
Abstract
The anterior cingulate cortex (ACC) is one of the critical brain areas for processing noxious information. Previous studies showed that peripheral nerve injury induced broad changes in the ACC, contributing to pain hypersensitivity. The neurons in layer 3 (L3) of the ACC receive the inputs from the mediodorsal thalamus (MD) and form the feedforward inhibition (FFI) microcircuits. The effects of peripheral nerve injury on the MD-driven FFI in L3 of ACC are unknown. In our study, we record the enhanced excitatory synaptic transmissions from the MD to L3 of the ACC in mice with common peroneal nerve ligation, affecting FFI. Chemogenetically activating the MD-to-ACC projections induces pain sensitivity and place aversion in naive mice. Furthermore, chemogenetically inactivating MD-to-ACC projections decreases pain sensitivity and promotes place preference in nerve-injured mice. Our results indicate that the peripheral nerve injury changes the MD-to-ACC projections, contributing to pain hypersensitivity and aversion.
Similar content being viewed by others


Introduction
The anterior cingulate cortex (ACC) regulates emotion, cognition, and action1,2,3. Neurons in the ACC process the noxious-related information and regulate sensory and affective pain4,5,6. Peripheral nerve damage easily impair the neuronal excitability and synaptic transmissions, and synaptic plasticity in the ACC4,7, the maladaptive changes in the ACC contribute to peripheral pain hypersensitivity7,8,9. Furthermore, neurons in the prefrontal cortex or hippocampus undergo morphological changes after nerve injury10,11,12,13, indicating the microcircuit reorganization in the pain-related brain regions14.
The feedforward inhibition (FFI) is one of the basic motifs of neuronal circuits, it includes an excitatory neuron, upon being activated, simultaneously sends signals to both a target neuron and an inhibitory interneuron, the inhibitory interneuron then suppresses the activity of the target neuron, modulating its response to the incoming excitatory signal15. The mediodorsal thalamus (MD) is a region of the thalamus that is intricately connected with the ACC, the information exchanges between the MD and the ACC are essential for modulating the sensory and emotional aspects of pain16. The MD thalamocortical projections excite the pyramidal neurons in layer III (L3)15,17 and layer V(L5)16,18 of the ACC and formed the FFI circuits. Recent studies showed that prolonged nerve injury generates silent synapses in the MD-to-ACC projections19. Furthermore, the peripheral nerve injury weakened the MD inputs to the L5 cingulate neurons, contributing to pain aversion16,18, indicating the involvement of MD-to-ACC projections in developing pathological pain20. L3 of the ACC is the primary layer to receive the MD inputs21,22,23,24 and construct the FFI microcircuits25. How the responses of L3 ACC neurons to MD inputs were changed and its effect on FFI remained unknown.
Here, we investigated the changes of MD projections to L3 of ACC after nerve injury. We examined the impairments of nerve injury on the E-I balance and temporal gating window within the feedforward circuits. Our results showed the enhanced thalamocortical synaptic transmissions in the L3 pyramidal neurons, but not the parvalbumin (PV) interneurons after nerve injury, which indicates the hyperactivation of FFI in the L3 of ACC. Furthermore, enhancing the MD-ACC inputs induced place aversion and decreased the paw withdrawal threshold (PWT) of naive mice. Inactivating the MD-to-ACC inputs generated place preference and elevated the PWTs of mice with nerve injury. Our results indicated that the peripheral nerve injury changed the MD-to-L3 projections, contributing to pain hypersensitivity and aversion.
Results
Activating of MD inputs drove the feedforward inhibition in L3 of the ACC
Previous studies showed that the MD sends dense projections to the ACC22,25,26,27. To confirm this point, we unilaterally injected AAV-retro-hSyn-mcherry into the ACC (Fig. 1A, B-a). We detected the expression of mCherry in the MD on day 21 after the injection (Fig. 1B-b), indicating the innervations from MD to the ACC.

A A representative coronary image in which the AAV-Retro-hSyn-mCherry virus was injected into ACC. B a: A representative image of retro virus expression in ACC. Scale bar: 200 μm. b: A representative image of retro virus in MD, scale bar: 200 μm; and the right was an enlarged diagram of the rectangle area, scale bar: 100 μm. C A coronal section of a mouse brain at Bregma −1.70 mm showed the site of ChR2-mCherry injection in the MD. D a: Fluorescent labeling of the targeted MD, scale bar: 200 μm. b: MD ChR2-mCherry fibers projected to ACC, scale bar: 200 μm; and the right was an enlarged view of the rectangle area, scale bar: 20 μm. E Schematic: recording current of ACC L3 pyramidal neurons in response to activation of ChR2 in MD terminals. F a–d: Sample traces of EPSC and IPSC in response to 1 ms light pulse stimulation at baseline, further with TTX, 4-AP, CNQX, and AP5. Scale bar: 20 ms/100 pA in figure a, b, d. The scale bar is 50 ms/100 pA in (E). G Left: Enlarged view of the circled area in (F-a), in which the two arrows indicated the onset of EPSC and IPSC. Right: Summary plots of the synaptic latency of EPSCs and IPSCs (Paired t-test, t = 12.20, df = 11, P < 0.001; EPSC/IPSC: n = 12 neurons/3 mice). H Histogram of photo-induced EPSC response amplitude at baseline with TTX and 4-AP (One-way ANOVA, Between columns, F(1.002, 3.006) = 17.95, P = 0.024; n = 4 neurons/3 mice). I Sample traces of IPSC response induced by 1 ms photostimulation at baseline, further with PTX, CNQX, and AP5, respectively, at voltage 0 mV. All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
The FFI is the basic pattern for the MD-to-ACC inputs25. The MD projections to the ACC innervate multiple layers22,25,26; previously, Delevich et al. showed that the MD inputs drive the FFI in L3 of the ACC25. In our study, we confirmed the FFI by combining the photostimulation and whole-cell patch-clamp recording approaches. We injected the AAV-CaMKIIα-ChR2-mCherry into the MD (Fig. 1C, D-a); similarly, we observed the mCherry expressing axons in L1, L3, L5 of the ACC on day 21 after the virus injection (Fig. 1D-b); we then performed the whole-cell patch-clamp recording on the pyramidal cells in L3 of the ACC (Fig. 1E). By photostimulating MD terminals in the ACC, we recorded the EPSC and IPSC with membrane potentials held at −70 mV and 0 mV, respectively (Fig. 1F-a). There was a significant difference in onset latency between the EPSCs (-70 mV) and outward currents (0 mV) (t = 12.20, P < 0.001, EPSC/IPSC: n = 12; Fig. 1G). The bath application of tetrodotoxin (TTX, 1 μM) blocked the activities of voltage-dependent sodium channels, and abolished both photostimulation-induced EPSCs and IPSCs (Fig. 1F-b), indicating that the photostimulation-induced EPSCs are action potentials dependent. With the application of TTX, further application of 4-Aminopyridine (4-AP, 100 μM), one potassium channel blocker, induced the EPSCs at −70 mV (Fig. 1F-c, H). Instead, an inward current was recorded at 0 mV (Fig. 1F-c), which was abolished by the application the cocktail of CNQX and AP5; this cocktail also abolished the EPSCs recorded at −70 mV (Fig. 1F-d), indicating that the involvements of AMPARs and NMDARs to the MD-to-ACC mono-synaptic inputs.
It also showed that the photostimulation-induced IPSCs were synaptic and mediated by the GABAARs25. We recorded the identical IPSCs by holding membrane potentials at 0 mV (Fig. 1I) and applied the picrotoxin (PTX) to confirm this point. Similarly, PTX blocked the activities of GABAARs, and we further recorded an inward current at 0 mV, which was subsequently blocked by the cocktail of CNQX and AP5 (Fig. 1I). Consistently, the onset latency of NMDARs mediated currents was shorter than that of IPSCs (Fig. 1I). Therefore, the MD inputs to the cingulate L3 drive FFI.
Peripheral nerve injury increased the MD-to-ACC excitatory synaptic transmissions
The nerve injury-enhanced excitatory synaptic transmissions have been broadly reported7,8, which indicates that the MD-to-ACC input in the FFI microcircuits may be changed. To examine the possible changes of FFI in L3 of ACC after nerve injury, we performed electrophysiological experiments on L3 pyramidal neurons on day 7 after nerve injury, the time point when nerve injury-induced neural plasticity and signaling changes have taken place in the ACC8, and recorded the AMPARs-mediated currents under varying intensities of photostimulation (Fig. 2A, B). We observed increased amplitudes of AMPAR currents corresponding to changes in photo intensity (Fig. 2C). Interestingly, the amplitudes of AMPARs-mediated currents recorded from the CPN ligation group were higher than those from the control group at higher light intensity (Two-way ANOVA, Interaction, F(5, 100) = 6.29, P < 0.01; Sham vs. CPN: F(1, 20) = 6.28, P = 0.02; n = 13/Sham, n = 9/CPN; Fig. 2D). Furthermore, we did not detect changes in the I–V curves of the recorded currents (Two-way ANOVA, Interaction, F(6, 120) = 1.38, P = 0.23; Sham vs. CPN: F(1, 20) = 0.13, P = 0.72; n = 9/Sham, n = 13/CPN; Fig. 2E, F); these observations indicate that the MD-to-ACC L3 inputs were enhanced after nerve injury.

A Schematic diagram of experimental design. B The whole-cell patch recording of ACC L3 pyramidal neurons. C Representative traces showing the light-evoked AMPAR current input-output relationship. D Summary plots of the AMPAR currents in (C) (Two-way ANOVA, Interaction, F(5, 100) = 6.29, P < 0.0001; Sham vs. CPN: F(1, 20) = 6.28, P = 0.02; n = 13 neurons/4 mice, n = 9 neurons/3 CPN mice). E Representative traces of light-evoked AMPAR current in Sham and CPN groups when the holding potentials were increased from −80 to 40 mV at a step of 20 mV. F Current–voltage plots of the light-evoked AMPAR currents in (E). Current amplitudes were normalized to that recorded at −80 mV (Two-way ANOVA, Interaction, F(6, 120) = 1.38, P = 0.23; Sham vs. CPN: F(1, 20) = 0.13, P = 0.72; n = 9 neurons/3 Sham mice, n = 13 neurons/4 CPN mice). G Representative traces show the light-evoked NMDAR current input-output relationship. H Summary plots of the NMDAR currents in (G) (Two-way ANOVA, Interaction, F(5, 90) = 7.19, P < 0.0001, Sham vs. CPN, F(1, 18) = 5.46, P = 0.03; n = 9 neurons/3 sham mice, n = 11 neurons/3 CPN mice). I Representative traces of light-evoked NMDAR current in Sham and CPN group when the holding potentials were increased from −80 to + 40 mV at a step of 20 mV. J Current–voltage plots of the light-evoked NMDAR current in (I). Current amplitudes were normalized to that recorded at +40 mV (Two-way ANOVA, Interaction, F(6, 174) = 0.41, P = 0.87; Sham vs. CPN, F(1, 29) = 0.47, P = 0.50; n = 13 neurons/4 Sham mice, n = 18 neurons/5 CPN mice). All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Additionally, we recorded NMDAR-mediated currents by holding different membrane potentials with the perfusion of a cocktail of PTX and CNQX; we did not observe the differences in the I–V curve of the NMDAR-mediated currents (Two-way ANOVA, Interaction, F(6, 174) = 0.41, P = 0.87; Sham vs. CPN, F(1, 29) = 0.47, P = 0.50; n = 13/Sham, n = 18/CPN; Fig. 2I, J). Holding the membrane potentials at + 40 mV, in line with the observations on AMPARs-mediated currents, we observed bigger NMDAR-currents in the CPN group (Two-way ANOVA, Interaction, F(5, 90) = 7.19, P < 0.01, Sham vs. CPN, F(1, 18) = 5.46, P = 0.03; n = 9/Sham, n = 11/CPN; Fig. 2G, H), indicating that the NMDARs-mediated synaptic transmissions were also increased by nerve injury. Therefore, CPN ligation led to higher MD-to-ACC inputs mediating by the AMPARs and NMDARs.
Peripheral nerve injury increased the excitability of intermediate (IM) cells but not the regular spiking (RS) cells
The pyramidal neurons are the major components of a FFI microcircuit15; the changes in intrinsic properties would affect the temporal integration of synaptic transmissions and, therefore, determine the circuit’s outputs17. We performed whole-cell patch-clamp recording on L3 pyramidal neurons from mice with sham or CPN ligation (Fig. 3A). After forming the whole-cell configuration, depolarizing pulse currents were injected into the soma and action potentials (APs) firing were recorded. Similar to previous studies, we identified two predominant action potential firing patterns based on the presence or absence of afterdepolarization (ADP) in individual action potential: regular spiking (RS) neurons without ADP (Fig. 3A), and intermediate firing (IM) neurons characterized by evident ADP.

A Upper: The whole-cell patch recording of RS neurons in ACC L3. Lower: Sample trace of RS neuron AP. B–D CPN ligation didn’t change RS neurons RMP, Rheo-base current, threshold, half-width (Two-tailed unpaired t-test in (B–D), in (B): t = 1.35, df = 16, P = 0.19; in (C): t = 0.23, df = 16, P = 0.82; in (D): t = 0.26, df = 16, P = 0.80; n = 10 neurons/3 Sham mice, n = 8 neurons/3 CPN mice). E Upper: Representative traces showed the voltage response to a series of 500 ms current pulses from 50 pA to 400 pA with 50 pA steps of RS neurons in the Sham and CPN group. Lower: the waveforms of the stimulation currents. F Quantification of AP number of RS neurons firing under current injection (Two-way ANOVA, Interaction, F(7, 168) = 1.15, P = 0.34; Sham vs. CPN: F(1, 24) = 1.58, P = 0.22; n = 14 neurons/4 Sham mice, n = 12 neurons/4 CPN mice). G Upper: The whole-cell patch recording of IM neurons in ACC L3. Lower: Sample trace of IM neuron AP. H–J CPN ligation didn’t change IM neurons RMP, Rheo-base current, threshold (Two-tailed unpaired t-test in (H–J), in (H): t = 0.02, df = 20, P = 0.99; in (I): t = 1.30, df = 20, P = 0.21; in (J): t = 0.34, df = 20, P = 0.74; n = 12 neurons/3 Sham mice, n = 10 neurons/3 CPN mice). K Upper: Representative traces showed the voltage response to a series of 500 ms current pulses from 50 to 400 pA with 50 pA steps of IM neurons in the Sham and CPN group. Lower: the waveforms of the stimulation currents. L Quantification of AP number of IM neurons firing under current injection (Two-way ANOVA, Interaction, F(7, 126) = 2.25, P = 0.03; Sham vs. CPN: F(1, 18) = 6.74, P = 0.02; n = 9 neurons/3 Sham mice, n = 11 neurons/3 CPN mice). All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
For RS neurons, there were no significant differences observed in the resting membrane potentials (RMP) (t = 1.35, P = 0.19; n = 10/Sham, n = 8/CPN; Fig. 3B), the rheobase (t = 0.23, P = 0.82; n = 10/Sham, n = 8/CPN; Fig. 3C), and the threshold of APs firing (t = 0.26, P = 0.80; n = 10/Sham, n = 8/CPN; Fig. 3D) between the two groups. The number of firing APs under different current intensities is essential in showing neurons’ excitability. In the RS neurons, we did not detect any difference in the firing numbers of APs between groups (Two-way ANOVA, Interaction, F(7, 168) = 1.15, P = 0.34; Sham vs. CPN: F(1, 24) = 1.58, P = 0.22; n = 14/Sham, n = 12/CPN; Fig. 3E, F), indicating that the nerve injury did not change the intrinsic properties of RS neurons.
For the IM neurons (Fig. 3G), we also did not detect changes in the RMP (t = 0.02, P = 0.99; n = 12/Sham, n = 10/CPN; Fig. 3H), the rheobase (t = 1.30, P = 0.21; n = 12/Sham, n = 10/CPN; Fig. 3I), the threshold (t = 0.34, P = 0.74; n = 12/Sham, n = 10/CPN; Fig. 3J) of APs firing between two groups. Instead, for the CPN group, the IM cells fired more APs than the control group (two-way ANOVA, Interaction, F(7, 126) = 2.25, P = 0.03; Sham vs. CPN: F(1, 18) = 6.74, P = 0.02; n = 9/Sham, n = 11/CPN; Fig. 3K, L), indicating that the nerve injury increased the excitability of IM neurons.
The excitatory signal input of MD to fast spiking (FS) neurons in ACC L3 did not change after nerve injury
Previous studies showed that the fast-spiking (FS) interneurons expresses parvalbumin (PV) mediates the FFI in L3 of the ACC25. To investigate potential changes in interneurons following nerve injury, we conducted whole-cell patch-clamp recording on the GFP-positive cells (GFP+) in the ACC of GAD67-GFP mice (Fig. 4A, B). We specifically examined the changes in MD inputs to the GFP+ neurons (Fig. 4B). Interestingly, we found that under the same stimulation intensity, the GFP+ neurons from both groups received the same amplitudes of EPSCs (t = 0.28, P = 0.78; n = 7/Sham, n = 8/CPN; Fig. 4C). The time constant of the EPSCs didn’t change (t = 1.087, P = 0.30; n = 7/Sham, n = 8/CPN; Fig. 4D). Among the recorded GFP+ cells, over 70% of cells fired APs at a very high frequency, indicating these are FS neurons. Additionally, we demonstrated through a combination of electrophysiological experiments and immunofluorescence staining that FS neurons are PV-positive neurons (Fig. 4E). Notably, the firing rates of FS neurons in response to high-intensity stimulation were comparable between the CPN and control groups (two-way ANOVA, Interaction, F(7, 112) = 9.57, P < 0.001; Sham vs. CPN, F(1, 16) = 1.49, P = 0.24; n = 9/3 Sham, n = 9/3 CPN; Fig. 4F, G). These findings suggest that nerve injury didn’t impair the excitability of FS cells in the ACC.

A Schematic diagram of experimental design. B Upper: The whole-cell patch recording of FS neurons in ACC L3. Lower: Representative traces of EPSC in Sham and CPN groups. C Histogram of EPSC amplitude values in Sham and CPN group (Two-tailed unpaired t-test, t = 0.28, df = 13, P = 0.78; n = 7 neurons/3 Sham mice, n = 8 neurons/3 CPN mice). D Histogram of EPSC Tau in Sham and CPN group (Two-tailed unpaired t-test, t = 1.087, df = 13, P = 0.30; n = 7 neurons/3 Sham mice, n = 8 neurons/3 CPN mice). E Examples showing that the recorded FS neurons are PV-positive neurons, scale bar: 20 μm. F Representative traces showed the voltage response of FS neurons to 200 pA current pulse in the Sham and CPN group. G Quantification of AP number fired under current injection (Two-way ANOVA, Interaction, F(7, 112) = 9.57, P < 0.001; Sham vs. CPN, F(1, 16) = 1.49, P = 0.24; n = 9 neurons/3 Sham mice, n = 9 neurons/3 CPN mice).
Peripheral nerve injury changed the temporal integration of feedforward inhibition microcircuits
Based on our observations of the pyramidal and FS neurons, we assumed that the nerve injury impaired the FFI driven by MD inputs. We recorded the EPSCs and IPSCs at −70 mV and 0 mV, respectively, on the pyramidal neurons (Fig. 5A, B). Interestingly, we detected the shorter onset latency of both EPSCs and IPSCs on day 7 after nerve injury (Two-way ANOVA, Interaction, F(1, 21) = 2.42, P = 0.13; Sham (EPSC) vs. CPN (EPSC): P = 0.02; Sham (IPSC) vs. CPN (IPSC): P < 0.001; Sham: EPSC vs. IPSC, P < 0.001; CPN: EPSC vs. IPSC, P < 0.001; n = 12/Sham, n = 11/CPN; Fig. 5C). FFI is also involved in the temporally related information process21,25,28. Thus, we further investigated potential changes in synaptic transmissions under train stimulations. We recorded dynamic changes in excitatory and inhibitory synaptic transmissions received by the pyramidal neurons in L3 of ACC following light activation of MD terminal fibers in ACC after nerve injury (Fig. 5D). The amplitude of EPSCs decayed under 10 Hz simulation in both sham and CPN group, with the CPN group exhibiting a slower decay process (Fig. 5D, E). Additionally, the latency of EPSCs in the CPN group was shorter than that in the sham group (Fig. 5F), indicating that the excitatory synaptic transmissions in the FFI were enhanced after peripheral nerve injury. Furthermore, the amplitudes and onset latency of IPSCs were also altered in the CPN group (Fig. 5G, I). The changes in onset latency narrowed the temporal windows between the EPSCs and IPSCs (Fig. 5I), suggesting the presence of temporal characteristics in MD-driven feed-forward inhibition micro loops.

A Schematic diagram of experimental design. B Upper: The whole-cell patch recording of pyramidal neurons in ACC L3. Lower: Example of light-evoked EPSC (eEPSC) and IPSC (eIPSC) recorded at −70 mV and 0 mV, respectively. C Statistics of EPSC and IPSC latency in Sham and CPN group (Two-way ANOVA, Interaction, F(1, 21) = 2.42, P = 0.13; Sham (EPSC) vs. CPN (EPSC): P = 0.02; Sham (IPSC) vs. CPN (IPSC): P < 0.001; Sham: EPSC vs. IPSC, P < 0.001; CPN: EPSC vs. IPSC, P < 0.001; n = 12 neurons/3 Sham mice, n = 11 neurons/3 CPN mice). D Example traces from representative cells in response to 10 Hz MD stimulation showed clear synaptic depression in Sham and CPN group. E Group synaptic dynamics of eEPSC in response to MD inputs (Two-way ANOVA, Interaction, F(9, 153) = 9.77, P < 0.001; Sham vs. CPN, F(1, 17) = 21.06, P < 0.001. n = 9 neurons from 3 Sham mice, n = 10 neurons from 3 CPN mice). F Statistics of eEPSC latency in response to MD inputs (Two-way ANOVA, Interaction, F(9, 153) = 0.45, P = 0.91; Sham vs. CPN, F(1, 17) = 14.05, P < 0.001; n = 9 neurons from 3 Sham mice, n = 10 neurons from 3 CPN mice). G Group synaptic dynamics of eIPSC in response to MD inputs (Two-way ANOVA, Interaction, F(9, 153) = 2.93, P < 0.01; Sham vs. CPN, F(1, 17) = 7.35, P = 0.01, n = 9 neurons from 3 Sham mice, n = 10 neurons from 3 CPN mice). H Statistics of eIPSC latency in response to MD inputs (Two-way ANOVA, Interaction, F(9, 153) = 3.07, P < 0.01; Sham vs. CPN, F(1, 17) = 18.14, P < 0.001, n = 9 neurons from 3 Sham mice, n = 10 neurons from 3 CPN mice). I Statistics of eEPSC and eIPSC latency delay in response to MD inputs (Two-way ANOVA, Interaction, F(9, 153) = 1.84, P = 0.06; Sham vs. CPN, F(1, 17) = 13.50, P < 0.01, n = 9 neurons from 3 Sham mice, n = 10 neurons from 3 CPN mice). All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Chemogenetic activation of the MD-to-ACC projections affected feedforward inhibition within the microcircuits
To investigate the activation of the MD-ACC pathway and its impact on FFI within this pathway, we examined whether chemogenetic activation influences synaptic activity in ACC neurons. We unilaterally injected AAV9-CaMKIIα-hM3Dq-mCherry virus into the MD. Then, we conducted in vitro whole-cell recordings of ipsilateral ACC L3 pyramidal neurons to measure spontaneous excitatory postsynaptic currents (sEPSCs) and spontaneous inhibitory postsynaptic currents (sIPSCs) at least 21 days after the virus injection (Fig. 6A–C). Interestingly, we observed a significant difference in the frequency of both sEPSCs and sIPSCs after adding 30 μM CNO to the ACSF for 10 min. Additionally, sEPSCs were significantly blocked by 20 μM CNQX (Fig. 6D, E), while sIPSC frequency and amplitude were reduced following the addition of 20 μM CNQX to the ACSF for 10 min (Fig. 6D, E). Hence, chemogenetic activation of MD-ACC pathway would affect the synaptic transmission of ACC neurons, thus influencing the FFI within the microcircuits.

A Schematic diagram of experimental design. B The whole-cell patch recording of ACC L3 pyramidal neurons. C Representative sEPSC traces recorded from ACC L3 pyramidal neurons. D Representative sIPSC traces recorded from ACC L3 pyramidal neurons. E Quantification of frequency of sEPSCs and sIPSC of ACC L3 pyramidal neurons (Two-way ANOVA, Interaction, F(2, 44) = 4.331, P = 0.0192; sEPSC, ACSF vs. CNO, P = 0.0054; ACSF vs. CNQX, P < 0.0001, CNO vs. CNQX, P < 0.0001; sIPSC, ACSF vs. CNO, P = 0.0469; ACSF vs. CNQX, P = 0.1789, CNO vs. CNQX, P = 0.0285; n = 14 neurons of sEPSC from 6 mice, n = 10 neurons of sIPSC from 5 mice). All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001. F Quantification of amplitude of sEPSCs and sIPSC of ACC L3 pyramidal neurons (Two-way ANOVA, Interaction, F(2, 44) = 49.53, P < 0.0001; sEPSC, ACSF vs. CNO, P = 0.9041; ACSF vs. CNQX, P < 0.0001, CNO vs. CNQX, P < 0.0001; sIPSC, ACSF vs. CNO, P = 0.0086; ACSF vs. CNQX, P < 0.0001, CNO vs. CNQX, P = 0.0744; n = 14 neurons of sEPSC from 6 mice, n = 10 neurons of sIPSC from 5 mice). All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Boosting the MD-to-ACC projections chemogenetically induced aversion and changed mechanical sensation
A multiple-channel recording experiment showed that the MD-to-ACC pathway conveys noxious information22. Ontogenetically activating the MD-to-ACC inputs induced aversion in nerve injury and chemotherapy-induced models of neuropathic pain28, which emphasized the role of ACC in regulating aversion. Previous studies showed that the cingulate neuronal activities hold mechanical sensation7,29,30. Here, we employed a chemogenetic approach to selectively activate the MD-to-ACC inputs. We bilaterally injected AAV2-retro-hSyn-cre-GFP into the ACC and AAV2-hSyn-DIO-hM3Dq-mCherry into the MD, the mice with the injection of AAV2-retro-hSyn-cre-GFP (ACC) and AAV2-hSyn-DIO-mCherry (MD) as control, to selectively activate the MD-to-ACC inputs, we locally administered CNO to the ACC (10 μM) (Fig. 7A, C). Immunofluorescence staining further revealed increased c-Fos expression in both L3 and L5 of the ACC, 90 min after intraperitoneal administration of CNO (4 mg/kg) to mice (Fig. 7D, E). CPA tests were conducted on day 21 after the virus injection (Fig. 7F, G). Similarly, CNO locally administration significantly induced place aversion (Two-way ANOVA, Interaction, F(1, 13) = 9.115, P = 0.0099; mCherry vs. hM3Dq (Test): P = 0.0339; In hM3Dq group, Pre vs. Test: P = 0.0118. mCherry: n = 7, hM3Dq: n = 8; Fig. 7H) and decreased the PWT in the mice with hM3Dq expression but not those with the control virus (Two-way ANOVA, Interaction, F(2, 24) = 5.45, P = 0.01; mCherry vs. hM3Dq (CNO): P = 0.01; In hM3Dq group: Baseline vs. CNO, P < 0.01; Saline vs. CNO, P = 0.02; mCherry: n = 7, hM3Dq: n = 7; Fig. 7I). Therefore, boosting the MD-to-ACC inputs regulated the aversion and mechanical sensation of pain in naive mice.

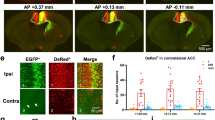
A Upper: Diagram to show the experiment design of mCherry and hM3Dq expression in MD and cannula implanted in ACC. Lower: The timeline of the experiment. B a, b: Representative images showed the expression of Retro-cre-GFP in ACC and the expression of mCherry and hM3Dq in MD, c: The canular planted in the ACC, scale bar: 200 μm. C Representative image showed the location of the cannula in ACC, scale bar: 500 μm. D Examples of c-Fos expression in different layers of ACC in mCherry or hM3Dq groups, scale bar: 200 μm. E The density of c-Fos positive neurons in different layers of ACC (two-way ANOVA, Sidak’s multiple comparisons test, L3: mCherry vs. hM3Dq, P = 0.0025, L5: mCherry vs. hM3Dq, P = 0.0334. n = 6). F CPA/CPP paradigm. G Representative traveling traces of the mCherry and hM3Dq mice in the CPA test. H The application of CNO to the hM3Dq group induced CPA (Two-way ANOVA, Interaction, F(1, 13) = 9.115, P = 0.0099; Sidak’s multiple comparisons test, mCherry vs. hM3Dq (Test): P = 0.0339; In hM3Dq group, Pre vs. Test: P = 0.0118. mCherry: n = 7, hM3Dq: n = 8). I The application of CNO decreased the hM3Dq group mechanical pain threshold not the mCherry group (Two-way ANOVA, Interaction, F(2, 24) = 5.45, P = 0.01; mCherry vs. hM3Dq (CNO): P = 0.01; In hM3Dq group: Baseline vs. CNO, P < 0.01; Saline vs. CNO, P = 0.02; mCherry: n = 7, hM3Dq: n = 7). All data were mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
Inactivating the MD-to-ACC induced the place reference and alleviated the mechanical allodynia
In the naive mice, boosting the MD-to-ACC inputs induced place aversion and decreased the PWTs; our electrophysiological recording results showed higher MD inputs after nerve injury, which may contribute to the pain hypersensitivity. To confirm this point, we employed the chemogenetic approach to further manipulate the MD inputs on the mice with a nerve injury. We bilaterally injected AAV2-retro-hSyn-cre-GFP into the ACC and AAV2-hSyn-DIO-hM4Di (Gi)-mCherry into the MD to selectively inactivate the MD-to-ACC inputs; we locally applied the CNO to the ACC (Fig. 8A–C). In line with our hypothesis, the application of CNO to the ACC induced CPP (Two-way ANOVA, Interaction, F(1, 18) = 8.64, P < 0.01; mCherry vs. hM4Di (Test): P = 0.04; In hM4Di group, pre vs. test: P = 0.03; mCherry: n = 11, hM4Di: n = 9; Fig. 8D, E) and increased the PWTs of mice with nerve injury (Two-way ANOVA, Interaction, F(1, 13) = 9.12, P = 0.01; mCherry vs. hM4Di (Test): P = 0.0339; In hM4Di group, Pre vs. Test: P = 0.0118. mCherry: n = 7, hM4Di: n = 8; Fig. 8F), indicating the alleviation of pain hypersensitivity in mice with nerve injury.

A Upper: Diagram to show the experiment design of mCherry and hM4Di expression in MD and cannula implanted in ACC. Lower: The timeline of the experiment. B a–c: Representative images showed the expression of Retro-cre-GFP in ACC and the expression of mCherry and hM4Di in MD, scale bar: 200 μm. C Representative image showed the location of the cannula in ACC, scale bar: 500 μm. D Representative traveling traces of the mCherry and hM4Di mice in the CPP test. E The application of CNO to the hM4Di group induced CPP (Two-way ANOVA, Interaction, F(1, 16) = 5.599, P = 0.0309; Sidak’s multiple comparisons test, mCherry vs. hM4Di (Test): P = 0.0271; In hM4Di group, pre vs. test: P = 0.042. mCherry: n = 9, hM4Di: n = 9). F The application of CNO increased the hM4Di group mechanical pain threshold, not the mCherry group (Two-way ANOVA, Interaction, F(3, 36) = 5.54, P < 0.01; mCherry vs. hM4Di (CPN + CNO), P = 0.03; mCherry: n = 6, hM4Di: n = 8).
Discussion
Here, we examined the possible changes of FFI in the L3 of ACC after nerve injury. Combining the photostimulation and whole-cell patch-clamp recording approaches, we confirmed that the MD-L3 of ACC formed the FFI; the peripheral nerve injury enhanced excitatory synaptic transmission to the pyramidal cells and increased excitability of IM cells, but not the FS neurons. Therefore, the nerve injury impaired the FFI in the L3 of the ACC. Chemogenetically activating the MD inputs induced place aversion and decreased the PWTs in naive mice. In line with this, inactivating the MD-to-ACC inputs induced place preference and increased the PWTs in mice with nerve injury. Our study indicated that the enhanced MD inputs to the L3 of ACC contributed to pain hypersensitivity and aversion in a mouse model of neuropathic pain.
Nerve injury enhanced the MD-L3 of ACC inputs
The excitatory synaptic transmissions along the somatosensory pathways convey the noxious information; as one of the significant parts of the pain matrix, the ACC has a crucial role in pain regulation5,8. Previous studies showed that the peripheral nerve injury enhanced the AMPARs-mediated currents in L3 of the ACC30; the maintenance of this long-term potentiation depended on the activities of PKMζ8. In contrast, the downregulation of Casp3 after nerve injury would affect the AMPAR’s internalization, therefore leading to the deficits of long-term depression7,9. Although the MD is the primary source of the harmful information to the ACC, the changes of MD to L3 of ACC after nerve injury were unknown. We combined the photostimulation, optogenetic, and whole-cell patch-clamp recording approaches specifically isolated the MD inputs to L3 of the ACC; we found the enhanced AMPARs and NMDARs-mediated MD inputs after the CPN ligation. Our results differed from the previous study showing that MD inputs to L5 of the ACC were weakened after nerve injury27. The difference may come from the layer difference31 and the subregion difference. Additionally, Wang et al., who noted an increase in silent synapses between MD and L3 cingulate neurons post-chronic constriction injury (CCI)19. These synapses gradually decreased in number over time following prolonged CCI. Moreover, the potential maturation of these synapses could lead to remodeling of the MD-to-ACC projection, creating new circuit connectivity and strengthening neurotransmission, which might contribute to the development of allodynia induced by nerve injury. Therefore, following neural injury, even with an increase in silent synapses, enhanced AMPAR currents may still be observed due to the presence of inactive AMPARs.
In line with the previous studies4,5, our results showed that the ACC regulated affective pain and mechanical sensation. We showed that chemogenetically activating the MD-to-ACC inputs sensitized the mechanical sensation and induced aversion in naive mice. In contrast, for the mice with nerve injury, which would have pain hypersensitivity and ongoing pain8,32, we showed that chemogenetically inactivating the MD-to-ACC inputs normalized the mechanical sensation and induced place preference, indicating the alleviation of continuing pain. Therefore, the peripheral nerve injury increased excitatory synaptic transmissions from MD-to-ACC, contributing to the abnormal pain.
Nerve injury impaired the feedforward inhibition in L3 driven by MD to the ACC
FFI is a basic pattern for somatic sensory information processing. Previously, it was shown that the FFI in the spinal cord regulates the noxious related information33,34. Additionally, the FFI also existed in projections from the PFC to the ventrolateral PAG region and other targets35. The MD drove FFI in both L325 and L5 of the rostral part of the ACC16,18. Furthermore, we described the same microcircuit in L3 of the ACC, our data clearly showed that the MD inputs to the L3 pyramidal neurons were enhanced by nerve injury; which is different from that in the L5 of the ACC16,18. Furthermore, we observed enhanced neuronal excitability in IM neurons36, since that there was no change in the excitability of FS neurons, the differences in MD inputs and IM excitability indicate the impairments of peripheral nerve injury on the FFI on L3 of ACC.
In line with this, we detect the changes in onset latency of the EPSCs and IPSC on pyramidal cells. Given that FFI mainly integrated information in a temporal domain, where IPSCs typically lag behind the EPSCs33,37, the synaptic strength a pyramidal neuron receives would mainly be determined by early-arriving excitatory synaptic inputs. Therefore, the enhanced MD inputs would impact the FFI, while the decreased E/I ratio may indicate heighten inhibition following excitation. Given that the FFI would control the temporal windows of information output25, the changes of L3-FFI may impair the activation of L3 neurons and lead to hyperactivations.
The MD has a critical role in cognition due to its reciprocal connections with the prefrontal cortex (PFC) and other cortical areas38. The MD-to-PFC connections are necessary for maintaining working memory39, and the activity-dependent plasticity happens in the FFI microcircuits40. Clinical studies and basic research on rodents or nonhuman primates showed that the dysfunction of thalamic-cortical communications underlines cognitive deficits23,41,42,43. Notably, much of the existing research has focused on male subjects, further studies should emphasize the importance of sex differences in thalamic-cortical communication and their impacts on cognitive function. Knowing the underlying mechanisms of thalamic-cortical communications would help treat cognitive deficits44.
Current study mainly showed that the pyramidal neurons with the soma located in the L3 of the ACC received the enhanced thalamic inputs, which lead to the hyperactivation of FFI microcircuits in the ACC. Due to the technical limitation, we were unable to selectively manipulate the FFI in L3. Therefore, the exact role of FFI in the L3 of ACC need to further studied.
Methods
Animals and CPN model
To rule out the effects of estrogen on the estrous cycle45, experiments were performed on adult male C57BL/6J mice and male GAD67-GFP mice (8–12 weeks old). The mice were housed in the experimental animal center of Zhejiang University and maintained on a 12-h light/dark cycle (light: 07:00–19:00) at room temperature (22–24 °C) with ad libitum access to food and water. We randomly tagged each mouse and then randomly assigned them to groups. Four or five mice were housed per cage. We have complied with all relevant ethical regulations for animal use. All mouse protocols were approved by the animal care and use committee of Zhejiang University (approval No. ZJU20220442)46.
The common peroneal nerve (CPN) ligation mouse model of neuropathic pain, as previously described, was utilized for the study7. Briefly, mice were anesthetized with isoflurane (1–3%, as needed, RWD, Shenzhen, China, R510-22). The left CPN between the anterior and posterior groups of muscles was gently ligated using a chromic gut suture 5-0 (Jinhuan, Shanghai, China, F503) until the digits began to twitch. The skin was sutured using a 5-0 silk suture and cleaned with povidone-iodine (Caoshanhu, Nanchang, China). Sham surgery was conducted similarly, but the nerve was not ligated. All animals were kept in a standard living chamber after surgery. The mice recovered for at least 7 days before the behavioral test.
Mechanical allodynia test
The von Frey behavioral test was performed using the up-down algorithm47. To determine reflex responses evoked by mechanical stimuli, animals were positioned on a raised mesh grid and enclosed within a clear plastic box for containment. Calibrated von Frey filaments were applied to the middle of the plantar surface of each paw until the filament bent. A brisk withdrawal or paw flinching was considered a positive response. Lifting of the paw due to normal locomotor behavior was ignored. Without a response, the filament of the next greater force was applied. Following a response, the filament of the next lower force was applied. The tactile stimulus producing a 50% likelihood of a withdrawal response was calculated and treated as the PWT.
Conditioned place preference and aversion test
Conditioned place preference and aversion (CPP/CPA) in adult mice was performed as described previously7,32,48. Briefly, mice were pre-conditioned for 2 days: they were allowed to explore the chamber freely for 15 min (min) every day; On the first day of the formal experiment, behaviors were recorded by a video camera for 15 min, and the time spent in each chamber was recorded; Over the subsequent two days, mice received the appropriate control (i.e. vehicle) paired with a randomly-selected chamber in the morning, and the appropriate drug treatment paired with the other chamber 4 h later for 30 min in the afternoon. Chamber pairings were counterbalanced. Twenty hours after the afternoon pairing, mice were placed in the CPP box with access to all chambers, and their behavior, recorded for 15 min, was analyzed for chamber preference. The preference index was calculated as the time spent in the drug-paired chamber minus the time spent in the vehicle-paired chamber.
Constructs, viral packaging, and stereotactic injection
AAV9-CaMKIIα-hM3Dq-mCherry (1.48 × 1013 vg/mL), AAV9-CaMKIIα-ChR2-mCherry (3.66 × 1013 vg/mL), AAV2-retro-hSyn-mCherry (7.99 × 1013 vg/mL), AAV9-hSyn-DIO-hM3Dq(Gq)-mCherry (8.44 × 1013 vg/mL), AAV9-hSyn-DIO-hM4Di(Gi)-mCherry (7.68 × 1013 vg/mL), AAV9-hSyn-DIO-mCherry (4.91 × 1013 vg/mL), and AAV2-retro-hSyn-cre-GFP (6.64 × 1013 vg/mL) were obtained from Vigene Biosciences (Shandong, China). For viral injection, mice were anesthetized with ketamine (100 mg/kg of body weight) and xylazine (8 mg/kg) by intraperitoneal injection and placed in a stereotactic frame. The coordinates of ACC injection were as follows: AP, +0.7 mm; ML, ±0.25 mm; DV, −1.70 mm (relative to bregma). The coordinates of MD injection were as follows: AP, −1.70 mm; ML, ±0.38 mm; DV, −3.60 mm (relative to bregma). For behavioral tests and immunostaining experiments, the mice received bilateral injections into the ACC (250 nl, per side) and the MD (100 nl, per side). For electrophysiological tests, the mice received unilateral injection into the right MD (100 nl). The viruses were injected using glass microelectrodes at a controlled rate (25 nl/min). The microelectrode was slowly retracted 10 min post-virus infusion. Experiments were started after at least 21 days for AAV after the injection. The injection sites were examined at the end of all the behavioral tests, and only data from animals with correct injections and correct cannula implanted were included. Brain slices of ACC were directly examined under a fluorescent microscope. The mechanical allodynia and CPP/CPA test were conducted in a double-blind manner. The experimenters were blinded to experimental groups.
Cannulation and microinjection
At least two weeks after virus injection, both control and test animals underwent a bilateral cannula implantation into the ACC. The cannula surgery and microinjection were performed as described previously49. Briefly, mice were anesthetized with isoflurane (1–3%, as needed, RWD, Shenzhen, China, R510-22) inhalation of 100% oxygen with a flow of 0.5 L/min delivered by facemask. The scalp was shaved and cleaned with iodine (Caoshanhu, Nanchang, China) and alcohol. The head of each mouse was fixed into a stereotaxic adapter mounted on a stereotaxic frame (RWD, Shenzhen, China, 68025), and eye ointment (Cisen, Jining, China) was applied to protect the eyes. An incision was made on the skull to expose the surface. A small hole was drilled above the ACC, and the dura was gently reflected. The double-lumen cannula was positioned at coordinates 0.7 mm anterior to the bregma, 0.0 mm lateral to the midline, and 1.20 mm ventral to the surface of the skull. Each sleeve is located 0.25 mm on either side of the midline. Mice were then left undisturbed for 3 to 7 days before undergoing behavioral testing.
For the microinjection, the mice were restrained in a plastic cone (RWD, Shenzhen, China, 68025). The dummy cannula was removed, and the microinjection cannula was inserted into the guide. An injection cannula was then inserted 0.5 mm below the guide. Clozapine (CNO, 5 μL, 0.1 ng/μl, MCE) was bilaterally administered at a rate of 0.1 μL/min using a syringe controlled by an infusion pump (Alcott, Shanghai, China, ALC-IP600). The volume produced was verified by observing the movement of the meniscus along calibrated polyethylene (PE10) tubing. After delivery to each side of the brain, the injection cannula was left in place for 2 min to prevent any solution from flowing back up the guide. 30 min after microinjection, the mechanical allodynia test or the CPP/CPA test was conducted.
Whole-cell patch-clamp recording
Coronal brain slices (300 µm) at the level of the ACC from mice after sham surgery or nerve injury were prepared. Then, slices were transferred to a submerged recovery chamber with oxygenated (95% O2 and 5% CO2) artificial cerebrospinal fluid (ACSF) containing (in mM) 124 NaCl, 2.5 KCl, 2 CaCl2, 1 MgSO4, 25 NaHCO3, 1 NaH2PO4, and 10 glucose at room temperature for at least 1 h. Experiments were performed in a recording chamber on the stage of a microscope equipped with infrared differential interference contrast optics for visualization. All recordings were made from L3 neurons located in the ACC contralateral to the nerve ligation surgery. Spontaneous and evoked excitatory postsynaptic currents (EPSCs) and inhibitory postsynaptic currents (IPSCs) were recorded from L3 neurons of the ACC with an Axon 700B amplifier.
α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR)-mediated EPSCs were induced by repetitive stimulation at 0.01 Hz, and neurons were voltage-clamped at –70 mV in the presence of AP5 (50 µM), and N-methyl-D-Aspartate receptor (NMDAR)-mediated currents were induced at + 40 mV in the presence of CNQX (20 µM). The recording micropipettes (3–5 MΩ) were filled with a solution containing (in mM) 115 CsMeSO4, 20 CsCl, 10 HEPES, 2.5 MgCl2, 4 Na-ATP, 0.4 Na2-GTP, 10 Na2phosphocreatine, 0.6 EGTA, 5 QX-314 (adjusted to pH 7.3 with CsOH). In these experiments, picrotoxin (PTX, 100 µM) was used to block γ-aminobutyric acid receptor-mediated inhibitory synaptic currents.
During recording the intrinsic property of neurons, the internal solution containing (in mM) 124 K-gluconate, 5 NaCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 MgATP, 0.1 Na3GTP, and 10 phosphocreatine disodium (adjusted to pH 7.2 with KOH). Biocytin (0.2%) was included in the pipette solution to label PV-positive neurons. The initial access resistance was 15–30 MΩ and was monitored throughout experiments. Data were discarded if the access resistance changed >15% during an experiment. Data were filtered at 1 kHz and digitized at 10 kHz.
Nissl staining
For the obtained frozen sections, rinsed the sections with distilled water for 2–5 min; added Nissl staining solution (Sangon Biotech, Cat# E607316) and incubated at room temperature for 10–30 min; washed twice with distilled water for 2 min each. Finally, samples were observed under microscopy.
Immunohistochemistry
Immunochemistry was referred to previous report49. Briefly, mice were deeply anesthetized, and then brains were removed, postfixed, and transferred to 30% sucrose in 0.1 M PB (pH 7.4) for 48–72 h. Coronal sections (25 μm) were cut on a cryostat (Leica CM3050 S) and stored in stocking buffer (50% PBS, 30% glycol and 20% glycerol). After washing with PBS, the sections were incubated in blocking buffer containing 5% normal goat serum and 0.5% Triton X-100/PBS for 1 h at room temperature and then with primary antibodies in blocking buffer overnight at 4 °C on a horizontal shaker. The primary antibodies used were c-Fos (1:1000, Synaptic System), PV (1:1000, CST). After three washing with PBS, sections were incubated with either Alexa Fluor 488 or Alexa Fluor 647-conjugated secondary antibodies at room temperature for 1 h. After washing three times in PBS, sections were then mounted on coverslips using Fluoroshield mounting medium with DAPI (Abcam), with immunofluorescence assessed using a laser confocal microscope (FV1000, Olympus). Cell counting was carried out manually using Fiji (NIH).
After electrophysiological recordings, slices containing biocytin-filled ACC PV neurons were fixed overnight in a cold solution containing 4% paraformaldehyde (PFA). The slices were then washed with PBS 3 times (5 min each time) at room temperature. Slices were then collected in 1% Triton X-100 (Sigma)/PBS at 4 °C for 24 h and then washed 3 times. The slices were then incubated in cy3-conjucted streptavidin diluted in 1:1000 in 0.3% Triton X-100/PBS at 4 °C for 24 h. After a final series of 3 washes with PBS, mount the slices on glass microscope slides.
Statistics and reproducibility
Before starting experiments, based on our previous behavioral studies, we estimated the sample size by using G*Power software. Offline analysis of whole-cell patch-clamp data was performed using Clampfit 10. 6 and MiniAnalysis 6.03. GraphPad 8.0 was used to plot and fit the data. An evaluator blinded to the group assignments performed the assessment and analyzed the data. Animals with correct virus injection and cannula placement were included in the analysis. Statistical tests were determined post-experiment. Statistical comparisons were made using the unpaired two-sample t-test, One-way or two-way ANOVA (the Student-Newman-Keuls test was used for post hoc comparison). All data were presented as the mean ± standard error of the mean (SEM). In all cases, n.s, not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Numerical source data for all figures are provided in Supplementary Data 1. Additional data supporting the findings of this study can be obtained from the lead corresponding author upon reasonable request.
References
Zhao, Y. et al. Anterior cingulate cortex in addiction: new insights for neuromodulation. Neuromodulation https://doi.org/10.1111/ner.13291 (2020).
Seamans, J. K. & Floresco, S. B. Event-based control of autonomic and emotional states by the anterior cingulate cortex. Neurosci. Biobehav. Rev. 133, 104503 (2022).
Kolling, N., Behrens, T. E. J., Wittmann, M. K. & Rushworth, M. F. S. Multiple signals in anterior cingulate cortex. Curr. Opin. Neurobiol. 37, 36–43 (2016).
Bliss, T. V. P., Collingridge, G. L., Kaang, B. K. & Zhuo, M. Synaptic plasticity in the anterior cingulate cortex in acute and chronic pain. Nat. Rev. Neurosci. 17, 485–496 (2016).
Xiao, X., Ding, M. & Zhang, Y. Q. Role of the anterior cingulate cortex in translational pain research. Neurosci. Bull. 37, 405–422 (2021).
Lindsay, N. M., Chen, C., Gilam, G., Mackey, S. & Scherrer, G. Brain circuits for pain and its treatment. Sci. Transl. Med. https://doi.org/10.1126/scitranslmed.abj7360 (2021).
Wang, Y., Liu, M., Wang, J. & Luo, J. Restoration of cingulate long-term depression by enhancing non-apoptotic caspase 3 alleviates peripheral pain hypersensitivity. Cell Rep. 33, 108369 (2020).
Li, X. Y. et al. Alleviating neuropathic pain hypersensitivity by inhibiting PKMζ in the anterior cingulate cortex. Science 330, 1400–1404 (2010).
Wang, J.-H., Wu, C., Lian, Y.-N., Liu, L. & Li, X.-Y. Targeting long-term depression of excitatory synaptic transmission for the treatment of neuropathic pain. FEBS J. 289, 7334–7342 (2022).
Metz, A. E., Yau, H.-J., Centeno, M. V., Apkarian, A. V. & Martina, M. Morphological and functional reorganization of rat medial prefrontal cortex in neuropathic pain. Proc. Natl Acad. Sci. USA 106, 2423–2428 (2009).
Kelly, C. J. & Martina, M. Circuit-selective properties of glutamatergic inputs to the rat prelimbic cortex and their alterations in neuropathic pain. Brain Struct. Funct. 223, 2627–2639 (2018).
Mitrić, M. et al. Layer- and subregion-specific electrophysiological and morphological changes of the medial prefrontal cortex in a mouse model of neuropathic pain. Sci. Rep. 9, 9479 (2019).
Tyrtyshnaia, A. A., Manzhulo, I. V., Konovalova, S. P. & Zagliadkina, A. A. Neuropathic pain causes a decrease in the dendritic tree complexity of hippocampal CA3 pyramidal neurons. Cells Tissues Organs 208, 89–100 (2019).
Li, X.-H. et al. Activation of the glutamatergic cingulate cortical-cortical connection facilitates pain in adult mice. Commun. Biol. 6, 1247 (2023).
Luo, L. Architectures of neuronal circuits. Science 373, eabg7285 (2021).
Zhang, Z. et al. Role of prelimbic GABAergic circuits in sensory and emotional aspects of neuropathic pain. Cell Rep. 12, 752–759 (2015).
Zhang, L. et al. Hyperactivated PTP1B phosphatase in parvalbumin neurons alters anterior cingulate inhibitory circuits and induces autism-like behaviors. Nat. Commun. 11, 1–16 (2020).
Meda, K. S. et al. Microcircuit mechanisms through which mediodorsal thalamic input to anterior cingulate cortex exacerbates pain-related. Neuron 102, 944–959.e3 (2019).
Wang, Y. Q. et al. Neuropathic pain generates silent synapses in thalamic projection to anterior cingulate cortex. Pain 162, 1322–1333 (2021).
Kaneko, M. et al. The role of N-methyl-D-aspartate receptor subunits in the rat thalamic mediodorsal nucleus during central sensitization. Brain Res. 1371, 16–22 (2011).
Lusk, N., Meck, W. H. & Yin, H. H. Mediodorsal thalamus contributes to the timing of instrumental actions. J. Neurosci. 40, 6379–6388 (2020).
Yang, J. W., Shih, H. C. & Shyu, B. C. Intracortical circuits in rat anterior cingulate cortex are activated by nociceptive inputs mediated by medial thalamus. J. Neurophysiol. 96, 3409–3422 (2006).
Parnaudeau, S., Bolkan, S. S. & Kellendonk, C. The mediodorsal thalamus: an essential partner of the prefrontal cortex for cognition. Biol. Psychiatry 83, 648–656 (2018).
Kuramoto, E. et al. Individual mediodorsal thalamic neurons project to multiple areas of the rat prefrontal cortex: a single neuron-tracing study using virus vectors. J. Comp. Neurol. 525, 166–185 (2017).
Delevich, K., Tucciarone, J., Huang, Z. J. & Li, B. The mediodorsal thalamus drives feedforward inhibition in the anterior cingulate cortex via parvalbumin interneurons. J. Neurosci. 35, 5743–5753 (2015).
Xue, M. et al. Mapping thalamic-anterior cingulate monosynaptic inputs in adult mice. Mol. Pain https://doi.org/10.1177/17448069221087034 (2022).
Meda, K. S. et al. Microcircuit mechanisms through which mediodorsal thalamic input to anterior cingulate cortex exacerbates pain-related aversion. Neuron 102, 944–959.e3 (2019).
Porter, J. T., Johnson, C. K. & Agmon, A. Diverse types of interneurons generate thalamus-evoked feedforward inhibition in the mouse barrel cortex. J. Neurosci. 21, 2699–2710 (2001).
Chen, T. et al. Top-down descending facilitation of spinal sensory excitatory transmission from the anterior cingulate cortex. Nat. Commun. 9, 1886 (2018).
Xu, H. et al. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J. Neurosci. 28, 7445–7453 (2008).
Ji, X. Y. et al. Thalamocortical innervation pattern in mouse auditory and visual cortex: laminar and cell-type specificity. Cereb. Cortex 26, 2612–2625 (2016).
Wang, Y. J. et al. Cingulate alpha-2A adrenoceptors mediate the effects of clonidine on spontaneous pain induced by peripheral nerve injury. Front. Mol. Neurosci. https://doi.org/10.3389/fnmol.2017.00289 (2017).
Zhang, Y. et al. Timing mechanisms underlying gate control by feedforward inhibition. Neuron 99, 941–955.e4 (2018).
Li, J. & Baccei, M. L. Neonatal injury alters sensory input and synaptic plasticity in GABAergic interneurons of the adult mouse dorsal horn. J. Neurosci. 39, 7815–7825 (2019).
Huang, J. et al. A neuronal circuit for activating descending modulation of neuropathic pain. Nat. Neurosci. 22, 1659–1668 (2019).
Cao, X.-Y. et al. Characterization of intrinsic properties of cingulate pyramidal neurons in adult mice after nerve injury. Mol. Pain. 5, 73 (2009).
Wang, Q. et al. Synaptic dynamics of the feed-forward inhibitory circuitry gating mechanical allodynia in mice. Anesthesiology 132, 1212–1228 (2020).
Perry, B. A. L., Lomi, E. & Mitchell, A. S. Thalamocortical interactions in cognition and disease: the mediodorsal and anterior thalamic nuclei. Neurosci. Biobehav. Rev. 130, 162–177 (2021).
Bolkan, S. S. et al. Thalamic projections sustain prefrontal activity during working memory maintenance. Nat. Neurosci. 20, 987–996 (2017).
Williams, L. E. & Holtmaat, A. Higher-order thalamocortical inputs gate synaptic long-term potentiation via disinhibition. Neuron 101, 91–102.e4 (2019).
Ferguson, B. & Delevich, K. Mediodorsal thalamus and prefrontal cortex: specialized partners in cognitive control. J. Neurosci. 40, 5515–5517 (2020).
Mitchell, A. S. The mediodorsal thalamus as a higher order thalamic relay nucleus important for learning and decision-making. Neurosci. Biobehav. Rev. 54, 76–88 (2015).
Pergola, G. et al. The regulatory role of the human mediodorsal thalamus. Trends Cogn. Sci. 22, 1011–1025 (2018).
Iwabuchi, S. J. et al. Medio-dorsal thalamic dysconnectivity in chronic knee pain: a possible mechanism for negative affect and pain comorbidity. Eur. J. Neurosci. 57, 373–387 (2023).
Knoedler, J. R. et al. A functional cellular framework for sex and estrous cycle-dependent gene expression and behavior. Cell 185, 654–671.e22 (2022).
Lian, Y. N. et al. Prediction of cell-cell communication patterns of dorsal root ganglion cells: single-cell RNA sequencing data analysis. Neural Regen. Res.19, 1367–1374 (2024).
Chaplan, S. R., Bach, F. W., Pogrel, J. W., Chung, J. M. & Yaksh, T. L. Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63 (1994).
Zuo, Z. X. et al. Huperzine A alleviates mechanical allodynia but not spontaneous pain via muscarinic acetylcholine receptors in mice. Neural Plast. https://doi.org/10.1155/2015/453170 (2015).
Wang, J. H. et al. Single-cell RNA sequencing uncovers the cell type-dependent transcriptomic changes in the retrosplenial cortex after peripheral nerve injury. Cell Rep. https://doi.org/10.1016/j.celrep.2023.113551 (2023).
Acknowledgements
We are thankful to Prof. Haohong Li for helpful suggestions on the manuscript. We thank Dr Sanhua Fang at the Core Facilities of Zhejiang University Institute of Neuroscience for technical assistance. This study was supported by the National Natural Science Foundation of China (32271042, 81571068, 31871062, 81801102, and 31771157), Youth Program of National Natural Science Foundation of China (No. 81801279), the Fundamental Research Funds for the Central Universities, the MOE Frontier Science Center for Brain Science & Brain-Machine Integration, Zhejiang University.
Author information
Authors and Affiliations
Contributions
Y.N.L. performed and analyzed the data for behavior tests and electrophysiology experiments. X.W.C. performed virus injection and behavior test; C.W, C.Y.P., and L.L. performed Nissl’s staining, and microscopy and analyzed the data; C.Z. and X.Y.L. designed the experiments and approved the draft; X.Y.L. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Thiago Cunha and Joao Valente [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lian, YN., Cao, XW., Wu, C. et al. Deconstruction the feedforward inhibition changes in the layer III of anterior cingulate cortex after peripheral nerve injury. Commun Biol 7, 1237 (2024). https://doi.org/10.1038/s42003-024-06849-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06849-4
