
Abstract
Colonizing in the gastrointestinal tract, Escherichia coli confronts diverse acidic challenges and evolves intricate acid resistance strategies for its survival. The lysine-mediated decarboxylation (Cad) system, featuring lysine decarboxylase CadA, lysine/cadaverine antiporter CadB, and transcriptional activator CadC, plays a crucial role in E. coli’s adaptation to moderate acidic stress. While the activation of the one-component system CadC and subsequent upregulation of cadBA operon in response to acid and lysine presence have been proposed, the molecular mechanisms governing the transition of CadC from an inactive to an active state remain elusive. Under neutral conditions, CadC is inhibited by forming a complex with lysine-specific permease LysP, stabilized in this inactive state by a disulfide bond. Our study unveils that, in an acidic environment, the disulfide bond in CadC is reduced by the disulfide bond isomerase DsbC, exposing R184 to periplasmic proteases, namely DegQ and DegP. Cleavage at R184 by DegQ and DegP generates an active N-terminal DNA-binding domain of CadC, which binds to the cadBA promoter, resulting in the upregulated transcription of the cadA and cadB genes. Upon activation, CadA decarboxylates lysine, producing cadaverine, subsequently transported extracellularly by CadB. We propose that accumulating cadaverine gradually binds to the CadC pH-sensing domain, preventing cleavage and activation of CadC as a feedback mechanism. The identification of DegP, DegQ, and DsbC completes a comprehensive roadmap for the activation and regulation of the Cad system in response to moderate acidic stress in E. coli.
Similar content being viewed by others

Introduction
The gastrointestinal tract represents a highly diverse and dynamic environment characterized by varying pH levels, contingent on factors such as luminal conditions, digestive states, and individuals’ age and health status1,2. Escherichia coli traverses this complex environment, encountering different acids produced by the host during digestion or by other intestinal bacteria. Within the small intestine, metabolic byproducts, including organic acids such as formic acid and propionic acid, contribute to acidic pressure on E. coli3,4,5,6. To thrive in this milieu, E. coli has evolved intricate acid tolerance mechanisms, which may aid the bacteria in adapting to the varying acidic conditions7,8,9.
Exposure to a non-lethal acidic environment prompts E. coli to employ multiple resistance mechanisms, including a membrane structure-dependent acid resistance barrier8,10, RpoS mediates acid resistance 1 (AR1) and amino acid-dependent acid resistance 2–5 (AR2-AR5)11,12. Specifically, the AR2-AR3 systems, relying on glutamate or arginine decarboxylation, are primarily utilized for survival in strongly acidic environments (pH 1.5-–4.0), while the AR4-AR5 systems, involving lysine or ornithine metabolism, aid E. coli in thriving within moderate acidic environments (pH 4.0–6.0). The amalgamation of multiple acid resistance systems equips E. coli to endure a broad spectrum of acidic conditions13,14,15,16,17,18,19.
The lysine-mediated decarboxylation (Cad) system serves as a pivotal mechanism for acid tolerance in organisms, comprising lysine decarboxylase CadA, lysine/cadaverine antiporter CadB, and pH-responsive transcriptional activator CadC. CadC, a membrane-integrated one-component system protein, features a cytoplasmic DNA-binding N-terminal domain, a transmembrane region, and a periplasmic pH-sensing C-terminal domain20. Under neutral conditions, CadC is inhibited by the lysine-specific permease LysP21. In acidic conditions with lysine presence, LysP imports lysine, alleviating CadC inhibition. This activation allows CadC to bind to the cadBA promoter, initiating the transcription of cadA and cadB. CadA contributes to pH homeostasis by consuming a proton and lysine, yielding the polyamine cadaverine, subsequently excreted from the cell via the lysine/cadaverine antiporter CadB. To maintain feedback inhibition, excessive cadaverine binds to the CadC periplasmic pH-sensing C-terminal domain, restraining its activity22.
Despite the proposed acid resistance pathway of Cad systems, the molecular mechanisms driving the transition of one-component CadC from an inactive to an active state remain unclear. At pH 7.6, a disulfide bond present in the periplasmic domain of E. coli CadC stabilizes its inactive state, reduced at pH 5.8 with unknown disulfide reductases, leading to CadC activation21. In Salmonella, CadC activation depends on proteolytic cleavage of its periplasmic domain during acid and lysine induction, with unknown periplasmic proteases performing these functions23. While CadA and CadB amino acid sequences share high homology between E. coli and Salmonella (98%), CadC exhibits lower homology (58.4%), suggesting the potential for different activation mechanisms. Therefore, the proteolytic cleavage of CadC in E. coli requires further verification24.
In this study, we validated the necessity of hydrolytic cleavage of periplasmic structural domain proteins and disulfide bond reduction during CadC activation in E. coli. Subsequently, we identified the periplasmic proteases DegP and DegQ, along with the disulfide bond isomerase DsbC, as crucial components for protein hydrolysis and disulfide bond reduction in the regulation of the Cad system, respectively.
Results
Activation of the lysine decarboxylation system relies on the proteolytic cleavage of CadC
The mRNA levels of cadC gene in E. coli MG1655 were assessed through qRT-PCR under six distinct conditions, encompassing pH 5.8, pH 7.0, pH 8.0 combined with or without 10 mM lysine. Consistent with previous findings24, cadC exhibited significant up-regulation within 0.5 hours at pH 5.8 in the absence of lysine. The presence of lysine induced a higher expression of cadC (9.46-fold for pH 5.8 and 32.76-fold for pH 5.8 with 10 mM lysine, Fig. 1a, b).

Transcription of cadC in E. coli MG1655 at pH 5.8, 7.0 and 8.0 without (a) or with (b) lysine, detected by qRT-PCR. Data are mean ± SD of n =3 biologically independent replicates. Transcription of cadA (c) and cadB (d) in E. coli MG1655, ΔcadC and ΔcadC-pCL1920cadC strains at pH 5.8 in the presence of lysine, detected by qRT-PCR. Data are mean ± SD of n = 3 biologically independent replicates. Different letters denote significant disparities among various samples at the same detection time, based on Duncan’s test (p < 0.05, n = 3). The protein level of CadC in E. coli MG1655 under different pH conditions. Corresponding Western blot results (e) and their gray values (f) are displayed. The full-length CadC (57.8 kDa) and its suspected cleavage fragment are marked in the diagram. DnaK serves as a loading control for calibrating the grayscale values of the corresponding samples.
To elucidate the regulation of cadBA operon by cadC, we quantified and compared the expression levels of cadA and cadB in wild-type E. coli (MG1655-pCL1920), cadC deletion strain (MG1655ΔcadC-pCL1920) and cadC complement strain (MG1655ΔcadC-pCL1920cadC) using qRT-PCR. The results indicated a tight correlation between cadC and cadBA operon. At pH 5.8 with the presence of 10 mM lysine, not only did the expression of cadC significantly increase, but also the cadA gene expression exhibited 145.3 and 183.6-fold up-regulation in wild-type and cadC complement strains, respectively, within 1 hour. Meanwhile, cadB transcription was up-regulated 167.4 and 153.0-fold in 0.5 hours, respectively. In contrast, the cadBA operon did not show a detectable response to the acidic stress in cadC deletion strain, indicating a dependence on cadC (Fig. 1c, d).
The alterations of CadC at the protein level were also measured using western blot analysis under different conditions. A smaller uniform band resulting from proteolytic cleavage of CadC was observed, which was thicker under acidic conditions, while the alkaline condition led to the weakest band (Fig. 1e, f). Notably, the full-length CadC band decreased gradually with time under acidic pH conditions, implying that proteolytic cleavage at specific sites leads to progressive degradation of the CadC protein. This result suggests that proteolytic cleavage may also be necessary for the activation of CadC, raising the question of which protease executes the cleavage.
DegP and DegQ are the crucial proteases in CadC cleavage for activation
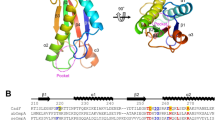
To figure out how CadC is activated, we constructed the expression plasmid of pET29bcadC (C-terminal His tag) for CadC expression in E. coli BL21. SDS-PAGE of purified CadC revealed two bands, with the approximately 55 kDa band representing the full-length CadC and the 35 kDa band corresponding to the C-terminal pH-sensing domain post CadC hydrolysis (Fig. 2a), which is consistent with the western blot analysis (in Fig. 1e, the supposed N-terminal DNA-binding domain fragment was about 20 kDa). N-terminal sequencing of the smaller protein fragments revealed a sequence of R-LPMSKS, indicating that CadC was cleaved on the carboxyl terminal of R184 (Fig. 2b and Figure. S1). The full-length CadC contains a pH-sensing periplasmic domain, a single transmembrane area (TM), and an intracellular DNA binding domain (DBD). R184 is located between the TM and pH-sensing domains, in close proximity to the TM region, which suggested the involvement of a periplasmic protease in CadC cleavage (Fig. 2b).

a Formaldehyde cross-linking results during CadC purification. b Pattern diagram illustrating the structural domains of CadC. The N-terminal (1-107) represents the DNA-binding structural domain, 160-183 is the transmembrane structural domain, and 189-512 is the pH-signal sensing structural domain. The arrow indicates the enzyme cleavage site, with N-terminal sequencing revealing R-LPMSKS, pinpointing the cleavage site between 184R-185L. c, Lysine decarboxylation (LDC) assay assessing the decarboxylation ability of MG1655 and its mutant strains. Transcription levels of cadA (d) and cadB (e) genes in MG1655 and its mutant strains post-acid and lysine stimulation, detected by qRT-PCR. Data are mean ± SD of n = 3 biologically independent replicates. Various letters signify significant differences among samples at the same detection time, as per Duncan’s test (p < 0.05, n = 3). f, g Western blot and corresponding gray scale results of CadC in MG1655, ΔdegP: degQ, ΔdegP, ΔdegQ. Samples were stimulated under pH 5.8 and lysine conditions over 0-1 hours. DnaK served as a control for immunoblotting analysis of the total protein. The results used for MG1655 strain in Fig. 2f are consistent with Fig. 1e.
To identify the protease responsible for CadC cleavage, we employed formaldehyde cross-linking during purification to capture interacting proteins for mass spectrometry analysis. Among the 14 proteins detected, only two, i.e., DegP and DegQ, were identified by mass spectrometry as proteases, designating them as candidate proteins (Fig. 2a and Table S1).
DegP and DegQ are conserved periplasmic bifunctional proteases in Gram-negative bacteria, known for their role in refolding or degrading misfolded proteins and contributing to bacterial stress resistance25,26,27,28. Both proteases contain a catalytic domain with a chymotrypsin-like fold and one or two carboxyl-terminal PDZ domains, which are well-characterized protein-protein interaction modules29,30. Although substrates such as lysozyme and casein have been reported for DegP and DegQ31,32, their in vivo substrates still lack clarity33. Furthermore, the association of DegP and DegQ family proteases with bacterial acid resistance has been suggested34,35, yet the exact mechanism remains elusive.
To assess the role of DegP and DegQ in lysine decarboxylation metabolism and the significance of cleavage at CadC R184, various strains were constructed, including ΔcadC, ΔcadC-pCL1920cadC, ΔcadC-pCL1920cadC R184Q, ΔdegP, ΔdegQ, and ΔdegP:degQ. These strains underwent lysine decarboxylation (LDC) assays to examine their respective capabilities. The LDC broth medium containing the cadC mutant exhibited a yellow color, indicating the loss of lysine decarboxylation ability. However, supplementary expression of full-length CadC, but not the R184Q mutant, resulted in a purple-colored medium, signifying the restoration of lysine decarboxylation. This observation underscores the necessity of proteolytic cleavage at R184 for CadC activation, essential for lysine decarboxylation (Fig. 2c). Intriguingly, both ΔdegP and ΔdegQ strains displayed impaired lysine decarboxylation ability, and the degP and degQ double mutation resulted in a complete loss of this ability (Fig. 2c), which suggests that DegP and DegQ collaborate in the lysine decarboxylation.
Furthermore, the activation of cadA and cadB genes over time in MG1655, ΔdegP, ΔdegQ, ΔdegP:degQ, and cadC strains was assessed in response to the acidic and lysine conditions using qRT-PCR. In the ΔdegP strains, cadA and cadB were moderately up-regulated. In contrast, in the ΔdegQ strain, both cadA and cadB genes were downregulated, which seems to imply that the DegQ protein plays a major role in the activation of cadBA. qRT-PCR was then used to examined the expression of the degQ gene in ΔdegP strain and degP gene in ΔdegQ strain. The results indicated that degQ was significantly upregulated but degP only slightly increased (Figure. S3). This was consistent with the cadBA operon transcription results. Intriguingly, only cadA expression was severely reduced. As for cadB, it was reduced to half of the wild-type level (Fig. 2e). The expression of cadA and cadB seems to be decoupled, which is reasonable, since CadB is an anti-transporter that may be needed to maintain cellular homeostasis. Additionally, the complementary expression of CadCR184Q mutant in MG1655 ΔcadC strain failed to restore the normal response to the acidic stress (Fig. 2d, e).
To delve deeper into the proteolytic cleavage of CadC under acidic stress, western blot analysis of CadC was conducted for ΔdegP:degQ, ΔdegP, and ΔdegQ strains. Single knockout of either degP or degQ moderately impaired CadC cleavage, but the cleavage was significantly compromised in the degP and degQ double mutant strain (Fig. 2f, g). These findings strongly indicate that DegP and DegQ play a crucial role in lysine decarboxylation by specifically targeting CadC for proteolytic cleavage.
CadC is degraded by DegP and DegQ in vitro
To elucidate the specific cleavage mechanism of CadC by DegP and DegQ, purified CadC, CadC R184Q, DegP, and DegQ were mixed in vitro to explore their interactions. Clear degradation of CadC by DegP or DegQ was observed within 3 hours, while CadC R184Q exhibited high resistance to both DegP and DegQ. Notably, the presence of 2 mM cadaverine significantly inhibited cleavage of CadC by DegP and DegQ (Fig. 3a).

a SDS-PAGE analysis illustrating the cleavage activity of DegP and DegQ on substrate CadC, comparing the effects on CadC R184Q and CadC with cadaverine. CadC fragment indicate the products post-cleavage, with CadC1-184 used for size calibration. Size-exclusion chromatography analysis of complexes formed between CadC and DegP (b) or DegQ (c). d The localization of CadC184 in cells was determined by subcellular fractionation experiments.
Results of size-exclusion chromatography demonstrated a pronounced forward shift in peak upon the addition of CadC for both DegP and DegQ, indicating successful protease assembly in complex with CadC (Fig. 3b, c). A subcellular fractionation assay was then performed to identified the localization of CadC1-184 in cells. The result indicated that CadC1-184 localized to the membrane23,32 (Fig. 3d).
Proteolytic cleavage of CadC results in an N-terminal fragment with enhanced cadBA promoter binding
In its role as a transcription factor, CadC is anticipated to possess DNA-binding capability, with stronger binding expected upon activation. To investigate this, the binding affinities of both full-length CadC and the CadC1-184 fragment to the cadBA promoter were assessed through electrophoretic mobility shift assay (EMSA). Complex formation occurred at a protein: DNA molar ratio of 10:1 for full-length CadC and 1:1 for CadC1-184, indicating an augmented binding affinity resulting from proteolytic cleavage (Fig. 4a, b). The addition of Zn2+ further strengthened the binding affinity (Fig. 4c, d).

EMSA results illustrate the interaction between the cadBA promoter and CadC1-184 fragment (a), full-length CadC (b), CadC1-184 with Zn2+ (c), and CadC with Zn2+ (d). FP assay assessing the binding affinity of cadBA promoter for CadC1-184 fragment and full-length CadC in the presence of Zn2+ (f) or absence of Zn2+ (e). Data are mean ± SD of n = 3 biologically independent replicates.
Quantitative measurements of these interactions were conducted using the fluorescence polarization (FP) method. The equilibrium dissociation constants (KD) were determined as 28.59 ± 3.9 μM for full-length CadC and 7.74 ± 0.27 for CadC1-184. In the presence of Zn2+, the KD values were 7.31 ± 0.26 μM for full-length CadC and 2.33 ± 0.11 μM for CadC1-184 (Fig. 4e, f). These findings suggest that proteolytic cleavage enhances the ability of CadC N-terminal fragments to tightly bind to the cadBA promoter, a crucial step in the regulatory mechanism.
DsbC activates the lysine decarboxylation metabolism by reducing the disulfide bond in CadC
Previous studies have emphasized the necessity of breaking the disulfide bond stabilizing the inactive state of CadC for its activation21. In contrast, LysP inhibits CadC through intramembrane and periplasmic contacts, crucial for stabilizing the inactive state of CadC21,36. This section delves into the intricate relationship between CadC hydrolytic activation, disulfide bond reduction, and LysP dissociation.
Six periplasmic disulfide isomerase enzymes, i.e., DsbA, DsbB, DsbC, DsbD, DsbE, and DsbG, have been identified in E. coli37. DsbA oxidizes sulfhydrygroups to form disulfide bonds37, while DsbB is responsible for the reoxidation of DsbA37. DsbC and DsbG reduce the abnormal disulfide bonds imported by DsbA37, and DsbD aids in the reduction of DsbC and DsbG37,38. In our pursuit of identifying enzymes that reduce disulfide bonds in CadC, our primary focus was on DsbG and DsbC, which are prominent enzymes in this context. To investigate, MG1655ΔdsbC, MG1655ΔdsbG, and MG1655ΔlysP strains were constructed, and their cadA and cadB expression in response to the acidic stress was assessed by qRT-PCR. cadA and cadB were moderately up-regulated in the dsbC mutant strain, the cadBA operon cannot be activated, suggesting that DsbC is responsible for reducing the disulfide bonds that stabilize the inactive CadC. Conversely, the dsbG mutant exhibited no impairment, suggesting a specific role for DsbC. Notably, CadC was unable to activate the cadBA operon in the ΔlysP strain. (Fig. 5a, b). LDC assays further confirmed the impaired lysine decarboxylation ability in the ΔdsbC and ΔlysP strains (Fig. 5c), emphasizing the role of DsbC and LysP in the decarboxylation process.

qRT-PCR analysis of cadA (a) and cadB (b) gene expression in MG1655ΔdsbC, ΔdsbG, and ΔlysP strains following acid and lysine stimulation. Data are mean ± SD of n = 3 biologically independent replicates. Different letters denote significant differences among various samples at the same detection time, according to Duncan’s test (p < 0.05, n = 3). c LDC assay was used to assess lysine decarboxylation capacity in MG1655, Δ dsbC, Δ dsbG, and Δ lysP strains. d qRT-PCR analysis of cadA and cadB gene expression in MG1655ΔdsbC, MG1655ΔdsbC-pcadC C208S, MG1655ΔdsbC-pcadC C272S, and MG1655ΔdsbC-pcadC C172S. Data are mean ± SD of n = 3 biologically independent replicates. e, f Western blot assay examining the protein levels of CadC in MG1655ΔdsbG, MG1655ΔdsbC, and MG1655ΔlysP strains. DnaK served as a control for immunoblotting analysis of total protein. g, qRT-PCR was used to detect the expression of cadBA gene in mutant strains MG1655ΔlysP: cadC-pcadC C208S and MG1655ΔlysP: cadC-pcadC C272S. Data are mean ± SD of n = 3 biologically independent replicates. h Western blot assay examining the protein levels of CadC in MG1655ΔlysP: cadC-pcadC C208S, MG1655ΔlysP: cadC-pcadC C272S strain. DnaK served as a control for immunoblotting analysis of total protein. i A spot assay on low pH agar is performed to evaluate the survival/growth of MG1655, MG1655ΔcadC-pcadC C208S, MG1655ΔcadC-pcadC C272S, MG1655ΔcadC, MG1655ΔcadC-pcadC R184Q, MG1655ΔcadC-pcadC; MG1655ΔdegP: degQ, MG1655ΔdegP, MG1655ΔdegQ, MG1655ΔlysP, MG1655ΔdsbC; MG1655ΔlysP: cadC-pcadC C208S, MG1655ΔlysP: cadC-pcadC C272S strains.
Wild-type CadC contains three cysteines, and previous research identified a disulfide bond between Cys208 and Cys272 in CadC21. Mutants of CadC C208S, CadC C272S, and CadC C172S were constructed. qRT-PCR results demonstrated that the activation of cadBA in CadC C208S and CadC C272S mutants is independent of DsbC under acidic conditions (Fig. 5d), confirming that DsbC catalyzes the reduction of the disulfide bond between cysteine 208 and 272 in CadC.
The proteolytic cleavage of CadC was examined in dsbG, dsbC, and lysP mutant strains (Figs. 5e, f). Western blot results unequivocally indicated that CadC almost did not undergo specific proteolytic cleavage in the dsbC mutant strain and was weakly cleaved in the lysP mutant strain. Compared with the wild-type protein, the degradation of CadC C208S and CadC C272S mutants was more significant and independent of external stimuli in ΔlysP:cadC strain (Fig. 5g). Meanwhile, the results of qRT-PCR showed in Fig. 5h that cadA and cadB genes were activated in ΔlysP:cadC -pcadC C208S and ΔlysP:cadC -pcadC C272S strains. We can speculate that during the absence of LysP, the intracellular lysine is too low to induce a conformational change in CadC, thus blocking its disulfide bond reduction. This suggests that cleavage-dependent CadC activation is also dependent on the presence of the LysP protein and the reduction of the disulfide bond.
Finally, ATR assay (a spot assay on low pH agar plates) was performed to evaluate the survival/growth of strains including MG1655, MG1655ΔcadC-pcadC C208S, MG1655ΔcadC-pcadCC272S, MG1655ΔcadC, MG1655ΔcadC-pcadCR184Q, MG1655ΔcadC-pcadC; MG1655ΔdegP: degQ, MG1655ΔdegP, MG1655ΔdegQ, MG1655ΔlysP, MG1655ΔdsbC; MG1655ΔlysP: cadC-pcadC C208S, MG1655ΔlysP: cadC-pcadC C272S. The results showed that mutant strains that lost the ability to reduce the disulfide bond all exhibited significant lower survival/growth, and that restoration of the ability to reduce the disulfide bond by complementary expression of the corresponding genes or cadC with cysteine mutation enabled the mutant strains to grow almost as well as the wild type strains (Fig. 5i).
Discussion
E. coli employs four distinct amino acid decarboxylase acid resistance (AR2-AR5) systems to enhance acid tolerance, namely glutamic acid, arginine, lysine, and ornithine-dependent acid resistance (GDAR, ADAR, LDAR, and ODAR, respectively) systems. Each AR system consists of two core components: amino acid decarboxylase and antiporter, along with various transcriptional regulatory factors16,39. These regulatory factors are predominantly located in the cytoplasm, governing the expression of their respective AR systems by detecting intracellular pH reductions7,40,41. Notably, the LDAR system undergoes direct regulation by the transmembrane transcription regulators CadC, allowing it to sense pH changes in the periplasmic space and promptly respond to environmental signals39. Consequently, LDAR activation occurs in a weak acid environment, while the GDAR and ADAR systems are activated in response to intracellular pH reductions induced by a strong acid environment. While the response to moderate acidic environments is underway, the activation mechanism of the ODAR system remains undisclosed16. In addition to E. coli, the LDAR system plays a crucial role in conferring weak acid tolerance in various bacteria16. Under neutral conditions, CadC forms a complex with LysP, maintaining an inhibitory state. It has been reported that the activation of CadC and, consequently, the lysine decarboxylation system necessitates both the proteolytic cleavage of CadC and the reduction of the disulfide bond stabilizing CadC in its inhibited state36. However, the identification of proteins responsible for these critical processes has long been elusive.
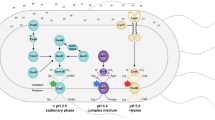
In this investigation, we have identified three proteins, i.e., DegP, DegQ, and DsbC, as integral components in the lysine decarboxylation system of E. coli, filling critical gaps in our understanding. Based on our discoveries, we propose a comprehensive model outlining the activation pathway of the lysine decarboxylation system in E. coli (Fig. 6). Under conditions of acidic pH within a lysine-rich environment, both CadC and LysP undergo a conformational change. This initial alteration exposes the disulfide bond, which stabilizes the inhibited CadC, allowing for its subsequent reduction by the disulfide bond isomerase DsbC. The reduction of the disulfide bond and the subsequent dissociation of CadC-LysP complex expose CadC residue R184. This exposed residue becomes susceptible to cleavage by the periplasmic protease DegP and DegQ, resulting in the breaking of the peptide bond between R184 and L185. Thus, the activated N-terminal DNA-binding domain (CadC1-184) binds to the promoter of the cadBA operon to initiate transcription of cadB and cadA. This activation marks the commencement of lysine decarboxylation and the reverse transport of cadaverine and lysine. Importantly, this process leads to the consumption of protons within the cytoplasm, ultimately alleviating the acidic stress. Over time, an increase in pH coupled with a deficiency of lysine results in the gradual accumulation of cadaverine in the periplasmic space. This accumulation induces the binding of cadaverine to the CadC pH-sensing domain, creating a feedback mechanism that prevents CadC’s cleavage and subsequent activation by DegP and DegQ. Furthermore, CadC is a multifunctional protein with a scope extending beyond the coordination of the lysine decarboxylation system. Previous studies have demonstrated that CadC exerts an influence on the arginine decarboxylation system as well39. Recent data from our investigations suggest that CadC might play a role in all AR2-AR5 systems (Figure. S2). The precise molecular mechanisms underlying these interactions remain to be elucidated in future studies.

When the pH value is ≥ 7, CadC activity is inhibited by LysP. Conversely, when the pH is < 7 and lysine is present, LysP initiates lysine transport (I). This process concomitantly triggers the opening of the intramolecular disulfide bond in CadC by DsbC (II). The exposed cleavage site allows proteases DegP and DegQ to cleave CadC. This results in the generation of an active N-terminal DNA-binding domain, activating the expression of cadBA. Upon activation, CadA decarboxylates lysine to produce cadaverine, transported extracellularly by CadB. As cadaverine accumulates to a threshold, feedback regulation inhibits the enzymatic digestion activation of CadC, maintaining cellular homeostasis (III).
One-component signal transduction systems represent a predominant class of signaling pathways in bacteria and archaea, typically located in cytoplasm42,43. CadC, a unique transmembrane transcription regulator (TTR), belongs to the family of one-component transcription regulators and has been identified in various species, including Vibrio spp., Escherichia spp., Salmonella spp., and Yersinia spp20,43,44,45,46,47,48. The abundance of TTRs in gut microbiota species, exemplified by the Eggerthella genus possessing over 117 TTRs per genome43, highlights their prevalence. Despite this, the functional mechanisms of many TTRs remain inadequately studied. When comparing the advantages of TTRs to classical one- and two-component systems, a common question arises: why localize the response regulator and DNA-binding domain to the cytoplasmic membrane43 ? Our research suggests that membrane-integrated systems facilitate rapid signaling transmission between the intracellular and the external environments. Additionally, the presence of DegP, DegQ, DsbC, and LysP provides further confirmation that TTRs localized to the cytoplasmic membrane enable more stringent regulatory control over their regulons. These findings align with the previous speculation that the signaling of TTRs is tightly regulated43.
CadC1-184 in the activated state has been determined to be localized on the membrane. It has been known that membrane-bound transcription factors can bind to DNA in three ways. Firstly, the DNA-binding domain is proteolytically cleaved, thus released into cytoplasm and bind to the target promotor in the same manner as a regular cytoplasmic transcription factor32. Secondly, co-transcriptional translation and protein translocation (“transertion”49,50) tethers the DNA locus to the membrane, and places the protein in the vicinity of its binding site on the DNA. Thirdly, the activated one-component system diffuses within the membrane and captures the DNA sequence brought to the membrane by conformational fluctuations (diffusion/capture mechanism)32. Our dada confirmed that CadC1-184 remains as a membrane protein following disulfide bond reduction and protease degradation, which suggests CadC1-184 adopts the diffusion and trapping mechanism.
Certain TTRs, including ToxR, VtrA, VtrrA, PsaE, MyfE, TcpP, and YqeI, possess associated proteins that contribute to TTR-mediated signal transduction, either by inhibiting TTR proteolysis or promoting heterodimerization43. In our study, we identified that periplasmic proteases DegP and DegQ exert an enzymatic cleavage activating effect on CadC, which is consistent with the recent report that DegP and DegQ are critical factors for bacterial acid resistance. While it was previously hypothesized that these proteases function by degrading acid-induced misfolded proteins34,51, our findings demonstrate that, in addition to this role, DegP and DegQ can recognize specific substrates for proteolytic cleavage, initiating a cascade of signal transduction52. Considering the associations of DegP and DegQ with various physiological functions of bacteria, it is plausible that other periplasmic signaling proteins, aside from CadC, could also serve as their substrates. Identification of these proteins may not only enhance our understanding of periplasmic protease function but also facilitate the discovery of new targets for antimicrobial drugs.
Methods
Bacterial strains, plasmids and culture conditions
Bacterial strains used in this study are listed in Table S2. The gene-deficient mutants were obtained through gene knockout by using the λ-Red mediated recombinase system described previously53. Primers used for gene knockout are listed in Table S3. For the construction of MG1655ΔcadC, MG1655ΔdegP and MG1655ΔdegQ, MG1655ΔdegQ: degP, MG1655ΔdsbC, MG1655ΔdsbG strains, the corresponding primers in addition to template plasmid PR6K-fliB-genta-LoxM54 were used to obtain linear DNA fragments with gentamycin gene cassettes flanked by Lox sites and homologous arms55. The polymerase chain reaction (PCR) products were transformed into MG1655 cells containing the helper plasmid pTKRED by electroporation. Positive clones were confirmed by PCR test (Table S3 for primers), and the resistance gene was removed by pCM157.
The plasmids used in this study were constructed using the Gibson assembly method56 and are listed in Table S4. PCR was performed using Phusion high-fidelity DNA polymerase (New England Biolabs) and its corresponding primers to amplify the linearized plasmid vector and the target overexpressed genomic DNA fragment (Table S5). The reaction mixture containing the linearized plasmid, genomic DNA fragment and Gibson assembly45 (New England Biolabs) was incubated at 42 ◦C for 40 minutes. The ligated plasmids were transformed into E. coli DH5α. Positive clones were further verified by DNA sequencing. Finally, plasmid DNA was extracted with Plasmid Mini Kit (Omega) and stored at -20◦C for future use.
The CadC R184Q, CadC C172S, CadC C208S and CadC C272S mutant plasmid was constructed using the modified Quick-change method57. Partially overlapping primers (Table S5), in addition to template plasmid pET29bcadC or pCL1920cadC, were used to obtain their mutated linear DNA fragments through PCR. After transformation, E. coli DH5α repaired the breaks to yield the plasmid with the desired mutation.
MG1655 was cultured in LB medium (1 L: 10 g tryptophan, 5 g yeast extract and 10 g NaCl) at 37 ◦C 200 rpm. If necessary, antibiotics were added at the following concentrations: ampicillin 100 μg/mL, kanamycin 50 μg/mL, and spectinomycin 50 μg/mL. Cells were diluted in fresh medium to OD600 value of 0.05 for growth. When the strain entered the mid-log phase, added 0.3 mM IPTG as an inducer. Cultures were then collected and used for subsequent experiments.
For acid stress experiments, the bacteria were grown to OD600 = 0.6 in LB medium (pH 7.0), and then transferred to LB medium of pH 5.8 and 10 mM L-lysine through centrifugal separation.
RNA isolation and real-time quantitative PCR (qRT-PCR)
Total RNA was extracted using the MiniBEST Universal RNA Extraction Kit (TaKaRa) according to the instructions of the kit. cDNA synthesis was performed using the PrimeScript RTKit (TaKaRa). Primers for qRT-PCR are listed in Table S6. Fluorescent quantitative PCR experiments were performed using the SYBR Premix Ex Taq™II kit purchased from TakaRa to normalize the expression of target genes with the expression of gapA mRNA. Relative transcript abundance was calculated using the 2-ΔΔCt method58.
Western blot
Bacterial total proteins were heated at 95 ◦C for 10 minutes and separated by SDS-PAGE. The proteins were transferred from gel onto a polyvinylidene fluoride (PVDF) membrane. After blocking with 5% milk in phosphate-buffered saline plus 0.1% Tween (PBST) at 16 ◦C for 2 h, the membrane was incubated with primary antibodies in PBST overnight at 4 ◦C. The membrane was then washed three times in PBST and incubated with secondary antibodies at 25◦C for 1 h. After three washes with PBST, protein on membrane was visualized with a chemiluminescent substrate and detected using a FluorChem imager. The protein levels were detected and quantified using ImageJ software. Primary antibodies against DnaK (1:2000 dilution), and CadC DBD domain (1: 2500 dilution) were used. The secondary antibodies were anti-mouse IgG antibodies (1:2000 dilution)19.
LDC assays
Colonies were inoculated in LDC broth medium supplemented with 0.1% l-lysine and bromocresol violet indicator. Sterile mineral oil was layered on the medium to keep the pH above 7 and incubated at 37 ◦C for 24 h. If glucose was fermented, yellow color was initially produced, but the medium gradually turned purple as the pH increased by the decarboxylase reaction.
Formaldehyde cross-linking mass spectrometry
For formaldehyde fixation, 37% formaldehyde was added to the medium to a final concentration of 1% and incubated at room temperature for 10 minutes. 2.5 mol/L glycine was added to the mixture of medium and formaldehyde to a final concentration of 125 mM and incubated at room temperature for 5 minutes to terminate the formaldehyde cross-linking reaction. Centrifuge the medium for 15 minutes (4200 × g) after the formaldehyde cross-linking reaction is terminated. The supernatant was discarded, and the cells were resuspended by adding resuspension buffer (25 mM Tris-HCl, pH 8.0, 500 mM NaCl, 3% glycerol), and then used for subsequent experiments.
The samples were re-solubilized with water after enzymatic digestion and analyzed under LC-MS/MS system59. Next, the proteomic database of MG1655 was searched in the database, and the data obtained from liquid-mass coupling were matched with the data in the database using Max Quant, Proteome Discoverer, and Peaks software. This process identified the proteins in the samples by finding the corresponding proteins in the database based on the masses of the primary and secondary fragments of the peptides.
Protein expression and purification
Full-length CadC and CadC1-184 were expressed in E. coli BL21 containing pET29b-cadC and pGL01-cadC1-184 respectively. The strains were cultured in LB medium containing appropriate antibiotics at 37 °C and 200 rpm until OD600 = 0.6, then cooled down to 16 °C for 1 h. Subsequently, the protein expression was induced by adding IPTG with a final concentration of 0.3 mM. After 16 h of induction, cells were collected by centrifugation at 4000 g for 10 min. Cell pellets were then resuspended in lysis buffer (25 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM PMSF, 20 g/mL DNase I) and homogenized by sonication. The lysate was then centrifuged at 3700 g for 10 min, the precipitate was discarded, and then the supernatant was centrifuged at 28,500 g for 50 min. The precipitate was resuspended in 25 mM Tris pH 8.0, 200 mM NaCl 3% glycerol, homogenized with a homogenizer, followed by a sonication. 0.1% DDM (n-dodecyl-β-D-maltoside) was added to the homogenate and mixed overnight for protein extraction. Afterwards the sample was centrifuged at 28,500 g for 50 min, the precipitate was discarded and the supernatant was loaded onto a Ni-NTA column (GE Healthcare) for purification. Target protein was eluted from the Ni-NTA column with elution buffer 25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.1% DDM (n-dodecyl-β-D-maltoside) and 250 Mm imidazole and further purified by size-exclusion chromatography in the buffer of 25 mM Tris-HCl, pH 8.0, 100 mM NaCl, 0.1% DDM (n-dodecyl-β-D-maltoside) on a Superdex 200 column (GE Healthcare). If necessary, his tag was removed using PPase protease. Purification of DegQ and DegP were expreesed in E. coli containing pGL01degQ and pET21bdegP respectively. The strains were grown in LB medium with appropriate antibiotics at 37 ◦C and 200 rpm until the OD600 = 0.6. Then, the final concentration of 0.3 mM of IPTG was added and protein expression was induced at 16 ◦C. After 16 h of induction, cells were collected by centrifugation at 4000 g for 10 min. Cell pellets were then resuspended in lysis buffer (25 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM PMSF, 20 g/mL DNase I) and homogenized by sonication. The lysate was centrifuged at 28500 × g for 50 minutes, and then the supernatant was loaded onto a Ni-NTA column (GE Healthcare) for affinity chromatography. Target protein was eluted from the Ni-NTA column with elution buffer (25 mM Tris-HCl, pH 8.0, 100 mM NaCl and 250 Mm imidazole) and further purified by size-exclusion chromatography in the buffer of 25 mM Tris-HCl, pH 8.0 and 100 mM NaCl on a Superdex 200 column (GE Healthcare). If necessary, his tag was removed using PPase protease. In all cases, the entire purification process was carried out at 4°C. Finally, the purity of the proteins was checked by SDS-PAGE.
In vitro digestion
CadC proteins were incubated with proteases DegP and DegQ in buffer (25 mM Tris-HCl, pH 8.0, 200 mM NaCl, 0.1% DDM), respectively, at 37°C. Samples were taken at 0 h, 3 h, and 6 h and examined by SDS-PAGE60.
Size exclusion chromatography
Proteins were prepared at 4 ◦C at concentrations between 100–200 μM. To characterize the interaction of CadC and DegP/DegQ, CadC was mixed with DegP and DegQ respectively with a molar ratio 1:1 and incubated at 4 °C for 10 minutes. CadC- DegP and CadC-DegQ complexes were then purified by size exclusion chromatography on a Superdex 200 column (GE Healthcare) equilibrated in buffer (10 mM Tris-HCl, pH 8.0 and 100 mM NaCl, 0.1% DDM). Subsequently, the elution peak of the complex was sampled out and subject to size exclusion chromatography side by side with CadC and DegP/DegQ under the same conditions to compare their peak positions. All images were processed using Origin 8.0 software (Originlab).
Subcellular fractionation of Escherichia coli
The method was performed as described in previous studies with minor modifications14.Cells were collected by centrifugation and resuspended in lysis buffer (25 mM Tris-HCl, pH 8.0, 200 mM NaCl, 1 mM PMSF, 20 g/mL DNase I) and homogenized by sonication. The lysate was then centrifuged at 3700 g for 10 min, the precipitate was discarded, and then the supernatant was centrifuged at 28,500 g for 50 min. The precipitate was resuspended in 25 mM Tris pH 8.0, 200 mM NaCl 3% glycerol, homogenized with a homogenizer, followed by a sonication. 0.1% DDM (n-dodecyl-β-D-maltoside) was added to the homogenate and mixed overnight, then sampled and examined by SDS-PAGE.
Electrophoretic Mobility Shift Assay (EMSA)
The FAM labeled double-stranded DNA of the cadBA promoter (TAAACATTAAATGTTTA) was used for EMSA. 25 nM DNA was incubated with different protein samples in a reaction buffer (10 mM Tris-HCl, pH 7.5, 1 mM MgCl2, 40 mM KCl, 0.1 mg/ml BSA, 5% (w/v) glycerol) at 37◦C for 15 minutes. If necessary, 10 mM ZnCl2 was added to the reaction buffer. The samples were then analyzed using natural 5% polyacrylamide gels in 0.5× TBE buffer (46 mM Tris base, 46 mM boric acid, 1 mM EDTA, pH 8.0) at 80 V for 80 minutes. Light was avoided during the experiment61. A Typhoon scanner (GE Healthcare) and Image quant software (GE Healthcare) were used for imaging and data analysis.
Fluorescence Polarization (FP)
Measurement of the fluorophore FAM-labeled cadBA promoter DNA was obtained in the same way as the DNA used in EMSA, using FP buffer. The 50 μM CadC, CadC1-184 protein gradient was diluted 15-fold and mixed with 50 μL of 2 nM FAM-DNA aliquots (10 mM Tris-HCl and 75 mM NaCl, pH 7.5) at 37 ◦C under protected light for 15 minutes61. If necessary, 10 mM ZnCl2 was added to the reaction buffer. FP measurements were performed using a Synergy 4 enzyme labeler (BioTek Synergy HT). All experiments were repeated three times. These curves were fitted using GraphPad Prism 5 software (GraphPad) to extrapolate the binding affinity.
ATR assay
ATR assay was carried out as reported previously with minor modifications 24. Each test strain was cultured overnight in LB broth, and then inoculated into fresh medium at a 0.1% dilution. The diluted culture was grown aerobically until OD600 reached 0.80 (4 × 108 CFU/ml), and then serially diluted and inoculated onto LB pH5.8 (10 mm lysine) agar plates to evaluate cell viability.
Statistics and reproducibility
Each set of data was characterized by three replicates. The data obtained were expressed as mean ± standard deviation (SD). Statistical analysis (mean and standard deviation) was performed using GraphPad Prism8 software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Data generated or analyzed in this study are included in this published article and its Supporting Information file. Uncropped images of SDS-PAGE gels and Uncropped Western Blot result plots have been uploaded as Supplementary Data 1. All figures from the main manuscript of the source data have been uploaded as Supplementary Data 2. Additional information is available from the corresponding author upon request.
References
Rychlik, I. & Barrow, P. A. Salmonella stress management and its relevance to behaviour during intestinal colonisation and infection. FEMS Microbiol. Rev. 29, 1021–1040 (2005).
Schwarz, J., Schumacher, K., Brameyer, S. & Jung, K. Bacterial battle against acidity. FEMS Microbiol. Rev. 46, fuac037 (2022).
Nicholson, J. K. et al. Host-gut microbiota metabolic interactions. Science 336, 1262–1267 (2012).
Schroeder, B. O. & Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 22, 1079–1089 (2016).
Audia, J. P. & Foster, J. W. Acid shock accumulation of sigmaS in Salmonella enterica involves increased translation, not regulated degradation. J. Mol. Microbiol. Biotechnol. 5, 17–28 (2003).
Foster, J. W. Escherichia coli acid resistance: tales of an amateur acidophile. Nat. Rev. Microbiol. 2, 898–907 (2004).
Kanjee, U. & Houry, W. A. Mechanisms of acid resistance in Escherichia coli. Annu. Rev. Microbiol. 67, 65–81 (2013).
Li, Z. et al. The role of bacterial cell envelope structures in acid stress resistance in E. coli. Appl. Microbiol. Biotechnol. 104, 2911–2921 (2020).
Lin, J. et al. Mechanisms of acid resistance in enterohemorrhagic Escherichia coli. Appl. Environ. Microbiol. 62, 3094–3100 (1996).
Seel, W., Flegler, A., Zunabovic-Pichler, M. & Lipski, A. Increased isoprenoid quinone concentration modulates membrane fluidity in Listeria monocytogenes at low growth temperatures. J. Bacteriol. 200, e00148-18 (2018).
Zhao, B. & Houry, W. A. Acid stress response in enteropathogenic gammaproteobacteria: an aptitude for survival. Biochem. cell Biol. 88, 301–314 (2010).
Tsai, M. F., McCarthy, P. & Miller, C. Substrate selectivity in glutamate-dependent acid resistance in enteric bacteria. Proc. Natl Acad. Sci. USA 110, 5898–5902 (2013).
Lund, P., Tramonti, A. & De Biase, D. Coping with low pH: molecular strategies in neutralophilic bacteria. FEMS Microbiol. Rev. 38, 1091–1125 (2014).
Mitchell, A. M. & Silhavy, T. J. Envelope stress responses: balancing damage repair and toxicity. Nat. Rev. Microbiol. 17, 417–428 (2019).
De Biase, D. & Lund, P. A. The Escherichia coli acid stress response and its significance for pathogenesis. Adv. Appl. Microbiol. 92, 49–88 (2015).
Mallick, S. & Das, S. Acid-tolerant bacteria and prospects in industrial and environmental applications. Appl. Microbiol. Biotechnol. 107, 3355–3374 (2023).
Wang, S., Yan, R., Zhang, X., Chu, Q. & Shi, Y. Molecular mechanism of pH-dependent substrate transport by an arginine-agmatine antiporter. Proc. Natl Acad. Sci. USA 111, 12734–12739 (2014).
Brameyer, S., Schumacher, K., Kuppermann, S. & Jung, K. Division of labor and collective functionality in Escherichia coli under acid stress. Commun. Biol. 5, 327 (2022).
Wang, W. et al. Host acid signal controls Salmonella flagella biogenesis through CadC-YdiV axis. Gut microbes 14, 2146979 (2022).
Kuper, C. & Jung, K. CadC-mediated activation of the cadBA promoter in Escherichia coli. J. Mol. Microbiol. Biotechnol. 10, 26–39 (2005).
Tetsch, L., Koller, C., Dönhöfer, A. & Jung, K. Detection and function of an intramolecular disulfide bond in the pH-responsive CadC of Escherichia coli. BMC Microbiol. 11, 74 (2011).
Eichinger, A., Haneburger, I., Koller, C., Jung, K. & Skerra, A. Crystal structure of the sensory domain of Escherichia coli CadC, a member of the ToxR-like protein family. Protein Sci. 20, 656–669 (2011).
Lee, Y. H., Kim, J. H., Bang, I. S. & Park, Y. K. The membrane-bound transcriptional regulator CadC is activated by proteolytic cleavage in response to acid stress. J. Bacteriol. 190, 5120–5126 (2008).
Lee, Y. H. et al. CadC has a global translational effect during acid adaptation in Salmonella enterica serovar typhimurium. J. Bacteriol. 189, 2417–2425 (2007).
Clausen, T., Southan, C. & Ehrmann, M. The HtrA family of proteases: implications for protein composition and cell fate. Mol. Cell 10, 443–455 (2002).
Kim, D. Y. & Kim, K. K. Structure and function of HtrA family proteins, the key players in protein quality control. J. Biochem. Mol. Biol. 38, 266–274 (2005).
Krojer, T., Sawa, J., Huber, R. & Clausen, T. HtrA proteases have a conserved activation mechanism that can be triggered by distinct molecular cues. Nat. Struct. Mol. Biol. 17, 844–852 (2010).
Xue, R. Y. et al. HtrA family proteases of bacterial pathogens: pros and cons for their therapeutic use. Clin. Microbiol. Infect. 27, 559–564 (2021).
Harris, B. Z. & Lim, W. A. Mechanism and role of PDZ domains in signaling complex assembly. J. cell Sci. 114, 3219–3231 (2001).
Krojer, T., Garrido-Franco, M., Huber, R., Ehrmann, M. & Clausen, T. Crystal structure of DegP (HtrA) reveals a new protease-chaperone machine. Nature 416, 455–459 (2002).
Malet, H. et al. Newly folded substrates inside the molecular cage of the HtrA chaperone DegQ. Nat. Struct. Mol. Biol. 19, 152–157 (2012).
Brameyer, S. et al. DNA-binding directs the localization of a membrane-integrated receptor of the ToxR family. Commun. Biol. 2, 4 (2019).
Zhang, S. et al. Degp degrades a wide range of substrate proteins in Escherichia coli under stress conditions. Biochem. J. 476, 3549–3564 (2019).
Fu, X. et al. DegP functions as a critical protease for bacterial acid resistance. FEBS J. 285, 3525–3538 (2018).
He, D., Zhang, M., Liu, S., Xie, X. & Chen, P. R. Protease-mediated protein quality control for bacterial acid resistance. Cell Chem. Biol. 26, 144–150.e3 (2019).
Schneider, J. & Wendisch, V. F. Biotechnological production of polyamines by bacteria: recent achievements and future perspectives. Appl Microbiol. Biotechnol. 91, 17–30 (2011).
Banaś, A. M., Bocian-Ostrzycka, K. M. & Jagusztyn-Krynicka, E. K. Engineering of the Dsb (disulfide bond) proteins - contribution towards understanding their mechanism of action and their applications in biotechnology and medicine. Crit. Rev. Microbiol. 45, 433–450 (2019).
Nakamoto, H. & Bardwell, J. C. Catalysis of disulfide bond formation and isomerization in the Escherichia coli periplasm. Biochim. et. Biophys. Acta 1694, 111–119 (2004).
Guan, N. & Liu, L. Microbial response to acid stress: mechanisms and applications. Appl. Microbiol. Biotechnol. 104, 51–65 (2020).
De Biase, D. & Pennacchietti, E. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: function, distribution and biomedical implications of the gadBC operon. Mol. Microbiol. 86, 770–786 (2012).
Krammer, E. M. & Prévost, M. Function and regulation of acid resistance antiporters. J. Membr. Biol. 252, 465–481 (2019).
Ulrich, L. E., Koonin, E. V. & Zhulin, I. B. One-component systems dominate signal transduction in prokaryotes. Trends Microbiol. 13, 52–56 (2005).
Demey, L. M., Gumerov, V. M., Xing, J., Zhulin, I. B. & DiRita, V. J. Transmembrane transcription regulators are widespread in bacteria and archaea. Microbiol. Spectr. 11, e0026623 (2023).
Rhee, J. E., Jeong, H. G., Lee, J. H. & Choi, S. H. AphB influences acid tolerance of Vibrio vulnificus by activating expression of the positive regulator CadC. J. Bacteriol. 188, 6490–6497 (2006).
Merrell, D. S. & Camilli, A. Regulation of vibrio cholerae genes required for acid tolerance by a member of the “ToxR-like” family of transcriptional regulators. J. Bacteriol. 182, 5342–5350 (2000).
Casalino, M. et al. Interference of the CadC regulator in the arginine-dependent acid resistance system of Shigella and enteroinvasive E. coli. Int. J. Med. Microbiol.: IJMM 300, 289–295 (2010).
Lee, Y. H. & Kim, J. H. Direct interaction between the transcription factors CadC and OmpR involved in the acid stress response of Salmonella enterica. J. Microbiol. 55, 966–972 (2017).
Hsieh, P. F., Lin, H. H., Lin, T. L. & Wang, J. T. CadC regulates cad and tdc operons in response to gastrointestinal stresses and enhances intestinal colonization of Klebsiella pneumoniae. J. Infect. Dis. 202, 52–64 (2010).
Roggiani, M. & Goulian, M. Chromosome-Membrane Interactions in Bacteria. Annu. Rev. Genet. 45, 115–129 (2015).
Woldringh, C. L. The role of co-transcriptional translation and protein translocation (transertion) in bacterial chromosome segregation. Mol. Microbiol. 43, 17–29 (2002).
Hansen, G. & Hilgenfeld, R. Architecture and regulation of HtrA-family proteins involved in protein quality control and stress response. Cell. Mol. Life Sci. 70, 761–775 (2013).
Sohn, J., Grant, R. A. & Sauer, R. T. Allosteric activation of DegS, a stress sensor PDZ protease. Cell 131, 572–583 (2007).
Datsenko, K. A. & Wanner, B. L. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl Acad. Sci. USA 97, 6640–6645 (2000).
Kuhlman, T. E. & Cox, E. C. Site-specific chromosomal integration of large synthetic constructs. Nucleic Acids Res. 38, e92 (2010).
Cherepanov, P. P. & Wackernagel, W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14 (1995).
Fritz, G. et al. Induction kinetics of a conditional pH stress response system in Escherichia coli. J. Mol. Biol. 393, 272–286 (2009).
Zheng, L., Baumann, U. & Reymond, J. L. An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 (2004).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408 (2001).
Wang, J. H. et al. Characterization of protein unfolding by fast cross-linking mass spectrometry using di-ortho-phthalaldehyde cross-linkers. Nat. Commun. 13, 1468 (2022).
Krojer, T. et al. Structural basis for the regulated protease and chaperone function of DegP. Nature 453, 885–890 (2008).
Zhang, F. et al. YdiV regulates Escherichia coli ferric uptake by manipulating the DNA-binding ability of Fur in a SlyD-dependent manner. Nucleic Acids Res. 48, 9571–9588 (2020).
Acknowledgements
We express our sincere gratitude to all study participants who invested their valuable time in our research. Special thanks to researcher Yin Kun of Shandong Institute of Parasitic Disease Control for providing the CadC antibody. This work was supported by the Youth Foundation of Shandong Natural Science Foundation [ZR202102230675], the National Natural Science Foundation of China [31970043], the China Postdoctoral Science Foundation [2021M701996], the Shandong Province Post-Doctoral Innovation Project Foundation [202101007], the National Natural Science Foundation of China [32170034], and the National Natural Science Foundation of China [81902038].
Author information
Authors and Affiliations
Contributions
M.C., F.Z. and L.G. conceived, designed and analyzed the experiments. M.C., W.C., and Y.S. participated in expression, purification of the CadC-DegP, CadC-DegQ complex and other proteins. M.C., J.Z. and X.W. performed functional assays of CadC and DegP, DegQ. M.C., F.Z., W.W. and B.L. participated in figure and manuscript preparation. H.W., K.Z., S.X., T.S., and H.D. involved in touching up articles. M.C., F.Z. and L.G. wrote the manuscript with inputs from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: Tobias Goris. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chen, M., Shang, Y., Cui, W. et al. Molecular mechanism of proteolytic cleavage-dependent activation of CadC-mediated response to acid in E. coli. Commun Biol 7, 1335 (2024). https://doi.org/10.1038/s42003-024-06931-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-06931-x
