 (C = O); 1H NMR (500 MHz, DMSO) δ/ppm: 2.77 (s, 3 H, CH3), 3.44 (d, 1H, J = 6.6 Hz, H-6′′), 3.48–3.50 (m, 2 H, H-6’, H-5′), 3.64–3.72 (m, 2 H, H-4′, H-3′), 3.96 (t, 1H, J = 9.3 Hz, H-2′), 4.63–4.67 (m, 2 H, 2OH), 5.00-5.04 (m, 1H, OH), 5.19–5.20 (m, 1H, OH), 5.43 (d, 1H, J = 9.8 Hz, H-1′), 5.90 (s, 2 H, CH2), 7.37 (t, 1H, J = 7.7 Hz, Ar-H), 7.47 (t, 1H, J = 7.7 Hz, Ar-H), 7.85 (d, 2 H, J = 6.3 Hz, Ar-H), 8.16 (s, 1H, triazole-H); 13C NMR (125 MHz, DMSO) δ/ppm: 27.99, 40.12, 60.53, 68.56, 69.43, 73.75, 78.57, 88.23, 112.14, 121.36, 122.32, 123.85, 126.03, 136.10, 141.11, 142.95, 145.49, 193.05; Analysis calcd. for C18H21N5O6 (403.40): C, 53.59; H, 5.25; N, 17.36. Found: C, 53.67; H, 5.18; N, 17.45%.
1-(1-((1-[β-D-Xylopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)ethan-1-one (8b)
Yield: 52%; m.p. 130–132 °C; IR (KBr) cm− 1, ν: 3462 (OH), 2918 (C-H), 2123 (N = N = N), 1675 (C = O); 1H NMR (500 MHz, DMSO) δ /ppm: 2.76 (s, 3 H, CH3), 3.24–3.29 (m, 2 H, H-5′′, H-5′), 3.42–3.43 (m, 1H, H-4′), 3.68–3.71 (m, 2 H, H-3′, H-2′), 5.13–5.14 (m, 1H, OH), 5.25–5.26 (m, 1H, OH), 5.33–5.34 (m, 1H, OH), 5.42 (d, 1H, J = 9.6 Hz, H-1′), 5.91 (s, 2 H, CH2), 7.37 (t, 1H, J = 7.6 Hz, Ar-H), 7.46 (t, 1H, J = 7.6 Hz, Ar-H), 7.83 (dd, 2 H, J = 16.5, 8.3 Hz, Ar-H), 8.18 (s, 1H, triazole-H); 13C NMR (125 MHz, DMSO) δ/ppm: 27.95, 40.14, 68.33, 69.13, 71.92, 77.10, 88.17, 112.09, 121.31, 122.66, 123.77, 125.97, 136.07, 141.08, 142.88, 145.41, 192.91; Analysis calcd. for C17H19N5O5 (373.37): C, 54.69; H, 5.13; N, 18.76. Found: C, 54.77; H, 5.03; N, 18.81%.
(E)-1-(1-((1-[β-D-galactopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)prop-2-en-1-one (9a)
Yield: 59%; m.p. 190–192 °C; IR (KBr) cm− 1, ν: 3431 (OH), 2918 (C-H), 2125 (N = N = N), 1650 (C = O), 1583 (C = C); 1H NMR (500 MHz, DMSO) δ/ppm: 3.43–3.50 (m, 3 H, H-6′′, H-6’, H-5′), 3.66 (t, 1H, H-4′), 3.72 (d, 1H, H-3′), 3.96 (t, 1H, J = 9.3 Hz, H-2′), 4.62–4.66 (m, 2 H, 2OH), 4.97-5.00 (m, 1H, OH), 5.17–5.18 (m, 1H, OH), 5.44 (d, 1H, J = 10.4 Hz, H-1′), 6.02 (s, 2 H, CH2), 7.34 (d, 2 H, J = 7.5 Hz, Ar-H), 7.41 (d, 1H, J = 7.1 Hz, CH = CH), 7.49–7.52 (m, 1H, Ar-H), 7.89–791 (m, 2 H, Ar-H), 7.94–798 (m, 3 H, Ar-H), 8.17 (d, 1H, J = 7.3 Hz, CH = CH), 8.21 (s, 1H, triazole-H); 13C NMR (125 MHz, DMSO) δ/ppm: 40.21, 60.47, 68.51, 69.40, 73.71, 78.54, 88.20, 112.12, F-Cortho: 116.22 (d, J = 22.1 Hz); 116.563 and 115.697, 121.37, 122.27, 122.82, 123.93, 126.13, F-Cmeta: 131.37 (d, J = 8.9 Hz); 131.539 and 131.249, 136.25, 141.28, 142.96, 146.21, F-C: 163.72 (d, J = 249.0 Hz); 165.319 and 161.845, 182.09; Analysis calcd. for C25H24FN5O6 (509.49): C, 58.94; H, 4.75; N, 13.75. Found: C, 59.00; H, 4.69; N, 13.85%.
(E)-1-(1-((1-[β-D-Xylopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)prop-2-en-1-one (9b).
Yield: 60%; m.p. 165–167 °C; IR (KBr) cm− 1, ν: 3431 (OH), 2922 (C-H), 2111 (N = N = N), 1639 (C = O), 1579 (C = C); 1H NMR (500 MHz, CDCl3) δ/ppm: 3.26–3.32 (m, 2 H, H-5′′, H-5’), 3.36–3.46 (m, 1H, H-4′), 3.77–3.82 (m, 2 H, H-3′, H-2′), 5.16–5.18 (m, 1H, OH), 5.27–5.32 (m, 1H, OH), 5.40 (s, 2 H, CH2), 5.51–5.58 (m, 1H, OH), 5.95 (d, 1H, J = 9.8 Hz, H-1′), 7.20 (d, 1H, J = 7.1 Hz, CH = CH), 7.33–7.50 (m, 4 H, Ar-H), 7.69–7.89 (m, 3 H, Ar-H), 8.09–8.19 (m, 1H, Ar-H), 8.24 (d, 1H, J = 7.4 Hz, CH = CH), 8.34 (s, 1H, triazole-H); 13C NMR (125 MHz, DMSO) δ/ppm: 40.00, 68.35, 69.12, 71.97, 77.08, 88.14, 111.41, 114.92, F-Cortho: 116.20 (d, J = 22.1 Hz); 116.761 and 115.684, 118.70, 119.64, 122.66, 127.23, 127.45, 128.13, 129.45, F-Cmeta: 131.31 (d, J = 8.9 Hz); 131.831 and 131.345, 136.26, 137.31, 141.35, F-C: 163.68 (d, J = 249.0 Hz); 165.413 and 161.941, 182.23; Analysis calcd. for C24H22FN5O5 (479.47): C, 60.12; H, 4.63; N, 14.61. Found: C, 60.01; H, 4.55; N, 14.53%.
1-(5-(1-((1-[β-D-galactopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)-4,5-dihydro-1 H-pyrazol-1-yl)ethan-1-one (10a)
Yield: 53%; m.p. 164–165 °C; IR (KBr) cm− 1, ν: 3431 (OH), 2918 (C-H), 2125 (N = N = N), 1650 (C = O); 1H NMR (500 MHz, DMSO) δ/ppm: 2.29 (s, 3 H, CH3), 3.22–3.27 (m, 1H, H-6′′), 3.47–3.51 (m, 2 H, H-6’, H-5′), 3.65–3.73 (m, 2 H, H-4′, H-3′), 3.98–4.09 (m, 2 H, 2OH), 4.64 (d, 2 H, CH2-diazole), 5.02–5.07 (m, 1H, OH), 5.18–5.20 (m, 1H, OH), 5.47 (t, 1H, CH-diazole), 5.57 (s, 2 H, CH2), 5.98-6.00 (m, 1H, H-2′), 6.11 (d, 1H, J = 10.2 Hz, H-1′), 7.15–7.18 (m, 2 H, Ar-H), 7.27–7.33 (m, 3 H, Ar-H), 7.37–7.40 (m, 1H, Ar-H), 7.71–7.72 (m, 1H, Ar-H), 7.82–7.83 (m, 1H, Ar-H), 8.15 (s, 1H, triazole-H); 13C NMR (125 MHz, DMSO) δ/ppm: 22.00, 43.69, 58.25, 60.43, 68.46, 69.37, 73.78, 78.50, 88.15, 111.23, F-Cortho: 115.47 (d, J = 22.4 Hz); 115.901 and 114.851, 119.84, 122.01, 122.92, 124.48, F-Cmeta: 127.83 (d, J = 7.7 Hz); 128.087 and 127.673, 136.19, 138.14, 142.31, 143.02, 143.75, 147.38, F-C: 161.65 (d, J = 221.9 Hz); 163.340 and 158.781, 168.24; Analysis calcd. for C27H28FN7O6 (565.56): C, 57.34; H, 4.99; N, 17.34. Found: C, 57.25; H, 5.05; N, 17.26%.
1-(5-(1-((1-[β-D-Xylopyranosyl]-1 H-1,2,3-triazol-4-yl)methyl)-1 H-benzo[d]imidazol-2-yl)-3-(4-fluorophenyl)-4,5-dihydro-1 H-pyrazol-1-yl)ethan-1-one (10b)
Yield: 51%; m.p. 153–155 °C; IR (KBr) cm− 1, ν: 3431 (OH), 2922 (C-H), 2211 (N = N = N), 1639 (C = O); 1H NMR (500 MHz, DMSO) δ/ppm: 2.28 (s, 3 H, CH3), 3.21–3.26 (m, 2 H, H-5′′, H-5′ ), 3.43–3.49 (m, 2 H, H-4′, H-3′), 3.76 (d, 2 H, CH2-diazole), 4.05 (t, 1H, CH-diazole), 5.15–5.16 (m, 1H, OH), 5.35 (s, 2 H, CH2), 5.44–5.46 (m, 1H, OH), 5.53–5.56 (m, 1H, OH), 6.00 (d, 1H, H-2′), 6.10 (d, 1H, J = 9.6 Hz, H-1′), 7.15–7.16 (m, 2 H, Ar-H), 7.28–7.38 (m, 4 H, Ar-H), 7.70–7.79 (m, 2 H, Ar-H), 8.19 (s, 1H, triazole-H); 13C NMR (125 MHz, DMSO) δ/ppm: 21.94, 40.01, 43.62, 58.18, 68.31, 69.09, 71.83, 77.11, 88.09, 111.20, 115.32, F-Cortho: 115.41 (d, J = 21.6 Hz); 116.039 and 114.801, 119.78, 122.26, 122.36, 122.85, 124.40, F-Cmeta: 127.82 (d, J = 7.7 Hz); 128.401 and 127.182, 136.16, 138.11, 142.28, 142.93, 143.69, 147.32, F-C: 161.39 (d, J = 242.3 Hz); 162.523 and 160.201, 168.18; Analysis calcd. for C26H26FN7O5 (535.54): C, 58.31; H, 4.89; N, 18.31. Found: C, 58.27; H, 4.93; N, 18.26%.
In vitro anticancer activity
Materials and methods
All data on the materials used are attached in the supplementary file.
Cell culture
All cell lines were purchased from the American Type Culture Collection (Rockville, MD, USA), and details were mentioned in supp. Inf. File.
Lactate dehydrogenase (LDH) assay
The lactate dehydrogenase (LDH) release assay was used to regulate the influence of every synthesized analog against membrane permeability in HepG-2, MCF-7 and HCT-116 cancer cell lines along with normal cell line (BJ-1)59,60, . The procedure is displayed briefly in the Supp. inf. file.
Docking studies on EGFR
For more details, check the supp. File.
Conclusion
The quest for novel EGFR inhibitors remains crucial in cancer research due to the significance of EGFR in promoting tumor growth and progression. In this pursuit, a series of glycosyl heterocyclic scaffolds (5a-10b) have been designed and synthesized, originating from 2-acetyl-1H-benzimidazole as a precursor of propargyl-derived nucleobases (2) and (3). Targeted 1,2,3-triazole glycosides 8a-10b bearing unprotected hydroxyl groups were generated through diverse glycol linkages. The new chemical entities were then subjected to cytotoxicity assessment against three various cancer cell lines such as HepG-2, MCF-7, and HCT-116, alongside a human normal cell line (BJ-1), utilizing the methodology of LDH assay. Among the tested series, derivatives 6b, 7b, 8b, 9a, 9b, 10a and 10b displayed potent activity, with compound 9a exhibiting the most promising efficacy against all targeted cell lines. Additionally, compound 9a demonstrated a favorable safety profile against BJ-1. Furthermore, compounds 8a-10b were evaluated for their inhibitory potential against EGFR compared to erlotinib, revealing that derivatives 9a and 9b are notable candidates with remarkable inhibitory effects, supported by molecular docking studies. These findings emphasize the potential of derivatives 9a and 9b as promising anti-cancer agents targeting EGFR kinase.
Data availability
All data generated or analyzed during this study are included in the manuscript and its supplementary information file.
5. References
dos Anjos, J. V. et al. Synthesis and cytotoxic profile of glycosyl-triazole linked to 1,2,4-oxadiazole moiety at C-5 through a straight-chain carbon and oxygen atoms. Eur. J. Med. Chem. 44 (9), 3571–3576 (2009).
Elganzory, H. H. et al. Design, synthesis, anticancer activity and molecular Docking of new 1,2,3-Triazole-Based glycosides bearing 1,3,4-Thiadiazolyl, Indolyl and arylacetamide scaffolds. Molecules 27 (20), 6960 (2022).
El-Sofany, W. I., El-sayed, W. A., Abd-Rabou, A. A. & El-Shahat, M. Synthesis of new imidazole-triazole-glycoside hybrids as anti-breast cancer candidates. J. Mol. Struct. 1270, 133942 (2022).
Jiang, N. et al. Synthesis and biological evaluation of novel 2-(2-arylmethylene)hydrazinyl-4-aminoquinazoline derivatives as potent antitumor agents. Eur. J. Med. Chem. 54, 534–541 (2012).
Mishra, B. B. & Tiwari, V. K. Natural products: an evolving role in future drug discovery. Eur. J. Med. Chem. 46 (10), 4769–4807 (2011).
Patil, D. Y. Click reactions and applications of Triazole in glycoscience: A review. Pharm. Reson. 3, 53–57 (2021).
Srour, A., Ahmed, N., El-Karim, A., Anwar, S. & El-Hallouty, M. Design, synthesis, biological evaluation, QSAR analysis and molecular modelling of new thiazol-benzimidazoles as EGFR inhibitors. Bioorg. Med. Chem. 28, 115657 (2020).
Debasis, D. et al. Discovery of new Quinazoline derivatives as irreversible dual EGFR/HER2 inhibitors and their anticancer activities – Part 1. Bioorg. Med. Chem. Lett. 29, 591–596 (2019).
Yarden, Y. & Pines, G. The ERBB network: at last, cancer therapy meets systems biology. Nat. Rev. Cancer. 12, 553–563 (2012).
Siegelin, M. D. & Borczuk, A. C. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab. Invest. 94, 129–137 (2014).
Shigeta, K. et al. Expression of epidermal growth factor receptor detected by cetuximab indicates its efficacy to inhibit in vitro and in vivo proliferation of colorectal cancer cells. PLoS One. 8 (6), e66302 (2013).
Garcia, R., Franklin, R. A. & McCubrey, J. A. Cell death of MCF-7 human breast cancer cells induced by EGFR activation in the absence of other growth factors. Cell. Cycle. 5 (16), 1840–1846 (2006).
Liu, Y. et al. Regulation of the EGFR/ErbB signaling by clathrin in response to various ligands in hepatocellular carcinoma cell lines. J. Cell. Mol. Med. 24 (14), 8091–8102 (2020).
Takahara, Y. et al. Early recurrence factors in patients with stage III non-small cell lung cancer treated with concurrent chemoradiotherapy. Thorac. Cancer. 13 (24), 3451–3458 (2022).
Kujtan, L. & Subramanian, J. Epidermal growth factor receptor tyrosine kinase inhibitors for the treatment of non-small cell lung cancer. Expert Rev. Anticancer Ther. 19, 547–559 (2019).
Zhanga, L. & Design Synthesis and biological evaluation of a novel series of Indole-3-Carboxamide derivatives for cancer treatment as EGFR inhibitors. Lett. Drug Des. Discov. 15, 70–83 (2018).
Manoharan, A. et al. Click chemistry: an emerging tool for developing a new class of structural motifs against various neurodegenerative disorders. ACS Omega. 8 (47), 44437–44457 (2023).
Kolb, H. C., Finn, M. G. & Sharpless, K. B. Click chemistry: diverse chemical function from a few good reactions. Angew Chem. Int. Ed. Engl. 40, 2004–2021 (2001).
Geng, Z., Shin, J. J., Xi, Y. & Hawker, C. J. Click chemistry strategies for the accelerated synthesis of functional macromolecules. J. Polym. Sci. 59 (11), 963–1042 (2021).
Hein, C. D., Liu, X. & Wang, D. Click chemistry, a powerful tool for pharmaceutical sciences. Pharm. Res. 25, 2216–2230 (2008).
Lokesh, K., Kashmiri, L., Ashwani, K., Avijit, K. P. & Anil, K. Pyrazoline tethered 1,2,3-triazoles: synthesis, antimicrobial evaluation and in Silico studies. J. Mol. Struc. 1246, 131154 (2021).
Tamer, M., Hany, F. N., Amira, A. E. S., Bahaa, A. H. & Wael, A. E. Design, synthesis, and antimicrobial activities of 1,2,3-Triazole glycoside clickamers. Molecules 25 (4), 790 (2020).
Li, L. T. et al. Facile synthesis of 1,2,3-triazole analogs of SGLT2 inhibitors by ‘click chemistry’. Bioorg. Med. Chem. Lett. 22 (1), 642–644 (2012).
Kumar, K. K., Kumar, R. M., Subramanian, V. & Das, T. M. Expedient synthesis of coumarin-coupled Triazoles via ‘click chemistry’ leading to the formation of coumarin-triazole-sugar hybrids. Carbohydr. Res. 345 (16), 2297–2304 (2010).
Olga, V. A. et al. Glycosides and glycoconjugates of the diterpenoid isosteviol with a 1,2,3-Triazolyl moiety: synthesis and cytotoxicity evaluation. J. Nat. Prod. 83 (8), 2367–2380 (2020).
Mohamed, A. M. et al. Anticancer activity of newly synthesized Triazolopyrimidine derivatives and their nucleoside analogs. Acta Pol. Pharm. Drug Res. 72, 307–318 (2015).
Srour, A. M., El-Bayaa, M. N., Omran, M. M., Sharaky, M. M. & El-Sayed, W. A. Synthesis and cytotoxic properties of new substituted Glycosides-Indole conjugates as apoptosis inducers in cancer cells. Anti-cancer Agents Med. Chem. 21, 1323–1333 (2021).
Halay, E., Ay, E., ¸Salva, E., Ay, K. & Karayıldırım, T. Syntheses of 1,2,3-triazolebridged pyranose sugars with purine and pyrimidine nucleobases and evaluation of their anticancer potential. Nucleosides Nucleotides Nucleic Acids. 36, 598–619 (2017).
Wolle, D. et al. Inhibition of epidermal growth factor signaling by the cardiac glycoside Ouabain in Medulloblastoma. Cancer Med. 3 (5), 1146–1158 (2014).
Seo, Y. H. et al. New sesquiterpene glycosides from the flowers of aster koraiensis and their Inhibition activities on EGF- and TPA-Induced cell transformation. Plants 12, 1726 (2023).
Tuan, H. L. et al. Degalactotigonin, a steroidal glycoside from solanum nigrum, induces apoptosis and cell cycle arrest via inhibiting the EGFR signaling pathways in pancreatic cancer cells. Biomed. Res. Int. 3120972. (2018).
Nofal, Z. M. et al. Novel benzimidazole derivatives as expected anticancer agents. Actapol Pharm. -drug Res. 68, 519–534 (2011).
Nofal, Z. M., Srour, A. M., El-Eraky, W. I. & Saleh, D. O. Girgis rational design, synthesis and QSAR study of vasorelaxant active 3-pyridinecarbonitriles incorporating 1H-benzimidazol-2-yl function. Eur. J. Med. Chem. 63, 14–21 (2013).
Yadav, S., Narasimhan, B. & Kaur, H. Perspectives of benzimidazole derivatives as anticancer agents in the new era. Anticancer Agents Med. Chem. 16 (11), 1403–1425 (2016).
Kalaria, P. N., Satasia, S. P., Avalani, J. R. & Raval, D. K. Ultrasound-assisted one-pot four-component synthesis of novel 2-amino-3-cyanopyridine derivatives bearing 5-imidazopyrazole scaffold and their biological broadcast. Eur. J. Med. Chem. 83, 655–664 (2014).
Ashok, D. et al. Microwave-assisted synthesis and in vitro antiproliferative activity of some novel 1,2,3-triazole-based pyrazole aldehydes and their benzimidazole derivatives. Med. Chem. Res. 29, 699–706 (2020).
Xu, G. Q. et al. Novel 1,2,3-Triazole erlotinib derivatives as potent IDO1 inhibitors: design, Drug-Target interactions prediction, synthesis, biological evaluation, molecular Docking and ADME properties studies. Front. Pharmacol. 13, 854965. https://doi.org/10.3389/fphar.2022.854965 (2022).
Rakesh, R. S. E. Sirassu Narsimha Dr. Synthesis and biological evaluation of benzo[d]thiazolyl-Sulfonyl-Benzo[4,5]isothiazolo[2,3-c][1,2,3]triazole derivatives as EGFR targeting anticancer agents. ChemistrySelect. ;8(6):e202204256. (2023). https://doi.org/10.1002/slct.202204256
Udayasree, N., Haridasyam, R. B., Palabindela, R., Krishna, T. M. & Narsimha, S. One-pot synthesis, anticancer, EGFR and caspases assays of novel fused [1,2,3]triazolo-pyrrolo[2,1-b]quinazolinones. J. Mol. Struct. 1320, 139570. https://doi.org/10.1016/j.molstruc.2024.139570 (2025).
Bandi, S. R. et al. Synthesis of novel pyrido[2,3-d]pyrimidine-thiazolidine-1,2,3-triazoles: potent EGFR targeting anticancer agents. J. Mol. Struct. 136451. https://doi.org/10.1016/j.molstruc.2023.136451 (2023).
Chu, B. et al. A benzimidazole derivative exhibiting antitumor activity blocks EGFR and HER2 activity and upregulates DR5 in breast cancer cells. Cell. Death Dis. 6 (3), e1686. https://doi.org/10.1038/cddis.2015.25 (2015).
Hagar, F. F., Abbas, S. H., Atef, E., Abdelhamid, D. & Abdel-Aziz, M. Benzimidazole scaffold as a potent anticancer agent with different mechanisms of action (2016–2023). Mol. Divers. (2), 1821–1849. https://doi.org/10.1007/s11030-024-10907-8 (2025).
Mirgany, T. O., Rahman, A. F. M. M. & Alanazi, M. M. Design, synthesis, and mechanistic evaluation of novel benzimidazole-hydrazone compounds as dual inhibitors of EGFR and HER2: promising candidates for anticancer therapy. J. Mol. Struct. 138177. https://doi.org/10.1016/j.molstruc.2024.138177 (2024).
Hashem, H. E., Amr, A. E. E., Nossier, E. S., Anwar, M. M. & Azmy, E. M. New Benzimidazole-, 1,2,4-Triazole-, and 1,3,5-Triazine-Based derivatives as potential EGFRWT and EGFRT790M inhibitors: Microwave-Assisted synthesis, anticancer evaluation, and molecular Docking study. ACS Omega. 7 (8), 7155–7171. https://doi.org/10.1021/acsomega.1c06836 (2022).
Ahmed, A. A., Mohammed, A. F., Almarhoon, Z. M., Bräse, S. & Youssif, B. G. Design, synthesis, and apoptotic antiproliferative action of new benzimidazole/1, 2, 3-triazole hybrids as EGFR inhibitors. Front. Chem. 12, 1541846. https://doi.org/10.3389/fchem.2024.1541846 (2025).
Kassem, A. F., Omar, M. A., Nossier, E. S., Awad, H. M. & El-Sayed, W. A. Novel pyridine-thiazolidinone-triazole hybrid glycosides targeting EGFR and CDK-2: design, synthesis, anticancer evaluation and molecular docking simulation. J. Mol. Struct. 136358, 1294 (2023).
Al-Blewi, F. et al. Design and synthesis of novel imidazole derivatives possessing Triazole pharmacophore with potent anticancer activity, and in Silico ADMET with GSK-3β molecular Docking investigations. Int. J. Mol. Sci. 22 (3), 1162 (2021).
Khattab, R. R. et al. Click chemistry based synthesis, cytotoxic activity and molecular docking of novel triazole-thienopyrimidine hybrid glycosides targeting EGFR. J. Enzyme Inhib. Med. Chem. 36, 504–516 (2021).
El-Sayed, W. A. et al. Novel 1,2,3-Triazole-coumarin hybrid glycosides and their tetrazolyl analogues: design, anticancer evaluation and molecular docking targeting EGFR, VEGFR-2 and CDK-2. Molecules, 27(7), p.2047. (2022).
Huang, M., Deng, Z., Tian, J. & Liu, T. Synthesis and biological evaluation of salinomycin Triazole analogues as anticancer agents. Eur. J. Med. Chem. 27, 900–908 (2017).
Lazrek, H. B. et al. Synthesis and anti-HIV activity of new modified 1,2,3-triazole acyclonucleosides. Nucleosides Nucleotides Nucleic Acids. 20 (12), 1949–1960 (2001).
Piperdi, B. & Perez-Soler, R. Role of erlotinib in the treatment of non-small cell lung cancer. Drugs 72, 11–19 (2012).
Ricciardi, S., Tomao, S. & de Marinis, F. Efficacy and safety of erlotinib in the treatment of metastatic non-small-cell lung cancer. Lung Cancer (Auckl). 2, 1–9 (2010).
Park, J. H., Liu, Y., Lemmon, M. A. & Radhakrishnan, R. Erlotinib binds both inactive and active conformations of the EGFR tyrosine kinase domain. Biochem. J. 448 (3), 417–423 (2012).
Park, J. H., Liu, Y., Lemmon, M. A. & Radhakrishnan, R. BIOVIA, Dassault Systèmes, [BIOVIA Discovery Studio Visualizer], [2021], San Diego: Dassault Systèmes, [2024].
Garrett, M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30 (16), 2785–2791 (2009).
Saman, E., Claeyssens, M., Kersters-Hilderson, H. & De Bruyne, C. K. Azido compounds as potential affinity labels for glycosidases. Carbohydr. Res. 30, 207–210 (1973).
Adesoye, O. G., Mills, I. N., Temelkoff, D. P., Jackson, J. A. & Norris, P. Synthesis of a D-Glucopyranosyl Azide: spectroscopic evidence for stereochemical inversion in the SN2 reaction. J. Chem. Educ. 89, 943–945 (2012).
Hanan, A. S., Ahmed, Y. M., Ahmed, E., Hanem, M. A. & Saad, S. E. Eco-friendly synthesis of amidochloroalkylnaphthols and its related Oxazepinones with biological evaluation. Monatshefte Für Chemie-Chemical Monthly. 147, 809–816 (2016).
Eman, M. F., Walaa, I. E., Hanem, M. A. & Mahmoud, E. First synthesis for bis-spirothiazolidine derivatives as a novel heterocyclic framework and their biological activity. Mini Rev. Med. Chem. 20 (2), 152–160 (2020).
Acknowledgements
National Research Centre, Cairo, Egypt. Princess Nourah bint Abdulrahman University Researchers Supporting Project Number (PNURSP2025R141), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
The authors extend their appreciation to the Deanship of Scientific Research at Northern Border University, Arar, KSA, for funding this research work through the project number (NBU-FFR-2025-144-04).
Author information
Authors and Affiliations
Contributions
Conceptualization, M.N. El-Bayaa and A.M. Srour; Methodology, M.N. El-Bayaa, A. L. Alanzy, A.M. Srour and H.M. Awad; Software, A. Temirak; Validation, M.N. El-Bayaa and A.M. Srour; Formal analysis, A. L. Alanzy, M.N. El-Bayaa and A.M. Srour; Investigation, M.N. El-Bayaa, M.G. Saleh, A.M. Srour, A. L. Alanzy, H.M. Awad and A. Temirak; Resources, A. Saleh, M.N. El-Bayaa and A.M. Srour, A. L. Alanzy, A. Saleh and W.A. El-Sayed; Data curation, M.N. El-Bayaa, M.G. Saleh and A.M. Srour; Visualization, M.N. El-Bayaa, and A.M. Srour; Supervision, M.N. El-Bayaa and A.M. Srour; Project administration, A. L. Alanzy and A. Saleh; Writing-original draft preparation, M.N. El-Bayaa, M.G. Saleh and A.M. Srour; Writing—review and editing, M.N. El-Bayaa, M.G. Saleh, A.M. Srour and W.A. El-Sayed; Funding acquisition, A. L. Alanzy, A. Saleh, M.N. El-Bayaa, A.M. Srour and W.A. El-Sayed; All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Srour, A.M., El-Bayaa, M.N., Temirak, A. et al. New benzimidazole-triazole glycoconjugates as anti-cancer agents and EGFR inhibitors. Sci Rep 15, 25514 (2025). https://doi.org/10.1038/s41598-025-96675-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96675-3
