
Abstract
Systematic assessment of the clinical applicability of cell-free RNAs (cfRNAs), which includes broader RNA categories beyond microRNAs, for patients with brain tumors remains largely unexplored due to the lack of sensitive profiling technologies. Our study endeavors to bridge this gap by utilizing an optimized cell-free transcriptome profiling technique that we have recently developed. We comprehensively profiled the cell-free transcriptome in plasma and cerebrospinal fluid (CSF) samples from a total of 85 patients with glioma, meningioma, or tumor-free central nervous system diseases. We identified 16 cfRNA signatures in CSF with robust performance in brain tumor detection (test set AUC = 0.94; validation set AUC = 1). The integration of CSF and plasma-derived cfRNAs outperformed individual analyses using either CSF or plasma candidates for the classification of glioma (test set AUC = 0.94; validation set AUC = 0.85) and meningioma (test set AUC = 0.92; validation set AUC = 0.83). Additionally, we identified 33 CSF and 3 plasma cfRNAs with prognostic significance for postoperative patient outcomes. Multivariate analysis showed that cfRNA-based risk scores (Hazard ratio=9.9) outperformed traditional risk factors in predicting recurrence-free survival. Importantly, our findings in liquid biopsies are consistent with results from primary tumor tissues. By delving into the diagnostic and prognostic implications of cfRNA signatures in CSF and plasma, our study paves the way for improved diagnostic precision and personalized therapeutic interventions for brain tumor patients.
Similar content being viewed by others


Introduction
The diagnosis and management of central nervous system (CNS) tumors present a formidable challenge due to the intrinsic complexity and heterogeneity of these diseases1. In the realm of brain tumors, glioma and meningioma represent two distinct entities with divergent prognostic outcomes. Glioma, as the predominant form of primary malignant brain tumor, is characterized by their aggressive nature and poor patient outcomes2. Meningioma, the most common primary tumor of the CNS, is generally more benign and better prognostic, but can unpredictably demonstrate malignant behaviors that complicate patient management2,3.
Current diagnostic protocols for brain tumors are contingent on neuroimaging techniques such as magnetic resonance imaging and computed tomography scans. While they are invaluable for tumor localization and assessment of structural abnormalities, they lack the capacity to provide molecular insights and may incur significant costs for longitudinal tracking3,4,5. Concurrently, the identification of genomic and epigenomic biomarkers within the tumor tissue has emerged as a cornerstone in oncology6,7,8,9. Tissue-derived signatures have illuminated pathways of oncogenesis, tumor evolution, and subtypes of brain tumors, offering vital insights into tumor biology and patient-specific therapeutic targets10,11,12,13,14,15. They have been shown to be reliable predictors of outcomes for patients with brain tumor in multiple retrospective and prospective studies16,17,18,19,20,21,22. While molecular characterization and histopathological analysis provide more definitive diagnoses, they rely heavily on the tissue samples obtained through a surgical procedure (craniotomy), which is limited by the risk of complications and the likelihood of sampling error due to tumor heterogeneity. Additionally, this tactic may not always be feasible for surgically inaccessible tumors and for patients with complex comorbidities. Furthermore, the prospect of multiple surgical interventions for ongoing assessment of these tumors is often impractical. Therefore, these limitations highlight the pressing need for minimally invasive techniques that can capture the dynamic molecular landscape of brain tumors.
Liquid biopsy technology has emerged as a promising tool to diagnosis and monitoring of brain tumor by enabling the analysis of circulating biomarkers in body fluids23,24,25,26,27,28. The study of mutations, copy number variations, and methylation signatures on cell-free DNAs (cfDNAs) has offered vital insights for classification, prognostication, and appropriate management of glioma29,30,31 and meningioma32,33. Despite the successes with cfDNAs, the potential of cell-free RNAs (cfRNAs) as diagnostic and prognostic biomarkers for brain tumors remains a frontier yet to be fully explored. cfRNAs represent a complementary resource that captures not only genetic alterations but also active transcriptomic changes, thereby offering a real-time reflection of gene expression and the functional state of the tumor34,35. However, the realization of cfRNA’s potential has been hampered by the absence of highly sensitive cfRNA profiling technologies capable of detecting the low abundance and highly fragmented nature of RNA molecules in circulation.
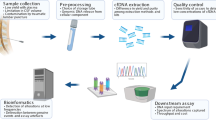
We have previously developed an optimized cfRNA profiling technique, termed SLiPiR-seq, which facilitates the effective identification and quantification of cfRNAs36. SLiPiR-seq has identified a set of cfRNA signatures which revealed robust performance in early cancer detection and classification. Given the success of SLiPiR-seq in plasma samples, we are now extending its application to cerebrospinal fluid (CSF) samples, with a focus on enhancing the understanding and management of brain tumors29. CSF is particularly valuable in neuro-oncology due to its direct contact with the brain and spinal cord, making it a more representative medium for detecting brain tumor biomarkers. In this study, we profiled the cell-free transcriptome in both plasma and CSF samples in parallel from patients with glioma, meningioma, and tumor-free CNS diseases. We delved into investigating the potential of cfRNA as a biomarker for the detection and classification of brain tumors, aiming to enhance diagnostic precision. Furthermore, we extended our research to evaluate the prognostic value of cfRNA by conducting a 3.5-year follow-up of patients’ post-surgical survival, thereby assessing the long-term implications of cfRNA expression patterns on patient outcomes. This comprehensive approach not only underscores the diagnostic capabilities of cfRNAs but also their significant prognostic potential in the context of brain tumor management. The study design workflow is shown in Fig. 1a.

a Schematic diagram showing study design workflow. b, c The ratio of clean reads that aligned to (b) tsRNA or (c) miRNA of different groups. The extremes of boxes define the upper and lower quartiles, and the center lines define the median. Whiskers indicate 1.5× interquartile range. d Principal component analysis on each group of samples using the top 20% of cfRNAs, ranked by their expression levels. e Transcriptome-wide Pearson’s correlation coefficients of the normalized sequencing reads of different types of RNAs between different groups.
Results
Characteristics of study cohort
We collected 159 liquid biopsies (77 CSF samples and 82 plasma samples) from 85 patients who were evaluated for a CNS disease through clinical diagnosis and neuroimaging. The patient cohort encompassed over three groups: glioma (GLI, n = 18), meningioma (MEN, n = 46) and tumor-free CNS diseases (control, n = 21) (Supplementary Table 1). All patients were followed up for a period of 3.5 years (42 months) or until their death. Among patients diagnosed with meningioma, 4 (8.7%) cases of tumor recurrence were documented, and no deaths occurred during the follow-up period. In contrast, among patients diagnosed with glioma, 8 (44.4%) cases of tumor recurrence were documented, and 7 (38.9%) deaths occurred during the follow-up period. Deaths among glioma patients were associated with tumor recurrence, underscoring the necessity for vigilant surveillance and therapeutic interventions. Participant demographics and clinicopathological features of patients included in this study are summarized within Table 1.
Cerebrospinal fluid and plasma transcriptome in CNS diseases
We acquired 16544299 median raw sequencing reads and 9675124 median clean reads after filtering out low quality reads. The median clean read ratio was 61.76% for CSF samples and 58.94% for plasma samples (Supplementary Fig. 1a–c, Supplementary Table 2, Supplementary Data 1). We annotated the clean reads to seven types of RNA, including long non-coding RNAs (lncRNAs), messenger RNAs (mRNAs), microRNAs (miRNAs), piwi-interacting RNAs (piRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), and transfer RNA-derived small RNAs (tsRNAs). Simultaneous profiling of cfRNAs in CSF or plasma samples revealed that the cell-free transcriptome of the two specimen types has different RNA compositions. We found that tsRNAs (CSF: 38.46%; plasma: 7.19%) and piRNAs (CSF: 5.48%; plasma: 1.57%) were more abundant in CSF than in plasma. Meanwhile, miRNAs (CSF: 0.26%; plasma: 18.77%), lncRNAs (CSF: 2%; plasma: 5.97%), and mRNAs (CSF: 0.99%; plasma: 2.8%) accounted for a higher proportion in plasma than in CSF (Fig. 1b, c, Supplementary Fig. 1d-f, Supplementary Table 2). Principal component analysis on the highest 20% of cfRNAs, ranked by expression levels detected across all samples, showed a clear separation between CSF and plasma samples regardless of their groups, underscoring the fundamental differences in their transcriptome profiles (Fig. 1d). PCA of the CSF samples demonstrated that the majority of glioma patients, meningioma patients, and tumor-free patients clustered on principal component one (PC1), except for patients with high-grade glioma (Supplementary Fig. 1g). PCA of the plasma samples showed no notable separation between three groups of patients (Supplementary Fig. 1h), suggesting a more uniform transcriptome profile in plasma possibly due to the influence of the blood-brain barrier, which restricts the exchange between brain fluids and the circulatory system. Transcriptome-wide comparison of the normalized sequencing reads between different groups showed high Pearson’s correlation coefficients (Fig. 1e), indicating the robustness and stability of the SLiPiR-seq technique for the profiling of cfRNAs in plasma and CSF samples.
Discriminative cfRNAs in cerebrospinal fluid
In the CSF samples, we identified 1083 differentially expressed (DE) cfRNAs between glioma patients and tumor-free patients and 115 DE cfRNAs between meningioma patients and tumor-free patients (Benjamini–Hochberg false discovery rate (BH-FDR) <0.1; absolute log2 fold change >0.5; mean normalized reads >5) (Fig. 2a, b, Supplementary Data 2). The two most dominant DE cfRNA species were mRNA (33.1%, n = 359 in glioma; 19.1%, n = 22 in meningioma) and tsRNA (31.2%, n = 338 in glioma; 45.2%, n = 52 in meningioma) (Supplementary Fig. 2a-b). We also performed differential expression analysis between glioma patients and meningioma patients, followed by a cross analysis to identify group-specific DE cfRNAs. We observed 840 overlapping DE cfRNAs (355 upregulated and 485 downregulated) in glioma patients compared to others. Only 10 overlapping DE cfRNAs (2 upregulated and 8 downregulated) were found when comparing meningioma patients with others. Moreover, we identified 75 overlapping DE cfRNAs (18 upregulated and 57 downregulated) in brain tumor (aka. glioma or meningioma) patients compared to tumor-free controls (Supplementary Fig. 2c). We then applied the Wilcoxon rank sum test (p <0.05) and the calculation of variance ( <10 in each group) for further selection of the overlapping cfRNAs, resulting in 179, 4, and 23 candidate cfRNAs for glioma, meningioma, and brain tumor, respectively.

a, b Volcano plots showing differentially expressed (DE) CSF cfRNAs from (a) glioma patients or (b) meningiomas patients versus tumor-free controls. Red dots indicate DE cfRNAs at Benjamini–Hochberg false discovery rate (BH-FDR) <0.1 (Wald test, two-sided p-values) and absolute log2 fold change >0.5. Blue dots represent DE cfRNAs with mean normalized counts <5. Gray dots represent non-significant cfRNAs. c Bar plots showing frequency of LASSO (left) and Boruta (right) feature selection out of 100 iterations for candidate cfRNAs that passed the inclusion criteria. cfRNA features selected for three sets of one-versus-other class comparisons are shown. Different colors represent different types of RNAs. d Heatmap of CSF cfRNA features (row) among each group of samples (column). Ward D2 clustering at Euclidean distance was performed for row and column. Z score normalization was performed on the normalized reads.
To discover discriminative CSF cfRNAs that can accurately classify glioma patients, meningioma patients and tumor-free CNS disease patients, we applied two feature selection algorithms (aka. LASSO and Boruta) on the three sets of candidate cfRNAs. Through this process, we pinpointed 17 glioma-specific cfRNAs (miR-9-3p, GALNT17, SGSM1, TMEM132B, CRACR2A, piR-28347, et al.), 4 meningioma-specific cfRNAs (PWRN4, ANGPTL6, DLGAP1, and piR-2325) and 16 cfRNAs common to both types of brain tumor (miR-193b-5p, ACSL3, SLC45A1, SYCP2L et al.) (Fig. 2c, Supplementary Fig. 3, Supplementary Data 3). The heat map of these discriminative cfRNAs showed clear group-specific expression pattern (Fig. 2d). Upon analyzing the nucleotide sequences of the tsRNA features for brain tumor and glioma, we found that 6 out of 11 contained the sequence ‘GGTTCGATTCCCCGACGGGG’ and correspond to the amino acid aspartic acid with an anticodon of GTC (AspGTC) (Supplementary Data 3)37. Compared with controls, these tsRNAs showed elevated expression in the CSF of brain tumor patients (Supplementary Fig. 3).
Discriminative cfRNAs in plasma
In the plasma samples, we performed differential expression analyses with identical criteria as above for the selection of DE cfRNAs. Interestingly, in contrast to the observations made in CSF, the number of DE cfRNAs in plasma was found to be higher in the ‘meningioma patients versus controls’ test (477 cfRNAs). However, this number is relatively low in the ‘glioma patients versus controls’ test (89 cfRNAs) (Fig. 3a, b, Supplementary Data 2). The two most prevalent DE cfRNA types for glioma were snRNA (53.9%, n = 48) and tsRNA (21.3%, n = 19), whereas tsRNA (30.8%, n = 147) and mRNA (23.5%, n = 112) were the predominant types for meningioma (Supplementary Fig. 4a-b). In addition, we performed intersection analyses and detected 5 overlapping cfRNAs (4 upregulated and 1 downregulated) in glioma patients compared to other groups, as well as 110 overlapping cfRNAs (9 upregulated and 101 downregulated) in meningioma compared to others. Furthermore, our analysis also revealed 70 overlapping cfRNAs (1 upregulated and 69 downregulated) in brain tumor patients against tumor-free controls (Supplementary Fig. 4c). Further selection of the overlapping cfRNAs via Wilcoxon rank sum test and variance resulted in 2, 19, and 18 candidate cfRNAs for glioma, meningioma, and brain tumor, respectively. To identify discriminative cfRNAs in plasma samples, we applied the same feature selection process on the candidate cfRNAs. As a result, 2 glioma-specific cfRNAs (miR-9-3p and miR-9-5p), 12 meningioma-specific cfRNAs (miR-151a-3p, miR-26b-5p, miR-574-3p, miR-93-5p, AOPEP, et al.) and 8 cfRNAs common to both types of brain tumor (RUVBL1, CCDC200, FGA, GSKIP, IFT88, et al.) were considered as candidates for subsequent machine learning analyses (Fig. 3c, d, Supplementary Fig. 5, Supplementary Data 3).

a, b Volcano plots showing DE plasma cfRNAs from (a) glioma patients or (b) meningiomas patients versus tumor-free controls. Red dots indicate DE cfRNAs at BH-FDR <0.1 (Wald test, two-sided p-values) and absolute log2 fold change >0.5. Blue dots represent DE cfRNAs with mean normalized counts <5. Gray dots represent non-significant cfRNAs. c Bar plots showing frequency of LASSO (left) and Boruta (right) feature selection out of 100 iterations for candidate cfRNAs that passed the inclusion criteria. cfRNA features selected for three sets of one-versus-other class comparisons are shown. Different colors represent different types of RNAs. d Heatmap of plasma cfRNA features (row) among each group of samples (column). Ward D2 clustering at Euclidean distance was performed for row and column. Z score normalization was performed on the normalized reads.
Machine learning classification of brain tumors
To evaluate the classification performance of the selected features, we carried out a series of one-versus-other class machine learning analyses using three algorithms (logistic regression (LR), random forest (RF), and support vector machine (SVM)). For brain tumor detection, 16 CSF cfRNA features exhibited robust performance in both the test set (LR Area Under Receiver Operating Characteristic (ROC) Curve (AUC) = 0.94 [0.91-1]; RF AUC = 0.94 [0.88-1]; SVM AUC = 0.94 [0.88-0.97]) and validation set (LR AUC = 1 [1-1]; RF AUC = 1 [1-1]; SVM AUC = 1 [1-1]) (Fig. 4a). Importantly, the high agreement of AUC scores across different algorithms underscores the reliability of CSF cfRNAs for brain tumor detection. For the classification of glioma, RF models trained with 17 CSF cfRNA features demonstrated high accuracy in the test set (RF AUC = 0.92 [0.86-0.97]) and modest accuracy in the validation set (RF AUC = 0.81 [0.78-0.86]) (Fig. 4b). Models trained using other algorithms also showed robust test set performance (LR AUC = 0.94 [0.89-0.97]; SVM AUC = 0.94 [0.86-0.97]) but performed poorly in the validation set (LR AUC = 0.64; SVM AUC = 0.67) (Fig. 4b). Conversely, models trained to differentiate meningioma from other classes using 4 CSF cfRNA features displayed suboptimal accuracy in both the test set (LR AUC = 0.82 [0.73-0.89]; RF AUC = 0.73; SVM AUC = 0.82 [0.75-0.88]) and validation set (LR AUC = 0.74; RF AUC = 0.76 [0.74-0.80]; SVM AUC = 0.74) (Supplementary Fig. 6a).

a–c AUC scores over 100 model iterations results in the training set, test set, and validation set of the study cohort using three different algorithms, including logistic regression (LR), random forest (RF), and support vector machine (SVM). The specimen type, binary classification issue, and the number of cfRNA features used for model training are shown in the plot titles. d Normalized sequencing reads of miR-9-3p and miR-9-5p identified via feature selection algorithms. Two-sided p-values were calculated via the Wilcoxon rank-sum test. The extremes of boxes define the upper and lower quartiles, and the center lines define the median. Whiskers indicate 1.5× interquartile range. e AUC scores in the test set and validation set over 100 model iterations in nine sets of RF analyses. Two-sided p-values were calculated using Welch’s t-test between single-specimen and dual-specimen models.
In contrast to the robust performance in CSF, models aiming to differentiate brain tumor patients from tumor-free individuals using 8 plasma cfRNA features demonstrated high accuracy in the test set (LR AUC = 0.92 [0.80-0.97]; RF AUC = 0.90 [0.82-0.95]; SVM AUC = 0.92 [0.74-1]) but inferior accuracy in the validation set (LR AUC = 0.69; RF AUC = 0.65; SVM AUC = 0.69) (Supplementary Fig. 6b). Although only 2 miRNAs (miR-9-3p and miR-9-5p) were used to discriminate glioma from other classes, SVM models achieved high AUC values in both the test set (SVM AUC = 0.92 [0.87-0.97]) and validation set (SVM AUC = 0.94 [0.81-0.94]) (Fig. 4c, d). Likewise, other algorithms performed well in the test set (LR AUC = 0.92 [0.86-0.96]; RF AUC = 0.90 [0.85-0.95]) but showed reduced accuracy in the validation set (LR AUC = 0.69; RF AUC = 0.78 [0.75-0.83]). The model trained for distinguishing meningioma from others using 12 plasma cfRNA features exhibited moderate performance in the test set (LR AUC = 0.81 [0.75-0.87]; RF AUC = 0.81 [0.76-0.87]; SVM AUC = 0.73) and validation set (LR AUC = 0.74; RF AUC = 0.72; SVM AUC = 0.67) (Supplementary Fig. 6c).
We further investigated if integrating cfRNA features from both CSF and plasma could enhance model performance. The RF classifier was used for the evaluation. For differentiating between brain tumor patients and tumor-free controls, combining CSF and plasma features provided no notable improvement in the model performance (test set AUC = 0.97 [0.88-1]; validation set AUC = 1 [1-1]) compared to using CSF features alone (test set p = 0.699; validation set p = 0.052) (Fig. 4e). For the classification of glioma, the models trained with combined features exhibited a substantial enhancement in test set accuracy (AUC = 0.94 [0.88-1]) compared to plasma (p = 8 × 10−5) features alone. The validation set accuracy (AUC = 0.85 [0.82-0.88]) also showed significant improvement compared to CSF (p = 0.0001) or plasma (p = 3.9 × 10−12) features alone (Fig. 4e). For the classification of meningioma, the combined analysis of CSF and plasma features offered the best performance in the test set (AUC = 0.92 [0.85-0.95]) and validation set (AUC = 0.83 [0.81-0.87]) compared to either CSF (test set p = 1.43 × 10−14; validation set p = 5.74 × 10−24) or plasma (test set p = 7.71 × 10−8; validation set p = 2.36 × 10−44) features alone (Fig. 4e). These findings suggested that the combination strategy is beneficial for the classification of glioma and meningioma, but not necessarily for the detection of brain tumors.
cfRNAs are indicative of tumor tissue of origin
To trace back the tissue of origin of DE cfRNAs, we carried out comprehensive in-silico analyses utilizing bulk RNA-seq data from the Genotype-Tissue Expression (GTEx, 34 tissue types), The Cancer Genome Atlas (TCGA, 6819 primary tumors across 16 distinct cancer types), and Gene Expression Omnibus (GSE10163838, 42 meningioma samples) data portal (Supplementary Data 4). We hypothesized that upregulated cfRNAs in patient body fluids are more likely to be derived from the disease site. Therefore, our analysis was focused on the 242 and 63 upregulated protein-coding transcripts in the CSF of glioma patients and the plasma of meningioma patients, respectively, as the number of DE cfmRNA in other sets are minimal. From the former set, we identified 98 genes that showed enhanced expressions in the brain and 65 genes with enriched expression in glioblastoma multiforme primary tumors (Supplementary Fig. 7). It is worth mentioning that the three machine learning features (aka. GALNT17, SGSM1, and TMEM132B) for glioma demonstrated brain-enriched expression pattern (Supplementary Fig. 8). TMEM132B also demonstrated glioblastoma multiforme-specific expression when compared to other cancer types. Expression clustering and correlation analysis indicated that TMEM132B was in the cluster of “neuronal signaling”, while SGSM1 was linked to the cluster of “synaptic signal transduction”. Furthermore, C10orf90 (oligodendrocytes - myelination), CTNND2 (neuronal signaling), and PCDH15 (visual perception) showed highly specific expression in both the brain and glioma primary tumor (Supplementary Fig. 8). From the latter set, we identified 20 genes that showed enhanced expressions in the brain. Of these, ADAP1 (oligodendrocytes - myelination), CRMP1 (synaptic signal transduction), LRRC4B, NCAN, PSD2 (all three related to neuronal signaling), and SRCIN1 (nervous system development) demonstrated high specificity to the brain (Supplementary Fig. 9). However, no gene was found to exhibit meningioma-specific expression. It is reasonable to infer that these brain or tumor-specific cfmRNAs are derived from the brain and the primary tumor as their expression levels in other tissues are low. These findings highlight the potential of cfRNAs as valuable biomarkers for indicating the tumor tissue of origin.
cfRNAs prognostic of post-surgical recurrence and survival outcomes
To determine whether the CSF or plasma samples collected prior to surgical resection of glioma are prognostic for postoperative recurrence-free survival (RFS) and overall survival (OS), we performed survival analyses focusing on the DE cfRNAs. The expression status (‘upregulate’ or ‘downregulate’) of cfRNAs in each glioma patient was determined based on the median expression level in the control group. With a 42.0-month minimum follow-up, we identified 27 cfRNAs whose ‘upregulate’ or ‘downregulate’ status was associated (hazard ratio (HR) > 1 and log-rank p-value <0.05) with disease recurrence in CSF samples (Fig. 5a & c, Supplementary Fig. 10, Supplementary Data 5). We identified 26 CSF cfRNAs whose dysregulation status was associated with worse postoperative survival (Fig. 5b, c, Supplementary Fig. 11, Supplementary Data 5). Notably, 20 CSF cfRNAs (CTC1, FOXC1, MAPK6, MT-ATP6, MT-CYB, MT-ND4, RPL13A, RPS8, SIX5, ST8SIA6, TFAP2D, et al.) were found to be prognostic of both RFS and OS. In the plasma samples, we found 3 cfRNAs that are prognostic of both RFS and OS (Supplementary Fig. 12). It is worth mentioning that although fewer prognostic cfRNAs were identified in plasma, the increased expression of tRF-21-RZ81JJ6R0 (RFS HR = 9.6; OS HR = 15.16) and tRF-20-RZ81JJ6R (RFS HR = 11.96; OS HR = 12.01) showed a strong association with poor patient outcomes.

a, b Forest plot showing the hazard ratio (HR) (with 95% confidence interval) and log-rank p-value of CSF cfRNAs that were associated with poorer recurrence-free survival (RFS, a) and overall survival (OS, b). c Kaplan-Meier plot that shows the association of MAPK6 and tRF-18-07QSNHD2 with post-surgical RFS (red) and OS (orange). d Oncoprint showing the expression status and frequency of the prognostic cfRNAs in glioma patients. The vital status, recurrence status, and WHO grade of each patient are shown on the top. The HR for RFS and OS of each cfRNA are shown on the right. e Forest plot showing the HR (with 95% confidence interval) and log-rank p-value of genes that have prognostic values in both the glioma primary tumor tissue and CSF in the TCGA cohort.
The downregulation of tRF-18-07QSNHD2 (68.8%), MAPK6 (62.5%), piR-hsa-3488 (56.3%) and FOXC1 (56.3%) and the upregulation of PPP6R3 (56.3%), tRF-22-F91SS2P44 (56.3%), tRF-27-QJ3KYU4RRW4 (56.3%), and tRF-21-RNLIK8980 (56.3%) were frequently detected in the CSF of glioma patients (Fig. 5d). However, alterations in the expression level of other cfRNAs are less frequent in the CSF and plasma samples. We observed no cancer recurrence or death among the patients with no change in MAPK6 and tRF-18-07QSNHD2 expression, resulting in an infinite HR value (Fig. 5c). In addition, patients with expression changes in CTC1, SIX5, TFAP2D, RPL13A, OAZ2, ENSG00000273112, SNORD26, tRF-18-YR6RIFD2, tRF-32-FZ9KR959NRZZP, and three mitochondrially encoded genes (MT-ND4, MT-ATP6 and MT-CYB) all relapsed or died during follow-up, warranting further investigation of their role in patient prognosis in a larger cohort. Of particular interest is tRF-32-FZ9KR959NRZZP, which has demonstrated both diagnostic and prognostic value.
To assess whether the prognostic cfmRNAs could also serve as predictors of post-surgical survival in primary tumor, we conducted in-silico survival analyses utilizing TCGA RNA-seq data from primary tumor tissues of 141 glioma cases (Supplementary Data 6). Surprisingly, lower expression of FOXC1 (HR = 1.44), MT-ATP6 (HR = 1.55), MT-ND4 (HR = 1.58), MT-CYB (HR = 1.58), RPL13A (HR = 1.77), RPS8 (HR = 2.14), and ST8SIA6 (HR = 1.61) in glioma primary tumor was associated with worse survival outcomes, with MT-ATP6 (log-rank p = 0.042), RPL13A (p = 0.0034), and RPS8 (p = 0.015) showing a particularly strong association (Fig. 5e, Supplementary Fig. 13). The concordance in survival analysis results between cfmRNA levels in CSF and mRNA expression levels in tumor tissue underscores the prognostic value of cfRNA, affirming its potential as a reliable biomarker for predicting patient outcomes after surgery.
We also examined the prognostic potential of DE cfRNAs in meningioma. Downregulation of LRRCC1 and ST3GAL2 in the CSF and downregulation of 9 cfRNAs (SNHG25, et al.) in the plasma were found to be linked to shorter RFS (Supplementary Fig. 14). Nevertheless, further validation is needed due to the minimal number of relapses in this group.
Multi-cfRNA-based risk scores in glioma prognosis
Glioma is a highly heterogeneous disease with considerable genetic diversity. Patients with glioma can have highly variable survival times depending on their molecular characteristics. This prompted us to look beyond a single biomarker to determine clinical outcomes. To this end, we developed an algorithm to calculate a risk score according to aberrations at the expression level of multiple prognostic cfRNAs for each patient (see Methods for details). We observed that patients with cancer recurrence and death during the follow-up have higher RFS and OS risk score than patients without such events (Fig. 6a). Kaplan–Meier curves showed that a high risk score in CSF samples at the pre-surgery time point was strongly associated with shorter disease-free survival (HR = 20.59, p = 0.00027) and poorer overall survival (HR = 8.67, p = 0.003) (Fig. 6b). The mean RFS time for patients with a high and low CSF risk score was 7.25 ± 1.81 months and 35.08 ± 3.52 months, respectively. The mean OS time for patients with a high and low CSF risk score was 16.60 ± 1.66 months and 36.73 ± 3.38 months, respectively. Importantly, the log-rank p-value of the multi-cfRNA-based RFS risk score was more significant than that of any individual cfRNA from CSF. This implies that the use of a multi-target approach could potentially address the issue of tumor heterogeneity.

a Risk scores based on prognostic CSF and plasma cfRNAs for each glioma patient. Icons indicates recurrence or death event. b Kaplan-Meier plot showing the association of post-surgical RFS (left) and OS (right) with risk scores calculated from CSF prognostic cfRNAs. c Forest plot showing the HR (with 95% confidence interval) of multivariate cox regression analysis integrating CSF risk score with survival-relevant risk factors (age, IDH mutation status, and 1p/19q codeletion status).
Next, we integrated prognostic cfRNAs from CSF and plasma to calculate risk scores. However, Kaplan–Meier curves for this combined dataset showed no notable difference for both RFS (HR = 11.19, p = 0.00073) and OS (HR = 8.67, p = 0.003) from using only the CSF-derived cfRNAs (Supplementary Fig. 15a). These findings demonstrated that CSF alone, collected before surgery, is adequate for prognostication of postoperative survival in glioma cases.
In consideration of known risk factors and genetic alterations linked to glioma prognosis, we performed further survival analyses incorporating age, WHO grade, isocitrate dehydrogenase (IDH) mutation status, 1p/19q codeletion status, and H3K27M mutation status (Supplementary Fig. 15b). Consistent with findings from previous studies, patients with younger age, lower grade, IDH mutation, 1p/19q codeletion, or H3K27M wildtype had favorable survival outcomes39,40. Multivariate survival analysis integrating the CSF cfRNA risk score (RFS HR = 9.9; OS HR = 2.5), age (RFS HR = 5.7; OS HR = 5.1), IDH mutation status (RFS HR = 1.5; OS HR = 2.4), and 1p/19q codeletion status (RFS HR = 1.1; OS HR = 1.7) demonstrated that cfRNAs outperformed traditional risk factors in predicting postoperative disease-free survival (Fig. 6c).
Discussion
The clinical utility of cfRNA signatures for brain tumors remained largely unexplored. While miRNAs in CSF have shown promise as brain tumor biomarkers, their investigation have mainly depended on low-throughput techniques such as qPCR41,42,43. Even high-throughput methods like Illumina small RNA sequencing fall short of capturing the entire cell-free transcriptome repertoire44. Our study introduces a groundbreaking approach for analyzing cfRNA in the liquid biopsies of patients with CNS tumor by utilizing SLiPiR-seq. SLiPiR-seq enables the profiling of cfRNAs independent of their 5’-end phosphorylation, thereby facilitating the identification of more unique cfRNA species and broader coverage of the cfRNA landscape. By integrating this sensitive cfRNA profiling technique, we provide a unique perspective for understanding the molecular dynamics of protein coding RNAs and six different types of non-coding RNAs in brain tumors. The findings of this study underscore the potential applications of cfRNAs in brain tumor diagnostics and prognostics.
The dual-sample approach, involving the concurrent analysis of cfRNA signatures from both the systemic circulation and the CNS microenvironment, facilitates a holistic examination of brain tumor biology. Transcriptome-wide profiling has revealed large differences in RNA composition and expression profiles between CSF and plasma. Consistent with a previous study in which ctDNA mutations in CSF that align with tumor tissues were absent in plasma29, our study also found that the DE cfRNAs observed in CSF were not mirrored in plasma. This discrepancy may be attributed to the selective permeability of the blood-brain barrier, which may impede molecular exchange between the CNS and peripheral circulation45. The high intra-group cfRNA expression variance among brain tumor patients renders their utilization in model construction challenging19. While single-specimen analysis showed modest performance in classifying glioma and meningioma, the combined analysis of cfRNA signatures from both CSF and plasma significantly enhanced model accuracy.
Previous studies interrogating the diagnostic potential of liquid biopsies for CNS tumors were primarily focused on genomic alterations and methylation aberrations in ctDNA. Mouliere et al. utilized a RF classifier to integrate copy number alterations with specific fragmentomics signatures, achieving highly accurate cancer classification with an AUC of 0.91 in glioma cases46. Lavon et al. reported the use of methylation at the MGMT promoter as a serum marker for brain tumors and demonstrated excellent specificity (100%) but modest sensitivity (59%)47, highlighting the limitations of a single-gene strategy. Sabedot et al. used bisulfite conversion of plasma-derived cfDNA followed by analysis on Epic array, coupled with a machine-learning classifier, to distinguish tumor from normal samples with robust performance (sensitivity: 100%, specificity: 97.78%)31. Similarly, Nassiri et al. applied bisulfite-free affinity-enrichment method to detect intracranial tumors using plasma cfDNA methylomes, achieving an AUC of 0.71 for brain tumors and 0.82 for glioma25. The findings of our study indicated that CSF cfRNA signatures exhibit superior diagnostic capabilities for brain tumor detection. The accuracy of cfRNA signatures has been shown to be comparable to that of previously reported ctDNA assays.
Our observations in liquid biopsy samples exhibit a substantial degree of concordance with the findings reported in previous studies that utilized tissue samples. Notably, the increased expression of miR-9-3p in both CSF and plasma in glioma patients, which was further qualified by the two feature selection algorithms we applied as a candidate biomarker for glioma detection, serves as a prime example. Consistently, previous studies have detected elevated levels of miR-9 in glioblastoma tissues48 and in extracellular vesicles derived from glioma stem cells49,50. The involvement of miR-9 in promoting glioma tumorigenesis and angiogenesis has also been reported51,52. Interestingly, our results showed no difference in miR-9 levels between meningioma patients and controls in either type of specimen, highlighting its specificity for glioma. Additionally, it has been reported that miR-193b-5p functions as a dual-strand tumor suppressor, with elevated levels of expression observed in glioma tissue and cells53.
The concurrent analysis of tissue-derived RNA-seq data has greatly enhanced our understanding of the biological relevance of cfmRNA signatures in glioma. The identification of certain cfmRNAs such as C10orf90, CTNND2, PCDH15, and TMEM132B that show concordantly elevated expression in glioma primary tumors, brain tissue, and CSF positions them as promising biomarkers for non-invasive glioma detection. C10orf90 is known as FATS (Fragile-site Associated Tumor Suppressor) because its co-localization with common fragile sites FRA10F54. C10orf90 plays an active role in regulating checkpoint functions following DNA damage and suppressing tumor growth55,56,57. Interestingly, while C10orf90 is downregulated or silenced in many cancers, it is upregulated in glioma due to its association with oligodendrocytes generation during myelin development58. CTNND2 inactivation has been identified as a pivotal genetic alteration that contributes to the aggressive mesenchymal phenotype characteristic of glioblastoma59. PCDH15 encodes protocadherin 15, a member of the cadherin superfamily of transmembrane proteins. PCDH15 is a critical regulator of oligodendrocyte progenitor cells proliferation and motility, which contributes to glioma pathogenesis60. Transmembrane protein TMEM132B is a GABAAR auxiliary subunit that promotes GABAAR expression and slows receptor deactivation61. Despite the relative obscurity of TMEM132, the TMEM proteins have been implicated in cell proliferation, migration, and invasion in many types of cancers62. The consistency of the expression of these cfmRNAs in the CSF with the brain and tumor tissue, coupled with the proximity of CSF to CNS tissues, highlight the feasibility of using cfRNAs for pinpointing the tumor tissue of origin.
In addition, the correlation between lower expression levels of specific cfmRNAs in glioma tissue-derived RNA-seq data and poorer post-surgical outcomes in glioma patients underscores their potential as prognostic markers. Particularly noteworthy are the strong associations of MT-ATP6, RPL13A, and RPS8 with survival. MT-ATP6 is a mitochondrially encoded gene that involves in the process of mitochondrial ATP synthesis coupled with proton transport. Single-cell RNA sequencing has revealed the preferential expression of MT-ATP6 in oligodendrocyte within glioblastoma samples63. Both RPL13A and RPS8 are ribosomal proteins, which are implicated in a variety of pathological processes, especially tumorigenesis and cell transformation64,65. The consistency of these findings in both CSF and tumor tissue reaffirms the robustness of cfRNA as a reliable biomarker for predicting patient’s post-surgical outcomes at baseline.
Nevertheless, it is crucial to acknowledge certain limitations of this study. Firstly, the sample size of our study cohort is relatively small. Obtaining both CSF and plasma samples from patients with CNS diseases presents significant challenges in clinical settings, due to the invasive nature of CSF collection and the associated risks. The small samples size resulted in the scarcity of IDH-mutated, 1p19q co-deleted, and H3K27M-mutated cases in our dataset, thereby limiting the generalizability of these widely used molecular signatures. Secondly, transcriptome analysis of matched tumor tissue was not performed. This limitation is mitigated by analyzing tissue-derived data from publicly available databases, which helps complement our findings. Lastly, the preliminary nature of our cfRNA signature findings also warrants attention. Compared to the extensively studied mechanisms of mRNA, lncRNA, and miRNA, the biological functions and biomarker potential of less-studied RNA species such as tsRNA, piRNA, snRNA, and snoRNA remain largely underexplored. These RNA categories have traditionally been overlooked in the realm of brain tumor research, yet they present a significant opportunity for unraveling complex biological mechanisms and elucidating disease etiology. Given the preliminary stage of these observations, it is essential to pursue additional research and validation in more varied and extensive cohorts. Such endeavors are crucial for firmly establishing the credibility and practicality of these novel biomarkers in the context of brain tumor diagnosis and prognosis.
In conclusion, this study provides proof of principle that cfRNAs, encompassing a broader spectrum of RNA categories beyond miRNAs, are diagnostics and prognostics of brain tumors. The identification of cfRNA signatures and the construction of machine learning models offer new pathways for improved diagnostic precision and personalized medicine for brain tumor patients.
Methods
Patient enrollment
Patients evaluated for a CNS disease (glioma, meningioma, and non-neoplastic diseases) were enrolled at Xiangya Hospital of Central South University from January 2021 to July 2021. Bio-specimen collections (CSF and plasma) were performed within ten days before surgical resection for patients with brain tumor. No patients received any pre-operative anti-cancer treatment. All patients provided written informed consent. This project was approved by the Institutional Review Boards at Xiangya Hospital of Central South University (IRB protocol #202103150).
All patients were followed up for a period of 3.5 years (42 months) or until their death. Events of tumor recurrence or death were recorded. RFS was calculated from the date of surgical resection to the date of tumor recurrence or the last follow-up date. OS was calculated from the date of sample collection to the date of death or the date of the last follow-up.
Sample processing
CSF and blood were drawn in the morning following an overnight fast and collected in EDTA anticoagulated vacutainer tubes. CSF was obtained from the lower part of the spinal column. CSF was centrifuged within 3 hours of collection at 1900g for 10 minutes at room temperature and then the supernatant was carefully separated. Plasma separation was conducted using a double-spin protocol as described previously36. Both types of specimens were then transported to Shenzhen University under cold-chain conditions and were preserved at −80 °C until downstream analyses.
cfRNA extraction, library preparation, and sequencing
The extraction of cfRNA from 400 μl CSF samples was conducted using the RNAiso Plus total RNA extraction reagent (TaKaRa), following the manufacturer’s protocol. For the plasma samples, cfRNA extraction was carried out on 200 μl aliquoted plasma utilizing the Apostle MiniMax™ High Efficiency cfRNA Isolation Kit (Apostle) in accordance with the manufacturer’s protocol. The preparation of cfRNA libraries for both sample types was conducted in strict accordance with the previously reported SLiPiR-seq protocol36. Libraries tagged with distinct sample barcodes were pooled and sequenced on the Illumina NovaSeq 6000 platform (HaploX, Shenzhen) to achieve a minimum of 10 million paired end reads for each sample.
Data processing, read calling, and normalization
The workflow and code for computational processing of raw sequencing data were consistent with previously described36. Read calling was performed for seven types of cfRNAs (aka. lncRNAs, mRNAs, miRNAs, piRNAs, snRNAs, snoRNAs, and tsRNAs). Quality controls were performed for the processed data of all samples. Low-quality samples were excluded based on the following exclusion criteria: (1) less than 3 million clean reads (reads >15nt in length); (2) clean reads ratio (clean reads / total reads) lower than 20%. As a result, 5 CSF and 1 plasma samples were excluded. Reads per million (RPM) normalization and log2 transformation were performed on the cfRNA expression matrix. Transcriptome-wide Pearson’s correlation analysis was performed on the normalized count matrix.
Differential expression analysis
Differential expression test was performed on three sets of comparisons for both CSF and plasma samples: (1) Glioma patients vs. Tumor-free controls; (2) Meningioma patients vs. Tumor-free controls; (3) Glioma patients vs. Meningioma patients. The count matrix was fitted to a negative binomial distribution using R package ‘DESeq2’66. Inclusion criteria for DE cfRNA candidates were: (1) BH-FDR <0.1; (2) absolute log2 fold change >0.5; (3) mean normalized counts across all samples >5. Cross analysis was applied to identify group-specific DE cfRNAs. Wilcoxon rank sum test (p <0.05) and variance calculation ( <10 in each group) were further applied to the overlapping cfRNAs to remove candidates with large dispersion within a group.
Feature selection
The study cohort was randomly partitioned into a discovery set and a validation set in a 80%–20% manner using R package ‘caret’67. The discovery set was further randomly partitioned into a training set and a test set, with a proportion of 3:1. The training set was then used for feature selection. The sample partition and feature selection process were repeated 100 times (bootstrapping). CSF and plasma samples were partitioned independently. Least absolute shrinkage and selection operator (LASSO) regularized binomial generalized linear model (R package ‘glmnet’68) and a decision tree-based strategy, Boruta (R package ‘Boruta’), were applied for feature selection. For LASSO, the parameter ‘lambda.1se’, identified through 10-fold cross-validation, was selected for its ability to yield a model that is both less complex and demonstrates minimal deviance. A cfRNA was considered as a discriminative cfRNA if it received a non-zero coefficient in at least 10 times out of 100 iterations. For Boruta, the maximum number of importance source runs was set to 500. Features that were rated as high importance in at least 50 of the 100 repetitions were considered as candidates.
Machine learning analysis
The study cohort was partitioned into a training set and a test set, and a validation set, as described above in the feature selection. Models were constructed on the training set and verified using both the test and independent validation sets. To minimize sampling bias, the partitioning, training, and validation were repeated 100 times. Three different machine learning algorithms, including Ridge regularized LR, RF, and SVM with Linear Kernel (svmLinear), were applied in this study with default parameters using the R packages ‘glmnet’, ‘randomForest’69, and ‘caret’, respectively. The performance of models was evaluated by AUC. The final prediction result was obtained from the median AUC values from all iterations (bootstrap aggregating).
Survival analysis
The categorized expression status of each DE cfRNA in a glioma patient was determined based on its position in the distribution of normalized sequencing reads among the tumor-free controls. For upregulated cfRNAs: “upregulated”, if higher than the third quantile; “non”, if lower then the third quantile. For downregulated cfRNAs: “downregulated”, if lower than the first quantile; “non”, if higher than the first quantile. The multi-target risk score of an individual was calculated by the sum of cfRNAs with aberrant expression (“upregulated” or “downregulated” counts as 1, “non” counts as 0). The risk score was dichotomized at its 60% quantile: “high risk” if above the 60% quantile value and “low risk” if below the 60% quantile value. Univariate cox proportional hazards regression was performed on the two groups of patients (“upregulated” vs. “non”; “downregulated” vs. “non”; “high risk” vs. “low risk”) using R package ‘survival’. Multivariate cox regression was conducted by integrating risk factors including cfRNA risk score, age, IDH mutation status, and 1p/19q codeletion status. Kaplan–Meier survival curves were plotted by R package ‘survminer’.
In-silico analysis
Tissue-derived RNA-seq data were obtained from TCGA (primary tumor tissues), GTEx (normal tissues), and GSE101638 (42 meningioma patients). Gene expression levels in Fragments Per Kilobase of transcript per Million mapped reads (FPKM) were calculated across 18 types of cancer (TCGA) and 34 kinds of tissues (GTEx). Survival analysis was performed on the normalized expression levels of glioma tumor tissue from 141 cases. Gene expression status was dichotomized by the maximally selected rank statistics from the ‘maxstat’ R package.
Data availability
The logarithmically transformed and Reads Per Million normalized read count matrix for all detected cfRNAs and all de-identified studied samples is available in Supplementary Data 1. Tissue-derived bulk RNA-seq data obtained from TCGA and GTEx for the genes of interest are available in Supplementary Data 4 (tissue of origin analysis) and Supplementary Data 6 (survival analysis). The processed sequencing data (high quality reads) for clinical samples reported in this paper have been deposited in the Genome Sequence Archive in National Genomics Data Center, China National Center for Bioinformation/Beijing institute of Genomics, Chinese Academy of Sciences [GSA-Human: HRA007247]. The sequencing data for clinical samples are under controlled access and are available upon request from the corresponding author to comply with the ethical regulations of Shenzhen University. Applicants should have obtained ethical approval from their ethics committees and submitted a research proposal for the data request. The timescale for granting access would be approximately one month and there are no restrictions on the duration of access. Reference genome for alignment and read calling: miRNA (miRbase, https://www.mirbase.org); piRNA (piRNABank, http://pirnabank.ibab.ac.in/); tsRNA (MINTbase, https://cm.jefferson.edu/MINTbase/); GRCh38 (hg38, https://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/); GENCODE (https://www.gencodegenes.org/human/stats_41.html).
Code availability
All computational analyses were performed using Linux shell, Python 3, or R 4.3. Scripts used to generate the findings in this study are available on Zenodo at https://doi.org/10.5281/zenodo.14869536.
References
Louis, D. N. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 23, 1231–1251 (2021).
Ostrom, Q. T. et al. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2016-2020. Neuro Oncol. 25, iv1–iv99 (2023).
Huang, R. Y. et al. Imaging and diagnostic advances for intracranial meningiomas. Neuro Oncol. 21, i44–i61 (2019).
Islim, A. I. et al. A prognostic model to personalize monitoring regimes for patients with incidental asymptomatic meningiomas. Neuro Oncol. 22, 278–289 (2020).
Verma, N., Cowperthwaite, M. C., Burnett, M. G. & Markey, M. K. Differentiating tumor recurrence from treatment necrosis: a review of neuro-oncologic imaging strategies. Neuro Oncol. 15, 515–534 (2013).
Ceccarelli, M. et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell 164, 550–563 (2016).
Olar, A. et al. Global epigenetic profiling identifies methylation subgroups associated with recurrence-free survival in meningioma. Acta Neuropathol. 133, 431–444 (2017).
Capper, D. et al. DNA methylation-based classification of central nervous system tumours. Nature 555, 469–474 (2018).
Parsons, D. W. et al. An integrated genomic analysis of human glioblastoma multiforme. Science 321, 1807–1812 (2008).
Harmanci, A. S. et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat. Commun. 9, 16215 (2018).
Clark, V. E. et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 339, 1077–1080 (2013).
Clark, V. E. et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat. Genet. 48, 1253–1259 (2016).
Brastianos, P. K. et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat. Genet. 45, 285–289 (2013).
Patel, A. J. et al. Molecular profiling predicts meningioma recurrence and reveals loss of DREAM complex repression in aggressive tumors. Proc. Natl. Acad. Sci. USA 116, 21715–21726 (2019).
Cancer Genome Atlas Research, N Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Sahm, F. et al. DNA methylation-based classification and grading system for meningioma: a multicentre, retrospective analysis. Lancet Oncol. 18, 682–694 (2017).
Nassiri, F. et al. DNA methylation profiling to predict recurrence risk in meningioma: development and validation of a nomogram to optimize clinical management. Neuro Oncol. 21, 901–910 (2019).
Maas, S. L. N. et al. Integrated Molecular-Morphologic Meningioma Classification: A Multicenter Retrospective Analysis, Retrospectively and Prospectively Validated. J. Clin. Oncol. 39, 3839–3852 (2021).
Nassiri, F. et al. A clinically applicable integrative molecular classification of meningiomas. Nature 597, 119–125 (2021).
Olar, A. et al. IDH mutation status and role of WHO grade and mitotic index in overall survival in grade II-III diffuse gliomas. Acta Neuropathol. 129, 585–596 (2015).
Choudhury, A. et al. Meningioma DNA methylation groups identify biological drivers and therapeutic vulnerabilities. Nat. Genet. 54, 649–659 (2022).
Bayley, J. C. T. et al. Multiple approaches converge on three biological subtypes of meningioma and extract new insights from published studies. Sci. Adv. 8, eabm6247 (2022).
Berzero, G., Pieri, V., Mortini, P., Filippi, M. & Finocchiaro, G. The coming of age of liquid biopsy in neuro-oncology. Brain 146, 4015–4024 (2023).
Mair, R. & Mouliere, F. Cell-free DNA technologies for the analysis of brain cancer. Br. J. Cancer 126, 371–378 (2022).
Nassiri, F. et al. Detection and discrimination of intracranial tumors using plasma cell-free DNA methylomes. Nat. Med. 26, 1044–1047 (2020).
Pages, M. et al. Liquid biopsy detection of genomic alterations in pediatric brain tumors from cell-free DNA in peripheral blood, CSF, and urine. Neuro Oncol. 24, 1352–1363 (2022).
Ignatiadis, M., Sledge, G. W. & Jeffrey, S. S. Liquid biopsy enters the clinic - implementation issues and future challenges. Nat. Rev. Clin. Oncol. 18, 297–312 (2021).
Heitzer, E., Haque, I. S., Roberts, C. E. S. & Speicher, M. R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 20, 71–88 (2019).
Miller, A. M. et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nature 565, 654–658 (2019).
Piccioni, D. E. et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 8, CNS34 (2019).
Sabedot, T. S. et al. A serum-based DNA methylation assay provides accurate detection of glioma. Neuro Oncol. 23, 1494–1508 (2021).
Herrgott, G. A. et al. Detection of diagnostic and prognostic methylation-based signatures in liquid biopsy specimens from patients with meningiomas. Nat. Commun. 14, 5669 (2023).
Ricklefs, F. L. et al. Diagnostic potential of extracellular vesicles in meningioma patients. Neuro Oncol. 24, 2078–2090 (2022).
Vorperian, S. K., Moufarrej, M. N., Tabula Sapiens, C. & Quake, S. R. Cell types of origin of the cell-free transcriptome. Nat. Biotechnol. 40, 855–861 (2022).
Larson, M. H. et al. A comprehensive characterization of the cell-free transcriptome reveals tissue- and subtype-specific biomarkers for cancer detection. Nat. Commun. 12, 2357 (2021).
Wang, J. et al. Terminal modifications independent cell-free RNA sequencing enables sensitive early cancer detection and classification. Nat. Commun. 15, 156 (2024).
Wang, C. et al. tRF-AspGTC Promotes Intracranial Aneurysm Formation by Controlling TRIM29-Mediated Galectin-3 Ubiquitination. Research-China 8, https://doi.org/10.34133/research.0574 (2025).
Vasudevan, H. N. et al. Comprehensive molecular profiling identifies FOXM1 as a key transcription factor for meningioma proliferation. Cell Rep. 22, 3672–3683 (2018).
Zhao, J., Ma, W. & Zhao, H. Loss of heterozygosity 1p/19q and survival in glioma: a meta-analysis. Neuro Oncol. 16, 103–112 (2014).
Han, S. et al. IDH mutation in glioma: molecular mechanisms and potential therapeutic targets. Br. J. Cancer 122, 1580–1589 (2020).
Baichurina, I., Valiullin, V., James, V., Rizvanov, A. & Mukhamedshina, Y. The study of cerebrospinal fluid microRNAs in spinal cord injury and neurodegenerative diseases: methodological problems and possible solutions. Int. J. Mol. Sci. 23, https://doi.org/10.3390/ijms23010114 (2021).
Baraniskin, A. et al. Identification of microRNAs in the cerebrospinal fluid as biomarker for the diagnosis of glioma. Neuro Oncol. 14, 29–33 (2012).
Akers, J. C. et al. A cerebrospinal fluid microRNA signature as biomarker for glioblastoma. Oncotarget 8, 68769–68779 (2017).
Kopkova, A. et al. Cerebrospinal fluid microRNA signatures as diagnostic biomarkers in brain tumors. Cancers (Basel) 11, https://doi.org/10.3390/cancers11101546 (2019).
Abbott, N. J., Patabendige, A. A., Dolman, D. E., Yusof, S. R. & Begley, D. J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 37, 13–25 (2010).
Mouliere, F. et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 10, https://doi.org/10.1126/scitranslmed.aat4921 (2018).
Lavon, I., Refael, M., Zelikovitch, B., Shalom, E. & Siegal, T. Serum DNA can define tumor-specific genetic and epigenetic markers in gliomas of various grades. Neuro Oncol. 12, 173–180 (2010).
Wu, Z. et al. Increased expression of microRNA-9 predicts an unfavorable prognosis in human glioma. Mol. Cell Biochem. 384, 263–268 (2013).
Geng, L. et al. Targeting miR-9 in glioma stem cell-derived extracellular vesicles: a novel diagnostic and therapeutic biomarker. Transl. Oncol. 22, 101451 (2022).
Zottel, A. et al. Analysis of miR-9-5p, miR-124-3p, miR-21-5p, miR-138-5p, and miR-1-3p in Glioblastoma Cell Lines and Extracellular Vesicles. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21228491 (2020).
Lucero, R., et al. Glioma-derived miRNA-containing extracellular vesicles induce angiogenesis by reprogramming brain endothelial cells. Cell Rep. 30, 2065–2074.e2064 (2020).
Madelaine, R. et al. MicroRNA-9 couples brain neurogenesis and angiogenesis. Cell Rep. 20, 1533–1542 (2017).
Zhu, M., Zhao, W., Zhao, H. & Zhang, J. Diagnostic and prognostic value of microRNA-193b in patients with glioma and its effect on tumor progression. Oncol. Lett. 18, 4882–4890 (2019).
Ma, K. et al. Common fragile sites: genomic hotspots of DNA damage and carcinogenesis. Int J. Mol. Sci. 13, 11974–11999 (2012).
Li, Z. et al. An HDAC1-binding domain within FATS bridges p21 turnover to radiation-induced tumorigenesis. Oncogene 29, 2659–2671 (2010).
Zhang, L. et al. Loss of fragile site-associated tumor suppressor promotes antitumor immunity via macrophage polarization. Nat. Commun. 12, 4300 (2021).
Zhang, X. et al. FATS is a transcriptional target of p53 and associated with antitumor activity. Mol. Cancer 9, 244 (2010).
Song, J. et al. Association study between C10orf90 gene polymorphisms and colorectal cancer. Front Oncol. 13, 1192378 (2023).
Frattini, V. et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat. Genet 45, 1141–1149 (2013).
Zhen, Y. et al. Protocadherin 15 suppresses oligodendrocyte progenitor cell proliferation and promotes motility through distinct signalling pathways. Commun. Biol. 5, 511 (2022).
Wang, G. et al. The TMEM132B-GABA(A) receptor complex controls alcohol actions in the brain. Cell 187, 6649–6668 e6635 (2024).
Herrera-Quiterio, G. A. & Encarnacion-Guevara, S. The transmembrane proteins (TMEM) and their role in cell proliferation, migration, invasion, and epithelial-mesenchymal transition in cancer. Front Oncol. 13, 1244740 (2023).
Yeo, A. T. et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat. Immunol. 23, 971–984 (2022).
El Khoury, W. & Nasr, Z. Deregulation of ribosomal proteins in human cancers. Biosci Rep 41, https://doi.org/10.1042/BSR20211577 (2021).
Shirakawa, Y. et al. Ribosomal protein S6 promotes stem-like characters in glioma cells. Cancer Sci. 111, 2041–2051 (2020).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Kuhn, M. Building Predictive Models in R Using the caret Package. Journal of Statistical Software, https://doi.org/10.18637/jss.v028.i05 (2008).
Friedman J. H. T., Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. Journal of Statistical Software 33, 1–22, https://doi.org/10.18637/jss.v033.i01 (2010).
Liaw, A. & W, M. Classification and Regression by randomForest. R. N. 2, 18–22 (2002).
Acknowledgements
This work was supported by Shenzhen Medical Research Fund (C2404001, B2302015) (DM.G.); Noncommunicable Chronic Diseases-National Science and Technology Major Project (2024ZD0526702) (DM.G.); National Natural Science Foundation of China (82241022, 82170070) (D.M.G.); National Key Research and Development Program of China (2022YFE0133400) (Q.L.);National Natural Science Foundation of China (82172834) (Q.L.); National Natural Science Foundation of China (82200067, 82470056) (J.W.); We thank the Instrument Analysis Center of Shenzhen University and the Public Service Platform for Large-Scale Instruments and Equipment of the College of Life Sciences and Oceanography for their assistance in instruments and equipment. We are grateful to all the participants who made this study possible through the donation of blood and cerebrospinal fluid samples.
Author information
Authors and Affiliations
Contributions
D.M.G. and Q.L. provided financial support and supervised the study. D.M.G., Q.L., J.Y.H., J.X.Z., and J.W. conceptualized the study. J.X.Z., H.Y.L., and C.W.W. collected and processed biospecimens and participants’ clinical information. J.X.Z. performed periodic follow-ups with patients. Y.Y.T., S.Y.Q., X.Y.L., and Q.W.G. executed cfRNA extraction and library preparation. J.Y.H., Y.Y.H, J.W., F.L.Z., K.K. and Y.Q.N. performed computational analyses. J.Y.H., L.Z., and Y.Y.H. formatted the layout of the figures. J.Y.H. wrote the original draft of the manuscript. J.Y.H., J.X.Z., Y.Y.H., and S.Y.Q. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
D.M.G, J.W., and J.Y.H. are listed as inventors on patents filed related to this work (China National Intellectual Property Administration, 202211648335.5, “A splint ligation based library preparation method for small RNAs”). No part of this paper may be used, reproduced, or distributed for commercial purposes without the express written permission of the patent holders. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, J., Zhou, J., Wang, J. et al. Diagnostic and prognostic potential of cell-free RNAs in cerebrospinal fluid and plasma for brain tumors. npj Precis. Onc. 9, 123 (2025). https://doi.org/10.1038/s41698-025-00909-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-025-00909-6
