
Abstract
Understanding the molecular mechanisms underlying genetic diseases is challenging due to environmental and genetic factors. Genome-wide association studies (GWAS) have identified numerous genetic loci, but their functional implications are largely unknown. Single-cell multiomics sequencing has emerged as a powerful tool to study disease-specific cell types and their relationship with genetic variants. However, comprehensive databases for exploring these mechanisms across different tissues are lacking. We present the Disease-Related Cell Type database (DRCTdb), integrating GWAS and single-cell multiomics data to identify disease-related cell types and elucidate their regulatory mechanisms. DRCTdb contains well-processed data from 16 studies, covering 4 million cells within 28 tissues. Users can browse relationships and regulatory mechanisms between SNPs of 42 genetic diseases and cell types based on GWAS and single-cell data. DRCTdb also offers data downloads and is available at https://singlecellatlas.top/DRCTDB.
Similar content being viewed by others


Introduction
Understanding the molecular basics mechanism of genetic diseases is a crucial question in modern biology1. However, the majority of mechanisms remain unknown, as they are believed to arise from a complex interplay between environmental and genetic factors. In recent years, multiple genome-wide association studies (GWAS) have revealed innumerable genetics loci related to genetic disease in the human genome while most of them lack a comprehensive explanation2. Statistics reveal that 90% of the genetic loci are in the non-coding regions of the genome, which further complicates understanding the mechanisms of these complex diseases3,4. Many quantitative trait loci (QTLs) mapping methods proposed to model these genetic variants to explain GWAS signals, such as the expression QTLs, splicing QTLs (sQTLs), and DNA methylation QTLs (mQTLs), but most of them are analyzed in bulk sample5,6,7. In the past decade, the rapid development of single-cell multiomics sequencing technology has provided unprecedented opportunities for us to measure transcriptome and epigenomics simultaneously at single cell resolution and continuous cell state and link cell types to specific diseases8,9,10. Based on the integration of GWAS and single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq) data, a statistical method called LD score regression (LDSC) has been developed9,11. This innovative approach aims to unravel the intricate connections between GWAS data of genetic disease and specific cell types derived from scATAC-seq data, by leveraging the single nucleotide polymorphisms (SNPs) associated with genetic diseases and the accessible chromatin regions specific to each cell type. Through the further analysis of single-cell multiomics data, it becomes possible to explain the intricate intercellular and intracellular regulatory mechanisms that exist between genetic diseases and cell types. This integrative analysis has not only allowed for the identification of disease-related cell types but has also enabled the explanation of the cell type-specific effects of SNPs on disease development and progression. However, there is no comprehensive database for users to effectively search and visualize the genetic disease-related cell types, as well as their underlying mechanisms, across various human tissues12,13.
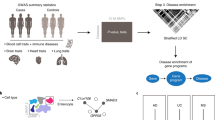
Here, we present the Disease Related Cell Type database (DRCTdb), a database that decodes relationships between genetic disease and cell types from single-cell multiomics data with a multi-functional and user-friendly interface. DRCTdb integrates data from 16 studies, encompassing 4 million well-processed single cells with transcriptomic and epigenetic information related to 42 genetic diseases (Fig. 1, Supplementary Table 1). Each single-cell multiomics dataset was manually normalized, embedded, and annotated with cell types. DRCTdb is implemented with a user-friendly web interface that allows users to explore the relationships between genetic disease and cell types from single-cell multiomics data. Through the web interface of DRCTdb, users can easily access information on genetic disease-related cell types across various human tissues. Furthermore, DRCTdb provides an extensive understanding of the underlying mechanisms that link these cell types to genetic diseases. Users can explore disease-related cell-cell interactions, cell types enriched with disease-related SNPs, and the intracellular gene regulatory network associated with disease-related cell types.

DRCTdb integrates single-cell multi-omics data, including scRNA-seq and scATAC-seq, to identify disease-related cell types (DRCTs). The workflow leverages GWAS datasets to map risk SNPs to cell-type-specific chromatin landscapes. Regulatory mechanisms such as transcription factor activity, cell-cell communication, and gene regulatory networks are analyzed to understand how these DRCTs contribute to disease.
Results
Overview of DRCTdb
The DRCTdb provides multiple single-cell multiomics datasets with nearly all available human tissues. We manually curated the data and added corresponding metadata information for every dataset, including cell type, tissue, and experiment type. We further performed several downstream analyses and provided profiles within the web interface, such as cell-cell communication, TF-activity, and gene regulatory network. In summary, the DRCTdb database provides 16 single-cell multiomics datasets across 28 tissues and encompassing 603 cell types (Fig. 2).

a Number of tissues summarized by different datasets. b Number of cell type enriched genetic diseases in each dataset, X axis represents dataset ID. c Number of cells in each dataset, color represents data modalities, X axis represents dataset ID.
Web design interface
DRCTdb provides a user-friendly web interface, allowing users to explore the relationships between cell types and genetic diseases in different human tissues. DRCTdb contains five main functional interfaces.
The ‘Home’ page gives a brief description of this database and statistics of data used in DRCTdb, including tissue number, cell number, and cell type number.
‘Search’ pages provide an interactive table for users to easily find their interested datasets. Users can search their interested datasets based on tissue and enriched diseases (Fig. 3a).

a Search page of DRCTdb, users are able to click on the column names (Study name, Disease, Stage, Tissue, PMID) in the interactive table in order to sort the table. b UMAP embedding plot of selected dataset in the result page. c LDSC analysis results of the selected dataset are presented in the interactive table. The table displays the genetic diseases that are enriched in specific cell types. d Differentially accessible regions (differential peaks) for each cell type. Column name ‘closed gene’ represents a gene associated with the peak and ‘region’ indicates peak annotation. e Top 10 transcription factors exhibiting the highest binding activity in each cell type.
After selecting datasets of interest, users have the option to explore the enrichment of genetic diseases in specific cell types, as well as the underlying mechanisms behind this association. On the left panel, users can access an overview of the selected datasets, including information about single cell embedding plot (Fig. 3b), statistical significance between cell types and genetic diseases (DRCT) (Fig. 3c), cell type-specific differential accessible regions (DAR) (Fig. 3d), and cell type-specific active transcription factors (TF activity) (Fig. 3e).
On the right panel, the DRCTdb frontend presents the results of downstream analysis to elucidate the underlying regulatory mechanisms linking genetic diseases and their enriched cell types. Users have the option to select a specific cell type and disease pair by using the ‘Choose Cell type’ and ‘Choose Disease’ buttons. Once both cell and disease types are selected, the exploration deepens with our four specialized modules. 1.’ Cell-Cell Communication’ module: A dynamic visualization of the intercellular interactions network between all cell types. By clicking on the “Figure” button, users are able to explore the detailed weights of the ligand-receptor interactions involved in these intercellular communications (Fig. 4a). 2. ‘SNP overlapped peaks’ module: This module showcases open chromatin regions in the selected cell type’s scATAC data that overlap with disease GWAS risk loci, highlighting the genetic vulnerabilities and resistance points in relation to the disease (Fig. 4b, Supplementary Fig. 1a). 3. ‘SNP overlapped genes’ module: Focusing on the scRNA data from the chosen cell type, this module identifies genes that overlap with disease GWAS risk loci, providing insights into the cellular mechanisms influenced by genetic risk factors (Fig. 4c, Supplementary Fig. 1b). 4. ‘Gene Regulatory Networks’ module: This gene regulatory network maps the transcription factor regulatory networks within the cell types related to a specific disease. Users can interactively explore the regulatory relationships between genes by dragging network nodes, offering a hands-on approach to understanding gene regulation (Fig. 4d, Supplementary Fig. 1c).

a Cell-Cell Communication among genetic diseases related cell types, colors represent cell types. b GO enrichment of genes which nearby SNPs overlapped with chromatin accessibility. c GO enrichment of genes which overlapped with SNPs. d Gene regulatory network in disease related cell types, blue represents genes, while each of the other colors represents a transcription factor.
For users who are interested in exploring the functional role of a set of SNPs related to a specific gene, DRCTdb provides an ‘Online Enrichment’ page. This page allows users to perform GO enrichment analysis for their selected set of genes. Users simply need to input an Excel format file that includes a column of gene names. Our ‘Online Enrichment’ tool will perform automatic calculations to determine the enrichment Gene Ontology terms associated with the provided genes (Supplementary Fig. 1d).
DRCTdb also enables users to download all well-processed data and analysis results in the ‘Download’ page (Supplementary Fig. 1e). This page provides an interactive table that facilitates the searching and downloading of all the processed data from this study. Users can conveniently search for their desired datasets based on either tissue or enriched disease options. Once identified, they can download the meticulously processed single-cell RNA-seq and single-cell ATAC-seq data, along with well-annotated metadata, in h5ad format.
The ‘Tutorial’ page provides documents about how to correctly browse and download the analysis result.
In summary, DRCTdb provides a user-friendly platform, enabling users to explore the integrative analysis result of GWAS and single-cell multiomics data.
Case study
We present a case study to illustrate the usage and capabilities of DRCTdb. We selected a single-cell multiomics dataset of human pancreatic islets for our demonstration14. This dataset contains 95,109 cells with both gene expression and chromatin accessibility. The following analysis will first reveal the diseases related cell types found in human pancreatic islets, and then identify the underlying regulatory mechanisms between the enriched genetic diseases and cell types.
Firstly, we identified cell types related to genetic diseases through LDSC analysis. This analysis integrates cell type-specific accessible regions and GWAS summarized statistics data to infer the association between genetic diseases and cell types. In order to conduct LDSC analysis, it is necessary to calculate cell-type specific accessible regions. We used the Wilcoxon rank-sum test to identify differentially accessible regions (DARs) in 9 cell types within the human pancreatic islets dataset. We identified a total of 5938 DARs across the nine cell types, with 2972 DARs located in the promoter region, 449 DARs in coding regions, and 2517 DARs in the intron and intergenic regions (Supplementary Data 1).
Next, we used scBasset to identify the transcription factors that have higher binding activity in these DARs. Our analysis revealed that both YY1 and REST genes exhibited the highest binding activity in the beta cell type. Previous studies have reported that these transcription factors play important roles in the growth and development of beta cells (Supplementary Data 1)15,16.
Third, We conducted LDSC analysis to identify genetic diseases enriched cell types. This analysis utilized the previously mentioned cell type-specific open chromatin regions as well as GWAS summarized statistics data. The analysis revealed a significant association between T2D (Type 2 Diabetes) and beta cells and delta cells, which aligned with previous research (Fig. 5a)13,17.

a The bar plot illustrates the relationship between T2D (Type 2 Diabetes) and cell types based on single-cell multiomics data. The dotted line represents the p-value cutoff. b Cell-Cell Communication among T2D related islet cell types, colors represent cell types c GO enrichment of gene which nearby T2D risk SNPs overlapped with beta cell chromatin accessibility (d) GO enrichment of gene which overlapped with T2D risk SNPs. e Gene regulatory network of risk gene in beta cells, cyan represents genes, while each of the other colors represents a transcription factor.
Several mechanisms may lead to the association between cell types and genetic diseases, including niche, transcriptome regulation, and epigenomic regulation. Therefore, we further investigate the underlying regulatory relationship between beta cells and delta cells in relation to T2D through the analysis of cell-cell communication and gene regulatory network (GRN). We then performed cell-cell communication analysis among T2D-enriched cell types (beta cells and delta cells). This analysis identified several significant ligand-receptor interaction pairs, including BMP5 and ACVR1, which are reported by previous studies that may regulate beta cell growth and development in T2D (Fig. 5b)18.
To further decode the underlying mechanisms between beta cells and T2D, we integrate scRNA-seq and scATAC-seq data of beta cells to construct a disease-related GRN. We initially select disease-related features (genes and accessible regions) to construct disease-related GRN (GWAS overlapped accessible regions and genes are disease-related features). We identified 21,148 accessible regions and 1870 genes overlapped with T2D-associated SNPs in beta cells. Then, we performed enrichment analysis for these genes and accessible regions (Supplementary Data 1). Our findings indicate that the SNPs overlapped genes are enriched for nucleobase-containing compound catabolic process and positive regulation of cellular catabolic process functions. SNPs overlapped accessible regions show enrichment for cell growth and small GTPase mediated signal transduction functions (Fig. 5c, d). By utilizing these 1870 genes and 21,148 accessible regions, we constructed a disease-related gene regulatory network to reveal the regulatory mechanisms between T2D and beta cells. Through the visualization of this gene regulatory network, we found that ATF3 and TCF4 as key nodes within the networks, indicating that they may have a regulatory role in beta cells leading to T2D (Fig. 5e)19. ATF3 and TCF4 were identified as risk genes for T2D in GWAS analysis20. Furthermore, previous studies have reported that ATF3 can induce beta cell stress, while TCF4 is known to cause maturity-onset diabetes of the young.21,22. These results demonstrate the reliability of our pipeline and the usefulness of the database.
Overall, this case study has revealed the genetic disease-related cell types and their underlying regulatory mechanisms. We have identified several ligand-receptor pairs and transcription factors that have previously been reported to regulate T2D. These findings demonstrate the reliability of the DRCTdb analysis pipeline and highlight the utility of the database. Consequently, we believe that DRCTdb will enhance our understanding of genetic diseases and assist in the identification of potential therapeutic targets for genetic disease screening and treatment.
Discussion
In this paper, we present DRCTdb, a database that decodes the relationships between SNPs of 42 genetic diseases and human cell types across 28 tissues, based on GWAS and single-cell multiomics data. By utilizing DRCTdb, users can easily determine which specific cell types in human tissues exhibit significant relationships with various genetic diseases and explore single-cell multiomics data. Furthermore, DRCTdb provides detailed explanations about the involvement of these cell types in genetic diseases, elucidating the connections at the cellular, transcriptional, genetic, and epigenetic levels.
We aim to continuously enhance the DRCTdb with advancements in single-cell multiomics. As new single-cell multiomics data becomes available, we will regularly update and upgrade the DRCTdb to ensure its relevance and comprehensiveness. In addition, we also plan to integrate other types of single-cell omics data, such as spatial transcriptomes and scBCR-seq, to further elucidate the underlying regulatory mechanisms of disease-related cell types23.
Methods
Data collection
For single-cell multiomics data, 16 scATAC-seq datasets were collected in this study (Supplementary Table 1) from the Gene Expression Omnibus (GEO), CATLAS, and EMBL-EBI databases. If a scATAC-seq dataset had its paired scRNA-seq dataset, the paired scRNA-seq dataset was selected as its corresponding scRNA-seq dataset. If a paired dataset was not available, we chose another publicly available scRNA-seq dataset with the same tissue and similar experimental conditions as its corresponding datasets. Only the datasets with cell type information will be considered for further analysis. For GWAS data, GWAS summarized statistics datasets of 42 known human genetic disease data were collected from the GWAS catalog and previous studies4. The preprocessed code of every dataset is available at https://github.com/jiang-junyao/DRCTdb.
Data preprocess
All datasets were preprocessed manually to ensure the data quality for the following analysis. For the scATAC-seq dataset with peak-cell matrix, we directly create the seurat (v.4.9) object and anndata (v.0.9.1) object24,25. For the scATAC-seq dataset without peak information, ArchR (v.1.0.2) was first used to create an ArchR object from a fragment file. Then, open chromatin regions were called for each cell type using macs2 (v. 2.2.9.1), based on the cell type information26,27. Then, we aggregate the peak count of cells from the same cell type. Finally, we export the peaks into bed file format. The scRNA-seq data was collected to create Seurat object and anndata object for downstream analysis. The preprocessing code for each dataset is available at https://github.com/jiang-junyao/DRCTdb.
LDSC analysis
LDSC (Linkage Disequilibrium Score Regression) analysis was utilized to identify cell types associated with disease, in order to elucidate the relationships between SNPs and these cell types11,13. LDSC command line tool can estimate heritability and genetic correlation with cell types using cell type-specific genomic regions and GWAS summary statistics data as input. The GWAS summarized statistics data was obtained from the GWAS catalog and previous study (Supplementary Table 2)4. We integrated open chromatin regions of each cell type and hg38 baseline-LD model v1.2 to estimate the coefficient p-value for each cell type. Then Benjamini-Hochberg method was used to correct for multiple tests. For datasets with reference genome hg19, we used liftover tools in R package rtracklayer (v.1.62.0) to convert cell type open chromatin regions into corresponding hg38 coordinate28.
Differential accessible regions and TF activity analysis
We calculated TF activity for each single cell from scATAC-seq data by python package scBasset (v.0.1), which is a deep learning method that leverages convolutional neural networks (CNNs) to predict single-cell chromatin accessibility from DNA sequences. The trained model could be used to perform dimension reduction analysis and TF activity analysis. Firstly, we identified differential accessible regions (DARs) with rank_genes_groups function from scanpy (v.1.9.1), DARs with |log2 fold change | > 1 were selected as significant regions. Then we annotated significant DARs with corresponding genomic features by R package ChIPseeker25,29. Then we trained the scbasset model to infer transcription factor activity with specific cell type labels based on the sequence’s information, hyperparameters ‘batch size’ was set to 256, and ‘epoch’ was set to 1000 epochs30.
Disease related cell-cell communication analysis
To construct a cell-cell communication network for each disease, firstly, we subset the scRNA-seq Seurat object to only include cell types that are significantly associated (LDSC p-value < 0.0001) with the disease in the corresponding scATAC-seq data. Next, R package CellChat (v.1.6.1) was used to perform cell-cell communication analysis for these significant cell types. CellChat is a computational tool that infers and analyzes intercellular communication networks from scRNA-seq data by leveraging a curated database of ligand-receptor interactions, enabling the identification of significant cell signaling pathways and interactions31. We employed the ‘computeCommunProb’ function to compute the communication between cell types. The parameter ‘k.min’ was set to 10, and ‘nboot’ was set to 100. Subsequently, we utilized the ‘computeCommunProbPathway’ function to infer significant ligand-receptor interactions. The parameter ‘thresh’ was set to 0.05.
Disease related peak and gene
Given the disease-related cell types, transcriptome data, and epigenomics data of disease related cell types, we employed the following criteria to identify cell-type-specific disease-related peaks: (I) a minimum fraction of cells exhibiting a count >2.5% in scATAC-seq data, (II) the presence of overlapping peak regions with disease-related SNPs, (III) peaks is in the promoter region. In addition, we identified cell-type-specific disease-related genes using the following criteria: (I) a minimum fraction of expressed cells exceeding 5%; and (II) the presence of overlapping gene TSS regions (1000 bp upstream and 500 bp downstream) with disease-related SNPs.
Enrichment analysis
The GO Enrichment of genes nearly or overlapped with disease-related SNPs was performed by R package ClusterProfiler (v.4.10.1) with the parameters “ont = ‘ALL’” and “qvalueCutoff = 0.05”32. The DRCTdb backend online enrichment analysis module is performed by the Enrichr (v.3.2) package due to its good performance33.
Construct basic DRCT gene regulatory network
We integrated scRNA-seq and scATAC-seq data to construct the DRCT gene regulatory network (GRN). The scRNA-seq data was utilized to infer the basic GRN, whereas the scATAC-seq data was employed to refine and enhance the accuracy of the basic GRN. We calculated the Pearson correlation for each gene pair based on scRNA-seq data. Then, only gene pairs that have an absolute value of the correlation coefficient >0.2 and contain at least one transcription factor from the TRANSFAC database (version 2018.3) were chosen as basic GRN. GRNs constructed from only scRNA-seq data contain lots of false positives, so we further used scATAC-seq data to refine the basic GRN. Based on the DRCT peaks, we first identified the binding motif (p < 5e-5) present in the peak region by R package motifmatchr (v.1.22.0)34. Position weight matrices of binding motifs used for matching motifs were from the TRANSFAC database (v. 2018.3). Then motif related transcription factors were selected as TF and peak related genes were selected as targets respectively as TF-target relationships. We selected gene pairs that exist in the TF-target relationships as DRCT GRN.
Statistics and reproducibility
Scripts associated with this study can be found at https://github.com/jiang-junyao/DRCTdb. The parameters of the tools used have not been specifically optimized. Users can download all raw data and freely adjust parameters according to the provided scripts.
Data availability
DRCTdb is a fully open-source database, where users can download the entire project and run it locally at https://github.com/jiang-junyao/DRCTdb. All processed datasets are available on DRCTdb (https://singlecellatlas.top/DRCTDB/) and Zenodo (https://zenodo.org/records/11362883). Codes for analyzing case study are available on GitHub (https://github.com/jiang-junyao/DRCTdb/tree/main/Reproduce_case) and Zenodo (https://zenodo.org/records/13362640)35.
Code availability
Processed code is available on https://github.com/jiang-junyao/DRCTdb.
References
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature 409, 860–921 (2001).
Tam, V. et al. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 20, 467–484 (2019).
Boix, C. A., James, B. T., Park, Y. P., Meuleman, W. & Kellis, M. Regulatory genomic circuitry of human disease loci by integrative epigenomics. Nature 590, 300–307 (2021).
Buniello, A. et al. The NHGRI-EBI GWAS Catalog of published genome-wide association studies, targeted arrays and summary statistics 2019. Nucleic Acids Res. 47, D1005–D1012 (2019).
Gaulton, K. J., Preissl, S. & Ren, B. Interpreting non-coding disease-associated human variants using single-cell epigenomics. Nat. Rev. Genet. 24, 516–534 (2023).
Yazar, S. et al. Single-cell eQTL mapping identifies cell type-specific genetic control of autoimmune disease. Science 376, eabf3041 (2022).
Zhang, F. et al. OSCA: a tool for omic-data-based complex trait analysis. Genome Biol. 20, 107 (2019).
Cuomo, A. S. E., Nathan, A., Raychaudhuri, S., MacArthur, D. G. & Powell, J. E. Single-cell genomics meets human genetics. Nat. Rev. Genet. 24, 535–549 (2023).
Bulik-Sullivan, B. et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015).
Ma, Y. et al. Polygenic regression uncovers trait-relevant cellular contexts through pathway activation transformation of single-cell RNA sequencing data. Cell Genom. 3, 100383 (2023).
Bulik-Sullivan, B. K. et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015).
Domcke, S. et al. A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020).
Zhang, K. et al. A single-cell atlas of chromatin accessibility in the human genome. Cell 184, 5985–6001.e19 (2021).
Wang, G. & Sander, M. A multi-omics roadmap of β-cell failure in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 17, 641–642 (2021).
Martins Peçanha, F. L. et al. The Transcription Factor YY1 Is Essential for Normal DNA Repair and Cell Cycle in Human and Mouse β-Cells. Diabetes 71, 1694–1705 (2022).
Martin, D. & Grapin-Botton, A. The Importance of REST for Development and Function of Beta Cells. Front Cell Dev. Biol. 5, 12 (2017).
Chiou, J. et al. Single-cell chromatin accessibility identifies pancreatic islet cell type- and state-specific regulatory programs of diabetes risk. Nat. Genet. 53, 455–466 (2021).
Jiang, Y., Fischbach, S. & Xiao, X. The Role of the TGFβ Receptor Signaling Pathway in Adult Beta Cell Proliferation. Int J. Mol. Sci. 19, 3136 (2018).
Boj, S. F. et al. Diabetes risk gene and Wnt effector Tcf7l2/TCF4 controls hepatic response to perinatal and adult metabolic demand. Cell 151, 1595–1607 (2012).
Mahajan, A. et al. Multi-ancestry genetic study of type 2 diabetes highlights the power of diverse populations for discovery and translation. Nat. Genet. 54, 560–572 (2022).
Nkonge, K. M., Nkonge, D. K. & Nkonge, T. N. The epidemiology, molecular pathogenesis, diagnosis, and treatment of maturity-onset diabetes of the young (MODY). Clin. Diab. Endocrinol. 6, 20 (2020).
Hartman, M. G. et al. Role for activating transcription factor 3 in stress-induced beta-cell apoptosis. Mol. Cell Biol. 24, 5721–5732 (2004).
Zhu, L., Peng, Q., Wu, Y. & Yao, X. scBCR-seq revealed a special and novel IG H&L V(D)J allelic inclusion rearrangement and the high proportion dual BCR expressing B cells. Cell Mol. Life Sci. 80, 319 (2023).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell 184, 3573–3587.e29 (2021).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018).
Granja, J. M. et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat. Genet. 53, 403–411 (2021).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Lawrence, M., Gentleman, R. & Carey, V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics 25, 1841–1842 (2009).
Yu, G., Wang, L.-G. & He, Q.-Y. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31, 2382–2383 (2015).
Yuan, H. & Kelley, D. R. scBasset: sequence-based modeling of single-cell ATAC-seq using convolutional neural networks. Nat. Methods 19, 1088–1096 (2022).
Jin, S. et al. Inference and analysis of cell-cell communication using CellChat. Nat. Commun. 12, 1088 (2021).
Wu, T. et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innov. (N. Y) 2, 100141 (2021).
Kuleshov, M. V. et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44, W90–W97 (2016).
Schep, A. N., Wu, B., Buenrostro, J. D. & Greenleaf, W. J. chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat. Methods 14, 975–978 (2017).
yuanlizhanshi & jiang_junyao. jiang-junyao/DRCTdb: Release for publicaiton. Zenodo https://doi.org/10.5281/zenodo.13362640 (2024).
Acknowledgements
This research was funded by the National Natural Science Foundation of China, 31601895. This work was supported by the Postgraduate Research & Practice Innovation Program of Jiangsu Province (KYCX22_3771).
Author information
Authors and Affiliations
Contributions
Yunhui Kong, Junyao Jiang, and Sheng Qin designed the research; Weikang Kong collected and downloaded public data; Yunhui Kong and Junyao Jiang analyzed data; Yunhui Kong drafted the initial manuscript. Yunhui Kong, Junyao Jiang, and Sheng Qin wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Sudhir Ghandikota, José M. Fernándezand the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editor: Aylin Bircan. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
kong, Y., jiang, J., Kong, W. et al. DRCTdb: disease-related cell type analysis to decode cell type effect and underlying regulatory mechanisms. Commun Biol 7, 1205 (2024). https://doi.org/10.1038/s42003-024-06833-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06833-y
