
Abstract
While gene drive strategies have been proposed to aid in the control of mosquito-borne diseases, additional genome engineering technologies may be required to establish a defined end-of-product-life timeline. We previously demonstrated that single-strand annealing (SSA) was sufficient to program the scarless elimination of a transgene while restoring a disrupted gene in the disease vector mosquito Aedes aegypti. Here, we extend these findings by establishing that complete transgene removal (four gene cassettes comprising ~8-kb) can be programmed in cis. Reducing the length of the direct repeat from 700-bp to 200-bp reduces, but does not eliminate, SSA activity. In contrast, increasing direct repeat length to 1.5-kb does not increase SSA rates, suggesting diminishing returns above a certain threshold size. Finally, we show that while the homing endonuclease Y2-I-AniI triggered both SSA and NHEJ at significantly higher rates than I-SceI at one genomic locus (P5-EGFP), repair events are heavily skewed towards NHEJ at another locus (kmo), suggesting the nuclease used and the genomic region targeted have a substantial influence on repair outcomes. Taken together, this work establishes the feasibility of engineering temporary transgenes in disease vector mosquitoes, while providing critical details concerning important operational parameters.
Similar content being viewed by others


Introduction
Aedes aegypti is a highly invasive mosquito vector that transmits arboviral pathogens such as Zika, yellow fever, dengue, and chikungunya viruses, representing a substantial threat to public health in developing countries in tropical and sub-tropical regions1,2,3. Due to the scarcity of highly effective, affordable vaccines or antiviral treatments and the mosquito’s increasing resistance to pesticides, the development of genetics-based control strategies, which include the generation of genetically engineered (GM) mosquitoes and their release to natural habitats, has been highlighted as a fundamentally preventive tool for mosquito-borne diseases4,5,6. Genetic control strategies such as the Release of Insects carrying a Dominant Lethal (RIDL) and gene drive have been proposed to reduce the wild species or inactivate their disease-transmitting ability [reviewed in refs. 7,8]. In particular, the emergence of CRISPR-based gene editing9 has significantly accelerated technical advances in homing gene drive approaches in Anopheles and Aedes mosquitoes that can rapidly spread beneficial transgenes into laboratory populations10,11,12,13,14,15,16,17,18,19,20.
While CRISPR-based homing gene drive is a potentially powerful tool for efficiently altering the genomes of disease vectors for the purposes of either population suppression or population replacement21,22, its anticipated highly invasive, self-propagating nature has also raised public concerns of unforeseen ecological risks23,24,25,26,27. This restriction has led to the development of gene drive with local confinement17, self-exhaustion28, and anti-CRISPR breaks such as CATCHA, e-CHACRs, ERACRs, and the anti-CRISPR AcrIIA4 protein29,30,31. We previously conceptualized a model for erasing gene drive transgenes from the genome and reversing the invasion back to the wild-type state32. Such a transgene elimination system was successfully engineered in the Aedes aegypti genome33, and was based on single-strand annealing (SSA), a eukaryotic DNA repair mechanism that triggers a deletion when a double-strand break (DSBs) occurs between two identical sequence motifs, known as direct repeats (DRs)34,35.
In this study, we demonstrate that transgene elimination and restoration of wild-type alleles can be triggered in cis, as well as in trans, an important next step in constructing a truly self-eliminating transgene. We also evaluated several parameters of the SSA mechanism for transgene elimination efficiency in Ae. aegypti. For this, we generated transgenic mosquito strains featuring various changes predicted to affect rates of SSA, such as the lengths of the DR sequence (0.2-kb; 0.7-kb; 1.5-kb), the distances between the DRs (spacer sizes: 3.7-kb; 7.9-kb), and the types of the homing endonuclease (I-SceI; Y2-I-AniI). Our organismal DNA repair assays reveal that various size ratios of Cargo-to-DR are feasible in the Ae. aegypti genome with context-dependent self-eliminating dynamics.
Results
Lengths of DR sequences can influence the rate of SSA in regard to removing transgenic cargo from the Aedes aegypti genome
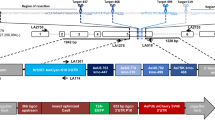
We previously demonstrated effective SSA utilizing a 0.7-kb direct repeat flanking two transgene cassettes33. To determine the influence of direct repeat (DR) length on the effectiveness of SSA, we generated a parallel transgenic strain at the same insertion site with a 0.2-kb DR and compared it to our previous construct (0.7-kb) in regard to SSA-mediated elimination of transgene components containing DsRED and EGFP (3.7-kb) integrated to exon 4 of Ae. aegypti kmo (Fig. 1a). In both cases, transgenic mosquitoes have marker phenotypes of white (Kmo−) eyes, DsRED+ eyes, and EGFP+ body, and the recognition sequence of the homing endonuclease I-SceI positioned in-frame next to the ATG translational start codon of DsRED. For 0.7-kb and 0.2-kb of DRs, kmo exons 2/3/4 and exons 3/4 were engineered in the cassettes, respectively (Fig. 1a, yellow bars), which created duplicates of the chromosomal counterparts (Fig. 1a, pink bars). Correct integration at the kmo locus was verified by PCR analysis using genomic DNAs obtained from kmoDR0.2-SceI and kmoDR0.7-SceI (Fig. 1b; Supplementary Data 1, Table S1).

a Schematic representation of pSSA-KmoDR0.2-SceI and pSSA-KmoDR0.7-SceI transgenes integrated into the Ae. aegypti genome resulting in kmoDR0.2-SceI and kmoDR0.7-SceI strains, respectively. Direct repeat (DR) sequences of kmo exons 3/2 (yellow bars) and chromosomal counterparts (pink bars); the I-SceI-induced DSB site at DsRED used to trigger the SSA pathway is indicated, along with locations of PCR primers utilized to confirm insertions (arrows). The DSBs induced at the I-SceI site are processed through NHEJ to disrupt DsRED (Kmo-; DsRED-; EGFP+) or SSA to restore kmo with the deletion of transgenes (Kmo+; DsRED−; EGFP−). b PCR-based verification of insertion site for each transgenic mosquito strain. c The schematic workflow of the SSA test in a split system, where kmoDR0.2-SceI or kmoDR0.7-SceI male mosquitoes were crossed with nuclease-providing females. F1 offspring females were outcrossed with kmo-/- males and individually oviposited in a 24-well tissue culture plate with 2% low melting point agarose. F2 larvae per female were scored for DNA repair event-dependent marker phenotypes. W, white eyes; G, EGFP+ body; R, DsRED+ eyes; Blk, black eyes. d DNA repair events were scored for F1 female founders (triplicate groups). Each bar represents percentage of F1 females that produced at least one NHEJ (WG, green bar) or SSA (Blk, orange bar) progeny. [F1 total (n)], the total number of F1 female founders; [marker (n or %)], the number or percentage of all F1 females that produced at least one NHEJ or SSA progeny; [SSA/NHEJ], the ratio of [marker (%)] (note that founders that produced both types of events would be counted in both). Tukey’s multiple comparisons test (1-way ANOVA): *P < 0.05; **P < 0.01; ****P < 0.0001. e The percentage of F2 offspring per individual F1 female for each DNA repair pathway-dependent marker phenotypes: WG for NHEJ [% WG/(WGR+WG+Blk)]; Blk for SSA [% Blk/(WGR+WG+Blk)]. Each dot represents rates of NHEJ and SSA events recovered in the progeny of a single F1 female. [F2 WGR (n)], the total number of F2 WGR mosquitoes; [marker (n or %)], the number or percentage of F2 progenies that have the NHEJ (green) or SSA (orange) phenotype; [SSA/NHEJ], the proportional ratio of [marker (%)]. Tukey’s multiple comparisons test (1-way ANOVA): *P < 0.05; ns not significant.
To determine the effect of DR length variation on transgene elimination, we performed the SSA test in a split system, where DR-flanked transgenes and the I-SceI nuclease (the SSA trigger) were provided from independent strains (Fig. 1c; Supplementary Data 1–2). F1 offspring females carrying both SSA components were outcrossed with kmo−/− males in triplicated experiments and allowed to individually oviposit. DNA repair events were scored in F2 progeny for each female. Following I-SceI DSB induction, NHEJ-driven indel mutations could frameshift the DsRED coding region, resulting in the phenotype WG (Kmo−; DsRED−; EGFP+). In contrast, repair by the SSA pathway would result in black eyes with no marker gene (Blk: Kmo+; DsRED−; EGFP−). These phenotypes were scored as: (i) the percentage of F1 females that produced at least one progeny with an NHEJ or SSA phenotype (Fig. 1d), (ii) the percent of F2 individuals per F1 mother that displayed an NHEJ or SSA phenotype (Fig. 1e). Both kmoDR0.2-SceI and kmoDR0.7-SceI had similar rates of NHEJ-mediated DSB repair when considering either F1 mothers (~20%) or the percentage of total F2 offspring (~2.5%). In contrast, kmoDR0.2-SceI showed significantly decreased rates of SSA (only 9.6% of F1 mothers and 1.4% of total F2 offspring), compared to kmoDR0.7-SceI (>40% of F1 mothers and 3.5% of F2 offspring). The ratio of SSA-to-NHEJ of kmoDR0.2-SceI was ~0.4, indicating that the DNA repair pathway selection was skewed toward NHEJ, while those of kmoDR0.7-SceI was ~1.5–2. We conclude that while 0.2-kb of DR was still capable of triggering SSA and could eliminate 3.7-kb of transgenic cargo, it was less efficient than the longer 0.7-kb repeat.
SSA-based transgene elimination is effective in cis as well as in trans
While we have demonstrated that SSA-mediated transgene elimination is possible in a split system, in order to provide temporal control over an invasive gene drive element it must also be effective in cis. That is, the source of the nuclease must be encoded within the DRs and would also be removed upon successful SSA. To determine if this was possible, we developed two additional integrations at the kmo locus consisting of the 1 piece-SSA transgene (7.9-kb), in which the homing endonuclease I-SceI gene (the SSA trigger) was engineered together with its target site and the DRs flanking a series of marker genes (Fig. 2a). These two SSA strains were designed to have 0.7-kb of kmo exons 2/3/4 or 1.5-kb of kmo exons 2/3/4 and intron 1 as the DR motifs, respectively for kmoDR0.7-nos-SceI and kmoDR1.5-nos-SceI (Supplementary Table S1). Both transgenic cargos include eye-specific DsRED with the I-SceI target site, eye-specific BFP, whole body EGFP, and a germline-specific nos promoter-controlled I-SceI. PCR analysis using genomic DNA and various primer pairs (Fig. 2a; Supplementary Table S2) verified correct integration of each transgene, including the size difference (0.8-kb) between the two transgene versions, the presence of nos-I-SceI, and chromosomal integration of transgenes to the kmo gene (Fig. 2b).

a Schematic representation of pSSA-KmoDR0.7-nos-SceI and pSSA-KmoDR1.5-nos-SceI integrated into genomes of kmoDR0.7-nos-SceI and kmoDR1.5-nos-SceI strains, respectively. The kmo exons 3/2 and exons 3/2-intron 1 sequences (yellow bars) were engineered for DR0.7 and DR1.5 motifs with chromosomal counterparts (pink bars), respectively. I-SceI-target site is shown, with I-SceI coding region under the control of the germline-specific nos gene promoter/3′UTR. b PCR-based verification of transgene insertion to verify insertion site, DR length gap between the two strains (KmoEx4-F + DsRED-5Ra), the presence of nos-I-SceI (NosPro-3Fa + SceI-5Ra), and the chromosomal integration of transgenes (SV40-R + In1-IR2). c The schematic workflow of the 1 piece-SSA test over multi-generations. For each generation, females (n = 20–25) carrying the intact transgene (WGR) as confirmed by HRMA were out-crossed with kmo-/- males. In the next generation, offspring larvae were scored for marker phenotypes to determine DNA repair events: W, white eyes; G, EGFP+ body; R, DsRED+ eyes; Blk, black eyes. Tukey’s multiple comparisons test (1-way ANOVA): ns not significant. d Each bar represents percentage of G10 females that produced at least one NHEJ (WG, green bar) or SSA (Blk, orange bar) progeny from three independent experimental groups. [G10 total (n)], the total number of G10 female founders; [marker (n or %)], the number or percentage of all G10 females that produced at least one NHEJ or SSA progeny; [SSA/NHEJ], the ratio of [marker (%)] (note that founders that produced both types of events would be counted in both). Tukey’s multiple comparisons test (1-way ANOVA): ns not significant. e Percent of G11 offspring per individual G10 female for DNA repair pathway-dependent marker phenotypes: WG for NHEJ [% WG/(WGR+WG+Blk)]; Blk for SSA [% Blk/(WGR+WG+Blk)]. Each dot represents rates of NHEJ and SSA events recovered in the progeny of a single G10 female. [G11 WGR (n)], the total number of G11 WGR mosquitoes; [marker (n or %)], the number or percentage of G11 progenies that have the NHEJ (green) or SSA (orange) phenotype; [SSA/NHEJ], the proportional ratio of [marker (%)]. Tukey’s multiple comparisons test (1-way ANOVA): ns not significant. f PCR analysis of genomic DNAs from white/EGFP+ eyed-mosquitoes using indicated primers from G11 mosquitoes. Identified large deletions are shown.
To analyze DNA repair outcomes, transgenic females (WGRB: Kmo−; EGFP+; DsRED+; BFP+) were outcrossed with kmo−/− males (W: Kmo−) in triplicate, and DNA repair pathway-dependent marker phenotypes were scored in offspring larvae for NHEJ (WGB: Kmo−; EGFP+; DsRED-; BFP+) and SSA (Blk: Kmo+; EGFP−; DsRED−; BFP−) (Fig. 2c; Supplementary Tables S3–S5, Supplementary Data 3). For each generation (G8, G9, G10), WGRB females (n = 25 per replicate) were screened by high-resolution melt analysis (HRMA) to eliminate any silent/in-frame indel mutation that occurred at the I-SceI target site, and only mosquitoes with the intact transgene were taken to be founding mothers for the next generation (Fn+1) (Supplementary Fig. S1). Both strains, kmoDR0.7-nos-SceI and kmoDR1.5-nos-SceI, were shown to maintain similar rates of SSA- or NHEJ-mediated repair for nuclease-induced DSBs throughout multiple generations (Fig. 2c). Similarly, the proportion of G10 females that produced at least one NHEJ or SSA event was not statistically different between the two strains (Fig. 2d; Supplementary Data 4), nor was the percentage of total progeny at G11 that exhibited NHEJ or SSA phenotypes (Fig. 2e; Supplementary Data 5) despite the appearance of a trend towards higher rates of SSA in the longer kmoDR1.5-nos-SceI strain (15.2% of mothers in G10 and 1.8% of progeny in G11) compared to those of kmoDR0.7-nos-SceI (5.3% of mothers at G10 and 0.6% of progeny in G11). Together, these results confirm that SSA-based transgene elimination can be programmed in cis, as well as in trans. Rates of SSA appeared lower than the in trans experiment, a phenomenon that may be due to the change in transgene configuration or simply the expanded distance between the DRs.
Unexpectedly, we found some mosquitoes displayed eye-specific EGFP fluorescence (WG-eye: Kmo−; EGFP+; DsRED−; BFP−) (Fig. 2f), which might have resulted from an aberrant, unidentified process of DNA repair. PCR analysis using the primer pair of Hsp70-F and GFP-3R in these individuals showed amplicons of 1.3-kb and 1.6-kb, smaller (about half) than predicted (Fig. 2a, >3-kb). Sequence analysis revealed that a range of sequences pertaining to [DsRED and SV40] and [downstream parts of the PUb promoter] were deleted, placing the EGFP gene close enough to be expressed by the eye-specific 3xP3 promoter. This aberrant conversion event was recovered at a frequency of ~0.1% in our assay (n = 6, out of >6000 WGRB mosquitoes).
Choice of homing endonuclease strongly impacts the efficiency of SSA DSB repair in Aedes aegypti
Previously, we determined that the LAGLIDADG-type homing endonucleases (LHEs) I-AniI, I-CreI, and I-SceI are effective at inducing DSBs for genome editing in Ae. aegypti36 when expressed transiently following embryonic injection. While I-SceI was successfully engineered to induce germline-specific DSBs to trigger SSA-based self-elimination of a kmo-integrated transgenic cargo33, I-AniI was actually much more efficient in our previous experiments36. To determine the impact of nuclease choice on DSB repair and SSA efficiency, we generated a new DSB trigger strain, nos-I-AniI, which expresses Y2-I-AniI37 (I-AniI, here in after) under the control of germline-specific nos promoter and 3′UTR (Supplementary Tables S1 and S6) and tested its activity to trigger DSB repair processing in the transgene of the Mariner Mos1-driven transgenic P5-EGFP strain36. In P5-EGFP, there are two groups of homing endonuclease target sites [sites-A] and [sites-B] and two gene cassettes [3xP3-DsRED-SV40] and [PUb-EGFP-SV40] (Fig. 3a). DSBs can trigger SSA by diverse pairs of DR elements such as [SV40], [loxP], and [individual HE sites] and are expected to remove [PUb-EGFP-SV40] from the transgene, resulting in the WR phenotype (Kmo−; EGFP−; DsRED+).

a Schematic representation of the SSA-capable P5-EGFP transgene integrated into the Ae. aegypti genome containing two marker genes (eye-specific DsRED and whole body-expressed EGFP) interspersed with two groups of homing endonuclease target sites (site-A and site-B) were engineered to flank the EGFP cargo. b Percentage of F2 larvae with phenotypic loss of the EGFP marker from triplicate experiments. The X-axis indicates the grandparent (F0)/parent (F1) contributing the nuclease gene. Tukey’s multiple comparison test was found to be significant (2-way ANOVA, P < 0.001), and statistically different groups are marked (a and b). c HRMA of the site-A for F2 mosquitoes scored as WGR (white and DsRED+ eyes; EGFP+ body) by using a primer pair of SV40-F and PUb-5R. d Sequence analysis of amplicons with altered melting curves from (c), compared to the baseline. Sequencing analysis revealed various numbers of nucleotide deletions occurred at site-A. The colors of IDs designate individual HRMA peaks shown in (c).
The P5-EGFP strain was reciprocally crossed with either nos-I-AniI or nos-I-SceI strains in triplicate, and DNA repair pathway-dependent marker phenotypes were scored in F2 offspring by each nuclease-providing lineage (Fig. 3b; Supplementary Table S7, Supplementary Data 6). Rates of SSA (WR phenotype) were substantially higher with I-AniI (~21%) than I-SceI (~0.4%) in F2 progeny from the female lineage. SSA events were substantially lower in the male-initiated lineages, likely due to the lack of maternally contributed nos-I-AniI. As NHEJ events were not directly scorable, we performed HRMA of PCR amplicons at [sites-A] using a primer pair of SV40-F and PUb-5R (shown in Fig. 3a) from F2 WGR progeny from nos-I-AniI (n = 73) and nos-I-SceI individuals (n = 80) (Fig. 3c). In sequence analysis, NHEJ at [sites-A] was identified to occur ~25% for nos-I-AniI; no such events were recovered for nos-I-SceI (Fig. 3d).
As WR phenotypes could be due to either SSA or to NHEJ following DSB induction at both sites-A and sites-B, with multiple DR features present in P5-EGFP, we further determined the rates of individual DR motif-dependent SSA events using high-throughput amplicon sequencing (Fig. 4). Genomic DNA was obtained from triplicate groups of F2 larvae with WR phenotypes (n = 100 per replicate) and utilized for PCR amplification of the processed region (Supplementary Fig. S2). The nanopore-based sequencing results (n = 794 reads, see Materials and Methods) were aligned with individual signature sequences (~60 to 100-bp, blue dotted lines in Fig. 4) of seven types of DNA repair end-products, which can be produced by diverse SSA- or NHEJ-mediated processes of DSB repair occurring at either [sites-A] or [sites-B] in P5-EGFP. SSA events were recovered most abundantly by SV40 DRs (>59%) (Fig. 4), potentially due to the larger size (~230-bp) of these sequences. Interestingly, just over 30% of events appear to be the result of SSA using the two I-CmoeI recognition sites as DRs, despite the small size (20-bp) of this sequence. Likewise, a small subset of SSA events occurred by loxP (2.5%) or I-SceI (2.9%) target sequences, but not by I-CreI or I-PpoI target sites, despite a similar length (Fig. 4). Finally, ~3% of events centered on the I-AniI site, and could have been generated either by SSA (following 1 DSB) or NHEJ (following 2 DSBs) (Fig. 4). Changing alignment sensitivity or performing de novo clustering of nanopore reads did not meaningfully affect the rates of each reported repair outcome (Supplementary Note 1).

Predicted end-products resulting from DR-associated DNA repair processes and their rate of occurrence in individuals that lost the P5-EGFP transgene. Blue dotted lines indicate signature sequences that are partially unique to each end-product. ‘Potential 2nd DSBs’ describe additional SSA or NHEJ processes that are predicted to occur on the I-AniI site when it remains intact after the initial DSB repair event.
We also noted that some of the possible repair outcomes we anticipated (Fig. 4) preserve an intact I-AniI target site, and thus themselves could be cleaved and potentially subject to a new round of SSA/NHEJ. For example, an initial SSA event mediated by SV40 DRs or between I-AniI target sites can be further subject to DSB induction for either NHEJ-driven indel formation or SSA-based repair via I-CmoeI, loxP, or I-SceI DRs (Fig. 4). These secondary cleavage events are likely to be abundant, as about half (n = 252, 48.2%) of the SSA products obtained via DRs of SV40 (n = 523) (Fig. 4), were shown to contain indel mutations at the I-AniI site (Supplementary Fig. S2). This might also explain the failure to obtain SSA events utilizing the I-PpoI or I-CreI recognition sites since both of those products can be re-cleaved and further processed with additional SSA, whereas events utilizing loxP, I-SceI or I-CmoeI sequences cannot be further processed. Taken together, we conclude that the frequency of SSA repair within the P5-EGFP is strongly influenced by the nuclease used (I-SceI vs. I-AniI), and can be as high as 25% of the total progeny.
I-AniI-induced DSBs are almost exclusively repaired via NHEJ-mediated processes for a transgene engineered at the kmo locus
Based upon the substantially higher levels of SSA obtained with I-AniI at the P5-EGFP locus, we attempted to maximize SSA-based elimination for the kmo-integrated transgene (~3.7-kb) by engineering 1.5-kb of DRs and the I-AniI recognition site (Fig. 5a) in a similar manner to our previous constructs. PCR analysis using genomic DNAs obtained from kmoDR0.7-SceI,33 and kmoDR1.5-AniI (Supplementary Table S1) and the primer pair of KmoEx4-F and DsRED-5Ra verified integration site and the anticipated size difference (0.5-kb) between the two DR integrations (Fig. 5b). Subsequent sequencing analysis confirmed the two DSB sites, I-SceI (5′-TAGGGATAACAGGGTAAT-3′) and I-AniI (5′-TTGAGGAGGTTTCTCTGTAAATA-3′), which were engineered in-frame next to the ATG translational start codon of DsRED in kmoDR0.7-SceI and kmoDR1.5-AniI strains, respectively.

a Schematic representation of pSSA-KmoDR0.7-SceI and pSSA-KmoDR1.5-AniI integrated into genomes of kmoDR0.7-SceI and kmoDR1.5-SceI strains, respectively. The I-SceI or Y2-I-AniI target site was engineered at DsRED to trigger the SSA pathway. b PCR-based verification of transgenic mosquitoes. The PCR primer pair of KmoEx4-F and DsRED-5Ra was used to confirm the identity of the nuclease site engineered in each strain (Bold); The underlined sequence designates the overhang resulting from nuclease cleavage. c Each bar represents the proportion of F1 females that produced at least one NHEJ (WG, green bar) or SSA (Blk, orange bar) progeny across three independent experiments. [F1 total (n)], the total number of F1 female founders; [marker (n or %)], the number or percentage of all F1 females that produced at least one NHEJ or SSA progeny; [SSA/NHEJ], the ratio of [marker (%)] (note that founders that produced both types of events would be counted in both). Tukey’s multiple comparisons test (1-way ANOVA): *P < 0.05; ****P < 0.0001; ns not significant. d Proportion of F2 offspring exhibiting either NHEJ or SSA phenotypes per individual F1 female: WG for NHEJ [% WG/(WGR+WG+Blk)]; Blk for SSA [% Blk/(WGR+WG+Blk)]. Each dot represents rates of NHEJ and SSA events recovered in the progeny of a single F1 female. [F2 WGR (n)], the total number of F2 WGR mosquitoes; [marker (n or %)], the number or percentage of F2 progenies that have the NHEJ (green) or SSA (orange) phenotype; [SSA/NHEJ], the proportional ratio of [marker (%)]. Tukey’s multiple comparisons test (1-way ANOVA): ***P < 0.0001; ns not significant.
kmoDR0.7-SceI or kmoDR1.5-AniI males were crossed with the nuclease-providing females, nos-I-SceI or nos-I-AniI in triplicate. F1 offspring females carrying both SSA components were outcrossed with kmo−/− males. As before, females oviposited individually, and repair outcomes were scored within each family. For kmoDR0.7-SceI, both NHEJ and SSA rates were similar to each other in F1 mothers (~25%, Fig. 5c; Supplementary Data 7) and F2 offspring (1.6–2%, Fig. 5d; Supplementary Data 8), making the SSA/NHEJ ratios close to 1, similar to previous results33, though slightly lower than that described in Fig. 1. In contrast, for kmoDR1.5-AniI, 97% of F1 mothers produced NHEJ-mediated DSB repair events, and ~50% of F2 offspring larvae showed NHEJ phenotypes. Accordingly, total SSA rates of kmoDR1.5-AniI were substantially lower than those of kmoDR0.7-SceI, especially in F1 mothers. Sequence analysis confirmed that WG phenotypes were indeed associated with indel mutations (−1, −2, and −4-bp) generated at the I-AniI site (Supplementary Fig. S4). Overall, repair of I-AniI-induced DSBs at the Ae. aegypti kmo locus was dramatically biased toward the NHEJ pathway rather than SSA, despite the increased length of DRs (1.5-kb).
The SSA-based transgene removal technology and genetic control approaches
Summarizing all SSA tests we performed at the kmo locus showed that there was little correlation between the ratio of cargo/spacer to DR (R) in terms of elimination of kmo-integrated transgenes (Fig. 6). For the 3.7-kb-sized cargo, 0.2-kb and 0.7-kb of DRs were engineered for R18.5 and R5.3, respectively. While R5.3 provided a relatively equal DNA repair pathway selection (SSA/NHEJ = 0.75 or 1.52), R18.5 resulted in a level of bias (SSA/NHEJ = 0.47). When the 7.9-kb-sized cargo was engineered with 0.7-kb (R11.3) or 1.5-kb (R5.3) of DRs, the average rate of SSA was higher in R5.3 (1.75%) than R11.3 (0.49%) while NHEJ rates were similar to each other. We conclude that the spacer length is relatively unimportant, while the DR length contributes only minimally to the selection of SSA. Both results are highly favorable to the development of SSA-based transgene removal approaches, allowing small repeats to potentially control large, complex transgene arrangements.

Transgenic mosquito strains were generated to contain various lengths of DR motifs (0.2-kb, 0.7-kb, or 1.5-kb) and the intervening cargo genes (3.7-kb or 7.9-kb), and their DNA repair dynamics were evaluated by crossing DSB trigger strains that express homing endonucleases (I-SceI or I-AniI) under the control of germline-specific nos promoter. One-piece SSA-featured mosquitoes were crossed with the kmo−/− strain in DNA repair tests. The [R] analysis presents the proportional values of cargo lengths (bp) divided by direct repeat lengths (bp) with respect to eliminating efficiency of kmo-integrated transgenes. Selection biases of DNA repair pathways were shown by the proportional values of SSA rates (%) normalized by NHEJ rates (%).
Discussion
Nuclease-based gene editing technology relies on eukaryotic DNA repair mechanisms, and their efficiency can be dependent on how the locally induced DSBs are processed in the genome of the targeted species38,39. In eukaryotic genomes, DSBs are processed mainly by two distinct, antagonistic pathways: homology-directed repair (HDR) and non-homologous end joining (NHEJ)40,41. HDR enables the conversion of a homologous sister chromosome to the damaged allele, leading to error-free recovery of the DSB. A homing gene drive cassette (GD) induces DSBs at a specific genomic locus and subsequently triggers the conversion of heterozygosity (geneGD/-) to homozygosity (geneGD/GD) in germline cells, which forces the encoding gene to be passed on to most or all of the resulting offspring. In contrast, NHEJ directly ligates the broken DNA ends and does not require a template sequence, which often leads to sequence errors with nucleotide insertions or deletions (indels), especially being error-prone if DSBs would be repetitively triggered by prolonged nuclease activity42. Moreover, indels that occur at the nuclease-targeted sequence may be resistant to further gene drive43,44. SSA is also a homology-based mechanism requiring resection of the DNA ends, but instead of a homologous chromosome or sister chromatid, it requires the recognition of identical sequences flanking the DSB site, which results in the elimination of the intervening segment of DNA45. Recently, we leveraged SSA to remove transgenic components and restore the function of the genetic target in the Aedes aegypti genome33, and we found this activity was elevated by disruption of core NHEJ factors46. In this study, we further extend these findings by demonstrating that transgene removal can be catalyzed in cis, a necessary step towards building a fully self-eliminating gene drive. We also show that cargo of approximately 8-kb comprising 4 distinct gene cassettes can be removed via SSA. As genetic control approaches often require a large transgenic cargo including CRISPR/Cas9, gRNAs, and effectors, our data suggest that transgene insertion size alone is not likely to be a barrier for SSA-based approaches.
In regard to unforeseen risks of genetically modified organisms (GMOs), other transgene removal systems have been developed in plants. First, the Cre-loxP system was employed for removing the marker gene from transgenic plants47. Within the transgene, the neomycin phosphotransferase II (nptII) gene was flanked by two loxP sites. The heat-inducible activation of the Cre-recombinase selectively excised the nptII gene and eliminated kanamycin-resistance from the transgenic lines, while preserving the effector-driven trait. This system enabled the complete excision of the marker gene while retaining the scar of a loxP site the targeted gene. More recently, homology-directed repair (HDR) was engineered for marker-free, transgenic rice48. The encoding sequences for hygromycin phosphotransferase (HPT) and CRISPR elements were flanked by two-separated portions of the GUS reporter gene (GU and US), which are converted into the GUS gene located in the sister chromosome, resulting in marker-excised progeny (>50% at T1), though in this case portions of the introduced transgene remained behind.
As a subtype of HDR, the SSA pathway requires extensive DNA end resection (the 5′-to-3′ nucleolytic process at the DSB site), generating long 3′-single-stranded DNA (3′-ssDNA) tails45. A competing complex can initiate the resection of one strand at both ends of the break49 and use the now single-stranded tails to perform a homology search for annealing between two identical sequence motifs50. As a critical factor, mammalian CtIP is recruited to the MRN complex and promotes the initial resection process of the broken ends by generating ssDNAs51. In Drosophila, in vivo DSB reporter assays showed that CtIP was critical for efficient SSA under short (~550 bp) and extensive (~3.6 kb) distance of resection52, suggesting that the initial generation of ssDNA may be a rate-determining process. Similarly, loss of Marcal1, an annealing factor that binds to ssDNA–dsDNA interfaces53,54, reduced SSA frequencies compared to wild type regardless of homology length55.
The threshold requirements for efficient SSA are likely to vary based on host organism, chromosomal loci, lengths and arrangements of direct repeats, and resection distances55,56,57,58,59,60. Intrachromosomal deletions mediated by the SSA pathway can result in deletions of at least 80-kb at high efficiency34 with the length of repeats and distance of separation strongly influencing this mode of repair in Drosophila melanogaster61, Saccharomyces cerevisiae34, and vertebrate cells35. In the yeast genome, SSA-mediated deletions occurred preferentially between the DR motifs closest to the DSB62, and its frequency was linearly dependent on the length of DRs57. Moreover, it was suggested that a competition can occur between two pairs of DRs, and a preference can be made based on their relative sizes and proximity to the DSB57. Another assay revealed that SSA rates were enhanced when 500-bp homologies were aligned to be the annealing target farther to the DSB site than closer55. In this work, we hypothesized that a larger DR motif may be more favorable for SSA-based transgene elimination. However, our multi-generational tests showed that increasing DR length from 0.7-kb to 1.5-kb did not result in a difference in SSA frequencies, implying that DR length may not be a rate-limiting factor for SSA-based transgene removal in Ae. aegypti, at least amongst the lengths tested here. Indeed, a ratio of 18.5 (~0.2-kb DR for ~3.7-kb cargo) was shown to be sufficient for triggering SSA, implying a potential that ~1-kb length of DR sequences may trigger SSA for a larger size of gene drive (typically >15-kb). For P5-EGFP, only ~20-bp of DRs were able to delete >2-kb of the transgenic cargo. Our current results indicate that SSA can occur with a spacer length of approximately 8-kb in the Aedes aegypti genome. Overall, various sizes of DRs and cargos are feasible for genetic control approaches, and characterizing the maximum size of the transgenic cargo that can be processed by SSA may depend more on the individual target gene context.
Our DNA repair tests showed that two types of homing endonucleases, I-AniI and I-SceI, were associated with very different repair outcomes depending on the target transgene. In the P5-EGFP strain, I-AniI was substantially more active than I-SceI as measured by the rates of F2 marker phenotypes associated with either SSA or NHEJ in F1 offspring, while maintaining a relatively similar ratio between NHEJ and SSA outcomes (~1:1). In stark contrast, DSB repair at kmoDR1.5-AniI was processed almost exclusively by NHEJ. While the genomic loci containing either the nos-I-AniI and nos-I-SceI transgenes may have some differential influence on expression levels (spatial/temporal/cell type), this is insufficient to explain such strong differences in repair outcomes due to the fact that the same drivers were used for both P5-EGFP and kmoDR crosses. Measuring rates of SSA at more sites and with a wider array of nucleases targeting regions with varied characteristics may shed additional light on critical factors that control repair outcomes.
To date, we have exclusively used the nanos promoter to express the homing endonucleases that trigger SSA and transgene removal. At the same time, various CRISPR-based homing gene drive cassettes engineered in Aedes aegypti have been developed under the control of regulatory sequences derived from exuperentia (exu, AAEL10097), zero population growth (zpg, AAEL6726), nanos (nos, AAEL012107), β2-tubulin (AAEL019894), benign gonial cell neoplasm (bgcn, AAEL004117), suppressor of defective silencing 3 (sds3, AAEL002084), and nup50 (AAEL005635)11,12,17,18,63. Each of these exhibited differences in the level and development stage of expression, subsequently affecting repair outcomes. Among them, the gene regulatory elements of sds3, whose gene function is critical in the development of testis and ovarian follicles64, was shown to lead to the highest level (~90%) of gene drive inheritance at the kmo locus11. The use of these alternative regulatory elements may also improve the selectivity of SSA over NHEJ. NHEJ-induced indels are an impediment to both homing gene drive43,44 and SSA-based transgene removal as described here. In the current work, we screened individual founders for the emergence of indels every generation to preserve SSA-compatible progeny that maintain the intact I-SceI site. Otherwise, DSB-resistant progeny may have accumulated within the test population and potentially halted the self-eliminating effect. While such an accumulation would have confounded the experiments described here, our previous mathematical modeling predicted that even with some NHEJ, low rates of SSA (~2% for maternal triggers and ~0.5% for paternal triggers) are sufficient for removing the gene drive mosquitoes from the field in ~60 generations with 5% fitness cost per transgene, or in ~35 generations if the strategy is engineered for the complete lethality cost33. That being said, the models suggest that further improvements to lower the NHEJ rate will increase the utility of SSA-based transgene removal approaches, for example by interfering with core NHEJ factors to bias DSB repair pathway toward homology-based repairs46. Thus, SSA-based self-eliminating transgenes should be further optimized with respect to potential interactions of individual components and DNA repair pathway choice in a target species.
Methods
Mosquito rearing
The Aedes aegypti Liverpool (Lvp) wild-type strain was maintained at 27 °C and 70% (±10%) relative humidity, with a day/night cycle of 14-h light and 10-h dark. Fertilized eggs were hatched in a pan filled with 2 L of distilled water and 200 mg of powdered fish food (TetraMin Tropical Flakes). At the L1 instar stage, larvae were manually counted to be ~500 per pan and replenished with fresh water and food every two days. Pupae were manually picked and separated with a size-based sorter and/or by identifying genitalia under the dissecting microscope. Adult mosquitoes were fed 10% sucrose solution and mated in a ♂:♀ ratio of 1:3. The mated females were fed on defibrinated sheep blood (Colorado Serum Company) using an artificial membrane feeder and oviposited on a wet-filter paper kept in a cup of ~30 ml of distilled water. The egg papers were air-dried and heat-sealed in polyethylene tubing for up to 4 months of storage at room temperature.
Generation of transgenic strains
CRISPR/Cas9-based site-specific integrations at the Ae. aegypti kmo site were obtained by microinjection into pre-blastoderm embryos65,66,67. For each strain, the injection mix included 400 ng/µl of Cas9 enzyme (PNA Bio), 100 ng/µl of sgRNA, and 300 ng/µl of donor plasmid and was microinjected into pre-blastodermic embryos of a recipient strain (Supplementary Table S1). G0 survivors were outcrossed with Lvp wild-type or kmo-null (kmo−/−) strain68, and transgenic mosquitoes were screened for their fluorescent marker expression at G1 larval phases. Chromosomal integration of the transgene at the kmo locus was confirmed by PCR analysis using genomic DNAs as the template and a primer set that is specific to both the transgene and kmo outside of the homology arm sequence (Supplementary Table S2).
To generate a transgenic strain expressing Y2-I-AniI, donor plasmid (500 ng/µl), pMOS-3xP3-BFP-nos-I-AniI was microinjected into pre-blastoderm embryos of the kmo−/− strain, along with the Mos1 helper plasmid (200 ng/µl), pKhsp82M69. For transgenic mosquitoes, transposon-chromosome junction sequences were amplified by inverse PCR using AluI-digested genomic DNA and Mos1 inverse repeat sequences-specific primers (Supplementary Table S2). Subsequently, the targeted chromosomal gene locus (AAEL010783 in Chr2) was identified by BLAST analysis through the VectorBase database70.
Subcloning
For kmo-targeted transgenic strains (Supplementary Table S1), donor plasmids were made by Golden Gate Assembly (NEB) with various combinations of three pieces of plasmid: (i) pGSP1 series provided homology arm 1 (HA1) and/or various sizes (0.2-kb, 0.7-kb, and 1.5-kb) of direct repeat sequences (DRs) at kmo; (ii) pGSP2 series provided homing endonuclease recognition sites (I-SceI or Y2-I-AniI) in-frame with the translational start codon (ATG) of the DsRED marker gene or the sgRNA-HybRED site for the introduction of SSA-featured DNA constructs; (iii) pGSP3 series provided HA2, the EGFP and/or BFP marker gene, and/or the nos promoter-driven I-SceI gene. For the details, (1) pSSA-KmoDR0.2-SceI (kmoDR0.2-SceI): pGSP1-KmoHA1-DR0.2 + pGSP2.3-DsRED-SV40 (SceI) + pGSP3.8 C#5-EGFP-KmoHA2; (2) pBR-KmoEx4#5 (kmoEGFP#5): pGSP1-KmoHA1 + pGSP2-REDh-SV40 + pGSP3.8 C#5-EGFP-KmoHA2; (3) pSSA-KmoDR1.5-AniI (kmoDR1.5-AniI): pGSP1-KmoHA1-DR1.5 + pGSP2.1-DsRED-SV40 (AniI) + pGSP3.8 C#5-EGFP-KmoHA2; (4) pBR-KmoEx4#5-nos-SceI (kmoEGFP#5-nos-SceI): pGSP1-KmoHA1 + pGSP2-REDh-SV40 + pGSP3.8 C#5-EGFP-nos-SceI-KmoHA2; (5) pSSA-KmoDR0.7-nos-SceI (kmoDR0.7-nos-SceI): pGSP1-KmoHA1-DR0.7 + pGSP2.3-DsRED-SV40 (SceI) + pGSP3.8 C#5-EGFP-nos-SceI-KmoHA2; (6) pSSA-KmoDR1.5-nos-SceI (kmoDR1.5-nos-SceI): pGSP1-KmoHA1-DR1.5 + pGSP2.3-DsRED-SV40 (SceI) + pGSP3.8 C#5-EGFP-nos-SceI-KmoHA2.
Some assembly plasmids were generated by modifying current or previously reported ones33. To generate pGSP1-KmoHA1-DR0.2, the KpnI-AgeI fragment of kmo exons 4/5 (HA1) and kmo intron 3 (0.2-kb of DR) was amplified by using a primer pair of KmoHA1-F-Kpn and KmoDR0.2-R-Age (Supplementary Table S2) and replaced the counterpart regions in pGSP1-KmoHA1-DR0.733. For pGSP1-KmoHA1-DR1.5, the KpnI-AgeI fragment of kmo exons 4/5 (HA1), kmo exons 2/3 (HA2, 0.6-kb of DR), and kmo intron 1 (0.9-kb of DR) was amplified by using a primer pair of KmoHA1-F-Kpn and In1-IR2-AgAv (Supplementary Table S2) and replaced the counterpart regions in pGSP1-KmoHA1-DR0.733. pGSP2.1-DsRED-SV40 (AniI) contains the synthesized DNA piece (GenScript) that encodes DsRED with the homing endonuclease Y2-I-AniI recognition sequence was engineered in-frame next to ATG translation start codon. To generate pGSP3.8 C#5-EGFP-KmoHA2, the DNA piece of PUb-EGFP-3′UTR#5 was amplified from a template plasmid pSLfa-PUb-EGFP-3′UTR#571 by using a primer pair of PUb-F and 3′UTR#5-R-BmBp (Supplementary Table S2). Subsequently, it replaced the counterpart regions by the flanking BlpI sites in pGSP3.8C-EGFP-KmoHA233. pGSP3.8 C#5-EGFP-nos-SceI-KmoHA2 was generated by adding the DNA piece that contains 3xP3-BFP-SV40 and nos-I-SceI-3′UTR into BamHI sites in pGSP3.8 C#5-EGFP-KmoHA2.
For pMOS-3xP3-BFP-nos-AniI (nos-I-AniI), the DNA piece containing the nanos promoter, Y2-I-AniI, and nanos 3′UTR sequences was synthesized (Epoch) and inserted to MluI and XhoI sites in pM2-3xP3-BFP, a Mariner Mos1-based plasmid backbone. Complete sequences of plasmid constructs used to generate transgenic mosquito lines are deposited in GenBank under accession numbers PP693225-PP693229.
The split-SSA test
Twenty adult males of individual SSA-featured strains (kmoDR0.2-SceI, kmoDR0.7-SceI, or kmoDR1.5-AniI) were crossed with 50 nuclease-providing females (nos-I-SceI or nos-I-AniI) in triplicated assays. F1 progenies were screened for marker phenotypes that were inherited from both parents (white, DsRED+, and BFP+ for eyes; GFP+ for the body). For each replicate, 50 F1 females were outcrossed with 20 kmo−/− males and individually oviposited by the EAgaL plate method72. DNA repair events per individual F1 females were determined by F2 larval phenotypes: (i) No DSB or complete repair, kmoDR-SceI/− (White eyes; DsRED+ eyes; EGFP+ body); (ii) NHEJ, kmoindel-DsRED/− (White eyes; DsRED- eyes; EGFP+ body); (iii) SSA, kmorestored+/− (Black eyes; DsRED- eyes; EGFP− body).
The SSA-featured P5-EGFP strain36 was reciprocally crossed with nuclease-providing females (nos-I-SceI or nos-I-AniI). Parental crosses were made with 30 ♂ and 100 ♀ per cage in triplicated assays. For F1 progenies (White eyes; DsRED+ eyes; EGFP+ body) in each replicate, 20 males and 50 females were outcrossed with 50 kmo−/− females and 20 kmo−/− males, respectively. Female mosquitoes were blood-fed two times, and all subsequent embryos were hatched and scored for F2 larval phenotypes.
The one piece-SSA test
For kmo-targeted transgenes carrying both direct repeats (SSA substrate) and the nuclease (SSA trigger), ~20–25 female mosquitoes (kmoDR0.7-nos-SceI; kmoDR1.5-nos-SceI) were outcrossed with 20 kmo−/− males in triplicated assays. For each replicate, ~500–800 offspring pupae were scored for DNA repair-associated marker phenotypes, as stated for the split-SSA test. In every generation, ~25 females per replicate that displayed intact marker phenotypes (White eyes; DsRED+ eyes; EGFP+ body) were further screened for silent indel mutation (an in-frame shift) in the DsRED gene by high-resolution melt analysis (HRMA). Only females with the active I-SceI site were outcrossed with kmo−/− males for the offspring generation. For the single-generation assay, HRMA-verified G10 females were allowed to individually oviposit, and progeny quantified by the EAgaL plate method72; G11 progeny per female were scored for DNA repair pathway-dependent marker phenotypes.
Sequencing analysis
To dissect the rates of individual DR motif-dependent processes within P5-EGFP, the split-SSA test was performed in triplicates. In F2 larval screening, L2 instar larvae (n = 100 per replicate) were collected for WR marker phenotypes (Kmo−; DsRED+), which resulted from the elimination of [PUb-EGFP-SV40]. Subsequently, genomic DNA was purified from each replicate pool as PCR templates, and a primer pair that is specific to 3′-DsRED (DsRED-3F1) and the Mos arm (MLF1) (Supplementary Table S2) were used for the amplification of SSA-processed regions (~0.7-kb to 0.9-kb), which were gel-extracted for Oxford Nanopore sequencing73. The initially obtained raw reads (n = 4449) were filtered by using ‘Amplicon_sorter’74 to isolate 1783 reads (40.1%) greater than or equal to the 0.5-kb cutoff and sort them into groups based on sequence similarity. These consensus groups were then aligned to seven types of signature sequences (~60–100 nucleotides), which represent the predicted end-products of SSA-based DSB processes occurred within P5-EGFP, by using Minimap275 enforcing a mapping quality of 20, with potential secondary alignments further filtered to obtain the successful alignment of 794 reads. Lastly, these alignments were sorted and indexed using SAMtools76 for subsequent visualization and analysis via the Broad Institute Integrative Genomics Viewer (IGV) web application77. Signature sequences of DSB repair end-products in P5-EGFP (FASTA) are described in Supplementary Note 2.
Statistics and reproducibility
As indicated, comparisons were performed ANOVA followed by either Tukey’s multiple comparison test (if data were normal) or the Kruskal–Wallis test (if normality test was failed) as implemented in Graphpad Prism V10.2.2. Thresholds for significance are described in each figure legend. All experiments were repeated a minimum of three times, number of mosquitoes screened was as large as possible given the realities of space and personnel available. Number of progeny scored was only limited by the reproductive capacity of each female (ie, all progeny were scored). Number of individuals genotyped (50–100) again as a compromise between depth and available resources. Replicates were either at the level of a pooled cross (all progeny from a group of founders) or from independent founders (when progeny were scored individually).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Plasmid sequences are deposited in GenBank (PP693225-PP693229) and amplicon sequencing data are available at NCBI Sequence Read Archive under BioProject ID PRJNA1101529. Raw data is available in Supplementary Data; uncropped images are available in Supplementary Fig. 5. All other data are available from the corresponding author (or other sources, as applicable) on reasonable request.
References
Norrby, R. Outlook for a dengue vaccine. Clin. Microbiol. Infect. 20, 92–94 (2014).
Powell, J. R. & Tabachnick, W. J. History of domestication and spread of Aedes aegypti-a review. Mem. Inst. Oswaldo Cruz 108, 11–17 (2013).
Wilder-Smith, A. et al. Epidemic arboviral diseases: priorities for research and public health. Lancet Infect. Dis. 17, e101–e106 (2017).
Bouzid, M., Brainard, J., Hooper, L. & Hunter, P. R. Public health interventions for Aedes control in the time of Zikavirus– A meta-review on effectiveness of vector control strategies. PLoS Negl. Trop. Dis. 10, e0005176 (2016).
Dahmana, H. & Mediannikov, O. Mosquito-borne diseases emergence/resurgence and how to effectively control it biologically. Pathogens 9, 310 (2020).
Moyes, C. L. et al. Contemporary status of insecticide resistance in the major Aedes vectors of arboviruses infecting humans. PLoS Negl. Trop. Dis. 11, e0005625 (2017).
Alphey, L. Genetic control of mosquitoes. Annu. Rev. Entomol. 59, 205–224 (2014).
Raban, R., William, A. C. G. & Omar, S. A. A perspective on the expansion of the genetic technologies to support the control of neglected vector-borne diseases and conservation. Front. Trop. Dis. 3, 999273 (2022).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Adolfi, A. et al. Efficient population modification gene-drive rescue system in the malaria mosquito Anopheles stephensi. Nat. Commun. 11, 5553 (2020).
Anderson, M. A. E. et al. Closing the gap to effective gene drive in Aedes aegypti by exploiting germline regulatory elements. Nat. Commun. 14, 338 (2023).
Verkuijl, S. A. N. et al. A CRISPR endonuclease gene drive reveals distinct mechanisms of inheritance bias. Nat. Commun. 13, 7145 (2022).
Carballar-Lejarazú, R. et al. Next-generation gene drive for population modification of the malaria vector mosquito, Anopheles gambiae. Proc. Natl. Acad. Sci. USA 117, 22805–22814 (2020).
Gantz, V. M. & Bier, E. The mutagenic chain reaction: a method for converting heterozygous to homozygous mutations. Science 348, 442–444 (2015).
Hammond, A. et al. Regulating the expression of gene drives is key to increasing their invasive potential and the mitigation of resistance. PLoS Genet 17, e1009321 (2021).
Kyrou, K. et al. A CRISPR–Cas9 gene drive targeting doublesex causes complete population suppression in caged Anopheles gambiae mosquitoes. Nat. Biotechnol. 36, 1062–1071 (2018).
Li, M. et al. Development of a confinable gene drive system in the human disease vector Aedes aegypti. Elife 9, e51701 (2020).
Reid, W. et al. Assessing single-locus CRISPR/Cas9-based gene drive variants in the mosquito Aedes aegypti via single-generation crosses and modeling. G3 Genes Genomes Genet. 12, jkac280 (2022).
Simoni, A. et al. A male-biased sex-distorter gene drive for the human malaria vector Anopheles gambiae. Nat. Biotechnol. 38, 1054–1060 (2020).
Smidler, A. L. et al. A confinable female-lethal population suppression system in the malaria vector, Anopheles gambiae. Sci. Adv. 9, eade8903 (2023).
Alphey, L. Can CRISPR-Cas9 gene drives curb malaria? Nat. Biotechnol. 34, 149–150 (2016).
Noble, C., Adlam, B., Church, G. M., Esvelt, K. M. & Nowak, M. A. Current CRISPR gene drive systems are likely to be highly invasive in wild populations. Elife 7, e33423 (2018).
Adelman, Z. et al. Rules of the road for insect gene drive research and testing. Nat. Biotechnol. 35, 716–718 (2017).
Akbari, O. S. et al. Safeguarding gene drive experiments in the laboratory: Multiple stringent confinement strategies should be used whenever possible. Science 349, 927–929 (2015).
James, S. et al. Pathway to deployment of gene drive mosquitoes as a potential biocontrol tool for elimination of malaria in sub-Saharan Africa: recommendations of a scientific working group. Am. J. Trop. Med. Hyg. 98, 1–49 (2018).
Kaebnick, G. E. et al. Precaution and governance of emerging technologies. Science 354, 710–711 (2016).
Oye, K. A. et al. Regulating gene drives. Science 345, 626–628 (2014).
Noble, C. et al. Daisy-chain gene drives for the alteration of local populations. Proc. Natl. Acad. Sci. USA 116, 8275–8282 (2019).
Taxiarchi, C. et al. A genetically encoded anti-CRISPR protein constrains gene drive spread and prevents population suppression. Nat. Commun. 12, 3977 (2021).
Wu, B., Luo, L. & Gao, X. J. Cas9-triggered chain ablation of cas9 as a gene drive brake. Nat. Biotechnol. 34, 137–138 (2016).
Xu, X. R. S. et al. Active genetic neutralizing elements for halting or deleting gene drives. Mol. Cell 80, 246–262 (2020).
Zapletal, J. et al. Making gene drive biodegradable. Philos. Trans. R. Soc. Lond. B Biol. Sci. 376, 20190804 (2021).
Chae, K. et al. Engineering a self-eliminating transgene in the yellow fever mosquito, Aedes aegypti. PNAS Nexus 1, pgac037 (2022).
Ivanov, E. L., Sugawara, N., Fishman-Lobell, J. & Haber, J. E. Genetic requirements for the single-strand annealing pathway of double-strand break repair in Saccharomyces cerevisiae. Genetics 142, 693–704 (1996).
Lin, F. L., Sperle, K. & Sternberg, N. Model for homologous recombination during transfer of DNA into mouse L cells: role for DNA ends in the recombination process. Mol. Cell. Biol. 4, 1020–1034 (1984).
Aryan, A., Anderson, M. A. E., Myles, K. M. & Adelman, Z. N. Germline excision of transgenes in Aedes aegypti by homing endonucleases. Sci. Rep. 3, 1603 (2013).
Takeuchi, R., Certo, M., Caprara, M. G., Scharenberg, A. M. & Stoddard, B. L. Optimization of in vivo activity of a bifunctional homing endonuclease and maturase reverses evolutionary degradation. Nucleic Acids Res. 37, 877–890 (2009).
Finney, M., Romanowski, J. & Adelman, Z. N. Strategies to improve homology‑based repair outcomes following CRISPR‑based gene editing in mosquitoes: lessons in how to keep any repair disruptions local. Virol. J. 19, 128 (2022).
Overcash, J. M., Aryan, A., Myles, K. M. & Adelman, Z. N. Understanding the DNA damage response in order to achieve desired gene editing outcomes in mosquitoes. Chromosom. Res. 23, 31–42 (2015).
Chang, H. H. Y., Pannunzio, N. R., Adachi, N. & Lieber, M. R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 18, 495–506 (2017).
Marini, F., Rawal, C. C., Liberi, G. & Pellicioli, A. Regulation of DNA double strand breaks processing: focus on barriers. Front. Mol. Biosci. 6, 55 (2019).
Bétermier, M., Bertrand, P. & Lopez, B. S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet 10, e1004086 (2014).
Hammond, A. M. et al. The creation and selection of mutations resistant to a gene drive over multiple generations in the malaria mosquito. PLoS Genet 13, e1007039 (2017).
KaramiNejadRanjbar, M. et al. Consequences of resistance evolution in a Cas9-based sex conversion-suppression gene drive for insect pest management. Proc. Natl. Acad. Sci. USA 115, 6189–6194 (2018).
Symington, L. S. & Gautier, J. Double-strand break end resection and repair pathway choice. Annu. Rev. Genet. 45, 247–271 (2011).
Chae, K. et al. CRISPR-based gene editing of non-homologous end joining factors biases DNA repair pathway choice toward single-strand annealing in Aedes aegypti. Curr. Res. Biotechnol. 5, 100133 (2023).
Roy, S. D. et al. Generation of marker free salt tolerant transgenic plants of Arabidopsis thaliana using the gly I gene and cre gene under inducible promoters. Plant Cell. Tissue Organ Cult. 95, 1–11 (2008).
Tan, J. et al. An efficient marker gene excision strategy based on CRISPR/Cas9-mediated homology-directed repair in rice. Int. J. Mol. Sci. 23, 1588 (2022).
Cannavo, E. & Cejka, P. Sae2 promotes dsDNA endonuclease activity within Mre11-Rad50-Xrs2 to resect DNA breaks. Nature 514, 122–125 (2014).
Bhargava, R., Onyango, D. O. & Stark, J. M. Regulation of single-strand annealing and its role in genome maintenance. Trends Genet 32, 566–575 (2016).
Sartori, A. A. et al. Human CtIP promotes DNA end resection. Nature 450, 509–514 (2007).
Yannuzzi, I., Butler, M. A., Fernandez, J. & Larocque, J. R. The role of drosophila ctip in homology-directed repair of dna double-strand breaks. Genes (Basel) 12, 1430 (2021).
Yusufzai, T. & Kadonaga, J. T. HARP is an ATP-driven annealing helicase. Science 322, 748–750 (2008).
Kassavetis, G. A. & Kadonaga, J. T. The annealing helicase and branch migration activities of Drosophila HARP. PLoS One 9, e98173 (2014).
Dewey, E. B., Korda Holsclaw, J., Saghaey, K., Wittmer, M. E. & Sekelsky, J. The effect of repeat length on Marcal1-dependent single-strand annealing in Drosophila. Genetics 223, iyac164 (2023).
Sugawara, N. & Haber, J. E. Characterization of double-strand break-induced recombination: homology requirements and single-stranded DNA formation. Mol. Cell. Biol. 12, 563–575 (1992).
Sugawara, N., Ira, G. & Haber, J. E. DNA length dependence of the single-strand annealing pathway and the role of Saccharomyces cerevisiae RAD59 in double-strand break repair. Mol. Cell. Biol. 20, 5300–5309 (2000).
Rothenberg, E., Grimme, J. M., Spies, M. & Ha, T. Human Rad52-mediated homology search and annealing occurs by continuous interactions between overlapping nucleoprotein complexes. Proc. Natl. Acad. Sci. USA 105, 20274–20279 (2008).
Kelso, A. A., Lopezcolorado, F. W., Bhargava, R. & Stark, J. M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet 15, e1008319 (2019).
Mendez-Dorantes, C., Bhargava, R. & Stark, J. M. Repeat-mediated deletions can be induced by a chromosomal break far from a repeat, but multiple pathways suppress such rearrangements. Genes Dev. 32, 524–536 (2018).
Do, A. T., Brooks, J. T., Le Neveu, M. K. & LaRocque, J. R. Double-strand break repair assays determine pathway choice and structure of gene conversion events in drosophila melanogaster. G3 Genes Genomes Genet 4, 425–432 (2014).
Hu, Q. et al. Break‐induced replication plays a prominent role in long‐range repeat‐mediated deletion. EMBO J. 38, e101751 (2019).
Li, M. et al. Germline Cas9 expression yields highly efficient genome engineering in a major worldwide disease vector, Aedes aegypti. Proc. Natl. Acad. Sci. USA 114, E10540–E10549 (2017).
Magnusson, K. et al. Transcription regulation of sex-biased genes during ontogeny in the Malaria vector Anopheles gambiae. PLoS One 6, e21572 (2011).
Aryan, A., Myles, K. M. & Adelman, Z. N. Targeted genome editing in Aedes aegypti using TALENs. Methods 69, 38–45 (2014).
Basu, S., Aryan, A., Haac, M. E., Myles, K. M. & Adelman, Z. N. Methods for TALEN evaluation, use, and mutation detection in the mosquito Aedes aegypti. Methods Mol. Biol. 1338, 157–177 (2016).
Kistler, K. E., Vosshall, L. B. & Matthews, B. J. Genome engineering with CRISPR-Cas9 in the mosquito Aedes aegypti. Cell Rep. 11, 51–60 (2015).
Aryan, A., Anderson, M. A. E., Myles, K. M. & Adelman, Z. N. TALEN-based gene disruption in the dengue vector Aedes aegypti. PLoS One 8, e60082 (2013).
Coates, C. J., Turney, C. L., Frommer, M., O’Brochta, D. A. & Atkinson, P. W. Interplasmid transposition of the mariner transposable element in non-drosophilid insects. Mol. Gen. Genet. 253, 728–733 (1997).
Amos, B. et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 50, D898–D911 (2022).
Chae, K., Valentin, C., Jakes, E., Myles, K. M. & Adelman, Z. N. Novel synthetic 3′-untranslated regions for controlling transgene expression in transgenic Aedes aegypti mosquitoes. RNA Biol. 18, 223–231 (2021).
Tsujimoto, H. & Adelman, Z. N. Improved fecundity and fertility assay for Aedes aegypti using 24 well tissue culture plates (EAgaL plates). J. Vis. Exp. 4, e61232 (2021).
Wang, Y., Zhao, Y., Bollas, A., Wang, Y. & Au, K. F. Nanopore sequencing technology, bioinformatics and applications. Nat. Biotechnol. 39, 1348–1365 (2021).
Vierstraete, A. R. & Braeckman, B. P. Amplicon_sorter: a tool for reference-free amplicon sorting based on sequence similarity and for building consensus sequences. Ecol. Evol. 12, e8603 (2022).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100 (2018).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Acknowledgements
We thank the research staff in the Adelman laboratory at Texas A&M University for excellent technical assistance for this study. This work was supported by the National Institute of Allergies and Infectious Diseases of the National Institutes of Health (NIH-NIAID) under award numbers (R01AI148787, R01AI137112). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
K.C. and Z.N.A. conceived the research and experimental design and wrote the manuscript. K.M.M. revised the manuscript. K.C. conducted the experiments. B.C. performed assays of transgene-eliminating dynamics and phenotypic screening. J.S.R. provided mass sequence alignment analysis. C.D. participated in the generation of transgenic strains. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
Z.N.A. and K.M.M. are listed as inventors on provisional patent TAMC:054USP2 related to self-eliminating transgenes.
Peer review
Peer review information
Communications Biology thanks Benjamin Matthews and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chae, K., Contreras, B., Romanowski, J.S. et al. Transgene removal using an in cis programmed homing endonuclease via single-strand annealing in the mosquito Aedes aegypti. Commun Biol 7, 660 (2024). https://doi.org/10.1038/s42003-024-06348-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06348-6
