
Abstract
Soft tissue sarcomas (STS), including rhabdomyosarcoma (RMS), exhibit significant heterogeneity and limited responsiveness to immune checkpoint blockade (ICB). Unsupervised tumor immune phenotype based on multi-omics expression profiling of STS has been less studied. To reveal the tumor immune phenotype of STS and identify promising therapeutic targets, multi-omics expression profiling across various subtypes of STS was investigated. Here, we established a novel molecular classifier based on immune cell subsets related to TGFβ1 and IFNγ to identify distinct immune phenotypes with higher or lower cytotoxic contents. Immune-high clusters demonstrated enriched immune cell infiltration, elevated IFNγ-related signatures, and favorable clinical outcomes. In contrast, immune-low clusters were enriched for immunosuppressive cell types and exhibited poor survival. CHEK1 emerged as a key node associated with immunosuppressive phenotypes and was significantly overexpressed in immune-low tumors. In situ analysis of independent validation cohorts revealed the significant correlation between CHEK1 and tumor-infiltrating immune cells. Collectively, our findings establish a novel risk assessment strategy for RMS and STS patients, and highlight the potential of CHEK1 as a promising therapeutic target in combination with immune checkpoint inhibitor therapy.
Similar content being viewed by others


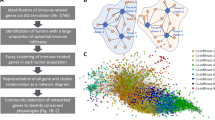
Introduction
Soft tissue sarcoma (STS) is a heterogeneous group of mesenchymal tumors encompassing more than 60 histological subtypes. Rhabdomyosarcoma (RMS) is one of the most common STS in children and adolescents, representing 5% of all childhood cancers1,2. RMS is differentiated from primitive mesenchymal stem cells, which cannot fully differentiate into skeletal muscle. It can occur anywhere in the human body, with the head and neck being the most common primary site3,4,5. RMS is divided into four subtypes based on its clinical and pathological characteristics: embryonic rhabdomyosarcoma (ERMS), alveolar rhabdomyosarcoma (ARMS), pleomorphic rhabdomyosarcoma (PRMS), and sclerosing rhabdomyosarcoma (SSRMS)6,7,8,9.
ERMS and ARMS are two major histological subtypes of RMS. ERMS occurs more commonly in younger children and has a more favorable prognosis. Histologically, ERMS showed primitive oval to spindle cells with minimal cytoplasm, resembling immature skeletal myoblasts. ERMS has a wide range of genetic alterations, termed PAX-fusion-negative or fusion-negative RMS7,10,11. ARMS occurs mainly in adolescents with poorer prognoses and characteristically exhibits an alveolar pattern with cells distributed around an open central space. The importance of the PAX-FOXO1 fusion has been highlighted in the ARMS pathomechanism12,13. Despite advances in multi-disciplinary treatment for RMS and STS, consisting of surgery, irradiation, chemotherapy, and targeted therapy, the clinical prognosis of patients has only improved slightly, and promising curative treatment remains a significant challenge14,15,16.
Transforming growth factor beta 1 (TGFβ1) is the most potent inhibitor of myogenic differentiation in RMS and is central to immune suppression within the tumor microenvironment4,17,18,19,20. Although immune checkpoint inhibition has demonstrated promise in improving clinical outcomes for certain cancers, soft tissue sarcomas remain limited in effectiveness in immune checkpoint blockade (ICB) based on current clinical trials21,22. The current revolution in understanding the molecular landscape clarifies that STS is ‘non-immunogenic’ with a low tumor mutation burden (TMB) and PD-L1 expression23. Interferonγ (IFNγ) is a cytokine pivotal in regulating PD-L1 expression and antitumor immunity24. Owing to the rarity and heterogeneity of STS, few studies have investigated the tumor microenvironment (TME) and tumor-infiltrating immune cells (TIIC) in different STS histologies, including RMS. A recent study used global gene expression data to define molecular immune signatures to stratify STS into distinct immune phenotypes and identified a subpopulation of patients with improved survival and a high response rate to PD1 inhibitor therapy25. In the interim, immune checkpoint blockade has few therapeutic benefits in STS patients. Therefore, it is urgent to explore combinations for a more efficient immunomodulator.
Here, we developed a novel molecular classification of RMS and STS based on immune cell subsets related to TGFβ1 and IFNγ expression, revealing distinct immune phenotypes. Moreover, we compared multi-omics expression profiles across subgroups of RMS and STS to identify CHEK1 as an unfavorable prognostic biomarker related to immunosuppressive phenotypes. Multiple immunohistochemistry (mIHC) staining assays were applied to assess the correlation between CHEK1 and tumor-infiltrating immune cells (Fig. 1).

Schematic overview of the integrative analysis pipeline used in this study. Transcriptomic, proteomic, and clinical data were collected from public databases (GEO and TCGA) and an RMS cohort. Differential expression analyses and immune deconvolution (CIBERSORTx) were performed to estimate tumor-infiltrating immune cell populations. Unsupervised clustering based on immune cell subsets defined distinct immune phenotypes. CHEK1 was identified as a candidate biomarker associated with immunosuppressive phenotypes and poor prognosis. Validation was conducted via immunohistochemistry (IHC), multiplex immunohistochemistry (mIHC), and survival analyses in independent cohorts.
Results
Distinct immune cell types related to TGFβ1 and IFNγ expression in RMS
We previously identified a role for activated TGFβ signaling in blocking the differentiation of human rhabdomyosarcoma26,27,28. The canonical TGFβ/SMAD signaling exerts significant functions in cancer progression by remodeling the architecture of the carcinomas and by suppressing antitumor immunity29,30. This study analyzed publicly available RNA sequencing (RNA-seq) data (GSE108022) from primary RMS samples. A total of 133 genes from the GSEAsig database (WP_TGFBETA_SIGNALING_PATHWAY) were collected, and the relative gene expression levels were presented in RMS subtypes. We identified 90 differentially expressed genes that were significantly upregulated in RMS as compared with muscle tissues (Supplementary Fig. 1A). The canonical TGFβ/SMAD signaling members were highly expressed in RMS regardless of the subtype (Supplementary Fig. 1B). IHC staining of RMS samples revealed that TGF-beta1 was highly expressed in the majority of primary tumors (n = 9/11 ERMS, n = 7/10 ARMS, and n = 8/9 PRMS) (Supplementary Fig. 1C), which was consistent with the findings of the previous studies26,27,28 and confirmed in an independent fraction of sarcomas within fibrosarcoma, gastrointestinal stromal tumors, and synovial sarcoma (Supplementary Fig. 1D–F). TGFβ, as an immunoregulatory master of the tumor microenvironment, has been less studied in soft tissue sarcomas, including RMS. IFNγ, an important cytokine, is critical for coordinating the antitumor immune response31. Activated IFNγ signaling upregulates PD-L1 expression and immune cell infiltration, which may improve the response to anti-PD-1 immunotherapy32.
Next, we analyzed the correlation between TGFβ1 and IFNγ across 33 tumor types from The Cancer Genome Atlas (TCGA) (Pan-cancer cohort) (Fig. 2A). A statistically significant positive correlation was observed in 24 TCGA cohorts, including TCGA-SARC. Only the TCGA-HNSC cohort revealed a negative association. Publicly available gene expression data (GSE108022) from primary RMS samples were analyzed using the CIBERSORTx deconvolution algorithm to assess the relative immune fraction scores of distinct immune cell subtypes. Statistically significant positive or negative associations between TGFβ1 (TGFB1) or IFNγ (IFNG) transcript levels and individual immune cell scores were assessed by Spearman correlation analysis. Our results revealed that two immune cell subtypes (naïve B cells and M1 Macrophages) had a significant positive correlation with TGFB1, and four immune cell subtypes (activated NK cells, monocytes, resting mast cells, and eosinophils) were negatively correlated with TGFB1. Meanwhile, we found that IFNγ (IFNG) expression level was positively correlated with three subtypes (naïve B cells, M1 Macrophages, and CD8+ T cells) and negatively correlated with three immune cell subtypes (monocytes, resting mast cells, and M0 Macrophages) (Fig. 2B).

A Spearman correlation analysis between TGFβ1 and IFNγ across 33 TCGA tumour types (Pan-cancer cohort, TCGA-SARC was marked in red). B Heatmap showed the significantly positive and negative Spearman correlation coefficients between either TGFβ1 (TGFB1) or IFNγ (IFNG) transcript levels and relative abundance of eight immune cell types assessed by the CIBERSORTx deconvolution algorithm. C Unsupervised hierarchical cluster analysis revealed two main RMS immune clusters and five subclusters. D, E Violin plots describe the transcript levels of TGFB1 and IFNG among the five subclusters. F Relative fraction scores of the selected eight immune cell types were compared between Cluster A and B2 in box-whisker plots. In all graphs, box-whisker plots show individual samples, group median, and min-max values. Two-sided t test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
All significantly relevant immune cell subtypes (CD8 + T cells, naïve B cells, M1 & M0 Macrophages, activated NK cells, resting Mast cells, Monocytes, and Eosinophils) were selected for further analyses. Unsupervised hierarchical cluster analysis of GSE108022 from primary RMS samples based on the eight selected immune cell subtypes revealed two RMS immune clusters, A and B (Fig. 2C). Cases in cluster A were enriched for CD8 + T cells, naïve B cells, and M1 Macrophages and had higher transcript levels of TGFB1 and IFNG (Fig. 2D, E). Cluster B was divided into four subclusters (B1, B2, B3, and B4) due to the significant differences in M0 Macrophages and activated NK cells. To evaluate whether stratification into molecular immune clusters A and B is also applicable to all sarcomas, transcriptome datasets of the TCGA-SARC cohort were analyzed using CIBERSORTx. Unsupervised hierarchical clustering revealed a similar pattern (Supplementary Fig. 2A). Cluster A was significantly correlated with higher TGFB1 and IFNG (Supplementary Fig. 2B, C). We compared cluster A cases with cluster B2 in the RMS cohort or cluster B3 in the TCGA-SARC cohort. This was particularly common evidence that CD8 + T cells and M1 macrophages were enriched in cluster A from both the RMS and TCGA-SARC cohorts (Fig. 2F, Supplementary Fig. 2D). The other selected immune cell subsets revealed more heterogeneous characterizations in the various subclusters. Interestingly, an inverse finding was observed in activated NK cells and monocytes among clusters A and B2 or B3, indicating a heterogeneous immune niche in the tumor microenvironment of the RMS and SARC cohorts.
Differences in immune gene signature and survival patterns related to immune phenotypes
Immune-related gene expression signatures are associated with immune cell infiltration and clinical response to immune checkpoint blockade (ICB) agents33. Hence, we evaluated the transcript levels of the immune checkpoint and IFNγ-related genes in the subclusters of the RMS and TCGA-SARC cohorts. Cases in cluster A from the RMS and SARC cohorts were enriched for the majority of the selected immune checkpoint and IFNγ-related genes. A 25-gene signature (CD274, PDCD1, PDCD1LG2, CTLA4, HAVCR2, LAG3, IDO1, CXCL10, CXCL9, HLA_DRA, STAT1, IFNG, CD3D, IL2RG, NKG7, CIITA, HLA_E, CD3E, CXCR6, CCL5, GZMK, TAGAP, CD2, CXCL13, and GZMB) was shown in the hierarchical clustering heatmap (Fig. 3A, B). The expression of each selected immune checkpoint gene and IFNγ immune signature scores were compared between clusters A and B2 or B3 in RMS and SARC cohorts (Fig. 3C, D). In the RMS dataset, the higher gene expression of CD274, PDCD1, PDCD1LG2, and CTLA4 was observed in cluster A tumors as compared to cluster B2. Similarly, PDCD1, PDCD1LG2, CTLA4, HAVCR2, and LAG3 were expressed at high levels in cluster A of the SARC cohort as compared to other clusters. Interestingly, CD274 (which encodes PDL1) was heterogeneously expressed in various clusters, which was also found in a previous study using another immune classification tool25. As expected, cluster A tumors have higher IFNγ immune signature scores in RMS and SARC cohorts. Recently, a “tumor inflammation signature” (TIS) was reported to predict the clinical benefit of anti-PD-1 therapy in several clinical trials34,35. TIS scores were also compared among the subclusters from the RMS and TCGA-SARC cohorts. A high TIS score was observed in cluster A as compared to B2 or B3 in RMS and SARC cohorts (Supplementary Fig. 3A, B). In terms of clinical relevance, patients in cluster A with available survival data (TCGA-SARC) exhibited a favorable overall survival as compared to cluster B3 and B4 (p = 0.048 and p < 0.0001, respectively) (Fig. 3E). In addition, patients in cluster A had better disease-free survival than patients in cluster B4 (p = 0.032) (Fig. 3F). These data suggested that our newly established stratification for distinct molecular immune phenotypes can predict clinical outcomes in a STS cohort.

Heatmaps show expression of a 25-gene signature related to immune checkpoints among distinct subclusters from the RMS cohort (A) and TCGA-SARC (B). C Relative expression scores of selected immune genes and signatures were compared between cluster A and B2 from the RMS cohort in box-whisker plots. D Relative expression scores of selected immune genes and signatures were compared among cluster A, B2, B3, and B4 from the TCGA-SARC cohort in box-whisker plots. Overall survival (E) and disease-free survival (F) of cluster A, B2, B3, and B4 from the TCGA-SARC cohort were estimated by Kaplan-Meier plots and two-sided log-rank test. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Differences in gene and protein expression related to the immune phenotype
To unravel relevant genetic alterations, we investigated the fraction of genome altered (FGA) and tumor mutational count (TMC) using the cBioportal web tool. The lowest FGA was found in cluster A as compared to the other clusters (p = 0.0012, Kruskal-Wallis Test, Supplementary Fig. 3C). The top 20 genes with the highest mutation frequency and most significant difference among the four clusters are listed (Supplementary Fig. 3D, F). Next, we identified 7485 and 5474 differentially expressed genes (DEGs) among clusters A and B2 or B3 in the TCGA-SARC and RMS cohorts, respectively. Gene set enrichment analysis (GSEA) was performed to evaluate the enrichment of predefined DEGs between immune subclusters. The top 10 positively enriched pathways are immune‑related. The up‑enrichment of cytokine/chemokine signaling, antigen presentation, complement, TLR/NOD pathways points to a heightened inflammatory or immune surveillance state, consistent with increased immune cell infiltration or activation (Supplementary Fig. 3E). In addition, 21 differentially expressed proteins were also screened in the TCPA-SARC database. In total, nine genes were observed differentially expressed at both the transcript and protein levels (Fig. 4A). Analysis of protein-protein interaction according to the STRING database indicated that these DEGs were closely related to each other (Fig. 4B). Significantly higher CHEK1 expression was found in subclusters B2 and B3 than in cluster A. The transcript level of CHEK1 was significantly elevated in both fusion-negative and fusion-positive RMS tumors compared to that in normal muscle tissue (Fig. 4C). CHEK1 (Checkpoint kinase 1) is a serine/threonine-specific protein kinase encoded in humans by the CHEK1 gene. The transcript level of CHEK1 was positively correlated with the protein levels in the TCGA-SARC cohort (Fig. 4D). Significantly higher CHEK1 transcript and protein expression were found in clusters B3 and B4 in the TCGA-SARC cohorts, which exhibited unfavorable overall survival (Fig. 4E, F). To further investigate the clinical relevance of CHEK1 in sarcoma, patients were divided into CHEK1 expression low and high groups. We performed Kaplan-Meier analysis for patients’ disease-free survival (PFS) and overall survival (OS). Our results revealed that high expression of CHEK1 served as an unfavorable prognostic biomarker at both the transcript and protein levels (Fig. 4G–J).

A Venn diagrams show the number of DEGs and DEPs between cluster A and subcluster B2 (RMS cohort) or B3 (SARC cohort). B Schematic presentation of protein-protein interaction network according to the STRING database. C Normalized expression for CHEK1 of normal muscle, Fusion N, and Fusion P RMS samples are compared in a box-whisker plot. D A dot plot illustrates the significantly positive correlation between CHEK1 protein and transcript levels in tumours of the TCGA-SARC cohort. E, F Box-whisker plots show the transcript and protein levels among cluster A, B2, B3, and B4 from the TCGA-SARC cohort. Kaplan-Meier plots show an unfavourable overall survival (G, I) and disease-free survival (H, J) for patients with higher transcript and protein levels of CHEK1 from the TCGA-SARC cohort. Survival analyses were performed with Kaplan-Meier estimates and two-sided log-rank tests. (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Association between CHEK1 and tumor immune microenvironment in soft tissue sarcomas
Next, we explored the associations between CHEK1 expression levels and tumor-infiltrating immune cells and immune-related genes in the TCGA-SARC and RMS cohorts. The Spearman’s ρ (rho) values of 22 immune cells with CHEK1 gene and protein expression were visualized by a heatmap (Fig. 5A, B). For CHEK1 transcript and protein levels, Spearman correlation analysis revealed nine immune cell subtypes with significant negative associations (M2 macrophages, Monocytes, resting CD4+ memory T cells, naïve B cells, CD8+ T cells, activated NK cells, gamma delta T cells, regulatory T cells, and M1 Macrophages). Three subtypes (M0 macrophages, Plasma cells, and activated dendritic cells) were found to have a significant positive correlation with CHEK1 transcript level. Regarding CHEK1 protein level, three subtypes (M0 macrophages, Plasma cells, and activated CD4+ memory T cells) were positively correlated. The transcript and protein levels of CHEK1 were also found to have statistically significant negative associations with the majority of selected IFNγ-related and expanded immune gene signatures (Fig. 5C, D). We divided the patients from the TCGA-SARC and RMS cohorts into low and high groups depending on the CHEK1 transcript or protein levels. In the RMS cohort, significantly higher scores for naïve B cells and follicular T helper cells were detected in the CHEK1 high group (Fig. 5E, F). Statistically significant low scores for resting CD4+ memory T cells and resting Mast cells were observed in the CHEK1 high group (Fig. 5G, H). In the TCGA-SARC cohort, the scores for six subtypes of immune cells (resting CD4+ memory T cells, regulatory T cells, gamma delta T cells, Monocytes, M1 Macrophages, and M2 macrophages) were significantly different between the CHEK1 transcript low and high levels (Fig. 5I). In terms of CHEK1 protein levels, nine immune cell subtypes (naïve B cells, memory B cells, resting CD4+ memory cells, regulatory T cells, activated NK cells, Monocytes, M1 Macrophages, M2 macrophages, and resting Mast cells) were significantly different between the CHEK1 low and high group (Fig. 5J). All relevant immune cell subtypes were enriched in the CHEK1 low group. We also compared the relative expression of IFNγ-related and expanded immune gene signatures between the CHEK1 low and high groups (Fig. 5K, L). The results revealed that patients with CHEK1 low expression had significantly higher IFNγ-related and expanded immune gene signature scores, which predict the clinical response to ICB therapy in various cancers.

Heatmap showing the Spearman correlation coefficients between CHEK1 transcript (A) and protein (B) expression levels and relative abundance of 22 immune cell types. Spearman’s ρ (rho) values were presented. Heatmap showing the Spearman correlation coefficients between CHEK1 transcript (C) and protein (D) expression levels with IFNγ-related genes as well as expanded immune gene signatures. Spearman’s ρ (rho) values were presented. E–H Violin plot showing the significant differences of four immune cell types between CHEK1 transcript low and high groups in the RMS cohort. Significant differences in immune cell types are compared between the CHEK1 low and CHEK1 high groups in transcript (I) and protein (J) levels. Violin plots showing the significant expression differences of IFNγ-related genes as well as expanded immune gene signatures between the CHEK1 low and CHEK1 high groups in transcript (K) and protein (L) levels. Two-sided t test (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
CHEK1 predicted clinical prognosis and molecular subtypes for RMS
To validate the expression and clinical relevance of CHEK1 for RMS in situ, we analyzed CHEK1 expression in an independent cohort of 33 cases by immunohistochemical staining. A heterogeneous staining pattern ranging from absence to moderate to prominent staining in tumor cells was observed in the RMS tissues (Fig. 6A). High CHEK1 protein levels were detected in the 14 patients. We did not observe any significant difference between CHEK1 low and high subgroups concerning histological types and clinicopathological characteristics, except for disease recurrent status (Fig. 6B, C). We performed Kaplan-Meier survival analysis of patients to confirm that the CHEK1 high expression subgroup was significantly associated with shorter relapse free survival (Fig. 6D). To complement the transcriptional analysis of the TIME between subgroups with different CHEK1 expressions, we analyzed the densities of CD4+, CD8+, T-regulatory cells (Foxp3+), tumor-associated macrophages (CD163 + TAM), and PD-L1 expression in RMS specimens. (Fig. 6E, Supplementary Fig. 4A, B) Using digital image analysis, we found higher densities of main tumor-infiltrating immune cells (CD4+, CD8+, T-reg cells) in the CHEK1 low tumor compared to the CHEK1 high expression. Similar to the immune-checkpoint gene CD274 detected through our analysis of publicly available data, mIHC analysis revealed that the mean PD-L1 expression was not significantly different between the subgroups with low and high CHEK1 expression (Fig. 6F). In line with a previous study36, TAMs predominated the sarcoma immune microenvironment with the highest intratumor density in the RMS specimens. However, we did not observe any significant differences between the CD163+ positive cells of the two RMS subgroups. We also discovered a very small population of CD4+ Foxp3+ Tregs in both subgroups of the RMS specimens. Together, these data suggest that CHEK1 expression serves as a potential prognostic biomarker and is correlated with tumor-infiltrating immune cells in RMS in our independent cohort.

A Representative image of immunohistochemical (IHC) staining for CHEK1 in RMS tumour tissues with the low and high Immunoreactivity. The distribution of histology types (B) and recurrence status (C) is compared between the CHEK1 expression low and high groups. D Kaplan-Meier plot shows an unfavourable relapse-free survival for patients with high CHEK1 expression from the Hefei-RMS cohort. E Multiplex IHC staining for RMS tumours with CHEK1 low and high expression, CD4 (green), CD8 (cyan), Foxp3 (orange), CD163 (red), PDL1 (yellow), DAPI staining is shown in blue. F Densities of immune cells and PD-L1 are compared between the CHEK1 expression low and high groups. Data are presented as mean values ± SEM. Two-sided t test (*p < 0.05, ns nonsignificant).
CHEK1 expression evaluation in soft tissue sarcomas and correlated with immune cell densities
To clarify more precisely the associations between CHEK1 expression and tumor-infiltrating immune cells in soft tissue sarcomas, we examined tumor samples of formalin-fixed paraffin-embedded (FFPE) tissue microarray (TMA) from 91 patients with STS. We observed a range of undetectable to prominent staining intensity in tumor cells by IHC staining (Fig. 7A). The CHEK1 staining intensity H-score was quantified using QuPath software for patients. Patients were divided into two subgroups according to the staining intensity with CHEK1high (n = 50) and CHEKlow (n = 41) for further analysis. We compared the distribution of CHEK1 expression across histological subtypes and found that the majority of synoviosarcoma tumors revealed higher CHEK1 expression (Fig. 7B). All available clinico-pathological features were compared between CHEKhigh and CHEKlow groups. In line with our results from the RMS cohort data, Chi-square analysis showed that high CHEK1 expression was significantly associated with advanced clinical stage (p = 0.006) (Fig. 7C). Regretfully, follow-up survival records of the TMA cohort were unavailable for further survival analysis. To complement the transcriptional analysis of associations between CHEK1 expression levels and tumor-infiltrating immune cells, we performed an Opal multiplex IHC tissue staining assay to characterize the soft tissue sarcoma TME (Fig. 7D). The densities of T cells (CD3+), CD4+ T cells (CD4+), CD8+T cells (CD8+), CD19 + B cells (CD19+), T-regulatory cells (Foxp3+), pan-macrophages (CD68+), M2-like macrophage (CD163+), and PD-L1 in each competition for tumor sections were calculated (Fig. 7E). A significant increase in the overall density of Foxp3, CD19+, CD68+, CD163+ and CD68 + CD163+ cells was observed in the CHEKlow group tumors. However, infiltration of T cells (CD3+, CD4+, and CD8+) and PD-L1 densities were not significantly different between the CHEKhigh and CHEKlow groups. Altogether, our digital image analysis strongly supports the immune deconvolution results that CHEK1 serves as an unfavorable prognostic biomarker related to diminished cytotoxic immune cell infiltration in STS.

A Representative images of Immunohistochemical (IHC) staining for CHEK1 in RMS tumour tissues with low and high Immunoreactivity. The distribution of distinct histology types (B) and clinical stages (C) is compared between CHEK1 expression low and high groups. D A representative multiplex IHC staining image for tissue microarrays. The densities (immune cells/mm2) of T cells (CD3+), CD4+ T cells (CD4+), CD8+ T cells (CD8+), CD19+ B cells (CD19+), T-regulatory cells (Foxp3+), pan-macrophage (CD68+), M2-like macrophage (CD163+), and PD-L1 in each compete for tumour sections were calculated. E Densities of immune cells and PD-L1 are compared between the CHEK1 expression low and high groups. Data are presented as mean values ± SEM. Two-sided t test (*p < 0.05, ns no significant).
Discussion
We previously reported that the TGFβ1 signaling pathway contributes to the growth and differentiation of RMS26. The immune cell subsets based on TGFβ1 and IFNγ expression in RMS have not been investigated. Here, utilizing a public oncogenic database, we revealed that TGFβ/SMAD signaling is highly expressed in human rhabdomyosarcoma. In addition, we established a new stratification model based on RNA expression profiling for RMS with distinct molecular immune phenotypes, which was validated in the TCGA-SARC cohort. Predefined immune gene signatures related to ICB responses were evaluated as statistically significant among distinct subgroups. Higher IFNγ-related and expanded immune gene signatures contribute to an improved prognosis. Importantly, the assessment of the variation in transcript and protein expression of DEGs with higher or lower cytotoxic immune phenotypes highlighted that CHEK1 served as an unfavorable biomarker and is related to reduced cytotoxic immune cell infiltration in soft tissue sarcomas. Finally, the density and distribution of tumor-infiltrating immune cells in CHEK1 low- and high-expression soft tissue sarcomas were evaluated using multiplex immunofluorescence staining. Elevated expression of CHEK1 in sarcomas was correlated with lower infiltrating immune cells, indicating that CHEK1 is a potential predictor of clinical prognosis and a potential novel adjuvant ICI therapeutic target for sarcoma.
TGFβ has three isoforms, TGFβ1, TGFβ2, and TGFβ3, which belong to a 33-member cytokine superfamily. TGFβ signaling has widespread and diverse effects on cell proliferation, differentiation, adhesion, migration, metabolism, and immune homeostasis. TGFβ1, as a modulator of RMS cell differentiation, plays a significant role in tumor growth and progression29,37,38. In the present study, RNA-seq data and IHC staining of tumor tissues from RMS revealed that canonical TGFβ/SMAD signaling was highly expressed in human RMS. The interaction between TGFβ1 and IFNγ plays a pivotal role in regulating antitumor host immunity. Activated IFNγ signaling upregulates PD-L1 expression and immune cell infiltration, which may improve the response to anti-PD-1 immunotherapy. RMS is a type of sarcoma with a heterogeneous group of soft-tissue tumors. Immunotherapy in RMS has limited effectiveness so far. Several previous studies have investigated the tumor microenvironment (TME) composition in different soft tissue sarcomas to understand the immune niche that maintains the tumor and how to mediate cancer immune escape. In 2020, Chen and colleagues36 delineated the immune characteristics of specific TMEs in RMS and undifferentiated pleomorphic sarcomas (UPS) and revealed an immunosuppressive TME dominated by tumor-associated macrophages. They speculated that in situ T-cell distribution in the STS TME could overcome the immunosuppressive niche and play a predominant role in ICB responsiveness. Interestingly, another study established an immune-based classification of TME composition using the microenvironment cell populations-counter method in sarcoma. They reported three main immune phenotypes in soft tissue sarcomas: immune-low, immune-high, and highly vascularized subgroups, with different clinical outcomes and response rates to ICB. Their study defined a subgroup of sarcoma patients who benefited from ICB, marked by a high density of B cells and the presence of tertiary lymphoid structures (TLS)25.
The present study differs from previous reports in that we conducted an integrative deconvolution analysis of multi-omics data from RMS to cluster the patients into subgroups depending on the immune cells related to TGFβ1 and IFNγ. In addition, we used the public dataset TCGA-SARC to validate our new stratification model and evaluate the prognostic outcomes of subgroups with lower or higher cytotoxic immune phenotypes. Consistently, the immune gene signature and tumor inflammation signature were enriched in cluster A, which is an accurate and independent predictive biomarker for ICB clinical outcome. We identified CHEK1 as a critical predictive biomarker in distinct TIME and as a potential therapeutic target to increase TILs and improve the clinical efficacy of ICB therapy in RMS and STS.
CHEK1 is a crucial mediator of cell cycle progression in response to DNA damage. The critical function of CHEK1 in normal and germinal center B-cell development, lymphomagenesis, and survival has been reported39,40. Therapeutic CHEK1 inhibition combined with BCR-signalling blockade in patients might improve the efficacy in eradicating B-cell lymphoma and leukemia cells40. Recently, several efforts have tested CHEK1 signaling drugs in combination with ICB in other human malignancies41,42,43. One study by Sato et al.44 highlights the critical role of CHK1 in regulating PD-L1 expression and immune response, indicating the translational application of CHEK1 agents in combination with ICB therapy. Most studies have demonstrated that the cell-cycle-related kinase function of CHEK1 is a promising therapeutic strategy for STS45,46,47,48. However, few studies reported the association between CHEK1 expression and immune cell infiltration in STS. Notably, the immune landscape of STS exhibits considerable heterogeneity across different histological subtypes, with implications for the treatment response and prognosis. By applying an integrative analysis of multi-omic expression profiling, our findings unraveled the heterogeneity and complexity of the immune microenvironment across distinct subclusters and significantly correlated with CHEK1 expression. In addition, we provide strong experimental evidence that highly infiltrating immune cells in STS were observed in the subgroup of patients with CHEK1 low expression, which revealed a favorable clinical outcome.
However, this study has some limitations. We analyzed only the quantitative relationship between CHEK1 expression and immune cell infiltration using tissue microarrays, without evaluating the spatial organization of immune cells within the tumor microenvironment. In future studies, we plan to expand the sample size and incorporate spatial proximity analysis of immune cell populations. By employing multiplex marker co-expression and high-resolution imaging, we aim to achieve in situ phenotypic characterization and visualization at the single-cell level. This approach will enable more refined classification of immune and tumor cell subtypes and allow for quantitative and spatial analyses of intercellular interactions within the tumor microenvironment. Moreover, multivariate prognostic Modeling of a large cohort should be applied to validate the prognostic association of CHEK1 and patients with RMS. Nevertheless, the prognostic significance of the immune cell density and spatial location in STS after ICB therapy is not completely understood. Therefore, a larger cohort with a more extended follow-up period, ideally from prospective clinical trials, is required to confirm our findings.
In conclusion, we established a novel molecular classifier based on immune cell subsets related to TGFβ1 and IFNγ expression to identify distinct immune phenotypes with high or low cytotoxic content. Additionally, we compared multi-omics expression profiles across subgroups of RMS and STS to identify CHEK1 as an unfavorable prognostic biomarker associated with immunosuppressive phenotypes. These findings provide new insights into the immune landscape of sarcomas and highlight the potential of CHEK1 as a promising therapeutic target in combination with immune checkpoint inhibitor therapy.
Methods
Patient cohorts and samples
A total of patients with RMS were enrolled, including 33 samples from our institute (titled the Hefei-RMS cohort). The tissue samples used in this study were obtained from patients with rhabdomyosarcoma diagnosed between 2016 and 2019, who were diagnosed with RMS according to the World Health Organization (WHO) guidelines. Paraffin-embedded RMS tissues were collected from the First Affiliated Hospital of the University of Science and Technology of China (USTC) for immunohistochemistry and immunofluorescence staining. Written informed consent was obtained from all participants or family members in the study. The research protocol was approved by the Ethics Committee of the First Affiliated Hospital of USTC (Ethic No 2024/RE256) in accordance with the Declaration of Helsinki.
Expression profiling and clinical datasets
RNA-seq data for 106 samples, including five normal muscles and 101 Rhabdomyosarcomas, were downloaded from the Gene Expression Omnibus (GSE108022). The TCGA-SARC cohort RNA, protein expression, and clinical data were downloaded from https://www.cbioportal.org/ in December 2022. The curated gene sets for TGFBETA_SIGNALING_PATHWAY were obtained from https://www.gsea-msigdb.org/gsea/msigdb. Immune checkpoint, IFNγ-related gene sets, and the tumor inflammation signature (TIS) were obtained from the published literature25,35,49. Fractional genomic alterations and mutation frequencies were analyzed using the cBioportal tool.
Histology and immunohistochemical staining
Tissue microarrays were purchased from Bioaitech Company (Xi’an, China), comprised 91 soft tissue sarcomas. RMS and soft tissue sarcoma tumors were fixed in 4% PFA, processed, and embedded in paraffin. Histological sections were stained with Hematoxylin and Eosin or immunohistochemical staining. The tissue sections were deparaffinized and rehydrated using the following steps: melting the wax at 65 °C for 2 h, 3 × 5 min xylene, 2 × 3 min 100% ethanol, 3 min 95% ethanol, 3 min 75% ethanol, and finally rinsed with water. Tissue sections were incubated with 10 mM sodium citrate buffer (pH 6.0) (Boster, Wuhan, China) in a microwave twice for 15 min each. After antigen retrieval, use 3% peroxidase solution to block endogenous enzymes (Chemical Technology, Jiangsu Yonghua, China) for 10 min, block with 5% BSA (Boster) for 20 min, and incubate the primary antibody overnight at 4 °C. The sections were incubated with biotinylated anti-rabbit secondary antibody (Boster) for 2 h. A solution of streptavidin-HRP (Boster) and peroxidase substrate (DAB) (MXB Biotechnologies, Fuzhou, China) was used to generate signals in tissue sections. CHEK1 staining scores were automatically determined using QuPath50 (version 0.3.2), reflecting positive cells and staining intensity. Automated cell detection was performed using QuPath’s built-in cell detection algorithm with hematoxylin channel thresholding, followed by positive cell classification based on DAB optical density. A supervised classifier was trained on representative areas to accurately distinguish CHEK1-positive and -negative tumor cells. Staining intensity was categorized into low, medium, and high based on preset DAB thresholds, and H-scores were calculated accordingly. Cases were stratified into CHEK1 high and CHEK1 low groups based on median H-score cutoffs. Cell density (cells/mm²) and percentage of positive cells were exported for statistical comparisons. Batch analysis was performed across all TMA cores to ensure consistent parameter application. Specific antibodies are indicated and outlined in Supplementary Table 1.
Immune cell scores deconvolution
Absolute immune cell infiltration levels from gene expression were predicted by CIBERSORTx using the website server (https://cibersortx.stanford.edu). The TPM-normalized expression matrix datasets from GSE108022 and TCGA-SARC were used as the input mixture files, and the relative levels for the 22 immune cells were computed by the LM22 gene signature.
Unsupervised hierarchical clustering
Transcriptome count data of genes were ln(x + 1)-transformed and clustered using correlation distance and average linkage. ClustVis, a web tool for visualizing multivariate data, was utilized for unsupervised hierarchical clustering and to visualize data in a heatmap51.
Spearman rank correlation analysis
All pairwise associations between continuous variables (e.g., CHEK1 expression vs. immune cell infiltration scores, or between gene expression levels) were assessed using Spearman’s rank-order correlation. Briefly, normalized expression data (log₂-transformed TPM or CPM values) and immune infiltration estimates (e.g., CIBERSORTx fractions) were imported into R (v4.2.1). For each variable pair, the cor.test() function (stats package) was run with method = “spearman” to compute the Spearman correlation coefficient (ρ) and two-sided p‑value. Multiple testing correction across all tested pairs was performed using the Benjamini–Hochberg procedure to control the false discovery rate (FDR), with adjusted p < 0.05 considered statistically significant.
Gene set enrichment analysis (GSEA)
Gene set enrichment analysis (GSEA) was performed to evaluate the enrichment of predefined gene sets between immune subclusters. Transcriptome-wide gene expression data were first ranked based on the signal-to-noise ratio between clusters of interest (e.g., Cluster A vs. Cluster B2 in RMS or Cluster A vs. Cluster B3 in SARC). The analysis was conducted using the GSEA software (v4.3.2, Broad Institute, Cambridge, MA) with 1000 phenotype permutations to assess statistical significance. Gene sets were retrieved from the Molecular Signatures Database (MSigDB v7.5), including Hallmark gene sets (H: hallmark gene sets), curated canonical pathways (C2: KEGG, Reactome), and immunologic signatures (C7). A false discovery rate (FDR) < 0.25 and a nominal p-value < 0.05 were considered statistically significant, in accordance with GSEA best practices. Enrichment plots and leading-edge subsets were used to identify core genes contributing to the enrichment signal.
Multiple Immunohistochemistry staining and image analysis
We designed a 5-plex immunofluorescence panel for RMS tissue and an 8-plex panel for sarcoma TMA to characterize the tumor immune microenvironment. Candidate commercial antibodies intended for mIF staining were first validated by IHC using RMS FFPE tissue to confirm optimal staining intensity, specificity, and signal-to-noise ratio. mIF was performed according to the Opal Multiplex IHC assay protocol (Akoya Biosciences) as previously described52. The antibody panel was then stained in the following order. Each primary antibody was incubated for 60 min, followed by 10-min incubation with a secondary antibody (Opal Polymer Anti-Rabbit HRP Kit, Akoya Biosciences), application of the Opal fluorophore (OPAL Fluor, Akoya Biosciences), and incubation for 10 min at room temperature. Detailed information is provided in the supplementary Table 1. mIF images were scanned using a Vectra Polaris automated quantitative pathology imaging system (Akoya Biosciences). The fluorescent images were unmixed and analyzed to quantify the mean fluorescent intensity for each marker using inForm Advanced Image software (inForm: 2.5.1, Akoya Biosciences).
Statistical analysis
All statistical analyses were performed using R software 4.2 and GraphPad Prism 10.2 (ID: GPS-1928733-EJSL-94BFE, San Diego, CA, USA). The two-tailed Wilcoxon-Mann-Whitney non-parametric test or Two-sided t test was performed to compare quantitative variables across two groups or subclusters. Kaplan-Meier estimation and log-rank tests were used for survival analysis. Differences between groups were compared using the chi-squared or Fisher’s exact tests for categorical variables. Correlations were evaluated using non-parametric Spearman analysis. Spearman’s ρ (rho) values were presented in the corresponding figure. Significance was calculated by a two‑tailed test. as* p values < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Data availability
The transcriptome data and survival information used in this study are publicly available from the GDC Portal TCGA-SARC (https://portal.gdc.cancer.gov/) and the GEO database (GSE108022). The proteomics data are publicly available from The Cancer Proteome Atlas (TCPA, http://tcpaportal.org/). All the other raw data are available from the corresponding authors upon reasonable request. The code used for statistical analysis and figure generation in this study can be accessed by the corresponding author upon reasonable request.
Code availability
The code used for statistical analysis and figure generation in this study can be accessed by the corresponding author upon reasonable request.
Abbreviations
- DEGs:
-
Differentially expressed genes
- DFS:
-
Disease-free survival
- ECL:
-
Electronic Chemistry Laboratory
- FGA:
-
Fraction of genome altered
- GSEA:
-
Gene set enrichment analysis
- ICB:
-
Immune checkpoint blockade
- IHC:
-
Immunohistochemistry
- IFNγ:
-
Interferon-γ
- GSEA:
-
Gene Set Enrichment Analysis
- mIHC:
-
multiple immunohistochemistry
- OS:
-
Overall survival
- RMS:
-
Rhabdomyosarcoma
- RNA-seq:
-
RNA sequencing
- STR:
-
Short Tandem Repeat
- STS:
-
Soft tissue sarcoma
- TGFβ1:
-
Transforming growth factor beta 1
- TIS:
-
Tumor inflammation signature
- TME:
-
Tumor microenvironment
- TMB:
-
Tumor mutation burden
- TMC:
-
Tumor mutational count
- TIIC:
-
Tumor-infiltrating immune cells
- UPS:
-
Undifferentiated pleomorphic sarcomas
- WHO:
-
World Health Organization
References
Skapek, S. X. et al. Rhabdomyosarcoma. Nat. Rev. Dis. Prim. 5, 1 (2019).
Dagher, R. & Helman, L. Rhabdomyosarcoma: an overview. Oncologist 4, 34–44 (1999).
Reilly, B. K. et al. Rhabdomyosarcoma of the head and neck in children: review and update. Int J. Pediatr. Otorhinolaryngol. 79, 1477–1483 (2015).
Bhushan, B. et al. Transforming growth factor Beta and Alveolar Rhabdomyosarcoma: A challenge of tumor differentiation and Chemotherapy Response. Int. J. Mol. Sci. 25, 2791 (2024).
Hettmer, S. & Wagers, A. J. Muscling in: Uncovering the origins of rhabdomyosarcoma. Nat. Med. 16, 171–173 (2010).
Leiner, J. & Le Loarer, F. The current landscape of rhabdomyosarcomas: an update. Virchows Arch.: Int. J. Pathol. 476, 97–108 (2020).
Kashi, V. P., Hatley, M. E. & Galindo, R. L. Probing for a deeper understanding of rhabdomyosarcoma: insights from complementary model systems. Nat. Rev. Cancer 15, 426–439 (2015).
Parham, D. M. & Barr, F. G. Classification of rhabdomyosarcoma and its molecular basis. Adv. Anat. Pathol. 20, 387–397 (2013).
Agaram, N. P. Evolving classification of rhabdomyosarcoma. Histopathology 80, 98–108 (2022).
Zhu, B. & Davie, J. K. New insights into signalling-pathway alterations in rhabdomyosarcoma. Br. J. Cancer 112, 227–231 (2015).
El Demellawy, D., McGowan-Jordan, J., de Nanassy, J., Chernetsova, E. & Nasr, A. Update on molecular findings in rhabdomyosarcoma. Pathology 49, 238–246 (2017).
Raze, T. et al. PAX-FOXO1 fusion status in children and adolescents with alveolar rhabdomyosarcoma: Impact on clinical, pathological, and survival features. Pediatr. Blood Cancer 70, e30228 (2023).
Anderson, J. R. et al. Fusion-negative alveolar rhabdomyosarcoma: modification of risk stratification is premature. J. Clin. Oncol. 28, e587–e588 (2010).
Zarrabi, A. et al. Rhabdomyosarcoma: Current therapy, challenges, and future approaches to treatment strategies. Cancers 15, 5269 (2023).
Martin-Broto, J., Moura, D. S. & Van Tine, B. A. Facts and Hopes in Immunotherapy of Soft-Tissue Sarcomas. Clin. cancer Res. 26, 5801–5808 (2020).
Chen, C., Dorado, Garcia, H., Scheer, M. & Henssen, A. G. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front. Oncol. 9, 1458 (2019).
Batlle, E. & Massague, J. Transforming growth factor-beta signaling in immunity and cancer. Immunity 50, 924–940 (2019).
Ali, S. et al. TGF-beta signaling pathway: Therapeutic targeting and potential for anti-cancer immunity. Eur. J. Pharm. 947, 175678 (2023).
Cerwenka, A. & Swain, S. L. TGF-beta1: immunosuppressant and viability factor for T lymphocytes. Microbes Infect. 1, 1291–1296 (1999).
Yoshimura, A. & Muto, G. TGF-beta function in immune suppression. Curr. Top. Microbiol. Immunol. 350, 127–147 (2011).
Panagi, M., Pilavaki, P., Constantinidou, A. & Stylianopoulos, T. Immunotherapy in soft tissue and bone sarcoma: unraveling the barriers to effectiveness. Theranostics 12, 6106–6129 (2022).
Birdi, H. K. et al. Immunotherapy for sarcomas: new frontiers and unveiled opportunities. J. Immunother. Cancer 9, e001580 (2021).
Smolle, M. A. et al. T-regulatory cells predict clinical outcome in soft tissue sarcoma patients: a clinico-pathological study. Br. J. cancer 125, 717–724 (2021).
Mandai, M. et al. Dual faces of IFNgamma in Cancer Progression: A role of PD-L1 induction in the determination of pro- and antitumor immunity. Clin. Cancer Res. 22, 2329–2334 (2016).
Petitprez, F. et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 577, 556–560 (2020).
Wang, S. et al. TGF-beta1 signal pathway may contribute to rhabdomyosarcoma development by inhibiting differentiation. Cancer Sci. 101, 1108–1116 (2010).
Sun, M. M. et al. TGF-beta1 suppression of microRNA-450b-5p expression: a novel mechanism for blocking myogenic differentiation of rhabdomyosarcoma. Oncogene 33, 2075–2086 (2014).
Sun, M. et al. Autoregulatory loop between TGF-beta1/miR-411-5p/SPRY4 and MAPK pathway in rhabdomyosarcoma modulates proliferation and differentiation. Cell Death Dis. 6, e1859 (2015).
Tauriello, D. V. F., Sancho, E. & Batlle, E. Overcoming TGFbeta-mediated immune evasion in cancer. Nat. Rev. Cancer 22, 25–44 (2022).
Chandiran, K. & Cauley, L. S. The diverse effects of transforming growth factor-beta and SMAD signaling pathways during the CTL response. Front Immunol. 14, 1199671 (2023).
Overacre-Delgoffe, A. E. et al. Interferon-gamma Drives T(reg) Fragility to Promote Anti-tumor Immunity. Cell 169, 1130–1141.e1111 (2017).
Chen, S. et al. Mechanisms regulating PD-L1 expression on tumor and immune cells. J. Immunother. Cancer 7, 305 (2019).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24, 1550–1558 (2018).
Danaher, P. et al. Pan-cancer adaptive immune resistance as defined by the Tumor Inflammation Signature (TIS): results from The Cancer Genome Atlas (TCGA). J. Immunother. Cancer 6, 63 (2018).
Damotte, D. et al. The tumor inflammation signature (TIS) is associated with anti-PD-1 treatment benefit in the CERTIM pan-cancer cohort. J. Transl. Med. 17, 357 (2019).
Chen, L. et al. The immunosuppressive niche of soft-tissue sarcomas is sustained by tumor-associated macrophages and characterized by intratumoral tertiary lymphoid structures. Clin. cancer Res. 26, 4018–4030 (2020).
Derynck, R. & Budi, E. H. Specificity, versatility, and control of TGF-beta family signaling. Sci. Signal 12, eaav5183 (2019).
Derynck, R. & Zhang, Y. E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425, 577–584 (2003).
Schuler, F. et al. Checkpoint kinase 1 is essential for normal B cell development and lymphomagenesis. Nat. Commun. 8, 1697 (2017).
Schoeler, K. et al. CHK1 dosage in germinal center B cells controls humoral immunity. Cell Death Differ. 26, 2551–2567 (2019).
Ngoi, N. Y. L., Peng, G. & Yap, T. A. A Tale of two checkpoints: ATR inhibition and PD-(L)1 blockade. Annu Rev. Med. 73, 231–250 (2022).
Sen, T. et al. Targeting DNA damage response promotes antitumor immunity through STING-mediated T-cell activation in small cell lung cancer. Cancer Discov. 9, 646–661 (2019).
Tang, Z. et al. ATR inhibition induces CDK1-SPOP signaling and enhances anti-PD-L1 Cytotoxicity in prostate cancer. Clin. Cancer Res. 27, 4898–4909 (2021).
Sato, H. et al. DNA double-strand break repair pathway regulates PD-L1 expression in cancer cells. Nat. Commun. 8, 1751 (2017).
Laroche-Clary, A. et al. CHK1 inhibition in soft-tissue sarcomas: biological and clinical implications. Ann. Oncol. 29, 1023–1029 (2018).
Dorado Garcia, H. et al. Therapeutic targeting of ATR in alveolar rhabdomyosarcoma. Nat. Commun. 13, 4297 (2022).
Yoshida, K. et al. Aberrant activation of cell-cycle-related kinases and the potential therapeutic impact of PLK1 or CHEK1 inhibition in Uterine Leiomyosarcoma. Clin. Cancer Res. 28, 2147–2159 (2022).
Jess, J. et al. Cell Context is the third axis of synergy for the combination of ATR inhibition and cisplatin in Ewing sarcoma. Clin. Cancer Res. 30, 3533–3548 (2024).
Ayers, M. et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Investig. 127, 2930–2940 (2017).
Bankhead, P. et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 7, 16878 (2017).
Metsalu, T. & Vilo, J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 43, W566–W570 (2015).
Locke, D. & Hoyt, C.C. Companion diagnostic requirements for spatial biology using multiplex immunofluorescence and multispectral imaging. Front Mol Biosci. 10, 1051491 (2023).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (82103121), the National Key Research and Development Program of China (2023YFB3810204), the Natural Science Foundation of Jiangsu Province (BK20200878), Key Projects of Students Academic Research Foundation of Soochow University (KY2023093A, KY2024273B), and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD). We thank all the patients and their families at The First Affiliated Hospital of University of Science and Technology of China (USTC) for their contributions.
Author information
Authors and Affiliations
Contributions
C.R., Y.L., and F.X. analyzed multi-omics data and performed statistical analyses. C.R. and S.W. conceived the study and designed the experiments. X.Z., J.Z., Z.X., J.W., L.C., Z.G., Z.Z., and J.A. performed the experiments. Y.L. and X.Y. collected clinical samples and information. C.R., Y.L., and F.X. wrote the manuscript; J.S. and J.H. contributed to manuscript review and conceptual advice. C.R., Q.Z., and S.W. directed the project and were responsible for funding acquisition. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rong, C., Liu, Y., Xiang, F. et al. Integrative bioinformatics analysis and experimental validation identify CHEK1 as an unfavorable prognostic biomarker related to immunosuppressive phenotypes in soft tissue sarcomas. npj Precis. Onc. 9, 268 (2025). https://doi.org/10.1038/s41698-025-01064-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41698-025-01064-8
