
Abstract
Essential genes are necessary for the survival or reproduction of a living organism. The prediction and analysis of gene essentiality can advance our understanding of basic life and human diseases, and further boost the development of new drugs. We propose a snapshot ensemble deep neural network method, DeEPsnap, to predict human essential genes. DeEPsnap integrates the features derived from DNA and protein sequence data with the features extracted or learned from four types of functional data: gene ontology, protein complex, protein domain, and protein-protein interaction networks. More than 200 features from these biological data are extracted/learned which are integrated together to train a series of cost-sensitive deep neural networks. The proposed snapshot mechanism enables us to train multiple models without increasing extra training effort and cost. The experimental results of 10-fold cross-validation show that DeEPsnap can accurately predict human gene essentiality with an average AUROC of 96.16%, AUPRC of 93.83%, and accuracy of 92.36%. The comparative experiments show that DeEPsnap outperforms several popular traditional machine learning models and deep learning models, while all those models show promising performance using the features we created for DeEPsnap. We demonstrated that the proposed method, DeEPsnap, is effective for predicting human essential genes.
Similar content being viewed by others

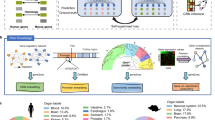

Introduction
Human beings have more than 20,000 genes, which form a redundant and highly fault-tolerant system. Among these genes, some are vital for the survival and reproduction of us, but others are not. These two groups of genes are named as essential genes and nonessential genes. Essential genes are a group of fundamental genes necessary for a specific organism to survive in a particular environment. Cell essential genes refer to a subset of genes that are indispensable for the viability of individual human cell types1,2 as opposed to genes required for the survival of a multicellular organism. Here we focus on predicting cell essential genes. These cell essential genes encode conservative functional elements which mainly contribute to DNA replication, gene translation, gene transcription, and substance transportation. The identification and analysis of essential genes is very important for understanding the minimal requirements of basic life, and it’s vital for drug-target identification, synthetic biology, and cancer research.
There are two ways to identify essential genes: wet lab experimental methods and computational approaches. For example, gene direct deletion and transposon-based randomized mutagenesis have been used to identify essential genes for bacteria and yeast in the genome scale3; microinject KO and nuclear transfer techniques have been used to identify essential genes in mice4; the CRISPR/Cas9 genome editing system has been used to identify essential genes from human cell lines1,2,5. Experimental methods are often costly, time-consuming, and laborious. The accumulation of essential gene datasets, gene and protein sequence data, as well as multiple functional data, enables researchers to explore the relationships between gene essentiality and different omics data and to develop effective models to predict gene essentiality. These computational methods can greatly reduce the cost and time involved in finding essential genes which further boosts our understanding of basic life and human diseases and helps to find new drug targets and develop drugs efficiently.
Computational methods can be classified into two categories. One focuses on designing a centrality measure to rank proteins/genes, while the other focuses on integrating multiple features using machine learning to predict gene essentiality. The most widely used centrality measures include degree centrality, betweenness centrality, closeness centrality, and eigenvector centrality, to name a few. These centrality measures have been found to have a relationship with gene essentiality in multiple model organisms and humans6,7. However, they can only differentiate a subset of essential genes from nonessential genes. A cause can be the incompleteness and false positive/false negative interactions in the protein-protein interaction (PPI) networks. Another reason might be the fact that gene essentiality relates to multiple biological factors rather than just topological characteristics. Due to these motivations, researchers have proposed more complicated centrality measures by integrating network topological properties with other biological information. For example, Zhang et al. proposed a method, CoEWC, to capture the common properties of essential genes in both date hubs and party hubs by integrating network topological properties with gene expression profiles, which showed significant improvement in prediction ability compared to those only based on the PPI networks6. An ensemble framework was also proposed based on gene expression data and the PPI network, which can greatly enhance the prediction power of commonly used centrality measures8. Luo et al. proposed a method, LIDC, to predict essential proteins by combining local interaction density with protein complexes9. Zhang et al. proposed OGN by integrating network topological properties, gene expression profiles, and orthologous information10. GOS was proposed by Li et al. by integrating gene expression, orthologous information, subcellular localization, and PPI networks11. These proposed integrated centrality measures have improved prediction power over the ones that are only based on PPI network topological properties. However, they still have limited prediction accuracy since gene essentiality relates to many complicated factors that are impossible to represent by a scalar score. At this point, machine learning is a good choice to fully utilize multiple features for predicting essential genes.
Many machine learning models and deep learning frameworks have been proposed and successfully applied in different fields. For example, a density-based neural network was used for pavement distress image classification12; a convolutional neural network (CNN) was used for drug-target prediction13; active learning and transductive k-nearest neighbors were used for text categorization14,15; Support Vector Machines (SVM)16 and graph attention networks (GAT)17 were used to predict essential genes. In the research field of essential gene prediction, many machine learning-based prediction methods have been proposed to integrate features from multiple omics data18. As shown in the review article by Zhang et al.18, traditional machine learning methods were used to predict gene essentiality, and most of them were evaluated on data from model organisms. In these methods, topological features together with features from sequence and other functional genomics data were extracted manually and then used to train the models. The approach to extracting informative features is very important and challenging, as it requires ample domain knowledge as well as the understanding of the relationship between gene essentiality and each omics data. Usually, we only know that an omics data would contribute to gene essentiality, but we are not sure which attributes of it have such an effect and how to represent it. This puts a limitation on traditional machine learning methods to obtain good prediction accuracy as they require manual feature engineering.
In recent years, deep learning techniques have been used to automatically extract features and to train a more powerful classification model for predicting essential genes. Grover et al. proposed a network embedding method based on deep learning, node2vec, to learn a low-dimensional representation for each node19. This method has been used to extract topological features from PPI networks for predicting essential genes, and these features are more informative than those obtained via some popular centrality measures7,20,21,22. CNN was used to extract local patterns from time-serial gene expression profiles from S. cerevisiae20 and Zeng et al. also used bidirectional long short-term memory (LSTM) cells to extract features from the same gene expression profiles21. The automatically learned features are combined with other manually extracted ones to train a deep learning model for human essential gene prediction7. A six-hidden-layer neural network was designed to predict essential genes in microbes by only using manually extracted features from sequence data23. Li et al. proposed a deep learning method for predicting cell line-specific essential genes based on sequence data24, which integrates a CNN, bidirectional LSTM, and a multi-head self-attention mechanism together, expecting to learn short- and long-range information from protein sequence and provide residue-level model interpretability. Since sequence data is not cell line-specific, the model can only be trained and tested for each cell line separately. Yue et al. proposed a deep learning model for predicting essential proteins by integrating the PPI network, subcellular localization, and gene expression profiles together, which is evaluated on the data of Saccharomyces cerevisiae25.
Recently, human essential genes have been identified in several human cancer cell lines by utilizing CRISPR-Cas9 and gene-trap technology1,2,26. These identified essential genes provide a clear definition of the requirements for sustaining the basic cell activities of individual human tumor cell types, and can be regarded as targets for cancer treatment. These essential gene datasets together with other available biological data sources enable us to test an important and interesting assumption that human gene essentiality can be accurately predicted using computational methods. In this paper, we propose a Deep learning-based Essential Protein prediction method using a novel snapshot ensemble mechanism, DeEPsnap, to predict human essential genes. DeEPsnap integrates features from five omics data, including features derived from nucleotide sequence and protein sequence data, features learned from the PPI network, features encoded using gene ontology (GO) enrichment scores, features from protein complexes, and features from protein domain data. The proposed snapshot ensemble mechanism is inspired by the work of Huang et al.27, which shows that cyclic learning rates can be effective for training CNNs. In this paper, we propose a new cyclic learning method for our essential gene prediction problem. We also show that DeEPsnap can predict human essential genes with high accuracy. The main contributions of this paper include: (1) extract useful features from multi-omics data and integrate them for predicting human gene essentiality. (2) propose a new snapshot ensemble mechanism to improve the prediction performance. (3) showcase the usefulness and effectiveness of features extracted from five omics data to gene essentiality prediction and the contributions of each.
Snapshot ensemble deep learning model
DeEPsnap consists of three main modules: input data module, feature extraction and feature learning module, as well as classification module. The flowchart of DeEPsnap is shown in Fig. 1. Using node2vec to learn the network embedding features from PPI data is very time-consuming. In order to improve the training efficiency, features from each omics data were first extracted or learned before training the classification model. According to the definition of end-to-end learning in AI, DeEPsnap is not an end-to-end learning model.

The Flowchart of DeEPsnap.
Input data module
In this paper, we mainly consider five omics data to explore their relevance and efficiency for predicting gene essentiality. As shown in Fig. 1, the input data module includes five biological data sources, which are sequence data (DNA and protein sequence), GO, PPI network, protein complex, and protein domain.
Feature extraction and feature learning module
The feature extraction and feature learning module uses different methods to extract or learn features from multiple omics data, as detailed below. Then the features are concatenated together as the input to the classification module. In this paper, we considered five types of features. More types of features can be easily integrated into our model to potentially further improve the prediction accuracy.
GO enrichment score encoded features
Features encoded with GO enrichment scores are calculated as follows. We choose the first 100 GO terms from the cellular component (CC) subcategory to encode the genes, where CC terms are ranked in descending order based on the number of essential genes involved in the terms. For each gene, we first obtain its direct neighbors from the PPI network to form a gene set consisting of itself and its neighbors, then perform GO enrichment analysis for this gene set against the 100 GO terms using a hypergeometric test. The enrichment score is calculated as \(-log_{10}\) (p-value) for each GO term. In this way, we get a 100-dimension feature vector for each gene. The GO enrichment score encoded features capture information from both the PPI network and subcellular localization of genes.
Features derived from the protein complex data
From protein complexes, we extracted two features for each gene. The first feature is the number of protein complexes the gene is involved in. The second feature is calculated as follows: For a gene, we first get the gene set N consisting of its direct neighbors in a PPI network. Suppose there are M neighbors of this gene involved in a protein complex. We calculate a score s = \(\frac{|M|}{|N|}\) as the ratio of its neighbors being involved in a protein complex. The second feature is the sum of the ratios across all the protein complexes. Therefore, the second feature also considers the information from the PPI network in addition to that from the protein complex data.
Features learned from the PPI network
Network features are learned based on a network embedding method: node2vec19. Each gene is represented by a 64-dimension feature vector learned from the PPI network. Previous studies showed that this low-dimension representation learned using node2vec is superior to the features calculated by popular centrality measures7,20,21.
Features derived from the sequence data
Sequence features consist of codon frequency, maximum relative synonymous codon usage (RSCUmax), codon adaptation index (CAI), gene length, GC content, amino acid frequency, and protein sequence length. There are 89 sequence features in total. For more details about how these features are calculated, please refer to Deephe7.
Features derived from the protein domain data
From the protein domain data, we extract three features for each gene/protein. The first feature is the number of domain types the protein has. The second feature is the number of unique domain types the protein has. The third feature is the sum of its inverse domain frequency (IDF). The frequency f of a domain is the number of proteins that have this domain. Its IDF is \(\frac{1}{f}\). Suppose a protein u has n domains, then the third feature, SIDF (sum of inverse domain frequency), of u is calculated as in Eq. (1).
Classification module
Baseline model
The classification module of DeEPsnap is based on a snapshot ensemble deep neural network. The baseline model here is a multilayer perceptron enhanced by several deep-learning techniques, and we name it DNN. It includes one input layer, three hidden layers, and one output layer. We use the rectified linear unit (ReLU) as the activation function for all the hidden layers, while the output layer uses the sigmoid activation function to perform discrete classification. The loss function in DeEPsnap is binary cross-entropy, as shown in Eq. (2), where N is the number of samples, \(y_i\) is the label (in our case, either 0 or 1), and \(p_i\) is the predicted probability of the label being 1. Each hidden layer is followed by a dropout layer to make the network less sensitive to noise in the training data and increase its generalization ability. To address the imbalanced learning issue inherent in the essential gene prediction problem, we utilize class weight to train a weighted neural network, which gives larger penalties when the model misclassifies an instance from the minority class. The use of class weight encourages the model to pay more attention to instances from the minority class than those from the majority class, leading to a more balanced and effective classifier.
Cyclic sine annealing
In machine learning, ensemble methods utilize multiple learning algorithms to obtain better predictive performance than could be obtained from any of the constituent learning algorithms alone. Likewise, ensembles of deep neural networks are known to be much more robust and accurate than individual networks. However, training multiple deep neural networks is computationally expensive. In this paper, we propose a snapshot ensemble mechanism to obtain multiple trained models without incurring extra training costs. The snapshot ensemble mechanism creates an ensemble of accurate and diverse models from a single training process. We expect such an optimization process which will visit several local minima before converging to a final solution. Model snapshots at these various minima are taken and their predictions are averaged at the test time.

Learning rate cycles for the snapshot ensemble models.
To converge to multiple local minima, we propose a cyclic annealing scheduler based on the sine function. The learning rate is decreasing at a very fast pace, encouraging the model to converge towards its first local minimum after as few as 5 epochs in the DeEPsnap training process. The optimization is then continued at a larger learning rate in order to perturb the model and dislodge it from the current local minimum. This process is repeated several times to obtain multiple convergences. Formally, the learning rate lr(t) has the form shown in Eq. (3), where t is the current training epoch number (0-based), C is the total number of epochs to be included in an annealing cycle, \(lr_0\) is the initial learning rate at the beginning of each cycle, while \(\%\) represents the modulo operation. If the number of the total training epochs is set as T, then the training process is split into \(M = \left\lfloor \frac{T}{C} \right\rfloor\) cycles. If T is not divisible by C, the remaining epochs will still be trained but will not contribute to the final ensemble model. Each cycle starts with a larger learning rate for exploration and faster convergence, which is annealed gradually to a smaller learning rate for more accurate positioning of the local minima. Finally, the M snapshot models will be saved at the end of their learning rate annealing cycles and used to make predictions during the testing stage. Figure 2 illustrates the learning rate cycles in DeEPsnap for the first 3 of the M snapshot models. In each learning rate cycle, the learning rate starts from 0.001 and anneals towards 0, but never reaches 0. The snapshot models, the time points at which they are saved are denoted by the dotted lines, form an ensemble at the end of training. The parameters of DeEPsnap in our experiments are shown in Table 1.
Results and discussion
Datasets
Human essential genes are downloaded from the DEG database28. There are 16 human essential gene datasets. We chose the genes that are contained in at least 5 of the datasets to be included in our essential gene dataset. Excluding the genes annotated as essential genes in any of the datasets in DEG, all other genes are considered nonessential genes.
The DNA sequence data and protein sequence data are downloaded from Ensembl29 (release 97, July 2019). We downloaded the PPI data from BioGRID30 (release 3.5.181, February 2020). Only physical interactions between human genes are used. After filtering out self-interactions and several small subgraphs, we obtain a PPI network with 17,762 nodes and 355,647 edges. This interaction network is used to learn embedding features for each gene. It also aids in computing some features from GO, protein complex, and protein domain.
GO data are downloaded from the Gene Ontology website31,32 and protein complex data are downloaded from CORUM33. Protein domain data are from the Pfam database34, and we collect this data via the Ensembl BioMart29. The genes having sequence features, network embedding features as well as GO enrichment scores are used for the following classification performance evaluation. In total, 2009 essential genes and 8414 nonessential genes are used for the following analysis.
Evaluation metrics
We use multiple metrics to evaluate the performance of DeEPsnap. The first metric is the area under the receiver operating characteristic (ROC) curve (AUROC). ROC plots represent the trade-off between sensitivity and specificity for all possible thresholds. The second metric is the area under the precision-recall curve (AUPRC). Precision-recall (PR) curves summarize the trade-off between the true positive rate and the positive predictive value using different probability thresholds. ROC curves are appropriate for balanced classification problems while PR curves are more appropriate for imbalanced datasets. Since essential gene prediction here is an imbalanced classification problem, the AUPRC metric is considered more important than AUROC. We further looked at the Matthews correlation coefficient (MCC) and F1 measure. In addition to these four comprehensive metrics, we also give the comparison results in terms of accuracy. The definitions of MCC, accuracy, and F1 are presented in Eqs. (4)–(6), where TP, TN, FP, and FN are the number of true positives, true negatives, false positives, and false negatives, respectively.
Performance evaluation
In the following experiments, we use the parameters for DeEPsnap as shown in Table 1. In order to cope with the imbalanced learning issue, we set the class weight to 4.5 for essential genes and 1 for nonessential genes. The stratified randomized 10-fold cross-validation is used to evaluate the performance of DeEPsnap. At each fold, \(10\%\) of the data are held out for testing, and the other \(90\%\) are used for training.

The ROC curves of DeEPsnap.
Figure 3 presents the ROC curve of the DeEPsnap across the 10-fold cross-validation. From Fig. 3, we can see that DeEPsnap reaches its best performance at folds 3, 4, 5, and 7, with AUROC = 0.97. The average AUROC of DeEPsnap across the 10-fold cross-validation is \(96.16\%\) with a standard deviation STD = \(0.59\%\), and the average AUPRC is \(93.83\%\) with STD = \(0.83\%\). In addition, the performance of DeEPsnap is quite stable since the difference is less than \(2.12\%\) between its best and worst AUROC scores across the 10-fold cross-validation. The worst AUPRC score is still above \(92.39\%\), indicating that DeEPsnap is very effective for predicting human essential genes. In addition to good scores of AUROC and AUPRC, its average accuracy, MCC, and F1 scores are \(92.36\%\), \(75.92\%\), and \(80.62\%\) respectively (Table 2).
Performance comparison with other machine learning models
To demonstrate the superiority of DeEPsnap, we also compare it with three popular traditional machine learning models-SVM, Random Forest (RF), and Adaboost-as well as a recent deep learning-based essential gene prediction model DeepHE, and two deep learning models (GAT and DNN). For a fair comparison, the three traditional machine learning models and DNN use the same input features that we created for DeEPsnap, so the only difference here is the classification method. GAT is based on the implementation and parameter settings in EPGAT17, while we use an early-stopping mechanism (patience = 200 epochs, \(10\%\) of the training data are held for validation) to save the best model for the testing stage and set the maximum epochs = 3000. Since GAT is a graph neural network that can capture the structure information of input graphs, we use the PPI network as the input graph and the features extracted from the other four omics data as node features. DNN is the baseline model used in DeEPsnap, while it uses a constant learning rate of 0.001 and an early stopping mechanism with patience of 15 epochs (\(10\%\) of the training data are used as validation data). For DeepHE, we use its original features and model structures7.
Table 2 shows the performance comparison between DeEPsnap and the other compared models across 10-fold cross-validation. From Table 2, we can see that the AUROC scores of all the compared models are above \(91\%\), which indicates that the features extracted/learned from the five omics data are very effective for predicting human gene essentiality. DeEPsnap outperforms all the compared models, especially DNN, for all the five measures, which tells us that the proposed snapshot ensemble mechanism is a useful technique for improving model performance without incurring extra training costs. The AUPRC scores of the three DNN-based models (DeEPsnap, DeepHE, DNN) are higher than those of the other compared models, which indicates that multilayer perceptron enhanced with deep learning techniques coupled with class weight can be more effective for coping with imbalanced learning problems. The comparison experiments also show that the training times of all the compared models, except GAT, are comparable-less than 16 minutes for a 10-fold cross-validation on a laptop using CPUs. However, GAT needs more than 9 hours in the same setting, which is 35 times longer than the time needed for DeEPsnap. While DeEPsnap only needs 50 epochs in training for each fold, GAT needs about 2500 epochs (the number of epochs needed for each fold varies from 1500 to 3000 epochs across the 10-fold cross-validation).
Ablation study
In order to comprehend the contribution of each type of feature used in DeEPsnap, we also evaluate it by removing one type of feature each time. The experiments use the same settings except for the input features. Table 3 gives the performance comparison results of DeEPsnap with different types of input features, which reveals that DeEPsnap with the integration of all the five types of features performs best which further confirms the contribution and complementarity of these five types of features. By eliminating one type of feature at a time, we can assess how each feature type contributes to the overall performance and how well it complements the other feature types.
From Table 3, we can see that the combination of N + G + C + D performs worse which might be due to the fact that three of the feature types (i.e. N, G, C) in this combination all utilize the PPI network topological information so that they have less complement effect. The other four combinations with sequence features perform only slightly worse than using all five types of features, which reveals that sequence features are highly complementary to the other types of features. As shown in Deephe7, when only using one type of feature, a deep learning model using network embedding features performs better than that using sequence features. Therefore, the above phenomenon doesn’t indicate that sequence features are superior to network features, but that sequence features are more complementary to the other three types of features.
Enrichment analysis for essential genes
The list of essential genes was submitted to Enrichr35 for analysis for GO Biological processes31,32 and Reactome pathway36 enrichment. The genes were highly statistically enriched for essential macromolecular biosynthetic processes, as shown in Fig. 4. These processes include translation, gene expression, ribosome biogenesis, mRNA processing, and more. The pathways enriched were similar to the GO term analysis with the exception that there is higher enrichment for cell cycle genes which is clearly an essential process. From this analysis, we conclude that our algorithm is selecting the types of genes and processes that would be expected for essential genes. This further confirms the fidelity of our method.

Biological process and pathway enrichment of the essential genes. (a) Bar plot of the top enriched BP terms; (b) Bar plot of the top enriched pathways in Reactome.
Conclusions and future directions
In this paper, we propose a snapshot ensemble deep learning method, DeEPsnap, to predict human essential genes. DeEPsnap integrates five types of features extracted/learned from sequence and functional genomics data. It utilizes multiple deep-learning techniques and a cyclic annealing mechanism to train an ensemble of cost-sensitive classifiers to enhance the prediction accuracy of gene essentiality. Our 10-fold cross-validation experiments demonstrate: (1) the proposed snapshot ensemble deep learning method, DeEPsnap, is superior to the traditional machine learning models and some deep learning models, which is more effective for predicting human essential genes; (2) the extracted features from the five omics data are effective and complementary to gene essentiality prediction; (3) the proposed snapshot ensemble mechanism is promising for improving a model’s prediction performance without incurring extra cost.
In the future, we are interested in how we can use deep learning to automatically learn features from different types of biological data as well as to explore the appropriate knowledge representations for different omics data. For example, learning a low-dimensional representation using all GO terms to encode genes instead of the subset of selected GO terms. It’s especially interesting to explore novel feature learning methods to extract more informative representation features from protein complex and protein domain data. In addition, exploring and integrating more biological data into the learning and classification model is another interesting direction, such as epigenomics data and gene expression profiles. Predicting cancer cell line-specific and tissue-specific essential genes by designing effective deep learning models and integrating cell line-specific and tissue-specific information would be very interesting, especially the prediction of essential genes across cell lines and tissues via useful transfer learning techniques. We are also interested in testing whether data editing37 and clustering-aided techniques38 are useful for imbalanced learning problems. Using cross-attention mechanisms to explore the interactions between different omics data so as to learn more informative features would be another interesting research direction.
Data availability
All data used in this study are third-party and freely accessible from public databases. PPI data are available from the BioGRID database at https://downloads.thebiogrid.org/File/BioGRID/Release-Archive/BIOGRID-3.5.181/BIOGRID-ALL-3.5.181.tab2.zip. Essential gene data are downloaded from the DEG database (Version 15.2, accessed in September 2020) at http://origin.tubic.org/deg/public/index.php/download. DNA sequence and protein sequence data are available at https://ftp.ensembl.org/pub/release-112/fasta/homo_sapiens/cds/Homo_sapiens.GRCh38.cds.all.fa.gz and https://ftp.ensembl.org/pub/release-112/fasta/homo_sapiens/pep/Homo_sapiens.GRCh38.pep.all.fa.gz. GO data are available at https://purl.obolibrary.org/obo/go/go-basic.obo. Protein complex data are available at https://mips.helmholtz-muenchen.de/corum/download/releases/old/corum_2018_09_03.zip. Protein domain data are downloaded via Ensembl BioMart at http://useast.ensembl.org/biomart/martview/e022fef50349a303d840afe580b0e487 (Attributes: at GENE tab, select “Gene Name” and at PROTEIN DOMAINS AND FAMILIES tab, select “Pfam ID”).
Code availability
The Python code of the model is freely available at https://github.com/wjxiao2020/DeEPsnap.
References
Wang, T. et al. Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. https://doi.org/10.1126/science.aac7041 (2015).
Hart, T. et al. High-resolution CRISPR screens reveal fitness genes and genotype-specific cancer liabilities. Cell 163, 1515–1526. https://doi.org/10.1016/j.cell.2015.11.015 (2015).
Grazziotin, A., Vidal, N. & Venancio, T. Uncovering major genomic features of essential genes in Bacteria and a methanogenic Archaea. FEBS J. 282, 3395–3411. https://doi.org/10.1111/febs.13350 (2015).
Liao, B. & Zhang, J. Mouse duplicate genes are as essential as singletons. Trends Genet. 23, 378–381. https://doi.org/10.1016/j.tig.2007.05.006 (2007).
Morgens, D., Deans, R., Li, A. & Bassik, M. Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat. Biotechnol. 34, 634–636. https://doi.org/10.1038/nbt.3567 (2016).
Zhang, X., Xu, J. & Xiao, W. A new method for the discovery of essential proteins. PLoS ONE 8, e58763. https://doi.org/10.1371/journal.pone.0058763 (2013).
Zhang, X., Xiao, W. & Xiao, W. J. DeepHE: Accurately predicting human essential genes based on deep learning. PLoS Comput. Biol. 16, e1008229. https://doi.org/10.1371/journal.pcbi.1008229 (2020).
Zhang, X., Xiao, W., Acencio, M. L., Lemke, N. & Wang, X. An ensemble framework for identifying essential proteins. BMC Bioinform. 17, 322. https://doi.org/10.1186/s12859-016-1166-7 (2016).
Luo, J. & Qi, Y. Identification of essential proteins based on a new combination of local interaction density and protein complexes. PLoS ONE 10, e0131418. https://doi.org/10.1371/journal.pone.0131418 (2015).
Zhang, X., Xiao, W. & Hu, X. Predicting essential proteins by integrating orthology, gene expressions, and PPI networks. PLoS ONE 13, e0195410. https://doi.org/10.1371/journal.pone.0195410 (2018).
Li, G. et al. Predicting essential proteins based on subcellular localization, orthology and PPI networks. BMC Bioinform. 17(Suppl 8), 279. https://doi.org/10.1186/s12859-016-1115-5 (2016).
Xiao, W., Yan, X. & Zhang, X. Pavement distress image automatic classification based on DENSITY-based neural network. In International Conference on Rough Sets and Knowledge Technology (RSKT), 686–692 (2006).
Ozturk, H., Ozgur, A. & Ozkirimli, E. DeepDTA: Deep drug-target binding affinity prediction. Bioinformatics 34, i821–i829. https://doi.org/10.1093/bioinformatics/bty593 (2018).
Zhang, X., Zhao, D., Chen, L. & Min, W. Batch mode active learning based multi-view text classification. In The Sixth International Conference on Fuzzy Systems and Knowledge Discovery, Vol. 7, 472–476 (2009).
Xiao, W. & Zhang, X. Active transductive KNN for sparsely labeled text classification. In The 6th International Conference on Soft Computing and Intelligent Systems, Jointly the 13th International Symposium on Advanced Intelligence Systems, 2178–2182 (2012).
Guo, F. et al. Accurate prediction of human essential genes using only nucleotide composition and association information. Bioinformatics 33, 1758–1764. https://doi.org/10.1093/bioinformatics/btx055 (2017).
Schapke, J., Tavares, A. & Recamonde-Mendoza, M. EPGAT: Gene essentiality prediction with graph attention networks. IEEE/ACM Trans. Comput. Biol. Bioinf. 19, 1615–1626. https://doi.org/10.1109/tcbb.2021.3054738 (2022).
Zhang, X., Acencio, M. L. & Lemke, N. Predicting essential genes and proteins based on machine learning and network topological features: A comprehensive review. Front. Physiol. 7, 75. https://doi.org/10.3389/fphys.2016.00075 (2016).
Grover, A. & Leskovec, J. node2vec: Scalable feature learning from networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, 855–864, https://doi.org/10.1145/2939672.2939754 (2016).
Zeng, M., Li, M., Wu, F. X., Li, Y. H. & Pan, Y. DeepEP: A deep learning framework for identifying essential proteins. BMC Bioinform. 20, 506. https://doi.org/10.1186/s12859-019-3076-y (2019).
Zeng, M. et al. A deep learning framework for identifying essential proteins by integrating multiple types of biological information. IEEE/ACM Trans. Comput. Biol. Bioinf. https://doi.org/10.1109/TCBB.2019.2897679 (2019).
Zhang, X., Xiao, W. & Xiao, W. A deep learning framework for predicting human essential genes by integrating sequence and functional data. bioRxiv https://doi.org/10.1101/2020.08.04.236646 (2020).
Hasan, M. A. & Lonardi, S. Deeplyessential: A deep neural network for predicting essential genes in microbes. bioRxiv https://doi.org/10.1101/607085 (2019).
Li, Y., Zeng, M., Zhang, F., Wu, F. X. & Li, M. DeepCellEss: Cell line-specific essential protein prediction with attention-based interpretable deep learning. Bioinformatics 39, btac779. https://doi.org/10.1093/bioinformatics/btac779 (2023).
Yue, Y. et al. A deep learning framework for identifying essential proteins based on multiple biological information. BMC Bioinform. 23, 318. https://doi.org/10.1186/s12859-022-04868-8 (2022).
Blomen, V. A. et al. Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. https://doi.org/10.1126/science.aac7557 (2015).
Huang, G. et al. Snapshot Ensembles: Train1, get M for free. In 5th International Conference on Learning Representations, ICLR 2017 (France, 2017).
Luo, H., Lin, Y., Gao, F., Zhang, C. T. & Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic Acids Res. 42, D574–D580. https://doi.org/10.1093/nar/gkt1131 (2014).
Ruffier, M. et al. Ensembl core software resources: Storage and programmatic access for DNA sequence and genome annotation. Database https://doi.org/10.1093/database/bax020 (2017).
Stark, C. et al. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 34, D535–D539. https://doi.org/10.1093/nar/gkj109 (2006).
Ashburner, M. et al. Gene ontology: Tool for the unification of biology. Nat. Genet. 25, 25–29. https://doi.org/10.1038/75556 (2000).
Consortium, T. G. O. The Gene Ontology knowledgebase in 2023. Genetics224, iyad031, https://doi.org/10.1093/genetics/iyad031 (2023).
Giurgiu, M. et al. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res. https://doi.org/10.1093/nar/gky973 (2018).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432. https://doi.org/10.1093/nar/gky995 (2019).
Kuleshov, M. V. et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucl. Acids Res. https://doi.org/10.1093/nar/gkw377 (2016).
Milacic, M. et al. The Reactome pathway knowledgebase 2024. Nucleic Acids Res. https://doi.org/10.1093/nar/gkad1025 (2024).
Zhang, X. & Xiao, W. Active semi-supervised framework with data editing. Comput. Sci. Inf. Syst. 9, 1513–1532. https://doi.org/10.2298/CSIS120202045Z (2012).
Zhang, X. & Xiao, W. Clustering based two-stage text classification requiring minimal training data. Comput. Sci. Inf. Syst. 9, 1627–1643. https://doi.org/10.2298/CSIS120130044Z (2012).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Nos. 61402423 and 51678282). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization: Xue Zhang, Wangxin Xiao. Data curation: Xue Zhang. Funding acquisition: Xue Zhang, Wangxin Xiao. Investigation: Xue Zhang, Wangxin Xiao. Methodology: Xue Zhang, Weijia Xiao. Software: Xue Zhang, Weijia Xiao. Supervision: Xue Zhang, Wangxin Xiao. Validation: Xue Zhang, Weijia Xiao, Brent Cochran. Visualization: Xue Zhang, Weijia Xiao, Brent Cochran. Writing - original draft: Xue Zhang, Weijia Xiao, Wangxin Xiao. Writing - review & editing: Xue Zhang, Weijia Xiao, Brent Cochran.
Corresponding authors
Ethics declarations
Competing interests
The authors have declared that no competing interests exist.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, X., Xiao, W., Cochran, B. et al. A deep ensemble framework for human essential gene prediction by integrating multi-omics data. Sci Rep 15, 26407 (2025). https://doi.org/10.1038/s41598-025-99164-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99164-9
