
Abstract
Glioma, a malignant intracranial tumor with high invasiveness and heterogeneity, significantly impacts patient survival. This study integrates multi-omics data to improve prognostic prediction and identify therapeutic targets. Using single-cell data from glioblastoma (GBM) and low-grade glioma (LGG) samples, we identified 55 distinct cell states via the EcoTyper framework, validated for stability and prognostic impact in an independent cohort. We constructed multi-omics datasets of 620 samples, integrating transcriptomic, copy number variation (CNV), somatic mutation (MUT), Microbe (MIC), EcoTyper result data. A scRNA-seq enhanced Self-Normalizing Network-based glioma prognosis model achieved a C-index of 0.822 (training) and 0.817 (test), with AUC values of 0.867, 0.876, and 0.844 at 1, 3, and 5 years in the training set, and 0.820, 0.947, and 0.936 in the test set. Gradient attribution analysis enhanced the interpretability of the model and identified key molecular markers. The classification into high- and low-risk groups was validated as an independent prognostic factor. HDAC inhibitors are proposed as potential treatments. This study demonstrates the potential of integrating scRNA-seq and multi-omics data for robust glioma prognosis and clinical decision-making support.
Similar content being viewed by others


Introduction
Gliomas are the most common primary malignant tumors of the central nervous system (CNS), accounting for 81% of all malignant CNS tumors. They primarily originate from glial cells or their precursor cells and further develop into astrocytomas, oligodendrogliomas, ependymomas, or oligoastrocytomas. According to the World Health Organization (WHO) classification, grades I and II are classified as low-grade gliomas (LGG), while grades III and IV are classified as high-grade gliomas, with grade IV glioblastoma (GBM), which is the most malignant form1. The incidence of gliomas varies across different regions and populations, with an annual incidence rate of approximately 3–4 per 100,000 individuals. Male patients are slightly more affected than females, and the incidence increases with age, particularly in the elderly population2. GBM accounts for approximately 57% of gliomas and 48% of primary malignant CNS tumors. GBM is characterized by its high invasiveness and recurrence rate, with a median survival period typically less than 15 months3. In contrast, LGG has a relatively better prognosis, with a slower progression and a median survival period of 8–10 years. However, LGG has the potential for malignant transformation and may eventually progress to GBM. Therefore, accurate early prognostic assessment is crucial for optimizing treatment decisions4.
Traditional prognostic models are primarily based on clinical features, such as the patient age, Karnofsky Performance Status (KPS) score, tumor size, and location5. While these models have been widely used in clinical practice, they fail to adequately capture the molecular heterogeneity of glioma. With the rapid development of high-throughput omics technologies, multi-omics data (such as genomics, transcriptomics, and microbiomics) has provided valuable information for exploring the molecular mechanisms of glioma. However, the high-dimensional characteristics and heterogeneity of multi-omics data pose significant challenges for modeling. Traditional statistical methods and machine learning algorithms often face overfitting issues when dealing with high-dimensional, low-sample-size data, limiting their application in clinical prognostic assessment6. Deep learning, with its powerful feature extraction capabilities, can automatically identify complex patterns in multi-omics data without requiring manual feature design. Its architectural design and regularization techniques effectively mitigate overfitting in high-dimensional, low-sample-size data, thereby enhancing model generalizability and applicability in clinical settings7. Moreover, deep learning can integrate multimodal data, such as genomics and imaging information, to achieve a more comprehensive patient profiling, thereby improving the accuracy of prognostic predictions. The scalability of deep learning models allows them to adapt to datasets of varying sizes, allowing rapid application to new data through transfer learning. Its automated learning process and ability to handle complex nonlinear relationships further enhance model performance. Therefore, deep learning offers a new solution for glioma prognostic assessment, with the potential to overcome the limitations of traditional methods8,9.
Tumor Microenvironment (TME) is a crucial factor in the occurrence, development, and metastasis of tumors, which comprises tumor cells, immune cells, stromal cells, and the extracellular matrix10. The composition and functional status of the TME may vary greatly depending on tumor type, intrinsic tumor characteristics, tumor stage, and patient-specific factors. Understanding the complex interplay between intrinsic tumor cells, extracellular factors, and systemic mediators of disease progression is essential for the development of effective anticancer therapies. In recent years, the emergence of single-cell omics technologies has brought new opportunities for tumor research. Single-cell omics technologies enable high-resolution analysis of individual cells, revealing cellular heterogeneity and complex intercellular interactions. Compared with traditional bulk sequencing methods, single-cell omics data can provide high-resolution cell type annotations, which are useful in identifying new tumor biomarkers and therapeutic targets. Moreover, they can capture the unique gene expression profiles of different cell types in the TME, thereby more accurately reflecting the molecular characteristics of tumors. This enables the identification of dynamic intercellular interactions and provides new perspectives on tumor invasion, metastasis, and resistance mechanisms. EcoTyper is an advanced machine learning framework designed to analyze single-cell gene expression data to characterize cell states and multicellular communities (also referred to as ecosystems) within the TME. This framework can transform bulk transcriptomic data from large cohorts into detailed information about cellular states and ecosystems identified from single-cell data. This enables precise mapping of tumor microenvironment features described at the single-cell level within bulk transcriptomic datasets.
For the first time, this study incorporates cell states and ecosystem data reflecting TME information based on the EcoTyper framework into the multi-omics prognostic model of glioma and combines an SNN to overcome the overfitting problem in modeling high-dimensional, low-sample-size data. In addition, the gradient attribution algorithm is employed to enhance the interpretability of the model. Our model not only provides accurate prognostic predictions but also reveals the impact of the complex ecosystem of the TME on prognosis, providing a scientific basis for personalized patient treatment. Furthermore, this study explores the application of single-cell omics data in glioma research, demonstrating its unique advantages in revealing TME information and offering new insights for glioma precision treatment.
Materials and methods
Data sources
The single-cell data were obtained from the Gene Expression Omnibus (GEO), specifically from the GSE182109 dataset, which includes data from primary LGG and GBM patients11. The bulk transcriptome data for the EcoTyper model were sourced from TCGA-LGG, TCGA-GBM, and the Chinese Glioma Genome Atlas (CGGA) datasets, including CGGA-693 and CGGA-32512. The copy number variation and somatic mutation data for glioma patients were downloaded from the XENA (xenabrowser.net). The microbiome data were obtained from the cBioPortal (www.cbioportal.org).
Preprocessing and analysis of scRNA-seq data
The single-cell datasets were processed using Seurat v413. Initially, low-quality cells were removed based on the following criteria: (1) cells with fewer than 500 or more than 10,000 gene expressions; (2) cells with fewer than 500 or more than 10,000 total mRNA molecules; (3) cells with a mitochondrial gene proportion exceeding 10%. Subsequently, the scRNA-seq dataset was normalized using the ‘NormalizeData’ function. The ‘FindVariableFeatures’ function was employed to identify the top 2000 most variable genes using the method=’vst’. The R package Harmony was utilized to remove batch effects between single-cell data samples14. The first 50 principal components were selected for dimensionality reduction and visualization. Clustering was performed using the ‘FindClusters’ function, and differential gene expression analysis was conducted between clusters using the ‘FindAllMarkers’ function to annotate each cluster with cell types. We referred to the marker genes of different cell types from related single-cell studies on glioma, and annotated our single-cell data finally.
Discovery of glioma cell States
Initially, 500 cells were randomly extracted from each cell type in the single-cell dataset. Genes with a Q-value greater than 0.05 were selected from each cell type (two-sided Wilcoxon test, with multiple hypothesis correction using the Benjamini–Hochberg method). Subsequently, the single-cell matrix was transformed using the Posneg method to meet the non-negativity requirement for Non-negative Matrix Factorization (NMF). NMF was applied to each transformed matrix for 2 to 20 clusters (cell states) using the Kullback–Leibler (KL) divergence minimization. To determine the number of cell states for each cell type, a heuristic method based on classification stability was applied, using the co-occurrence coefficient for measurement. For each cell type, the number of cell states was selected by identifying the last occurrence where the co-occurrence coefficient dropped below 0.95 within the range of 2 to 20. The number of cell states closest to 0.95 was chosen immediately adjacent to this intersection point. Cell states with fewer than 10 marker genes were removed as low-quality states. Additionally, an adaptive false-positive index was implemented to eliminate spurious cell states driven by negative features resulting from the Posneg transformation before NMF. A total of 55 distinct cell states were identified, with each cell type having 2 to 8 cell states, which were included in downstream analyses15,16.
Discovery of glioma ecosystems
We implemented EcoTyper to identify ecotypes in glioma patients, defined by the co-occurrence of cell states within glioma samples. Each sample was assigned the most abundant cell state for each cell type, and a binary matrix was constructed with cell states as rows and samples as columns. The Jaccard index matrix was calculated for all pairwise combinations of cell states, and a hypergeometric test was run to test the null hypothesis of no overlap. Under the null hypothesis of no overlap between two cell states, a hypergeometric test was conducted for each pair of cell states. If the P-value was greater than 0.05, the Jaccard index was set to 1; otherwise, it was set to 0. Finally, unsupervised clustering of the Jaccard matrix was performed using the ‘hclust’ function from the R package stats. The optimal number of clusters was determined by maximizing the silhouette width. The glioma ecotype network was visualized as a network graph in Cytoscape, with edge thickness representing the Jaccard index between cell states assigned to each sample.
Survival analysis
Survival analysis and Kaplan-Meier curve generation were performed using the R package “survfit,” with P-values less than 0.05 considered significant.
Functional analysis of cell States
To elucidate the biological functions of cell states, marker genes for each cell state were first identified using EcoTyper. Subsequently, functional GO and KEGG enrichment analyses were conducted using the clusterProfiler package17,18,19. In our analysis, biological processes from Gene Ontology (GO) were considered. Pathways with P < 0.05 and adjusted P < 0.05 were deemed significant.
Cell communication analysis
To investigate the interactions between cell states within the glioma ecosystems, we utilized CellChat for cell communication analysis. The cell communication network within the ecosystems and the interaction relationships between cell states were inferred, with the strongest signaling pathways visualized20.
Multi-omics data integration
In our study, we exclusively utilized primary tumor samples across all omics datasets. Specifically, the transcriptome dataset comprised 670 samples, the copy number variation dataset included 1,069 samples, the mutation dataset contained 901 samples, and the microbiome dataset encompassed 673 samples. To integrate these datasets, we performed an intersection analysis, retaining only the samples common to all datasets, resulting in a final cohort of 620 samples.
Subsequently, we conducted univariate Cox regression analyses on the features derived from RNA expression, CNV, MUT, and microbiome data, selecting features with a significance level of p < 0.05. The final multi-omics dataset was constructed by merging these significant features with the results from EcoTyper analysis. For clarity, we defined CNV and MUT data collectively as DNA data, and the EcoTyper results as ECO data. After integration, the RNA data comprised 3,704 features, the DNA data comprised 3,804 features, the MIC data comprised 560 features, and the ECO data comprised 62 features. In integrating multi-omics data, we primarily adopted an early integration method. Specifically, we combined the multi-omics data into a single matrix, which was then fed into our SNN model.
Construction of the ScRNA-seq enhanced multi-omics prognostic model
In learning scenarios with hundreds or thousands of features and relatively few training samples, feedforward networks are prone to overfitting. Compared to other neural network architectures such as CNNs, where weights are shared, feedforward networks are more sensitive to training instability caused by perturbations and regularization techniques such as stochastic gradient descent and Dropout. To mitigate overfitting in high-dimensional, low-sample-size genomic data and adopt more robust regularization techniques for training feedforward networks, we employed the regularization layer from Klambauer et al.‘s Self-Normalizing Networks. In the Self-Normalizing Network (SNN), the ReLU activation was replaced by the Scaled Exponential Linear Unit (SeLU) to drive the output after each layer towards zero mean and unit variance. Combined with the improved regularization technique (Alpha Dropout) that maintains this self-normalizing property, we were able to train well-regularized feedforward networks that would otherwise be unstable due to gradient vanishing or explosion21. Our network architecture consisted of four fully connected layers, followed by Exponential Linear Unit (ELU) activation and Alpha Dropout to ensure the self-normalizing property.
We divided the patient data into training and test sets at a ratio of 80% and 20%, respectively. In the training set, we used five-fold cross-validation and optimized the model using the C-index as the primary evaluation metric. We also assessed the final model’s performance on the test set. In addition to the C-index, we calculated the area under the curve (AUC) for predicting 1-, 3-, and 5-year survival rates. We compared the SNN model with four classical machine learning models and one deep learning model, and evaluated the impact of integrating multi-omics data versus using single-omics data on model performance.
Interpretation of the multi-omics model
To interpret our model, we employed the Integrated Gradients algorithm, which calculates the gradient of the model output with respect to the input features to assess the contribution of each feature to the model’s decision-making process, thereby characterizing the importance and influence direction of the features22. For our multi-omics cohort, we visualized the distribution of feature attributions across all patients (also known as global interpretability), with each point representing the attribution and feature value of a single feature for an individual patient sample. The global interpretability plot and terminology were derived from the decision plot in Shapley Additive Explanations-based methods.
Multi-layer network
Multilevel interaction networks based on microbiota-metabolite-mRNA/protein were constructed using OmicsNet (https://www.omicsnet.ca/OmicsNet/). On the OmicsNet homepage, there are six panels corresponding to six different input types: gene/protein, transcription factor, miRNA, metabolite, microbiome, and graph text. The top 20 genes or proteins identified by gradient attribution and the list of microbe were input separately into the website. Based on the website’s construction, a multilevel interaction network was built. KEGG enrichment analysis was also performed separately for mRNA/proteins and metabolites within the network23.
Drug treatment response and drug sensitivity analysis
Drug sensitivity analysis was conducted using the R package ‘Oncopredict’ to assess the sensitivity to common chemotherapeutic drugs in different risk groups24. Differential genes between high- and low-risk groups and normal samples were calculated and input into the Open Cancer Therapeutic Discovery (OCTAD; http://octad.org) to mine potential drugs for treating high- and low-risk groups. The Reversed Gene Expression Score (RGES), a quantitative measure of the degree to which a compound reverses the gene signature, was calculated. This algorithm first ranks genes based on their expression values in each drug profile and uses the Kolmogorov–Smirnov test to estimate the enrichment of upregulated/downregulated disease genes in the ranked drug profile. When the number of genes exceeds 100, the top 100 upregulated or downregulated disease genes were selected. A compound may have multiple available expression profiles because they were tested in different cell lines, drug concentrations, treatment durations, and even different replicates, resulting in multiple RGES for a drug-disease prediction. Multiple RGES were summarized into sRGES based on previously proposed simple statistics. An sRGES threshold of -0.01 was used to identify compounds that effectively reverse the disease signature. Differential genes between high- and low-risk groups and normal samples were calculated using limma, with p < 0.05 as the screening criterion, and the differential genes for high- and low-risk groups were input into OCTAD to obtain potential drugs for each group25.
Comparison of model prediction capability with traditional clinical features
Univariate and multivariate Cox proportional hazards regression analyses were conducted to test whether the risk stratification of the multi-omics SNN prognostic model is an independent prognostic factor. Prognostic nomograms and calibration curves were established to assess the survival probability of patients at 1, 3, or 5 years using the rms R package. Age, sex, subtype, IDH mutation status, ATRX mutation status, and high/low-risk groups were used as independent parameters.
Statistical analysis
Data analysis and result generation were performed using R (4.3.3). The Wilcoxon rank-sum test was used to compare continuous variables between two groups. A significance level of p < 0.05 was considered the statistical threshold.
Results
Single-cell enhanced multi-omics glioma prognostic model analysis overview
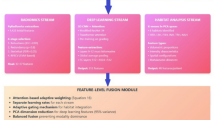
We collected single-cell data for glioma patients from the GEO database. Utilizing the EcoTyper framework, we analyzed 122,902 high-quality single cells from 11 distinct cell types to discover their cell states and ecosystems (Fig. 1A). We collected multi-omics data (RNA, DNA, CNV, MIC) from LGG and GBM patients and used the transcriptomic data to recover the cell states and ecosystem information we previously identified, thereby obtaining the ECO data for LGG and GBM patients (Fig. 1B). Based on this, we integrated the multi-omics data of glioma to construct an enhanced single-cell multi-omics SNN prognostic model (Fig. 1C). We employed gradient attribution algorithms to identify key features for each omics within the prognostic model and built multi-level networks to interpret the key features in the microbiome (Fig. 1D). Additionally, we analyzed the survival differences between high- and low-risk groups in glioma patients using Kaplan–Meier curves. Finally, we assessed the differential drug sensitivities of the high- and low-risk groups and sought potential therapeutic agents for glioma patients (Fig. 1E).

Schematic diagram of the study design.
The human glioma single-cell atlas
We utilized 122,902 high-quality cells from 26 primary GBM and LGG samples from GSE182109 for further analysis (Fig. 2A, Table S1). After principal component analysis, we identified 24 distinct cell clusters (Fig. 2B). Based on the marker genes of different glioma cell types, the 122,902 high-quality cell data was annotated into 11 cell types, namely: NK/T cells (CD3D, CD3E), Microglia (P2RY12, CX3CR1, TMEM119), OPC (BCAN, SCRG1), Astrocyte (AQP4, GFAP), Macrophage (CD163, CD68), NPC (HIST1H4C, PCLAF), Fibroblast (DCN, COL1A1), Oligodendrocyte (PLP1, TF/PPP1R14A), Neuron (STMN2, SOX4), Endothelium (PECAM1, VWF), and B cell (CD79A, CD19) (Fig. 2C). The proportion of cells in different samples is shown in Fig. 2D. We observed strong heterogeneity of Microglia cells across different samples, with their proportion far exceeding other cell types in some samples (Fig. 2E). These single-cell data served as the discovery dataset for subsequent EcoTyper analysis.

Single-cell atlas of low-grade and high-grade glioma. (A) t-distributed stochastic neighbor embedding (t-SNE) plot of 122,902 cells colored by sample classification. (B) T-SNE plot of 122,902 cells clustered at a resolution of 0.4. (C) T-SNE plot displaying the cell types of 122,902 cells. (D) Heatmap showing the distribution of marker gene expression across 11 cell types. (E) Proportion of each cell type in the 26 samples.
Landscape of cell states in the TME of glioma
We input the well-defined single-cell data into the EcoTyper framework. The EcoTyper framework is capable of identifying distinct cell states within each cell type based on the input single-cell expression data and cell type annotations. A total of 55 distinct cell states were identified across 11 cell types, with each cell type comprising 2 to 8 cell states (Fig. 3A–D). To verify the cell states in glioma, we used four independent cohorts to recover and evaluate the identified cell states. We found that in the TCGA and CGGA cohorts, 89–100% of patients’ cell states were recovered, and most of the identified cell states were significantly detected (Fig. S2), indicating that the identified cell states are stable and reliable.

Landscape of cell states in glioma. (A–D) Heatmaps depicting the cell states of Astrocyte, Macrophage, OPC, and Microglia identified from single-cell glioma data. (E–H) Kaplan-Meier plots showing differences in overall survival between patients with specific cell states of Astrocyte, Macrophage, OPC, and Microglia and those with other cell states. (I) Results of GO enrichment analysis for the four cell states. (J) Results of KEGG enrichment analysis for the four cell states. The threshold for enrichment analysis was set at adjust p value<0.05.
To further investigate the prognostic value of different cell states in glioma, we performed survival analysis on the TCGA-LGG and TCGA-GBM cohorts after recovering the cell states. The results revealed that multiple cell states have prognostic value for glioma, such as NPC_S01, Astrocyte_S01, Astrocyte_S06, and Microglia_S03 (Fig. 3E–H). Specifically, NPC_S01 and Astrocyte_S01 were associated with poor prognosis in glioma, while Astrocyte_S06 and Microglia_S03 were associated with favorable prognosis. To further explore the mechanisms by which these cell states influence glioma, we performed GO and KEGG enrichment analyses based on the marker genes of the four cell states (Fig. 3I, J). NPC_S01 and Astrocyte_S01 were both enriched in pathways related to nervous system development, suggesting that these cell states affect glioma prognosis through their impact on nervous system development within the tumor microenvironment. Microglia_S03 was enriched in immune regulation signaling pathways and lymphocyte proliferation, which are related to immune regulation. This indicates that Microglia_S03 exhibits strong immune regulatory and activation capabilities within the glioma microenvironment. It helps balance the immune response in the central nervous system, enhances immune surveillance and tumor clearance, and thereby improves patient prognosis26.
Identification and functional analysis of distinct ecosystems in glioma TME
Tumors are complex ecosystems, characterized not only by the diversity of their components but also by the dynamic interactions between different cell types. To gain a deeper understanding of the glioma TME, we systematically identified multicellular communities using the EcoTyper tool. Through this approach, we identified seven distinct multicellular communities, termed tumor ecosystems, in glioma (Fig. 4A). Each patient is typically assigned to a dominant ecotype, while the tumor is constituted by multiple ecosystems. The ecosystem recovery rate exceeded 85% in four independent cohorts, indicating that the identified ecotypes are highly stable and consistent (Fig. S1).

Landscape of ecotypes in glioma. (A) Abundance patterns of cell states in the single-cell glioma data, with cell states categorized into seven ecotypes (E1–E7) and tumor samples (columns), sorted by the most abundant ecotype in each sample. (B) Distribution of cell states across the seven ecotypes. (C) Percentage of LGG and GBM within each ecotype in the glioma validation cohort.
Further analysis revealed significant differences in the cellular composition and cell states within different ecosystems. For example, the most complex ecosystem E4 comprises nine different cell states, while ecosystems E6 and E7 contain only three cell states (Fig. 4B). By analyzing the proportion of LGG and GBM patients within each ecosystem, we found that E4 consists almost entirely of LGG patients, while E2 is primarily composed of GBM patients. Since LGG patients generally have a better prognosis than GBM patients, these results suggest that E2 may be linked to a poorer prognosis, while E4 is associated with a better prognosis (Fig. 4C).
To validate this hypothesis, we further examined the survival rates of patients associated with different ecosystems. Our results indicated that the survival outcomes of E2 and E4 were consistent with our predictions: patients with E2 exhibited significantly worse prognosis, while those with E4 had a better prognosis (Fig. 5A, C). This further confirmed the prognostic value of ecosystems.

Functional mechanisms of glioma ecotypes. (A,C) Kaplan-Meier plots showing differences in overall survival between patients with ecosystem E2 or E4 and those with other ecosystems in the cohort. (B,D) Interaction frequency and strength between different cell states within ecosystem E2 or E4. (E,F) Overview of outgoing and incoming signaling pathways in ecosystem E2 or E4. (G) Heatmap showing interactions of the COLLAGEN signaling pathway across different cell states in ecosystem E2. (H) Heatmap showing interactions of the SPP1 signaling pathway across different cell states in ecosystem E4.
We performed single-cell level cell communication analysis on these two ecosystems using CellChat, aiming to reveal the complex interactions between cell states within the ecosystems (Fig. 5B,D). The results showed that in the E2 ecosystem, the COLLAGEN signaling pathway had the highest cumulative sending and receiving strength (Fig. 5E). Specifically, Fibroblast_S07 was the main senders of this signaling pathway, while four cell states, excluding Neuron_S01, were the main receivers (Fig. 5G). Moreover, the interaction between Fibroblast_S07 and Macrophage_S03 was the strongest. In the TME, tumor-derived factors may activate resident fibroblasts to transform into cancer-associated fibroblasts (CAFs), which produce and deposit extracellular matrix (ECM) components. Meanwhile, CAFs can recruit monocytes and macrophages through chemokines and other chemoattractants (such as C3a). They also promote the polarization of tumor-associated macrophages (TAMs), thereby creating a microenvironment favorable for tumor growth27.
In contrast, the SPP1 signaling pathway exhibited the highest activity in the E4 ecosystem (Fig. 5F). The heatmap of the SPP1 signaling pathway indicated that Macrophage_S02, Microglia_S03, and Oligodendrocyte_S03 were the main senders of this signaling pathway, while multiple cell states, excluding Neuron_S04, were the receivers (Fig. 5H). Previous studies have demonstrated that OPN/SPP1 in the brain not only acts as a damage sensor but also induces the production of neuroprotective signals28. This finding provides an explanation for why the E4 ecosystem is associated with a better prognosis.
scRNA-seq enhanced multi-omics prognostic model improves survival prediction accuracy
We have demonstrated the significant prognostic value of glioma cell states and ecosystems using single-cell data and the EcoTyper framework. These findings indicate that incorporating these features into a prognostic model could enhance prediction accuracy. Therefore, we integrated transcriptomic, copy number variation, somatic mutation, and microbiome data from TCGA-LGG and TCGA-GBM with ecotype and cell state data to construct a multi-omics dataset comprising 620 samples (Fig. 6A, Table S2). To address the scenario where the number of features far exceeds the number of training samples, we employed a SNN to build the prognostic model. The results showed that the multi-omics model achieved a C-Index of 0.822, significantly outperforming single-omics models and other algorithms (Random Survival Forests, Survival Support Vector Machines, CoxBoost, Gradient Boosting Machine and DeepHit). The model maintained a high C-Index of 0.817 on the independent test set (Fig. 6B). Furthermore, the model exhibited excellent performance in both short-term and long-term prognosis, with AUC values of 0.867, 0.876, and 0.844 for 1, 3, and 5 years, respectively, in the training set, and 0.820, 0.947, and 0.936 in the test set (Fig. 6C,D). These results not only demonstrate the benefits of multi-omics data in comprehensively characterizing tumor heterogeneity but also highlight the superior performance of the SNN model in handling high-dimensional complex data.

Results of the multi-omics prognostic model for glioma. (A) Schematic diagram of the multi-omics cohort of glioma, with the horizontal axis representing glioma patients. A multi-omics cohort data set of 620 patients was ultimately obtained. (B) Heatmap showing the C-index results of the SNN model and five other models. (C,D) ROC curves for glioma patients at 1, 3, and 5 years in the training and test sets.
Interpretability of the scRNA-seq enhanced multi-omics prognostic model
To further elucidate and validate our multi-omics SNN prognostic model, we introduced interpretability methods based on attention and gradients, and utilized SHAP-style attribution decision plots to visualize the attribution weights and directions of different omics features (Fig. 7A–C, Fig. S5). These features were identified as key factors influencing glioma prognosis. Through gradient attribution analysis, we quantified the contribution of each feature to the prognosis and reveal the direction of its positive or negative impact. An overview of the glioma multi-omics cohort (Fig. 7D) displayed information on these key features, which aids in understanding the interrelationships between different omics data.

Interpretability of the multi-omics SNN prognostic model. (A) Global interpretation of the top 10 features from the transcriptome in the multi-omics SNN prognostic model. (B) Global interpretation of the top 10 somatic mutation features in the multi-omics SNN prognostic model. (C) Global interpretation of the top 10 microbiome features in the multi-omics SNN prognostic model. (D) Overview of clinical, RNA, MUT, CNV, and MIC features for 620 glioma patients, with all samples in the same order.
The patient cohort was divided into high-risk and low-risk groups based on the median risk score calculated by the model. The results showed that most GBM patients were classified into the high-risk group, a proportion significantly exceeding that of LGG patients (Fig. S5B). This confirmed that GBM patients exhibit a higher risk level than LGG patients and reflected the high credibility of our model in risk stratification.
In terms of MUT, the low-risk group exhibited more FUBP1 mutations, while the high-risk group had more PDGFRA mutations (Fig. S5B). FUBP1 is a DNA-binding protein primarily involved in regulating gene expression, especially during cell proliferation and differentiation. Studies have shown that FUBP1 mutations are associated with various diseases, particularly in low-grade gliomas such as oligodendrogliomas. In contrast, PDGFRA mutations are more common in a GBM subtype with shorter survival and poorer response to chemotherapy and radiotherapy29,30. Even with more than three subsequent chemotherapy cycles, there was no significant improvement in patient survival.
In CNV analysis, we found that KCNMB2 exhibited more mutations in the high-risk group (Fig. S5B). Although current research on KCNMB2 and glioma is limited, Previous studies have indicated that KCNMB2 is associated with various neuropsychiatric disorders such as autism, hippocampal sclerosis, and depression. This suggests that KCNMB2 may contribute to gliomagenesis and warrants further investigation31,32.
To further elucidate the impact of the microbiome on tumor metabolism and the immune microenvironment, we constructed an interaction network of proteins, metabolites, and microbes based on the top 20 key features from both the transcriptome and microbiome data (Fig. 8A). Metabolite enrichment analysis revealed that central carbon metabolism, branched-chain amino acid metabolism, and the metabolic pathways of glycine, serine, and threonine play a central role in tumor metabolic reprogramming (Fig. 8B). Amycolatopsis, a genus of bacteria belonging to the phylum Actinobacteria, is widely distributed in soil and is well known for its ability to produce a variety of secondary metabolites, including antibiotics, anti-tumor drugs, and other bioactive compounds33,34. The enrichment of these pathways suggests that microbes such as Amycolatopsis may influence energy metabolism pathways through their secondary metabolites, thereby supporting the rapid proliferation and survival of tumor cells. These microbes not only provide tumor cells with ample energy but also enhance their adaptation to environmental changes. Alterations in central carbon and amino acid metabolism can promote energy acquisition via glycolysis in tumor cells and regulate amino acid synthesis, further supporting tumor cell growth under adverse conditions. Moreover, the direct metabolites of Amycolatopsis, such as antibiotics, may directly influence tumor initiation and progression35.

Interpretation of key features of the microbiome in a multi-layer network. (A) Multi-layer network of Microbe-Metabolite-mRNA/Protein interactions constructed using the top 20 important features from the microbiome and transcriptome. (B) Results of KEGG enrichment analysis for metabolites. (C) Results of KEGG enrichment analysis for mRNA/Protein. The threshold for enrichment analysis was set at adjust pvalue < 0.05.
Additionally, the potential impact of the microbe on glioma prognosis is also reflected in its promotion of immune evasion. In our protein enrichment analysis, we found enrichment of the PD-1/PD-L1 checkpoint pathway, indicating that tumors may resist immune system attacks by activating this pathway (Fig. 8C). The activation of the PD-1/PD-L1 pathway is crucial in the tumor microenvironment, inhibiting T cell function and promoting tumor immune evasion. Microbial communities may promote immune evasion ability of tumor cells by regulating gut immune responses and affecting the state of host immune cells36,37. Moreover, the enrichment of ABC transporters indicates that tumor cells may enhance their drug resistance by regulating drug efflux systems, thereby affecting treatment outcomes38.
Model evaluation of patient prognosis, immune characteristics, and drug treatment
Our multi-omics SNN prognostic model effectively stratifies patients into high-risk and low-risk groups, demonstrating significant prognostic differences in both the training and test sets (Fig. 9A, Fig. S6). Further analysis revealed that the model successfully identifies two distinct subtypes among LGG patients, each with markedly different characteristics (Fig. 9B).

Evaluation of patient prognosis, immune characteristics, and drug treatment. (A) Kaplan-Meier plot showing differences in overall survival between high-risk and low-risk groups in the training set. (B) Kaplan-Meier plot showing differences in overall survival between two subtypes of LGG. (C) Forest plot of univariate Cox analysis for age, sex, subtype, IDH mutation status, ATRX mutation status, and high/low-risk groups in 620 patients. (D) Forest plot of multivariate Cox analysis for age, sex, subtype, IDH mutation status, ATRX mutation status, and high/low-risk groups in 620 patients. (E) Nomogram constructed using age, sex, subtype, IDH mutation status, ATRX mutation status, and high/low-risk group features. (F) Heatmap shows the differences in immune checkpoint expression between high-risk and low-risk groups. The heatmap scales the data by columns. (G) Differences in the abundance of immune cell-related states between high-risk and low-risk groups. (H) Estimated sensitivity to four common drugs (Temozolomide, Carmustine, Vorinostat, and Erlotinib) and comparison between high-risk and low-risk groups. Wilcoxon test was used, with *p < 0.05; **p < 0.01; ***p < 0.001.
To independently evaluate the prognostic impact of model risk stratification, we performed univariate and multivariate Cox analyses on age, sex, subtype, IDH mutation status, ATRX mutation status, and high/low-risk groups36. Our results indicated that the SNN model risk stratification was a significant risk factor in univariate Cox regression analysis (HR = 6.54, p < 0.001, Fig. 9C). In multivariate analysis, even when considering other potential confounding factors, the SNN model risk stratification continued to demonstrate its independent prognostic significance (HR = 2.72, p < 0.001, Fig. 9D). To further quantify risk for glioma patients, a nomogram was generated using risk stratification along with other clinical features (Fig. 9E). Calibration curves for the nomogram’s 1-, 3-, and 5- year survival probabilities indicated high accuracy of the nomogram (Fig. S6B).
To assess the immune characteristics between the high- and low-risk groups, we compared the expression levels of immune checkpoints. The results indicated that the high-risk group exhibited higher expression levels across nearly all immune checkpoints, including PD-1, PD-L1, and CTLA-4 (Fig. 9F). This finding suggests that patients in the high-risk group may experience more intense immune responses, indicating a potentially heightened immune-activated state within the tumor microenvironment.
To further validate this hypothesis, we analyzed the abundance of previously defined cell states and found that the high-risk group had significantly higher abundance of immune cell-related states compared to the low-risk group (Fig. 9G). This further confirmed the increased immune infiltration in the high-risk group, likely reflecting more complex immune interactions and a greater risk of immune evasion within the tumor microenvironment.
To evaluate the model’s potential in predicting drug treatment efficacy, we conducted sensitivity analyses for several common glioma therapeutic drugs. The results showed that the high-risk group was more sensitive to Temozolomide, while the low-risk group exhibited greater sensitivity to Carmustine, Vorinostat, and Erlotinib (Fig. 9H). Additionally, we identified HDAC inhibitors as potential therapeutic targets for glioma treatment, with their specific mechanisms warranting further investigation (Table 1)39.
Discussion
This study innovatively integrates multi-omics data and incorporates tumor microenvironment information from single-cell data to construct a glioma prognostic model based on a SNN. The model successfully stratifies patients into high-risk and low-risk groups, demonstrating significant prognostic differences in both the training and test sets. It not only identifies two distinct subtypes among LGG patients but also reveals the contributions of different omics features to prognosis, particularly in terms of immune characteristics and drug sensitivity. These findings provide new insights and potential therapeutic targets for the personalized treatment of glioma.
Traditional prognostic models are often based on single-omics data (such as transcriptomics or mutation data), but such single-omics approaches often fail to fully capture the complexity of tumors. By integrating transcriptomics, copy number variations, somatic mutations, microbiome data, and single-cell tumor microenvironment information, we have constructed a multi-dimensional prognostic model. This multi-omics integration strategy not only captures the complex information from different omics levels but also reveals potential biological mechanisms that cannot be identified by single-omics analyses. The results show that the multi-omics model significantly outperforms single-omics models and other machine learning algorithms, indicating the great potential of multi-omics data in improving the accuracy of prognostic models40,41,42.
Nevertheless, our study has certain limitations. Although we have integrated various omics data, the interactions between different omics data have not been fully elucidated. Future research should further investigate the dynamic relationships between different omics data, especially the synergistic effects between gene expression, copy number variations, mutations, and the microbiome. Addressing these questions will help to more comprehensively understand the biological mechanisms of glioma and provide theoretical support for the development of new therapeutic strategies.
In summary, this study integrates multi-omics data to construct an efficient glioma prognostic model and reveals the contributions of different omics features to prognosis through interpretability methods based on attention and gradients. Our study shows that patients in the high-risk group have more intense immune responses and higher immune infiltration, and are more sensitive to certain drugs. These findings not only provide new ideas for the personalized treatment of glioma but also lay the foundation for future clinical applications. Future research will further explore the in-depth mining of multi-omics data, immune evasion mechanisms, optimization of personalized treatment, and external validation of the model, promoting the development of precision medicine for glioma.
Data availability
The transcriptomic, CNV, and MUT data for TCGA-LGG and TCGA-GBM were obtained from the XENA database (xenabrowser.net). The MIC data were retrieved from the cBioPortal database (https://www.cbioportal.org/). The remaining glioma transcriptomic data were acquired from the Chinese Glioma Genome Atlas datasets (CGGA, https://www.cgga.org.cn/). The single-cell data were obtained from the GEO database (https://www.ncbi.nlm.nih.gov/geo/) with the accession number GSE182109.
References
Louis, D. N. et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol. 23 (8), 1231–1251 (2021).
Ostrom, Q. T. et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the united States in 2015–2019. Neuro Oncol. 24 (Suppl 5), v1–v95 (2022).
Wen, P. Y. et al. Glioblastoma in adults: a society for Neuro-Oncology (SNO) and European society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 22 (8), 1073–1113 (2020).
Tan, A. C. et al. Management of glioblastoma: state of the Art and future directions. CA Cancer J. Clin. 70 (4), 299–312 (2020).
Lamborn, K. R. & CHANG S M PRADOS M D. Prognostic factors for survival of patients with glioblastoma: recursive partitioning analysis. Neuro Oncol. 6 (3), 227–235 (2004).
He, X. et al. Artificial intelligence-based multi-omics analysis fuels cancer precision medicine. Semin Cancer Biol. 88, 187–200 (2023).
Topol, E. J. High-performance medicine: the convergence of human and artificial intelligence. Nat. Med. 25 (1), 44–56 (2019).
Wekesa, J. S. & Kimwele, M. A review of multi-omics data integration through deep learning approaches for disease diagnosis, prognosis, and treatment. Front. Genet. 14, 1199087 (2023).
Lin, C. A. & Berger, M. S. Advancing neuro-oncology of glial tumors from big data and multidisciplinary studies. J. Neurooncol. 146 (1), 1–7 (2020).
De Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell. 41 (3), 374–403 (2023).
Abdelfattah, N. et al. Single-cell analysis of human glioma and immune cells identifies S100A4 as an immunotherapy target. Nat. Commun. 13 (1), 767 (2022).
Zhao, Z. et al. Chinese glioma genome atlas (CGGA): A comprehensive resource with functional genomic data from Chinese glioma patients. Genomics Proteom. Bioinf. 19 (1), 1–12 (2021).
Macosko, E. Z. et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell 161 (5), 1202–1214 (2015).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with harmony. Nat. Methods. 16 (12), 1289–1296 (2019).
Luca, B. A. et al. Atlas of clinically distinct cell States and ecosystems across human solid tumors. Cell 184 (21), 5482–5496 (2021).
Steen, C. B. et al. The landscape of tumor cell States and ecosystems in diffuse large B cell lymphoma. Cancer Cell. 39 (10), 1422–1437 (2021).
Kanehisa, M. et al. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–D677 (2025).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. Protein Sci. 28 (11), 1947–1951 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Jin, S. et al. Inference and analysis of cell-cell communication using cellchat. Nat. Commun. 12 (1), 1088 (2021).
Chen, R. J. et al. Pan-cancer integrative histology-genomic analysis via multimodal deep learning. Cancer Cell. 40 (8), 865–878 (2022).
Sundararajan, M., Taly, A. & Yan, Q. Axiomatic attribution for deep networks. In Proceedings of the 34th International Conference on Machine Learning—Volume 70 3319–3328 (JMLR.org, 2017).
Zhou, G. et al. OmicsNet 2.0: a web-based platform for multi-omics integration and network visual analytics. Nucleic Acids Res. 50 (W1), W527–W533 (2022).
Maeser, D., Gruener, R. F. & Huang, R. S. OncoPredict: an R package for predicting in vivo or cancer patient drug response and biomarkers from cell line screening data. Brief. Bioinform. 22 (6), 1 (2021).
Zeng, B. et al. OCTAD: an open workspace for virtually screening therapeutics targeting precise cancer patient groups using gene expression features. Nat. Protoc. 16 (2), 728–753 (2021).
Borst, K., Dumas, A. A. & Prinz, M. Microglia: immune and non-immune functions. Immunity 54 (10), 2194–2208 (2021).
Buechler, M. B., Fu, W. & Turley, S. J. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity 54 (5), 903–915 (2021).
Yim, A., Smith, C. & Brown, A. M. Osteopontin/secreted phosphoprotein-1 harnesses glial-, immune-, and neuronal cell ligand-receptor interactions to sense and regulate acute and chronic neuroinflammation. Immunol. Rev. 311 (1), 224–233 (2022).
Jiao, Y. et al. Frequent ATRX, CIC, FUBP1 and IDH1 mutations refine the classification of malignant gliomas. Oncotarget 3 (7), 709–722 (2012).
Verhaak, R. G. et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 17 (1), 98–110 (2010).
Bergen, S. E. et al. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry. 17 (9), 880–886 (2012).
Xia, L. et al. Genome-wide association analysis of autism identified multiple loci that have been reported as strong signals for neuropsychiatric disorders. Autism Res. 13 (3), 382–396 (2020).
Kisil, O. V., Efimenko, T. A. & Efremenkova, O. V. Looking back to amycolatopsis: history of the antibiotic discovery and future prospects. Antibiot. (Basel). 10 (10), 1 (2021).
Song, Z. et al. Secondary metabolites of the genus amycolatopsis: structures, bioactivities and biosynthesis. Molecules 26 (7), 1 (2021).
Martínez-Reyes, I. & Chandel, N. S. Cancer metabolism: looking forward. Nat. Rev. Cancer. 21 (10), 669–680 (2021).
Tang, Q. et al. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 13, 964442 (2022).
Ai, L., Xu, A. & Xu, J. Roles of PD-1/PD-L1 pathway: signaling, cancer, and beyond. Adv. Exp. Med. Biol. 1248, 33–59 (2020).
Nobili, S. et al. Role of ATP-binding cassette transporters in cancer initiation and progression. Semin Cancer Biol. 60, 72–95 (2020).
Chen, R. et al. The application of histone deacetylases inhibitors in glioblastoma. J. Exp. Clin. Cancer Res. 39 (1), 138 (2020).
Akhoundova, D. & Rubin, M. A. Clinical application of advanced multi-omics tumor profiling: shaping precision oncology of the future. Cancer Cell. 40 (9), 920–938 (2022).
Vandereyken, K. et al. Methods and applications for single-cell and Spatial multi-omics. Nat. Rev. Genet. 24 (8), 494–515 (2023).
Baysoy, A. et al. The technological landscape and applications of single-cell multi-omics. Nat. Rev. Mol. Cell. Biol. 24 (10), 695–713 (2023).
Funding
This study was funded by the National Key R&D Program of China [2022YFF1203303] and the Ningbo Science and Technology Innovation Yongjiang 2035 Project [2024Z229].
Author information
Authors and Affiliations
Contributions
Xuan Fan: Formal analysis and article design; Zihao He: Formal analysis and article design; Jing Guo: Formal analysis; Dechao Bu: Methodology; Dongchen Han: Formal analysis; Xinchi Qu: Visualization; Qihang Li: Visualization; Sen Cheng: Methodology; Aiqing Han: Project administration and Supervision; Jincheng Guo: Methodology, Validation and Project administration.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All co-authors have consented the version of manuscript for publication.
Ethical approval
This study does not involve human participants, animal subjects, or personal data, and therefore did not require ethics approval. All data analyzed were obtained from publicly available databases or previously published datasets, which were used in accordance with the terms of use specified by the data providers.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Fan, X., He, Z., Guo, J. et al. Leveraging TME features and multi-omics data with an advanced deep learning framework for improved Cancer survival prediction. Sci Rep 15, 14282 (2025). https://doi.org/10.1038/s41598-025-98565-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98565-0
