
Abstract
PHD2 is essential in modulating HIF-1α levels upon oxygen fluctuations. Hypoxia, a hallmark of uterus, and HIF-1α have recently emerged as opposing regulators of mesendoderm specification, suggesting a role for PHD2 therein. We found that PHD2 expression initially covered the epiblast and gradually receded from the primitive streak, which was identical to hypoxia and exclusive to HIF-1α. The investigations performed in mESCs, embryoids, and mouse embryos together demonstrated that PHD2 negatively regulated mesendoderm specification. Single-cell RNA sequencing revealed that PHD2 governed the transition from epiblast to mesendoderm. The downstream effect of PHD2 relied on the HIF-1α regulated Wnt/β-catenin pathway, while it was regulated upstream by miR-429. In summary, our research highlights PHD2’s essential role in mesendoderm specification and its interactions with hypoxia and HIF-1α.
Similar content being viewed by others


Introduction
Prolyl Hydroxylase Domain-Containing Protein 2 (PHD2, also known as EGLN1) along with PHD1 and PHD3 belong to the PHD protein family in mammals, which catalyzes the hydroxylation of the α subunits of hypoxia-inducible factors (HIF-α). Hydroxylated HIF-α proteins were then recognized by the von Hippel Lindau protein and subsequently subjected to ubiquitin-proteasome protein degradation1,2. Among the three PHDs, PHD2 is the most evolutionarily conserved isoform and is widely regarded as the primary regulator of HIF-1α levels, which is the essential factor mediating hypoxia responses3. As the hydroxylation process was oxygen dependent, the PHD2 triggered HIF-1α degradation serves to sense oxygen concentration and dynamically adjusts the effects of hypoxia and HIF-1α. Apart from oxygen levels, the expression level of PHD2 was another determinant for its catalytic activity, which was tightly controlled at multilayers. In particular, loss of PHD2 improved the stability of HIF-1α factors and generated a so called “pseudohypoxia” status despite oxygen concentrations4,5. The biological significance of PHD members was previously determined via genetic knockout. As a result, the global depletion of PHD2 led to embryonic lethality in mice around E12.5-14.5 stages because of the defects of placenta and cardiac developments, whereas the mice with depletions of either PHD1 or PHD3 could normally survive, implying an indispensable role of PHD2 in mice embryo development6. Besides these, conditional knockouts of PHD2 in mesendoderm-derived lineages, such as vascular endothelial and smooth muscle, all displayed remarkable defects, implying an essential role in mesendoderm development7. Nevertheless, the detailed regulatory role and mechanism of PHD2 in embryo development, especially mesendoderm development, were uninvestigated.
Mesendoderm is the common precursor for both the mesoderm and endoderm lineages, and its formation is fundamental to gastrulation and the establishment of the body axis8. Mesendoderm formed as the posterior epiblast ingressed through the primitive streak, while the anterior epiblast differentiated into neuroectoderm. The development of mesendoderm was precisely controlled by multiple signaling pathways, transcription factors, and environmental conditions within the uterus. Hypoxia as a typical feature of the uterus widely and profoundly affects embryo development and variable processes of embryonic stem cells (ESCs) of mammals9,10. In most of these studies, HIF-1α served as a faithful mediator of the effects of hypoxia and thus exhibited a synergistic phenotype with hypoxia11,12,13. In contrast, our recent study discovered hypoxia and HIF-1α exerted opposite functions in regulation mesendoderm via the Wnt/β-catenin pathway14. However, the mechanism mediating such contradictory functions of hypoxia and HIF-1α remained unclear. Considering that PHD2 regulates the stability of HIF-1α in response to oxygen changes, we speculated that PHD2 manipulated hypoxia/HIF-1α axis and regulated the mesendoderm development. Moreover, the Wnt/β-catenin pathway, the mutual downstream axis for hypoxia and HIF-1α, is stimulated by exogenic BMP factors secreated from extraembryonic parts and initiated the mesendoderm development program in mouse embryos15. Numerous in vitro and in vivo studies have demonstrated that activation of the Wnt/β-catenin pathway efficiently induces and boosts mesendoderm development16,17. However, it is still unknown whether PHD2 regulates the Wnt/β-catenin pathway.
In this study, we investigated the effect of PHD2 on mesendoderm development in multiple systems, including mESCs, mouse embryos, and embryoids. The results consistently indicated that moderate PHD2 safeguards mesendoderm development. Furthermore, we employed single-cell RNA sequencing to validate this finding and discover the specific function window of PHD2. Our research also explored the potential interplay between hypoxia, HIF-1α, and PHD2, revealing that PHD2 might mediate pseudohypoxia rather than authentic hypoxia in regulating HIF-1α. The regulatory role of PHD2 in mesendoderm development hinges on its control over HIF-1α and the downstream Wnt/β-catenin pathway.
Results
PHD2-mediated pseudohypoxia was closely associated with mesendoderm development
Mouse embryos developed in a relatively hypoxic uterine environment. Our recent study revealed that hypoxia inhibited mesendoderm development, while HIF-1α promoted this process14. PHD2 catalyzes the hydroxylation of HIF-1α, thereby regulating the dynamic changes of hypoxia and HIF-1α. However, an integrated overview of hypoxia, HIF-1α, and PHD2 in early embryogenesis was lacking. Through immunostaining for hypoxia and OCT4 in mouse embryos, we observed that hypoxia was present throughout the whole embryos, including the inner cell mass (ICM) at the E3.5 stage. By the E6.0 stage, hypoxia was exclusively localized to the OCT4-positive epiblast and was only present in the extraembryonic parts. Similarly, at the E7.5 stage, hypoxia was clearly detected in extraembryonic structures but was absent in the T-positive primitive streak (corresponding to mesendoderm in terms of germ layers). (Supplementary Fig. 1a) These results support our previous finding that hypoxia suppresses mesendoderm specification.
We then conducted co-immunostaining of PHD2, HIF-1α, and hypoxia in mouse embryos. At the E4.5 stage, hypoxia, HIF-1α, and PHD2 were co-localized throughout the entire mouse embryos. From E5.5 to E7.5, hypoxia was restricted to the extraembryonic parts and primordial germ cells (PGCs). PHD2 was initially expressed in the entire embryos but gradually reduced in the epiblast, with HIF-1α becoming progressively more pronounced in the epiblasts where PHD2 was not expressed. (Fig. 1a, Supplementary Fig. 1b) To validate the patterns of hypoxia and HIF-1α in embryos, we analyzed RNA sequencing (RNA-seq) data reported previously14. The RNA-seq data was consisted of the following three groups: (1) wildtype AB2.2 mESCs subjected to mesendoderm differentiation under hypoxia versus normoxia; (2) HIF-1α knockdown AB2.2 mESCs subjected to mesendoderm differentiation under hypoxia versus normoxia; (3) Dox-inducible HIF-1α overexpressing AB2.2 mESCs subjected to mesendoderm differentiation under normoxia treated with Dox versus non-Dox control. (Supplementary Fig. 1c) In this analysis, we found that F10 and Cldn2 were induced under the influence of hypoxia independent of HIF-1α, whereas Nkx1-2 and Notum were induced by HIF-1α rather than hypoxia. (Supplementary Fig. 1d) By examining spatial transcriptomics data from previous reports18, we determined that F10 and Cldn2 were mainly expressed in extraembryonic parts, while Nkx1-2 and Notum were primarily expressed in the primitive streak of E7.5 embryos, resembling the patterns of hypoxia and HIF-1α at this stage. (Supplementary Fig. 1e) Moreover, HIF-1α was more enriched in the posterior side than the anterior at the E7.5 stage, suggesting a potential role in mesendoderm development. (Fig. 1b) In summary, these data demonstrate that PHD2 might restrict the enrichment of HIF-1α in mesendoderm despite oxygen abundance, a state known as pseudohypoxia.

a The patterns of hypoxia, HIF-1α, and PHD2 at E7.5 stage. The dashed line represents the primordial germ cells (PGCs). (n = 3). b The relative intensity of HIF-1α from anterior to posterior of the epiblast. c The expression pattern of PHD2 at E7.5 stage (n = 3), and d the relative intensity of PHD2 from anterior to posterior of the epiblast. e The schematic diagram showing how mESCs were clustered into the PHD2low and PHD2high groups using a genetical EGFP knock-in to the end of the PHD2 locus. f The mRNA levels of mesendoderm markers after the mesendoderm differentiation of the PHD2low and PHD2high mESC, respectively. (n = 4). g The immunostaining against T and (h) the flow cytometry to determine the T-positive cell ratios after the mesendoderm differentiation of the WT and PHD2-KO mESCs, respectively. Blue region represents gating negative cells and red region represents treated cells. (n = 3). i Western blots verification of PHD2 inductions with variable Dox treatments in Dox-inducible PHD2 overexpressing mESCs (n = 3). j The T-positive cell ratios determined by flow cytometry (k) and the mRNA levels of T and Mixl1 after the mesendoderm differentiation of the Dox-inducible PHD2 overexpressing mESCs treated with variable concentrations of Dox (n = 4). l The mRNA levels of T and Mixl1, the immunostaining against T in (m), and (n) the T-positive percentages after the mesendoderm differentiation of the WT and PHD2-KO mESCs that were treated with either DMSO or IOX4. (n = 4) Scale bars, 100 μm, BF bright field, WT wild type, KO PHD2-KO, A anterior, P posterior, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns not significant.
In vitro differentiation of mouse embryonic stem cells (mESCs) is a widely used platform for studying developmental processes. In our study, we conducted mesendoderm differentiation of mESCs and evaluated the expressions of PHD2 and HIF-1α throughout the process. We observed that the protein level of PHD2 decreased on differentiation day 2, coinciding with the increase in HIF-1α levels. (Supplementary Fig. 1f, g) The mRNA expression pattern of Phd2 mirrored that of its protein, while Hif-1α mRNA, in contrast to HIF-1α protein, remained stable at a relatively low level throughout the differentiation process. (Supplementary Fig. 1h) The disparities between Hif-1α mRNA and protein levels suggested that the regulation of HIF-1α primarily occurs at the protein level. Additionally, we compared PHD2 with other members of the PHD family and found that, unlike Phd2, the expressions of Phd1 and Phd3 did not show an adverse correlation with HIF-1α, suggesting that PHD2 plays a central role in regulating HIF-1α. Based on these findings, we embarked on a study to investigate whether PHD2 regulates mesendoderm development by modulating HIF-1α.
PHD2 suppressed mesendoderm specification of mESCs
The expression patterns of PHD2 during mouse embryo development exhibited high dynamics. PHD2 was notably expressed in the ICM of E4.5 mouse embryos. As development progressed, PHD2 gradually downregulated within the epiblast, and it became predominantly enriched in the extraembryonic regions by the E7.5 stage. It’s worth noting that at the E7.0 stage, PHD2 was observed in the anterior side of the epiblast but not the posterior side. (Fig. 1c, d, Supplementary Fig. 1i) Immunostainings at the end of mesendoderm differentiation of mESCs revealed exclusive expression of T and PHD2. (Supplementary Fig. 1j) These findings suggested that proper mesendoderm development may have necessitated the downregulation of PHD2.
Subsequently, we investigated the lineage tendencies of mESCs with varying levels of PHD2 expression. We generated an mESC line called PHD2-EGFP, which integrated a P2A-EGFP cassette at the stop codon of Phd2 gene (Supplementary Fig. 2a). Using FACS, we divided PHD2-EGFP cells into PHD2 high-expressing and low-expressing mESCs (PHD2high and PHD2low), and detected the expression of PHD2 and HIF-1a, respectively. As expected, the PHD2low group exhibited lower PHD2 and higher HIF-1a expression compared to the PHD2high group. (Fig. 1e, Supplementary Fig. 2b, c) By the end of the differentiations, both the expression levels of mesendoderm markers (T, Eomes, Gsc, and Mesp1) and the ratios of T-positive cells were significantly increased in the PHD2low group, suggesting that low PHD2 expression might enhance mesendoderm differentiation (Fig. 1f, Supplementary Fig. 2d).
To validate this concept, we established a PHD2 knockout mESC line (PHD2-KO), which was subjected to mesendoderm differentiation along with its control wild type (WT) mESCs. (Supplementary Fig. 2e) The expression of mesendoderm markers and the proportion of T-positive cells were both markedly increased in the PHD2-KO group. (Fig.1g, h, Supplementary Fig. 2f) Similarly, we generated two PHD2 knockdown mESC lines (shPHD2-1 and shPHD2-2). (Supplementary Fig. 2g, h) In comparison to the scramble control, both PHD2 knockdown mESCs exhibited enhanced mesendoderm differentiation, as indicated by the upregulation of mesendoderm markers. Furthermore, the effects of PHD2 were dose-dependent, as evident from the expression levels of PHD2 and mesendoderm markers in the shPHD2-1 and shPHD2-2 groups. (Supplementary Fig. 2i) This dose-dependent effect was further confirmed by experiments conducted on a Doxycycline (Dox)-inducible PHD2 overexpression mESC line. By varying the doses of Dox, we induced two different levels of PHD2 overexpression. (Fig. 1i) As a result, the expressions of T and Mixl1, as well as the proportion of T-positive cells, were significantly reduced compared to the non-Dox control and displayed an inverse correlation with the PHD2 levels (Fig. 1j, k).
Since the mesendoderm differentiations mentioned above were carried out in AB2.2 background mESCs, we further confirmed the effects of PHD2 in R1 mESCs. Similarly, PHD2 knockout and overexpression led to significant upregulation and downregulation of mesendoderm differentiation, as indicated by the expression levels of mesendoderm markers, respectively. (Supplementary Fig. 2j–m) Additionally, to investigate whether the effect of PHD2 depended on its enzymatic activity, we introduced a PHD enzyme inhibitor, IOX419, during the mesendoderm differentiation of WT and PHD2-KO mESCs. Compared to the DMSO control, IOX4 treatment significantly enhanced the expression of T and Mixl1 in the WT group. However, in the PHD2-KO group, the effect of IOX4 was negligible (Fig. 1l–n). These results suggested that downregulation of PHD2 promoted mesendoderm specification, while upregulation of PHD2 hindered this process, and these effects were contingent on its catalytic activity.
PHD2 depletion led to excessive mesendoderm development
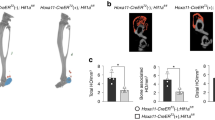
We achieved PHD2 knockout mice by genetically removing the 2nd and 3rd exons of Phd2 using the CRISPR/Cas9 method. We set up intercrossing of PHD2+/− mice and obtained wild-type (PHD2+/+), PHD2 heterozygous knockout (PHD2+/−), and PHD2 homozygous knockout (PHD2−/−) embryos (Fig. 2a). Through immunostaining against T at the E7.5 stage, we found that mesendoderm development was similar between the PHD2+/+ and PHD2+/− embryos, although PHD2+/− embryos exhibited a slightly expanded T-positive region. In contrast, PHD2−/− embryos displayed significantly excessive mesendoderm formation (Fig. 2b, Supplementary Fig. 2n). The T fluorescence signal spectrum, from anterior to posterior, also revealed a more extensive T-positive area in PHD2−/− embryos compared to PHD2+/+ and PHD2+/− embryos (Fig. 2c). Similarly, T-positive region ratios were markedly increased in PHD2−/− embryos when compared to PHD2+/+ and PHD2+/− embryos (Fig. 2d). Meanwhile, we assessed the hypoxic regions within these three types of embryos and found that PHD2 knockout had little impact on hypoxia distributions and the T-positive mesendoderm regions were all exclusive to hypoxia (Fig. 2b).

a The schematic diagram showing the mating strategy. b The immunostaining against T and hypoxia probe in PHD2+/+ (n = 3), PHD2+/− (n = 8), and PHD2−/− embryos (n = 4) at the E7.5 stage. c The fluorescence signal spectrum and d positive percentages of T in (b) were measured. Scale bars, 100 μm; A anterior, P posterior, +/+ PHD2+/+, +/− PHD2+/−, −/−, PHD2+/−, ***P < 0.001, ****P < 0.0001, ns not significant.
Following this, we employed an embryonic–inducible trophoblast–inducible extraembryonic endoderm (EiTiX) embryoid formation system to investigate the influence of PHD2 on mesendoderm differentiation. The EiTiX-embryoid formation protocol was previously reported20,21 and comprised three types of mESCs: Dox-inducible CDX2 overexpressing mESCs (iCdx2) for trophoblast production, Dox-inducible GATA4 overexpressing mESCs (iGata4) for extraembryonic endoderm (Supplementary Fig. 2o), and candidate mESCs for generating the epiblast or germ layers. We conducted EiTiX-embryoid formation assays with WT, PHD2 knockout (PHD2-KO), and PHD2 overexpression (PHD2-OE) mESCs as candidate mESCs, respectively. The EiTiX-embryoid formation process spanned six days, resulting in embryoids that resembled the E7.5 stage of embryos. (Fig. 3a) Through immunostaining against T, we observed that the PHD2-KO group exhibited expanded mesendoderm formation compared to the WT group, while the PHD2-OE group hardly showed any T-positive regions. (Fig. 3b) The expressions of T and Mixl1 were significantly upregulated in the PHD2-KO group and downregulated in the PHD2-OE group in comparison to the WT group. (Supplementary Fig. 2p)

a The schematic diagram showing the overall design of embryoid formation with different mESC lines. b The immunostaining against T on the embryoids formed with the WT (n = 4), PHD2-KO (n = 4), and PHD2-OE mESCs (n = 4), respectively. c The schematic diagram showing the overall design of embryoid formation with a mixture of the WT and PHD2-KO mESCs that were labeled with GFP and mCherry, respectively. d The immunostaining against GFP and mCherry on the embryoids (n = 6); The dashed line represents the exclusion of extraembryonic endoderm tissue from the whole embryo. Scale bars, 100 μm; BF bright field, A anterior, P posterior, WT wild type, KO PHD2-KO, OE PHD2-OE, iCdx2 inducible CDX2 expression, iGata4 inducible GATA4 expression.
Subsequently, we generated GFP-labeled WT mESCs and mCherry-labeled PHD2-KO mESCs. These two cell types were mixed at a 1:1 ratio and used to form EiTiX-embryoids alongside iCdx2 and iGata4 cells. (Fig. 3c) We observed that the mCherry-labeled PHD2-KO cells tended to localize at primitive streaks, whereas the WT mESCs contributed to the entire inner-embryo regions (Fig. 3d). These results suggested that PHD2 depletion enhances the potential of mESCs to contribute to mesendoderm.
PHD2 depletion facilitated the differentiation from epiblast to mesendoderm
We conducted mESC differentiations on both WT and PHD2-KO mESCs using a high-serum medium to avoid lineage bias. On differentiation day 4, we performed single-cell RNA sequencing (scRNA-seq) on cells from these two groups. The scRNA-seq dataset included 18,552 genes and 16,048 cells (comprising 8841 cells from the WT group and 7207 cells from the PHD2-KO group). We grouped all cells into nine clusters (clusters 0–8) and assigned them to four cell subgroups: undifferentiated ES (undiff. ES), epiblast, mesendoderm, and ectoderm, based on canonical cell markers. (Fig. 4a, b, Supplementary Fig. 3a) Notably, we did not identify primitive endoderm and trophoblast cells in our data, which was consistent with previous reports22,23 indicating that both primitive endoderm and trophoblast are not typically generated under normal differentiation conditions. The proportions of ectoderm and undifferentiated ES were similar between the WT and PHD2-KO groups. However, the mesendoderm proportion was significantly increased in the PHD2-KO group, while the epiblast proportion decreased (Fig. 4c). Correspondingly, the T-positive cell ratio was elevated in the PHD2-KO group (Fig. 4d).

a The UMAP plot showing the cell subgroups in the scRNA-seq data. b The UMAP plot showing the cells belonging to the WT and PHD2-KO groups, respectively. c The ratios of each cell subgroup and d the T-positive cell ratios in the WT and PHD2-KO groups, respectively. e The differentiation trajectory of the scRNA-seq data. f The pseudotime distributions of each cell subgroup of the WT and PHD2-KO groups using the Wilcoxon Rank Sum Test. g The mRNA levels of Nanog, Esrrb, Otx2, Dnmt3b, T, and Mixl1 along the mesendoderm differentiation of the Ctrl and PHD2-OE groups (n = 4). h The NES and significance of the GSEA terms related to developmental processes. i The dot plot showing the NES and p-values of the GSEA related to the development-related pathways. The GSEA curves showing the changes of (j) the canonical Wnt pathway and (k) the HIF-1 signaling pathway with PHD2-KO. T- T negative, T+ T positive, NES normalized enrichment score, WT wild type, KO PHD2-KO, Ctrl control, OE PHD2-OE, ****P<0.0001, ns, not significant.
We also analyzed the differentiation trajectory (Fig. 4e). The pseudotime distribution was notably increased in the mesendoderm subgroup of PHD2-KO, while it remained unchanged in the other three subgroups. (Fig. 4f) The pseudotime distributions of the markers used for cell annotations aligned with their expected expression patterns during differentiation. (Supplementary Fig. 3b) These findings suggested that PHD2-KO led to a precocious and enhanced transition from epiblast to mesendoderm. Consistently, we conducted RT-qPCR to assess the expression of markers representing various developmental stages. We found that the markers for naïve (Nanog and Esrrb) and primed (Otx2 and Dnmt3b) states remained unchanged in the PHD2-OE group compared to the WT group. However, mesendoderm markers (T and Mixl1) were significantly downregulated, resulting in a delayed transition (Fig. 4g).
Since PHD2 was found to be involved in the transition from epiblast to mesendoderm, we focused on the epiblast subgroup extracted from the scRNA-seq data to understand the changes in gene expressions, biological processes, and signaling pathways associated with PHD2-KO. A significant upregulation of most mesendoderm markers (T, Fgf8, Mixl1, Tdgf1, Mesp1, and Gsc) was observed in the epiblast subgroup due to PHD2-KO. In contrast, ectoderm markers were less affected by PHD2-KO, suggesting that PHD2 has a relatively minor impact on ectoderm specification, which aligns with the finding that the ectoderm cell ratio remained unchanged with PHD2-KO. (Supplementary Fig. 4a) Furthermore, through gene set enrichment analysis (GSEA), we determined that cell fate commitment, gastrulation, and developmental processes related to mesendoderm, mesoderm, and endoderm were significantly promoted by PHD2-KO, while ectoderm development was not significantly affected (Fig. 4h).
We then proceeded to examine the impact of PHD2-KO on signaling pathways related to early embryo development, including the canonical and non-canonical Wnt, BMP, FGF, Nodal, and Notch pathways. Through GSEA of processes associated with these pathways, the canonical Wnt signaling pathway, also known as the Wnt/β-catenin pathway, emerged as the top-ranked pathway in terms of both normalized enrichment scores (NES) and significance. (Fig. 4i, j, Supplementary Fig. 4b) Several components of the Wnt/β-catenin pathway, including positive regulators (Wnt3, Wnt8a, Fzd10, Ctnnb1, Tcf7, Lef1) and downstream targets (Sp5 and Axin2), were significantly upregulated in response to PHD2-KO, while Tcf7l1, a negative regulator of the Wnt/β-catenin pathway, was significantly downregulated. (Supplementary Fig. 4c) These findings strongly indicate that the Wnt/β-catenin pathway is significantly promoted by PHD2 depletion. Furthermore, considering PHD2’s essential role in regulating HIF-1α, we also examined the changes in the HIF-1 signaling pathway with PHD2-KO. The results showed that PHD2-KO significantly enhanced the HIF-1 pathway, agreeing with the previous finding that loss of PHD2 led to pseudohypoxia. This enhancement was confirmed by the significant upregulation of downstream targets of the HIF-1 pathway, such as Pgk1, Ldha, and Aldoa. (Fig. 4k, Supplementary Fig. 4d) In light of our earlier discovery that HIF-1α significantly promotes mesendoderm specification by facilitating the Wnt/β-catenin pathway, we speculate that PHD2-KO may enhance the Wnt/β-catenin pathway by modulating HIF-1α, consequently promoting mesendoderm specification. Additionally, we investigated changes in biological processes within the mesendoderm subgroup induced by PHD2-KO. (Supplementary Fig. 4e) The processes related to gastrulation, germ layer formation, mesendoderm development, and their subsequent processes were significantly promoted, while cell proliferation and certain metabolism-related processes were inhibited. These findings further indicated that PHD2 depletion plays an important role in mesendoderm development.
HIF-1α mediated the effects of PHD2 on mesendoderm specification
We proceeded to investigate whether PHD2 regulated mesendoderm specification by targeting HIF-1α. To begin, we examined the expression patterns of HIF-1α in PHD2+/+, PHD2+/−, and PHD2−/− embryos through immunostaining at the E7.5 stage. In PHD2+/+ embryos, HIF-1α was specifically expressed in the epiblast, with relatively higher expression at the posterior side, which corresponds to its known role in promoting mesendoderm development. In contrast, HIF-1α was extensively expressed throughout the embryos in the PHD2−/− group. HIF-1α expression in PHD2+/− embryos fell between the PHD2+/+ and PHD2−/− groups. (Fig. 5a–c) It’s worth noting that the mesendoderm parts in all these types of embryos were normoxic (Fig. 2b). Therefore, the upregulation of HIF-1α was due to the pseudohypoxia caused by PHD2 loss.

a The immunostaining against HIF-1α and PHD2 in PHD2+/+ (n = 4), PHD2+/− (n = 6), and PHD2−/− embryos (n = 3) at E7.5 stage. b The fluorescence signal spectrum and (c) positive percentages of HIF-1α in (a) were measured. d Western blots showing the protein expressions of HIF-1α in the WT and PHD2-KO mESCs that were treated with either DMSO or IOX4 (n = 4). e The relative levels of HIF-1α in (d) were quantified. f The mRNA levels of Pgk1 and Ldha after the mesendoderm differentiation of the WT and PHD2-KO mESCs that were treated with either DMSO or IOX4. (n = 4). g Western blots showing the protein expressions of HIF-1α and PHD2 in the mESCs of the Ctrl, HIF-1α, and HIF-1α + PHD2 groups, respectively. (n = 4). h The immunostaining against T and i the mRNA levels of mesendoderm markers after the mesendoderm differentiation of mESCs of the Ctrl, HIF-1α, and HIF-1α + PHD2 groups, respectively (n = 4). Scale bars, 100 μm; +/+ PHD2+/+, +/− PHD2+/−, −/− PHD2+/−, A anterior, P posterior, WT wild type, KO PHD2-KO, DMSO DMSO treatment, IOX4 IOX4 treatment, Ctrl control, HIF-1α HIF-1α overexpression; HIF-1α + PHD2, HIF-1α/PHD2 dual overexpressions; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns not significant.
As demonstrated earlier, IOX4, an inhibitor of PHD catalytic activity, promoted mesendoderm differentiation in WT mESCs but had no effect when PHD2 was depleted. Similarly, IOX4 treatment significantly increased HIF-1α levels in WT mESCs but did not change them in PHD2-KO mESCs. This effect was also observed in downstream targets of HIF-1α, Pgk1, and Ldha. (Fig. 5d–f) To further explore the role of HIF-1α in this context, we generated HIF-1α and HIF-1α/PHD2 dual overexpression mESCs and performed mesendoderm differentiation with these cells, along with WT control mESCs. Mesendoderm differentiation was significantly enhanced in both the HIF-1α-OE and HIF-1α/PHD2-OE groups compared to the WT control, with the enhancement being partially compromised in the HIF-1α/PHD2-OE group compared to the sole HIF-1α group, as indicated by the expressions of mesendoderm markers. (Fig. 5g–i) We also tested if the overexpression of HIF-1α in PHD2-OE mESCs could rescue the suppressed mesendoderm differentiation. As a result, HIF-1α overexpression was able to boost the expressions of mesendoderm markers. (Supplementary Fig. 4f) These combined results suggested that HIF-1α mediates the function of PHD2 in mesendoderm differentiation.
The PHD2/HIF-1α axis regulated mesendoderm development through the Wnt/β-catenin pathway
In our previous study, we established that HIF-1α promotes mesendoderm differentiation through the Wnt/β-catenin pathway. In this research, the scRNA-seq data suggested that PHD2 depletion enhances the Wnt/β-catenin pathway. Furthermore, we’ve shown that HIF-1α plays a pivotal role in mediating the function of PHD2 in regulating mesendoderm differentiation. Therefore, we hypothesized that the PHD2/HIF-1α axis regulates mesendoderm differentiation through the Wnt/β-catenin pathway. To test this hypothesis, we used an inhibitor of WNT production-2 (IWP2) to block the Wnt/β-catenin pathway in mesendoderm differentiations of both WT and PHD2-KO cells. The results showed significant downregulations of T and Mixl1, indicating that the Wnt/β-catenin pathway plays a crucial role in mesendoderm differentiation in both WT and PHD2-KO cells. (Supplementary Fig. 4g).
We performed TOPflash assays and found that PHD2-KO significantly promoted the Wnt/β-catenin pathway, and this enhancement was partially compromised by ectopic PHD2 expression. (Fig. 6a) Similarly, HIF-1α overexpression significantly promoted the Wnt/β-catenin pathway, which was partially suppressed by the addition of PHD2. (Fig. 6b) These findings were further confirmed by western blots, where both β-catenin and p-GSK3β (S9) showed trends consistent with the TOPflash results. (Fig. 6c-f) Additionally, the downstream targets of the Wnt/β-catenin pathway, Axin2 and Sp5, were upregulated with PHD2-KO and HIF-1α overexpression but slightly inhibited with PHD2 overexpression. (Supplementary Fig. 4h-i).

a The relative TOPflash activities in the WT, PHD2-KO/Ctrl, and PHD2-KO/PHD2-OE groups (n = 4). b The relative TOPflash activities in the Ctrl, HIF-1α, HIF-1α + PHD2 groups. (n = 4). c Western blots showing the protein expressions of key components of the Wnt/β-catenin pathway in the WT, PHD2-KO/Ctrl, and PHD2-KO/PHD2-OE mESCs, respectively. (n = 4). d The relative levels of β-catenin and p-GSK3β(S9) in (c) were quantified. e Western blots showing the protein expressions of key components of the Wnt/β-catenin pathway in the Ctrl, HIF-1α, HIF-1α + PHD2 mESCs, respectively. (n = 4). f The relative levels of β-catenin and p-GSK3β(S9) in (e) were quantified. g The peaks of the ChIP-seq against HIF-1α, H3K4me3, H3K27ac, H3K9me2, and H3K27me3 in mESCs at the locus of Ctnnb1. h The immunostaining against β-catenin in PHD2+/+ (n = 3), PHD2+/− (n = 5), and PHD2−/− embryos (n = 3) at E7.5 stage. i The fluorescence signal spectrum and j positive percentages of β-catenin in (h) were measured. Scale bars, 100 μm; WT, wild type; KO, PHD2-KO; OE, PHD2-OE; Ctrl, control; HIF-1α, HIF-1α overexpression; HIF-1α + PHD2, HIF-1α/PHD2 dual overexpressions; +/+ PHD2+/+, +/− PHD2+/−, −/− PHD2+/−, A anterior, P posterior; *P < 0.05; **P < 0.01; ****P < 0.0001.
To understand how HIF-1α regulates the Wnt/β-catenin pathway, we analyzed previously reported ChIP-seq data against HIF-1α in mESCs11. HIF-1α peaks were notably identified at the promoter region of Ctnnb1, the gene encoding β-catenin. The patterns of transcription-favorable histone modifications (H3K4me3 and H3K27ac) and inhibitory modifications (H3K9me2 and H3K27me3) suggested that HIF-1α can promote the transcription of Ctnnb1. (Fig. 6g) Through RT-qPCR, we observed that both PHD2-KO and HIF-1α overexpression enhanced the expressions of Ctnnb1, and these improvements were suppressed by PHD2 overexpression. (Supplementary Fig. 4j, k).
Furthermore, in WT E7.5 embryos, β-catenin displayed similar expression patterns to HIF-1α and was primarily enriched in the primitive streak. However, like HIF-1α, β-catenin was extensively expressed throughout the entire embryos in PHD2+/− and PHD2−/− embryos. (Fig. 6h, i) Notably, the ratios of β-catenin-positive regions increased in the order of WT, PHD2+/−, and PHD2−/− embryos. (Fig. 6j) These findings suggested that PHD2 might negatively regulated β-catenin through HIF-1α-mediated transcriptional regulation.
miR-429 is an upstream regulator of PHD2 in mesendoderm specification
We next determined the upstream regulator of PHD2 in mesendoderm differentiation. The expression patterns of Phd2 mRNA and protein were similar, indicating the regulation of PHD2 might be at least partially at the RNA level. (Supplementary Fig. 1f-h) Previous studies have discovered that the expression of the miR-200 families, including miR-200b and miR-429, were involved in epithelial-mesenchyme-transition (EMT) and controlled the cell fate determinations towards mesendoderm of ESCs24,25,26. miR-200b and miR-429 were both predicted to target the 3’UTR of Phd2. (Supplementary Fig. 5a) As shown in Fig. 1a, the expression of PHD2 mostly overlapped with hypoxia in mouse embryos. We next determined if these microRNAs changed in response to oxygen alteration and thereby regulated PHD2. As a result, miR-429 was significantly repressed by hypoxia, whereas miR-200b did not display such a pattern. (Fig. 7a, Supplementary Fig. 5b) Moreover, the expression of miR-429 was not affected by HIF-1α knockdown, which orchestrated the exclusive distributions of HIF-1α and PHD2 in embryos. (Supplementary Fig. 5c) Along mesendoderm differentiation, the expression of miR-429 was just opposite to Phd2. (Fig. 7b) By western blots, PHD2 expressions were decreased with miR-429 overexpression. (Fig. 7c, Supplementary Fig. 5d) We then performed luciferase assays to examine if miR-429 targeted Phd2 and found miR-429 overexpression could target WT Phd2 3’UTR but not the 3’UTR with the mutated binding site of miR-429. (Fig. 7d) These results implied that miR-429 might be an upstream regulator of PHD2. Subsequently, we explored if miR-429 manipulated the function of PHD2 in mesendoderm differentiation. miR-429 overexpression resulted in the upregulation of mesendoderm markers (T, Eomes, Mesp1, and Gsc) and T-positive cell ratios. (Fig. 7e, f, Supplementary Fig. 5e) When we simultaneously overexpress miR-429 and PHD2, whose transcription region contained only open reading frame (ORF) sequences but no miR-429 binding site, the mesendoderm differentiation was like the outcome of sole PHD2 overexpression, indicated by repressed expressions of mesendoderm markers (T, Eomes, Mesp1, and Gsc) and T positive cell ratios. (Fig. 7g–k, Supplementary Fig. 5f) By RT-qPCR, Sp5 and Axin2 were found to be repressed as well. (Supplementary Fig. 5g) These results demonstrated that miR-429 could target Phd2 and thus regulated mesendoderm specification in the presence of abundant oxygen. (Fig. 7l).

a The RNA levels of miR-429 along the mesendoderm differentiation of mESCs under normoxia and hypoxia, respectively. (n = 4). b The RNA levels of miR-429 and Phd2 along the mesendoderm differentiation. (n = 4). c Western blots showing the PHD2 expression change with miR-429 overexpression. (n = 3). d The luciferase assays were performed to prove miR-429 targeting Phd2 mRNA. (n = 4). e The immunostaining against T and f the T-positive cell ratios after the mesendoderm differentiation of the ctrl and miR-429-OE mESCs. Blue region represents gating negative cells and red region represents treated cells. (n = 4). g The schematic diagram showing the construction of Dox-inducible miR-429/PHD2 dual overexpression (429-P-OE) mESCs. h The overexpression of PHD2 and i miR-429 in 429-P-OE were verified. j The immunostaining against T and k the T-positive cell ratios after the mesendoderm differentiation of the Ctrl and miR-429-OE mESCs (n = 4). l In the presence of abundant oxygen, a working model of miR-429, PHD2, HIF-1α, and the Wnt/β-catenin pathway in regulating mesendoderm specification. Scale bars, 100 μm; ctrl/Ctrl no inducible control; miR-429-OE miR-429 overexpression, 429-P-OE miR-429/PHD2 dual overexpressions; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001; ns not significant.
Discussion
Unveiling the underlying regulatory mechanism of mesendoderm development is fundamental to understanding the whole embryo development process and directionally inducing the derived lineages of mesoderm and endoderm for cell-based therapy. In this study, we discovered that PHD2 was expressed in the epiblast of blastocysts, and its withdrawal was essential to ensure the modest specification of mesendoderm.
PHD2 is an enzyme that regulates protein stability through hydroxylation. HIF-α factors, including HIF-1α, HIF-2α, and HIF-3α, are all candidate targets of PHD227. Depletion of HIF-1α led to embryonic lethality at approximately the E9.5 stage. In contrast to HIF-1α, the depletion of either HIF-2α or HIF-3α did not result in marked defects in early embryogenesis28. Ectopic expression of HIF-2α could not rescue the defects caused by HIF-1α knockout in mouse embryos29. These data suggest a more critical role of HIF-1α in early embryogenesis. Hydroxylated HIF-1α, catalyzed by PHD2, is subsequently recognized by an E3 ligase, pVHL, and subjected to proteasome-mediated protein degradation. On the contrary, loss of PHD2 improved the stability of HIF-1α and thus upregulated the steady levels of HIF-1α despite normoxic conditions, termed pseudohypoxia5,30. Furthermore, our recent work has revealed that hypoxia and HIF-1α exhibit opposing functions in regulating mesendoderm differentiation14. It is reasonable to hypothesize that PHD2 mediates the functions of hypoxia and HIF-1α in mesendoderm development. By investigating the dynamic distribution of hypoxia, PHD2, and HIF-1α, we found that they coexist in the ICM or epiblast of pre- or peri-implantation embryos. This may indicate that hypoxic pathways are involved in preimplantation blastocyst formation, which is consistent with recent reports31,32. However, hypoxia and PHD2 were together withdrawn from the epiblast and are only detected in extraembryonic regions, such as the visceral endoderm and trophoblast. This agreed with previous reports that hypoxia promoted the PHD2 expression4,33. Nonetheless, there are differences between the distributions of hypoxia and PHD2. Firstly, we observed specific hypoxia in the primordial germ cells (PGCs) region of E7.5 mouse embryos, where we did not see the expression of PHD2. This corresponds with a previous study that suggested hypoxia plays an important role in PGC development34. Secondly, we noted that the retreat of PHD2 from the epiblast was uneven. We observed an even distribution of PHD2 in the entire epiblast at the E5.5 stage, while the expression of PHD2 was only detected in the anterior side of the epiblast at the E7.0 stage. By the E7.5 stage, the expression of PHD2 was completely undetectable in the epiblast. However, we did not observe a similar pattern for hypoxia. These results suggested that the expression pattern of PHD2 mostly aligned with that of hypoxia, although some differences existed. Furthermore, PHD2-mediated hydroxylation is oxygen-dependent, which may seem contradictory to the identical patterns of hypoxia and PHD2 in embryos. Recent studies have provided a possible explanation, suggesting that oxygen within cells could be redistributed to enhance regional oxygen concentration, thereby enabling PHD2 to function, especially in cells under chronic hypoxia35,36. In such cases, oxidative phosphorylation is suppressed while glycolysis is enhanced. This could explain the patterns of PHD2, hypoxia, and HIF-1α in mouse embryos and how PHD2 operates under hypoxia. Therefore, for embryos developing in the uterus under chronic hypoxia, HIF-1α is tightly regulated by PHD2 rather than by hypoxia. This might partially explain why HIF-1α exhibits opposing functions to hypoxia rather than synergetic effects, as widely accepted.
The primary pathway for sensing oxygen concentrations and regulating HIF-1α levels is through PHD2-triggered HIF-1α degradation. This study reveals the exclusive distributions and opposing functions of PHD2 and HIF-1α in mesendoderm development, aligning with the degradation working model. However, we cannot rule out the possibility that they may co-express at certain time points, such as during dynamic changes in hypoxia patterns as embryos develop. These moments are highly dynamic and may escape our observations. Furthermore, we believe that the pattern of HIF-1α expression is primarily regulated by PHD2 and is not directly associated with hypoxia, as suggested by immunostaining results in embryos. These expression patterns support our previous findings that hypoxia and HIF-1α oppositely regulate mesendoderm differentiation, potentially indicating a decoupling of HIF-1α from hypoxia in embryo development. In this study, both the withdrawal of PHD2 from mesendoderm in normal embryogenesis and PHD2 knockout resulted in upregulated HIF-1α expression, even in the absence of hypoxia, underscoring the role of PHD2 in pseudohypoxia. In our previous work, hypoxia treatment during mesendoderm differentiation of mESCs resulted in more significant changes in both the range and amplitude of gene expression compared to sole HIF-1α overexpression14. Moreover, the HIF-1α overexpression caused a more specific and promotive influence on mesendoderm differentiation. These results collectively suggest that mesendoderm development requires pseudohypoxia, a consequence of PHD2 downregulation leading to HIF-1α stabilization, rather than actual hypoxia.
The upstream and downstream regulations on the PHD2/HIF-1α axis was also studied. The Wnt/β-catenin pathway, a crucial pathway in mesendoderm development37,38, was found to mediate the effects of the PHD2/HIF-1α axis. Previous reports have demonstrated that HIF-1α could promote the Wnt/β-catenin pathway partially by physically interacting with β-catenin39,40. In this study, by utilizing ChIP-seq data of HIF-1α, we discovered that β-catenin was a transcriptional target of HIF-1α as well, thereby influencing the Wnt/β-catenin pathway in mESCs. This notion is further supported by the expanded expression of β-catenin in PHD2-knockout embryos compared to wild-type embryos. However, it’s worth noting that the patterns of T and β-catenin expression differ in PHD2-knockout embryos, with β-catenin being broadly expressed throughout the embryos, including extraembryonic regions. As is known to all, the Wnt/β-catenin pathway is not solely regulated by β-catenin levels, and mesendoderm development is also not solely controlled by the Wnt/β-catenin pathway41,42. Thus, the global upregulation of β-catenin resulting from PHD2 depletion does not necessarily lead to the overall activation of the Wnt/β-catenin pathway and mesendoderm formation. As to the upstream regulation, we revealed that miR-429 directly targeted PHD2 and thus regulated mesendoderm specification. While previous studies have suggested that PHD2 may be a transcriptional target of HIF-1α4, the exclusive distributions of PHD2 and HIF-1α did not support this notion in our study. Meanwhile, our study revealed that miR-429 was downregulated under hypoxia during mesendoderm differentiation. This finding is consistent with other studies. In human endothelial cells, prolonged hypoxia led to a decrease in miR-429 expression, facilitating the transition from HIF-1α to HIF-3α43,44. A possible mechanism underlying hypoxia-induced downregulation of miR-429 is hypoxia-triggered epigenetic remodeling. Hypoxia can induce extensive changes in the epigenetic landscape, such as histone modifications and DNA methylation, which can influence miRNA expression45. A previous study identified CpG islands upstream of the miR-429 locus, and the methylation dynamics of these CpG islands were closely associated with miR-429 expression46. Furthermore, a long non-coding RNA, H19, which is upstream regulated by histone acetylation, controlled the expression of miR-429 in hepatocellular carcinoma47. Additionally, miR-429 expression was significantly repressed following the treatment with HDAC inhibitors in triple-negative breast cancer48. Nevertheless, the relationship between epigenetics and miR-429 under hypoxic conditions warrants further investigation.
Our study here established an essential role of PHD2 in mesendoderm specification. Moreover, we observed that PHD2 was withdrawn from the epiblast and restricted to extraembryonic regions, suggesting its possible involvement in placental development. This was supported by the finding that the germline depletion of PHD2 results in embryonic lethality around the E12.5 to E14.5 stage, with evident defects in placental angiogenesis6. Additionally, conditional knockout of PHD2 in endothelial and smooth muscle cells leads to defective arterial remodeling and pathologies in the pulmonary and cardiovascular systems, respectively7. These tissues belong to mesendoderm-derived lineages, suggesting a possible function of PHD2 in the further development of mesendoderm-derived lineages. The promising roles of PHD2 in the development of placental and mesendoderm-derived lineages warrant further investigations.
Last but not least, we employed a so-called embryoid technique in our study. The embryoids were synthetic embryos constructed with in vitro cultured cells49,50,51. With the advancement of embryoid culture procedures, embryoids could eventually be developed up to gastrula-like stages (also called gastruloids)52,53. With the combined advantages of convenience as in vitro stem cell culture and reliability as in vivo embryos, the embryoid platform allows researchers to mimic embryo development under precise manual control. By comparing the embryoids constructed with WT and PHD2-KO mESCs to the PHD2+/+ and PHD2−/− embryos, respectively, we validated the role of PHD2 in mesendoderm specification and reliability of our embryoid formation system. Furthermore, we conducted competition assays by equally mixing WT and PHD2-KO mESCs to assess their tendencies for lineage contributions, which was technically difficult for in vivo embryo studies. As expected, WT mESCs contributed to the entire epiblast at the E7.5 stage. However, PHD2-KO mESCs specifically contributed to mesendoderm. These results were in line with other works in mESCs and mouse embryos. Thus, embryoids provide a convenient and reliable platform for studying regulatory mechanisms in addition to classical cell culture and mouse embryos.
Methods
Mice
The mice used in our experiments were bred and housed at the Laboratory Animal Center of Anhui Normal University. All experiments involving these mice were conducted with the approval of the Institutional Animal Care and Use Committee of Anhui Normal University.
Mice with PHD2 knockout were purchased from GemPharmatech and obtained by the genetic removal of exon2 and exon3 of Phd2 using CRISPR/Cas9 technology. These mice were subsequently maintained in the C57BL/6 genetic background. Wild-type (WT) C57BL/6 and ICR mice were purchased from the Qing Long Shan Laboratory Animal Center. Genotyping was performed by PCR on the genomic DNA extracted from mouse tails or embryos. The sequences of genotyping primers can be found in Supplementary Table 1.
Embryo collection
PHD2 homozygous knockout (PHD2−/−), PHD2 heterozygous knockout (PHD2+/−), and their littermate WT (PHD+/+) embryos were obtained through mating between PHD2+/− mice. To exclusively obtain WT embryos, mating involved only WT mice. Embryo collection involved different procedures based on the developmental stage: Pre-implantation embryos (E3.5 or E4.5) were gently washed out and retrieved from the uterus using M2 medium (Merck, Cat No. M7167). Post-implantation embryos (E5.5-E7.5) were carefully isolated from the decidua under an anatomical microscope. In particular, when assessing the distribution and intensity of hypoxia in embryos, pregnant mice received a tail intravenous injection of a hypoxia probe, Pimonidazole Hydrochloride (60 mg/ml, Hypoxyprobe, Cat No. HP-100 mg), 2 h before embryo collection.
Plasmids and cell lines
The pcDNA3 mHIF-1α MYC (P402A/P577A/N813A) plasmid, encoding oxygen-resistant HIF-1α, was purchased from Addgene. The oxygen-resistant HIF-1α gene fragment was ligated into the pCW57-MCS1-2A-MCS2 plasmid to produce a doxycycline (Dox)-inducible oxygen-resistant HIF-1α expression plasmid. Plasmids for labeling mESCs with EGFP and mCherry (pCDNA3-EGFP and pCDNA3-mCherry) were developed by inserting EGFP and mCherry gene fragments into the pCDNA3 vector, respectively. Phd2, Gata4, and Cdx2 genes were synthesized by the GenScript Biotech Corporation, with the Phd2 gene sequence codon-optimized. The PHD2 overexpression plasmid (PHD2-OE) was generated by ligating the PHD2 gene fragment into the pCDNA3 vector. Dox-inducible PHD2, GATA4, and CDX2 plasmids were produced by ligating the Phd2, Gata4, and Cdx2 gene fragments into the pCW57-MCS1-2A-MCS2 vector, respectively. miR-429 was amplified from the mouse genome and ligated into the pCW57-MCS1-2A-MCS2 vector. The PHD2 knockout plasmid was constructed by ligating the Phd2 gRNA fragment into the lentiCRISPR v2 vector. PHD2 knockdown plasmids were constructed by ligating two designed shRNA against Phd2 into the pll4.0 vector. HIF-1α knockdown and scramble plasmids were obtained as previously reported14. The Phd2 3’UTR luciferase plasmid was constructed by inserting the 3’UTR region of Phd2 into the pmirGLO vector. The Phd2 3’UTR-mut plasmid was constructed by mutating the predicted miR-429 targeting site on the Phd2 3’UTR luciferase plasmid.
The EGFP knock-in at Phd2’s stop codon in the genome was achieved using CRISPR/Cas9-mediated homologous recombination. First, a gRNA was designed to target the site adjacent to the stop codon of Phd2 gene and then ligated into the lentiCRISPR v2 vector. Next, 1000 bp sequences upstream and downstream of the targeting site were amplified and placed upstream and downstream of positioned on either side of a P2A-EGFP coding sequence on the PUC19 plasmid. These two plasmids were co-transfected into mESCs to generate the PHD2-EGFP knock-in cell line. The primer sequences were listed in Table S1.
To construct cell lines, the relevant plasmids were transferred into cells using the Lipofectamine 3000 (Invitrogen, Cat No. L3000001), following the manufacturer’s instructions. Stable transfection was achieved by subjecting the cells to puromycin (1 μg/ml) or G418 (400 μg/ml) selection, as indicated by the selection markers present on the plasmids. Drug selection was initiated 48 h after the transfection. Specifically, knockout and knock-in cell lines were obtained from single-cell isolation and cloning, followed by PCR genotyping for confirmation.
Cell culture and differentiation
AB2.2 and R1 mESCs were cultured with Knock Out DMEM (Gibco, Cat No. 10829018) supplemented with 15% fetal bovine serum (FBS, Excell Bio, Cat No. FND500), 1x MEM Non-Essential Amino Acids Solution (NEAA, Gibco, Cat No. 11140050), 2 mM L-Glutamine (Gibco, Cat No. 25030081), 50 U/ml Penicillin, 50 μg/ml Streptomycin (Gibco, Cat No. 15240062), 0.1 mM 2-Mercaptoethanol (Merck, Cat No. M3148), 3 μM CHIR99021(MCE, Cat No. HY-10182), 1 μM PD0325901 (MCE, Cat No. HY-10254), and 103 units of LIF (Millipore, Cat No. ESG1107) on 0.1% Gelatin (Sigma, Cat No. G1890)-coated cell culture dishes. HEK293T cells were cultured with DMEM (Hyclone, Cat No. SH30022.01), 10% FBS, 50 U/ml Penicillin, and 50 μg/ml Streptomycin.
All differentiation assays were conducted by hanging drop culture of mESCs (2 × 104/ml) to generate embryonic bodies14. The differentiation for single-cell RNA sequencing (scRNA-seq), which was the non-lineage tendency, was performed with Knock Out DMEM supplemented with 20% FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, and 0.1 mM 2-Mercaptoethanol. All other differentiation towards mesendoderm was performed with 75% IMDM (Gibco, Cat No. 12440053) and 25% F12 medium (Gibco, Cat No. 11765047) supplemented with 2 mM L-glutamine, 0.25% BSA (Merck, Cat No. V900933), 1x B-27 (Gibco, Cat No. 17504044), 0.5x N-2 (Gibco, Cat No. 17502048), 50 U/ml penicillin, 50 μg/ml streptomycin, 0.45 mM 1-thioglycerol (Merck, Cat No. M6145), and 10 ng/ml Activin A (R&D, Cat No. 338-AC). All differentiations were performed with the AB2.2 background mESCs except for the verification assays performed with R1 backgroud mESCs. The entire differentiation assays spanned 4 days, with sample collection occurring on day 4, unless otherwise specified. To inhibit PHD2 enzyme activity, 5 μM IOX4 (MCE, Cat No. HY-120110) was introduced for 24 h starting from differentiation day 2. 3 μM IWP2 (MCE, Cat No. HY-13912) was supplied throughout the entire differentiation to inhibit the Wnt/β-catenin pathway.
EiTiX-embryoid formation
EiTiX-embryoid formation was performed as previously reported20,21,50. To create Dox-inducible GATA4 and CDX2 overexpressing mESCs (iGata4 and iCdx2), AB2.2 mESCs were stably transfected with Dox-inducible GATA4 and CDX2 plasmids, respectively. iCdx2 and iGata4 were initially treated with 1 μg/ml Dox for 24 h. Subsequently, a mixture was prepared containing 6 × 103 cells/ml of mESCs, 6 × 103 cells/ml of iGata4, and 3.84 × 104 cells/ml of iCdx2 in the FC medium (DMEM supplemented with 15% FBS, 0.1 mM 2-Mercaptoethanol, 1% penicillin/streptomycin, 2 mM Glutamax (Gibco, Cat No. 35050061), 1x NEAA, and 1 mM sodium pyruvate (Gibco, Cat No. 11360070)). To this mixture, 1 μg/ml heparin (Selleck, Cat No. S1346), 25 ng/ml Fgf4 (R&D, Cat No. 7486-F4-025), and 10 nM ROCK inhibitor (Y-27632, MCE, Cat No. HY-10071) were added, and the cells were then seeded onto AggreWell plates (STEMCELL, Cat No. 34450). The AggreWell plates were then spined at 100 g for 3 min. Culture medium was changed daily. For the first two days, the EiTiX-embryoid culture medium consisted of the fresh FC medium. On day 3, the culture medium was replaced with peri-implantation medium (80% advanced DMEM/F12 (Gibco, Cat No. 12,634-010) supplemented with 20% FBS, 2 mM GlutaMax, 1% penicillin–streptomycin, 1x insulin-transferrin-selenium-ethanolamine (Gibco, Cat No. 51,500-056), 8 nM β-estradiol (Merck, Cat No. E8875), 200 ng/mL progesterone (MCE, Cat No. HY-N0437-1G), and 25 μM N-acetyl-L-cysteine (Merck, Cat No. A7250-10G)). Starting from day 4, EiTiX-embryoids were collected into ultra-low attachment plates (LABSELECT, Cat No. 11218), and the culture medium was changed to a modified peri-implantation medium that contained 30% FBS instead of 20% FBS. On day 5, 30% KnockOut Serum Replacement (KSR, Gibco, Cat No. 10828028) was added to the modified peri-implantation medium instead of 30% FBS. EiTiX-embryoids were collected on day 6.
Single-cell RNA sequencing (scRNA-seq) analysis
Non-lineage tendency differentiation was performed on WT and PHD2-KO mESCs, respectively. On differentiation day 4, the cells from both groups were sent to the Novogene Co, Ltd. and underwent the standard scRNA-seq procedures. The scRNA-seq data was initially analyzed using the Seurat package (version 4.3.0) in R54. The scRNA-seq data was filtered using the following criteria: the cells should express more than 200 and less than 8000 genes; the ratio of mitochondrial genes should be less than 5%; the ratio of ribosomal genes should be more than 10%; the genes should be expressed in no less than 3 cells. This resulted in a dataset of 18,552 genes and 16,048 cells (8,841 for the WT group and 7,207 for the PHD2-KO group). Subsequently, doublet removal was performed on the scRNA-seq data using the DoubletFinder package (version 2.0.3) in R55. Data from both groups were integrated using the harmony package (version 0.1.1) in R56. Data was normalized with the built-in NormalizeData function in the Seurat package. The top altered 3000 genes were selected for principal component analysis (PCA), and the top 40 principal components were used for Uniform Manifold Approximation and Projection (UMAP). With a resolution parameter set at 0.6, all cells were divided into 9 clusters. Cell clusters were annotated using previously reported markers: Pou5f1 (Oct4), Nanog, Sox2, and Zfp42 for undifferentiated ESCs (undiff. ES); T, Mixl1, Mesp1, Eomes, and Gsc for mesendoderm; Btg2, Crip2, Cd24a, Nkx2-9, and Snai3 for ectoderm. Cells with equivalent Pou5f1 expression but lower Nanog and Sox2 expressions compared to undiff. ES were annotated as epiblast. Differentially expressed genes (DEGs) were determined with the FindMarkers function in the Seurat package. Next, the differentiation trajectories of WT and PHD2-KO mESCs were explored using the monocle3 package (version 1.3.1) in R57. The undiff. ES subgroup was selected as the root. All visualizations of the scRNA-seq results were generated using the built-in functions of the Seurat and monocle3 packages, as well as the ggplot2 package (version 3.4.2)58.
Chromatin immunoprecipitation sequencing (ChIP-seq) and spatial transcriptomics analysis
The HIF-1α and histone modification ChIP data (H3K27ac, H3K9me2, H3K4me3, H3K27me3) were filtered and trimmed using Trim Galore! (version 0.6.7). Subsequently, the trimmed reads were aligned to the mm10 mouse genome using Bowtie 2 (version 2.3.5.1)59. The resulting alignment files in SAM format were sorted, duplicates were removed, and the files were indexed. Finally, these SAM files were converted into BAM files using the samtools software (version 1.7)60. The BAM files were further processed to create bigWig files with the bamCoverage function within the deeptools package (version 3.5.1)61. These bigWig files were then used for visualization in the IGV software (version 2.9.4)62.
For spatial transcriptomics analysis of E7.5 embryos, data were obtained from an online platform, JingLab’s Geo-seq data (http://egastrulation.sibcb.ac.cn/). To display the spatial patterns of gene expression, corn plots were generated.
Immunostaining for embryos and EiTiX-embryoids
Embryos and EiTiX-embryoids were fixed at room temperature with 4% paraformaldehyde (PFA) for 1 h. Following fixation, embryos were permeabilized with 0.5% Triton X-100 in PBS at room temperature for 30 min and subsequently blocked with a blocking buffer (consisting of 5% goat serum, 1% BSA, and 0.1% Triton X-100 in PBS) at 4 °C overnight. On the next day, embryos were incubated with primary antibodies diluted in the blocking buffer at 4 °C overnight. On the third day, embryos were washed with the PBST buffer (0.1% Triton X-100 in PBS) and blocked again in the blocking buffer for 3 h. Subsequently, embryos were incubated with secondary antibodies diluted in the blocking buffer at 4 °C overnight. On the fourth day, embryos were washed with PBST and stained with 1 μg/ml DAPI in the PBST buffer at room temperature for 20 min. After DAPI staining, embryos were washed with the PBST buffer and transferred to the PBS buffer for observation. Embryo images were captured using a Leica DMi8 fluorescence microscope. Fluorescence intensity along the anterior-posterior (A-P) axis was quantified using the Plot Profile analysis in ImageJ (version 1.53t) software63. In addition, the proportion of fluorescence region was determined using the area analysis in the ImageJ software, with the area of DAPI serving as a control. The following antibodies were used: anti-T mAb (1:100, Abcam, Cat No. ab209665), anti-HIF1α pAb (1:100, Novus, Cat No. NB100-479.), anti- Pimonidazole mAb (1:100, Hypoxia probe, Cat No. HP1-100kit), anti-β-catenin mAb (1:100, Cell signaling, Cat No. 8480), anti-OCT4 mAb (1:100, Abcam, Cat No. ab181557), anti-PHD2 mAb (1:100, Proteintech, Cat No. 66589-1-Ig), anti-PHD2 conjugated 488 (1:100, Proteintech, Cat No. CL488-66589), anti-GFP mAb (1:100, Invitrogen, Cat No. MA5-15256), anti-mCherry pAb (1:100, Abcam, Cat No. ab167453), Goat anti-rabbit Alex Fluor 488-conjugated IgG (1:500, Invitrogen, Cat No. A11008), Goat anti-mouse Alexa Fluor Plus 555-conjugated IgG (1:500, Invitrogen, Cat No. A32727), Goat anti-rabbit Alexa Fluor 405-conjugated IgG (1:500, Abcam, Cat No. ab175652), Goat Anti-rabbit IgG H&L Alexa Fluor 555-conjugated IgG (1:500, Abcam, Cat No. ab150078), Goat anti-mouse Alexa Fluor Plus 488-conjugated IgG (1:500, Invitrogen, Cat No. A-11001).
Immunostaining for cultured cells
Cell immunostaining was conducted as described previously14. Briefly, cultured cells were fixed with 4% PFA and then blocked with a cell blocking buffer composed of 10% normal goat serum and 0.1% Triton X-100 in PBS. Following blocking, the cells were incubated with primary antibodies diluted in the cell blocking buffer overnight. On the next day, the cells were incubated with the secondary antibodies diluted in PBS for 90 min. Subsequently, the cells were washed with PBST and incubated with 1 μg/ml DPAI diluted in ddH2O for 5 min. Post DAPI staining, the cells were washed with PBST and kept in PBS for observation. Images were captured with a Leica DMi8 fluorescence microscope. To quantify the proportion of the fluorescent region, area analysis in ImageJ software was employed, with the area of DAPI serving as a reference for control.
Western blots
Protein extraction was carried out using a lysis buffer that included protease inhibitors (Roche, Cat No. 5892970001) and phosphatase inhibitors (Beyotime, Cat No. P1082). Protein concentrations were determined using a BCA protein assay kit (Biosharp, Cat No. BL521A) and adjusted to be equal across samples prior to SDS-PAGE electrophoresis. SDS-PAGE electrophoresis was performed as described previously14. Subsequently, proteins were transferred from gels to polyvinylidene fluoride (PVDF, Millipore, Cat No. IPVH00010) membranes. The membranes were then blocked with the 5% milk for 1 h and incubated with the primary antibodies overnight. On the following day, the membranes were incubated with the secondary antibodies for 90 min and subjected to a chemiluminescent substrate reaction (Biosharp, Cat No. BL520B). Signals were detected using a Tanon 5200 Imaging system. The following antibodies were used: anti-PHD2 mAb (1:1000, Santa Cruz Biotechnology, Cat No. sc-271835), anti-β-catenin mAb (1:1000, Cell Signaling, Cat No. 8480), anti-GAPDH pAb (1:2000, Biosharp, Cat No. BL006B), anti-p-GSK3β(S9) mAb (1:1000, Cell Signaling, Cat No. 5558), anti-GSK3β mAb (1:1000, Invitrogen, Cat No. MA5-15109), anti-HIF1α mAb (1:1000, Cell Signaling, Cat No. 14179), goat anti-mouse HRP secondary antibody (1:2000, Biosharp, Cat No. BL001A), and donkey anti-rabbit HRP secondary antibody (1:2000, Invitrogen, Cat No. 31458). Gel band intensities were quantified using the ImageJ software.
Flow cytometry and fluorescence-activated cell sorting (FACS)
Cultured cells were trypsinized into single cells and washed with PBS. Subsequently, the cells were fixed with 4% PFA for 15 min and incubated with 90% methanol at −20 °C overnight. On the next day, the cells were incubated with primary antibodies for 1 h. A control group, without primary antibodies, was used to establish gating criteria for flow cytometry. Following primary antibody incubation, the cells were incubated with secondary antibodies for 30 min. The cells were then analyzed using a BD FACSCanto II Flow Cytometer. In the case of FACS targeting GFP, the cells were trypsinized into single cells and subjected to sorting by a BD FACSMelody Cell Sorter. Sorted cells were collected and re-plated onto cell culture dishes for subsequent experiments, while cells lacking GFP expression were used as gating controls.
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted using the Total RNA Isolation Reagent (Biosharp, Cat No. BS259A). For reverse transcription, mRNAs and miRNAs were processed separately using the FastKing kit (TIANGEN, Cat No. KR118) and the miRNA First Strand Synthesis cDNA kit (TIANGEN, Cat No. KR211-02), respectively. Quantitative PCR (qPCR) was conducted using the Powerup SYBR Master Mix (ABI, Cat No. A25778) for mRNAs and the miRNA Fluorescence Quantitative Detection Kit (TIANGEN, Cat No. FP411-02) for miRNAs, following the respective manufacturer’s protocols. Gapdh and U6 were utilized as normalization controls for mRNA and miRNA quantification, respectively. The primers used for RT-qPCR were listed in Supplementary Table 2.
Luciferase assays
Luciferase assays were conducted using the dual-luciferase assay kit (Beyotime, Cat No. RG027) according to the manufacturer’s instructions. For TOPflash luciferase assays, HEK293T cells were co-transfected with plasmids encoding specific factors, PRL-TK plasmids, and TOPflash or FOPflash plasmids. Samples were collected at 48 h post-transfection. The TOP and FOP activities were determined by normalizing the firefly luciferase activities of TOPflash and FOPflash to the activities of Renilla luciferase, which was encoded by the PRL-TK plasmid. For each experimental setup, the relative TOPflash activity = TOP activity/FOP activity. For microRNA luciferase assays, the miR-429 Dox-inducible overexpression plasmid was co-transfected along with Phd2 3’UTR and Phd2 3’UTR-mut luciferase plasmids, respectively. The induction of miR-429 overexpression was achieved by supplying 1 μg/ml doxycycline. All firefly luciferase activities were normalized to Renilla luciferase.
Statistics and reproducibility
No statistical method was utilized to pre-determine sample size. Heterozygous mice were used to generate embryos, which were blindly subjected to immunofluorescence staining, however, other experiments were not blindly performed. For the analysis of immunostaining and flow cytometry data, representative images were selected, and these were obtained from random fields or cell fractions. All experiments performed at least three independent biological replications or technical replications, as indicated in the figure legends. Two-group comparisons were assessed using two-sided Student’s t tests, while for comparisons involving three or more groups, a one-way analysis of variance (ANOVA) was applied, if not otherwise specified. P < 0.05 was considered statistically significant. The data are presented as means ± standard deviation (SD).
Data availability
The data generated in this study are available from the corresponding author upon reasonable request. The scRNA-seq data generated in this study have been deposited in the Gene Expression Omnibus (GEO) database under accession number (GSE245439). The ChIP-seq data related to HIF-1α and histone modifications in mESCs were retrieved from the GEO using the accession numbers GSE195545 and GSE60204, respectively. Additionally, the spatial transcriptomics data was directly obtained from an online platform, jingLab’s Geo-seq data, which can be accessed at (http://egastrulation.sibcb.ac.cn/). All source data in this study for bar graphs in figures were provided in Supplementary Data file. The gating strategy were shown in Supplementary Fig. 6 and Uncropped blots are shown in Supplementary Fig. 7.
Code availability
Codes generated on this work used for bioinformatics analysis are available at (https://github.com/xpshen-ahnu/scRNA).
References
Taylor, C. T. & Scholz, C. C. The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 18, 573–587 (2022).
Baik, A. H. & Jain, I. H. Turning the oxygen dial: balancing the highs and lows. Trends Cell Biol. 30, 516–536 (2020).
Strocchi, S., Reggiani, F., Gobbi, G., Ciarrocchi, A. & Sancisi, V. The multifaceted role of EGLN family prolyl hydroxylases in cancer: going beyond HIF regulation. Oncogene 41, 3665–3679 (2022).
Li, L. et al. Searching for molecular hypoxia sensors among oxygen-dependent enzymes. Elife 12, https://doi.org/10.7554/eLife.87705 (2023).
Hayashi, Y., Yokota, A., Harada, H. & Huang, G. Hypoxia/pseudohypoxia-mediated activation of hypoxia-inducible factor-1alpha in cancer. Cancer Sci. 110, 1510–1517 (2019).
Takeda, K. et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell Biol. 26, 8336–8346 (2006).
Elamaa, H. et al. PHD2 deletion in endothelial or arterial smooth muscle cells reveals vascular cell type-specific responses in pulmonary hypertension and fibrosis. Angiogenesis 25, 259–274 (2022).
Suppinger, S. et al. Multimodal characterization of murine gastruloid development. Cell Stem Cell 30, 867–884 e811 (2023).
Dunwoodie, S. L. The role of hypoxia in development of the mammalian embryo. Dev. Cell 17, 755–773 (2009).
Houghton, F. D. Hypoxia and reproductive health: hypoxic regulation of preimplantation embryos: lessons from human embryonic stem cells. Reproduction 161, F41–F51 (2021).
Lopez-Anguita, N. et al. Hypoxia induces an early primitive streak signature, enhancing spontaneous elongation and lineage representation in gastruloids. Development 149, https://doi.org/10.1242/dev.200679 (2022).
Pimton, P. et al. Hypoxia enhances differentiation of mouse embryonic stem cells into definitive endoderm and distal lung cells. Stem Cells Dev. 24, 663–676 (2015).
Mazumdar, J. et al. O2 regulates stem cells through Wnt/beta-catenin signalling. Nat. Cell Biol. 12, 1007–1013 (2010).
Shen, X. et al. Hypoxia is fine-tuned by Hif-1alpha and regulates mesendoderm differentiation through the Wnt/beta-Catenin pathway. BMC Biol. 20, 219 (2022).
Zinski, J., Tajer, B. & Mullins, M. C. TGF-beta family signaling in early vertebrate development. Cold Spring Harb. Perspect. Biol. 10, https://doi.org/10.1101/cshperspect.a033274 (2018).
Yaman, Y. I. & Ramanathan, S. Controlling human organoid symmetry breaking reveals signaling gradients drive segmentation clock waves. Cell 186, 513–527 e519 (2023).
Blassberg, R. et al. Sox2 levels regulate the chromatin occupancy of WNT mediators in epiblast progenitors responsible for vertebrate body formation. Nat. Cell Biol. 24, 633–644 (2022).
Chen, J. et al. Spatial transcriptomic analysis of cryosectioned tissue samples with Geo-seq. Nat. Protoc. 12, 566–580 (2017).
Philip, M. et al. Metabolic studies of hypoxia-inducible factor stabilisers IOX2, IOX3 and IOX4 (in vitro) for doping control. Drug Test Anal. 13, 794–816 (2021).
Lau, K. Y. C. et al. Mouse embryo model derived exclusively from embryonic stem cells undergoes neurulation and heart development. Cell Stem Cell 29, 1445–1458 e1448 (2022).
Tarazi, S. et al. Post-gastrulation synthetic embryos generated ex utero from mouse naive ESCs. Cell 185, 3290–3306 e3225 (2022).
Niwa, H. et al. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell 123, 917–929 (2005).
Fujikura, J. et al. Differentiation of embryonic stem cells is induced by GATA factors. Genes Dev. 16, 784–789 (2002).
Du, Z. W., Ma, L. X., Phillips, C. & Zhang, S. C. miR-200 and miR-96 families repress neural induction from human embryonic stem cells. Development 140, 2611–2618 (2013).
Rosati, J. et al. Smad-interacting protein-1 and microRNA 200 family define a nitric oxide-dependent molecular circuitry involved in embryonic stem cell mesendoderm differentiation. Arterioscler. Thromb. Vasc. Biol. 31, 898–907 (2011).
Li, Z., Gou, J., Jia, J. & Zhao, X. MicroRNA-429 functions as a regulator of epithelial-mesenchymal transition by targeting Pcdh8 during murine embryo implantation. Hum. Reprod. 30, 507–518 (2015).
Semenza, G. L. The genomics and genetics of oxygen homeostasis. Annu. Rev. Genom. Hum. Genet. 21, 183–204 (2020).
Jiang, Y., Duan, L. J. & Fong, G. H. Oxygen-sensing mechanisms in development and tissue repair. Development 148, https://doi.org/10.1242/dev.200030 (2021).
Covello, K. L. et al. HIF-2alpha regulates Oct-4: effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 20, 557–570 (2006).
Losman, J. A., Koivunen, P. & Kaelin, W. G. Jr 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer 20, 710–726 (2020).
Sozen, B. et al. Reconstructing aspects of human embryogenesis with pluripotent stem cells. Nat. Commun. 12, 5550 (2021).
Heidari Khoei, H. et al. Generating human blastoids modeling blastocyst-stage embryos and implantation. Nat. Protoc. 18, 1584–1620 (2023).
Jaskiewicz, M. et al. The transition from HIF-1 to HIF-2 during prolonged hypoxia results from reactivation of PHDs and HIF1A mRNA instability. Cell Mol. Biol. Lett. 27, 109 (2022).
Lopez-Iglesias, P. et al. Hypoxia induces pluripotency in primordial germ cells by HIF1alpha stabilization and Oct4 deregulation. Antioxid. Redox Signal. 22, 205–223 (2015).
Lee, P., Chandel, N. S. & Simon, M. C. Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 (2020).
Hagen, T., Taylor, C. T., Lam, F. & Moncada, S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science 302, 1975–1978 (2003).
Anand, G. M. et al. Controlling organoid symmetry breaking uncovers an excitable system underlying human axial elongation. Cell 186, 497–512.e423 (2023).
Karvas, R. M. et al. 3D-cultured blastoids model human embryogenesis from pre-implantation to early gastrulation stages. Cell Stem Cell 30, 1148–1165.e1147 (2023).
Boso, D. et al. HIF-1alpha/Wnt signaling-dependent control of gene transcription regulates neuronal differentiation of glioblastoma stem cells. Theranostics 9, 4860–4877 (2019).
MacColl Garfinkel, A. et al. Mitochondrial leak metabolism induces the Spemann-Mangold Organizer via Hif-1alpha in Xenopus. Dev. Cell, https://doi.org/10.1016/j.devcel.2023.08.015 (2023).
Liu, J. et al. Wnt/beta-catenin signalling: function, biological mechanisms, and therapeutic opportunities. Signal Transduct. Target. Ther. 7, 3 (2022).
Mukherjee, S., Luedeke, D. M., McCoy, L., Iwafuchi, M. & Zorn, A. M. SOX transcription factors direct TCF-independent WNT/beta-catenin responsive transcription to govern cell fate in human pluripotent stem cells. Cell Rep. 40, 111247 (2022).
Bartoszewska, S. et al. The hypoxia-inducible miR-429 regulates hypoxia-inducible factor-1alpha expression in human endothelial cells through a negative feedback loop. FASEB J. 29, 1467–1479 (2015).
Janaszak-Jasiecka, A. et al. miR-429 regulates the transition between Hypoxia-Inducible Factor (HIF)1A and HIF3A expression in human endothelial cells. Sci Rep. 6, 22775 (2016).
Kindrick, J. D. & Mole, D. R. Hypoxic regulation of gene transcription and chromatin: cause and effect. Int. J. Mol. Sci. 21, https://doi.org/10.3390/ijms21218320 (2020).
Davalos, V. et al. Dynamic epigenetic regulation of the microRNA-200 family mediates epithelial and mesenchymal transitions in human tumorigenesis. Oncogene 31, 2062–2074 (2012).
Zhang, L. et al. Epigenetic activation of the MiR-200 family contributes to H19-mediated metastasis suppression in hepatocellular carcinoma. Carcinogenesis 34, 577–586 (2013).
Mekala, J. R. et al. Epigenetic regulation of miR-200 as the potential strategy for the therapy against triple-negative breast cancer. Gene 641, 248–258 (2018).
Dupont, C. et al. Efficient generation of ETX embryoids that recapitulate the entire window of murine egg cylinder development. Sci. Adv. 9, eadd2913 (2023).
Amadei, G. et al. Embryo model completes gastrulation to neurulation and organogenesis. Nature 610, 143–153 (2022).
Oldak, B. et al. Complete human day 14 post-implantation embryo models from naive ES cells. Nature 622, 562–573 (2023).
Liu, L. et al. Modeling post-implantation stages of human development into early organogenesis with stem-cell-derived peri-gastruloids. Cell 186, 3776–3792.e3716 (2023).
Zhai, J., Xiao, Z., Wang, Y. & Wang, H. Human embryonic development: from peri-implantation to gastrulation. Trends Cell Biol. 32, 18–29 (2022).
Stuart, T. et al. Comprehensive integration of single-cell data. Cell 177, 1888–1902.e1821 (2019).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8, 329–337.e324 (2019).
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods 16, 1289–1296 (2019).
Trapnell, C. et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat. Biotechnol. 32, 381–386 (2014).
Wickham, H. in Use R!, 1 Online resource (XVI, 260 pages 232 illustrations, 140 illustrations in color (Springer International Publishing, 2016).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Ramirez, F., Dundar, F., Diehl, S., Gruning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–191, (2014).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Acknowledgements
We thank the platforms and resources provided by Anhui Provincial Key Laboratory of Molecular Enzymology and Mechanism of Major Metabolic Diseases, Anhui Provincial Engineering Research Centre for Molecular Detection and Diagnostics, Anhui Provincial Key Laboratory of Biodiversity Conservation and Ecological Security in the Yangtze River Basin, and Key Laboratory of Biomedicine in Gene Diseases and Health of Anhui Higher Education Institutes. This work was supported by the National Natural Science Foundation of China (No. 31701289), Anhui Provincial Natural Science Foundation (Nos. 2208085MH209, 1808085QH234), Fund for Excellent Young Scholars in Higher Education of Anhui Province of China (No. 2022AH030022), the Outstanding Innovative Research Team for Molecular Enzymology and Detection in Anhui Provincial Universities (2022AH010012), Anhui Provincial Funding Scheme to Outstanding Innovative Programs by Returned Scholars (No. 2019LCX003), Anhui Provincial Key Laboratory of Molecular Enzymology and Mechanism of Major Diseases (Nos. fzmx202001, fzmx202013), Educational Commission of Anhui Province of China (Nos. 2022AH050199, 2022xscx038), Wuhu City Application and Fundamental Research Project (No. 2022jc08), and Funds from the Anhui Normal University (start-up funds to XS). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
Conceptualization, X.S. and M.L.; Methodology, M.L., H.J., and Y.Z.; Software, X.S. and M.L.; Investigation, X.S., M.L., H.J., Y.Z., G.Z., Y.L., H.L. and X.S.; Writing – original draft, M.L.; Writing – review and editing, X.S.; Visualization, X.S. and M.L.; Supervision, X.S.; Project administration, X.S.; Funding acquisition, X.S., M.L. All authors contributed to the article and approved the submitted version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Kaliya Georgieva
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Li, M., Jin, H., Zhao, Y. et al. PHD2 safeguards modest mesendoderm development. Commun Biol 7, 1100 (2024). https://doi.org/10.1038/s42003-024-06824-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06824-z
