
Abstract
Myostatin (MSTN) is a negative regulator of muscle growth, and its relationship with the gut microbiota is not well understood. In this study, we observed increase muscle area and branched-chain amino acids (BCAAs), an energy source of muscle, in myostatin knockout (MSTN-KO) cattle. To explore the link between increased BCAAs and rumen microbiota, we performed metagenomic sequencing, metabolome analysis of rumen fluid, and muscle transcriptomics. MSTN-KO cattle showed a significant increase in the phylum Bacteroidota (formerly Bacteroidetes), particularly the genus Prevotella (P = 3.12e-04). Within this genus, Prevotella_sp._CAG:732, Prevotella_sp._MSX73, and Prevotella_sp._MA2016 showed significant upregulation of genes related to BCAA synthesis. Functional enrichment analysis indicated enrichment of BCAA synthesis-related pathways in both rumen metagenomes and metabolomes. Additionally, muscle transcriptomics indicated enrichment in muscle fiber and amino acid metabolism, with upregulation of solute carrier family genes, enhancing BCAA transport. These findings suggest that elevated rumen Prevotella in MSTN-KO cattle, combined with MSTN deletion, synergistically improves muscle growth through enhanced BCAA synthesis and transport.
Similar content being viewed by others


Introduction
The gut microbiota refers to the diverse community of microorganisms inhabiting the gastrointestinal tract of animals. This community includes bacteria, viruses, fungi, and other organisms that play crucial roles in various physiological processes. In recent years, there has been increasing interest in understanding the potential connection between the gut microbiota and muscle function1. Several studies have suggested that the composition and diversity of the gut microbiota may influence the development and maintenance of muscle mass and strength2,3. The gut microbiota constantly influences the metabolic balance of the host. For instance, germ-free mice fed a high-fat diet exhibit a lean phenotype4. The gut microbiota has also been implicated in regulating fat storage in multiple organs, with obesity being associated with a distinct composition of the gut microbiota compared to the lean phenotype5.
There are several mechanisms by which the gut microbiota may influence muscle function. One possibility is through the production of short-chain fatty acids (SCFAs) in the gut. SCFAs are produced by certain gut bacteria during the fermentation of dietary fiber and have been shown to have a number of beneficial effects on the body, including reducing inflammation and improving insulin sensitivity6. In ruminants, SCFAs are mainly produced by the microbial fermentation of dietary fiber in the rumen and serve as an important energy source for ruminants, contributing to overall energy metabolism7,8. SCFAs, especially butyrate, have been shown to promote muscle protein synthesis and regulate muscle cell differentiation through histone deacetylase inhibition and G-protein coupled receptor activation pathways9,10. These processes are crucial for muscle development and repair, suggesting that SCFAs may have a direct impact on muscle growth and function in cattle. Another potential mechanism involves the production of other gut-derived metabolites, such as creatine, which is involved in energy metabolism and is important for maintaining muscle function11. Some studies have suggested that the gut microbiota composition may influence the production of creatine and other metabolites, which in turn could have an impact on muscle function12.
Myostatin is a protein that plays a critical role in regulating muscle mass. It acts by inhibiting the growth and differentiation of muscle cells, thereby limiting the amount of muscle an individual can develop. Research has shown that individuals with lower myostatin levels typically have greater muscle mass than those with higher levels. Myostatin and the gut microbiota are both important factors in muscle function and growth. However, the relationship between these elements remains poorly studied.
This study explored the potential relationship between myostatin and the rumen microbiota and suggested that rumen microbiota-produced BCAAs may interact synergistically to influence the signaling pathways involved in muscle protein synthesis and contribute to an increased muscle mass phenotype. However, to fully understand the relationship between these factors and how they interact to affect muscle function and growth, further research is needed. In conclusion, the association between the rumen microbiota and myostatin may aid in identifying specific rumen microbial compositions or rumen-derived metabolites that could help improve muscle mass and function.
Results
MSTN-KO cattle increased muscle fiber area and decreased intermuscular fat
Gut microbes can regulate host energy homeostasis, lipid metabolism, and glucose metabolism via the gut-brain axis, gut-liver axis, gut-lipid axis, and gut-immune axis13,14,15,16. As the rumen is the most important microbial fermentation organ in ruminants, we wanted to explore the potential link between skeletal muscle and rumen microbes in cattle after myostatin gene editing. We aimed to explore this linkage by analyzing changes in muscle morphological structure and nutrient composition as well as rumen microbes and metabolites in MSTN-KO cattle (Fig. 1A). In the gastrocnemius muscles, a typical skeletal muscle significantly influenced by the MSTN gene, histological analysis revealed that both MSTN-KO cattle and WT cattle had properly aligned muscle fibers. However, MSTN-KO cattle displayed a denser arrangement of muscle fibers and reduced connective tissue between them. The cross-sectional area of muscle fibers in MSTN-KO cattle was significantly enlarged, indicating increased thickness (Fig. 1B, C). Additionally, MSTN-KO cattle exhibited smaller intermuscular fat widths (Fig. 1B, D).

A Methodological process of the study. B—a–c HE-stained histological images of WT cattle gastrocnemius muscle at 40×, 100×, and 200× magnification under a light microscope. B—d–f HE-stained histological images of MSTN-KO cattle gastrocnemius muscle at 40×, 100×, and 200× magnification under a light microscope. C Comparison of the muscle fiber area between the MSTN-KO and WT groups. The statistical analysis involved measuring 100 fibers in each gastrocnemius muscle sample. D Comparison of the intermuscular fat width between the MSTN-KO and WT groups. The statistical analysis involved measuring 100 intermuscular fat widths in each gastrocnemius muscle sample. Data: mean ± SD. N = 100. *P < 0.05, **P < 0.01, ***P < 0.001.
Leucine, isoleucine, and valine are considered essential BCAAs that are pivotal for muscle growth and development17. In the muscles of MSTN-KO cattle, the levels of leucine, isoleucine, and valine were significantly increased, as were the levels of other amino acids, such as threonine, aspartic acid, and phenylalanine (Table 1). However, the overall composition of muscle tissue, including energy, ash (%), protein (%), moisture (%), and fat (%), did not significantly change (Supplementary Information-Table S1). These findings suggest that the notable increase in BCAAs in MSTN-KO cattle muscle tissue contributes to remarkable myostatin-mediated muscle growth.
Prevotella is a key bacterium for microbiological changes in the rumen of MSTN-KO cattle
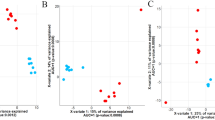
The regulation of host energy metabolism is influenced by many metabolites produced by the gut microbiota18. To verify whether the high levels of BCAAs in skeletal muscles depend on the rumen-muscle axis for their transfer, We analyzed the rumen microbiota, which is the largest microbial fermentation organ in cattle. Metagenomic analysis yielded a total of 2,537,168 unigenes. At the phylum level, the dominant bacterial phyla included Bacteroidota, Bacillota (formerly Firmicutes), and Fibrobacterota (formerly Fibrobacteres). The microbial composition of the MSTN-KO group differed from that of the WT group, with a greater proportion of Bacteroidota in the rumen of the MSTN-KO group (234,140.5 ± 27,697.61 vs. 137,913.89 ± 8,635.76, P = 0.001, Fig. 2A, B, Supplementary Information—Table S2).

A Composition of rumen microorganisms at the phylum level. B Relative abundances of Bacteroidota, Bacillota, and Fibrobacterota. C Shannon and Simpson indices of the rumen Bacteroidota. D Analysis of the differential abundance of the top ten genera in the rumen microbiota of Bacteroidota. Data: mean ± SD. MSTN-KO (N = 5), WT (N = 3).*P < 0.05, **P < 0.01.
Therefore, we conducted further analysis of the microbial diversity of the Bacteroidota phylum in the two groups. There were no significant differences were found in the Shannon or Simpson indices of the genera belonging to the Bacteroidota phylum (Fig. 2C). Similarly, we also observed differences in the whole rumen microbiota between the MSTN-KO and WT groups at the genus level (Supplementary Information—Fig S1A). These differences were particularly evident in terms of species richness, with the MSTN-KO group exhibiting significantly lower richness than the WT group (Richness index, P = 0.001), while no differences were observed in terms of evenness (Shannon and Simpson, Supplementary Information—Fig. S1B). In addition, we found that the differences in the Bacteroidota phylum were mainly attributed to the Prevotella genus (P = 3.12 × 10−4, Fig. 2D), which also exhibited the most prominent differences among all rumen microbial genera (Supplementary Information—Fig. S1C). These findings suggest that the rumen microbiota of MSTN-KO cattle undergoes significant changes compared to that of WT cattle, with a marked alteration in the relative abundance of the genus Prevotella.
An increase in Prevotella improves rumen BCAA metabolism
To further explore the specific differences in species within the Prevotella genus, differential analysis was conducted on the 111 identified species belonging to the Prevotella genus. Almost all Prevotella species were produced at higher levels in the MSTN-KO group (Fig. 3A). According to linear discriminant analysis effect size (LEfSe), bacterial biomarkers revealed that Prevotella was a key dominant genus, with Prevotella_bryantii and Prevotella_oralis being the most crucial species belonging to the Prevotella genus (Fig. 3B).

A Heatmap of all Prevotella species in the rumen of the MSTN-KO and WT groups. B LEfSe of all Bacteroidota. C Scatterplot of BCAA metabolism-related unigenes. D Analysis of the correlation between differentially abundant Prevotella genera and metabolites. MSTN-KO (N = 5), WT (N = 3). *P < 0.05, **P < 0.01.
By annotating the rumen metagenome, 307 unigenes related to BCAA biosynthesis were identified, of which 227 unigenes had differential ratio information. These 227 unigenes were further analyzed for differential sorting, and among the top five upregulated unigenes, three belonged to the Prevotella genus, including unigene19544 belonging to Prevotella_sp._CAG:732, unigene876208 belonging to Prevotella_sp._MSX73, and unigene741138 belonging to Prevotella_sp._MA2016 (Fig. 3C, Table 2). These results indicated that the increased abundance of the Prevotella genus in the MSTN-KO group is pivotal in BCAA synthesis, particularly through Prevotella_sp._CAG:732, Prevotella_sp._MSX73, and Prevotella_sp._MA2016.
Since most of the Prevotella species demonstrated similar abundance patterns, we analyzed the potential metabolic patterns of the microbiota of the Prevotella genus. It was found that traumatin, droperidol, and linoleic acid were significantly positively correlated, while acetic acid, LysoPS 20:4, LysoPE 17:1, and LysoPG 16:0 were significantly negatively correlated with Prevotella (Fig. 3D). These findings suggest that MSTN-KO cattle harbor multiple Prevotella species with high abundance and that the BCAA metabolism may significantly influence host BCAA metabolic pathways.
Enhanced metabolic function and increased absorption of BCAAs in the rumen of MSTN-KO cattle
The above results demonstrated that the Prevotella genus is the key microorganism influencing BCAA synthesis, and we then analyzed the significance of these changes in the broader rumen microbiota. We performed a functional analysis based on differential unigene abundance. GO functional enrichment analysis revealed significant changes in the valine biosynthetic process, and pathway enrichment analysis also revealed significant alterations in the leucine and isoleucine biosynthesis pathways (Fig. 4A, B). Similarly, nontargeted metabolomics pathway analysis confirmed the significant changes in valine, leucine, and isoleucine biosynthesis (Fig. 4C). As expected, the increased levels of BCAAs in the skeletal muscle of MSTN-KO cattle can be attributed to significant alterations in the metabolism of BCAAs by the rumen microbiota, and an increase in the rumen population of Prevotella is thought to be one of the factors contributing to the enhanced synthesis of BCAAs.

A Differential microorganisms subjected to GO enrichment analysis. B KEGG pathway analysis of differentially abundant microorganisms. C Pathway analysis of differentially abundant metabolites in the rumen. D Percentage of unigenes categorized by eggNOG. E Histological examination of rumen tissue using HE staining. a–c WT cattle, d–f MSTN-KO cattle.
We used the eggNOG database to compare the functional profiles of the rumen microbiota in MSTN-KO and WT cattle. Our analysis revealed that amino acid transport and metabolism were more enriched in MSTN-KO cattle, whereas extracellular structures were more enriched in WT cattle. Additionally, MSTN-KO cattle exhibited greater enrichment of defense mechanisms (Fig. 4D). To further explore the potential impact of microbial shifts on nutrient absorption and metabolism, we compared carbohydrate-active enzymes (CAZymes) encoded in the rumen metagenomes of MSTN-KO and WT cattle. While the diversity of carbohydrate enzymes in the rumen microbiome of MSTN-KO cattle appeared greater than in WT cattle, this difference was not statistically significant (Supplementary Information—Fig. S2A, B). Similarly, trends suggesting higher abundance of polysaccharide lyases, glycosyl transferases, and carbohydrate esterases in the rumen of MSTN-KO cattle were observed (Supplementary Information—Fig. S2C), although these differences also did not reach statistical significance. These findings highlight potential functional differences in the rumen microbiota of MSTN-KO cattle in utilizing plant carbohydrates, but further studies are required to confirm these observations.
Furthermore, considering that host gastrointestinal epithelial cells can derive nutrients from microbial metabolism, the thickened rumen muscle layer aligns with similar observations in skeletal muscle (Fig. 4E). This physiological adaptation may better accommodate the diversity of ruminal microbial metabolism and enhance nutrient absorption19,20.
Increased efficiency of amino acid transport increased the expression of SLC family genes in MSTN-KO cattle skeletal muscles
The results above indicate that significant changes occurred in the BCAA pathway within the rumen of MSTN-KO cattle, providing a molecular basis for the higher BCAA levels observed in MSTN-KO cattle skeletal muscles. Furthermore, we wanted to determine whether stronger BCAA absorption capacity. Transcriptomic analysis of the gastrocnemius muscle revealed significant changes in gene expression following myostatin gene knockout (Fig. 5A), with a total of 2669 genes showing significant differential expression, including 1192 upregulated genes and 1477 downregulated genes, using a fold change greater than 2 and an adjusted p-value of less than 0.05. The upregulated genes were associated with myofiber development, muscle system processes, and fiber contraction (Fig. 5B), primarily attributed to alterations in genes related to lipid and amino acid metabolism (Fig. 5C).

A Volcano plot analysis of differentially expressed genes (DEGs) between the MSTN-KO and WT groups. B GO enrichment analysis of highly expressed genes in the MSTN-KO group. C KEGG pathway classification of highly expressed genes in the MSTN-KO group. D Expression analysis of SLC family genes. Data: mean ± SD. N = 3. *P < 0.05, **P < 0.01, ***P < 0.001.
Notably, solute carrier (SLC) family proteins play important roles in the BCAA metabolic network, with SLC25A44 being responsible for transporting BCAAs into the mitochondria for breakdown21,22. In this study, we identified a total of 58 SLC family genes and selected the top 10 highly expressed genes for analysis (Fig. 5D). Seven genes, particularly SLC25A6, SLC25A4, and SLC25A21, which belong to the SLC25 family along with SLC25A44, were highly expressed in the skeletal muscles of MSTN-KO cattle and exhibited significant upregulation compared to wild-type cattle. These findings indicate that MSTN-KO cattle muscle tissue has enhanced BCAA metabolism, consistent with upregulated muscle development-related genes.
Discussion
The relationship between the gut microbiota and the host is closely intertwined, and changes in the host genotype can alter the gut microbiota composition23. In this study, it was found that MSTN-KO cattle skeletal muscles have a lower fat content, which may be related to the high abundance of Bacteroidota in the rumen (Fig. 1B, Fig. 2B)24. Generally, the host and gut microbiota maintain a dynamic equilibrium25, and knocking out the MSTN gene appears to establish a new balance, collectively reducing host fat accumulation. Studies have shown that the composition of the gut microbiota can influence the efficacy of dietary interventions. In overweight individuals, a higher Prevotella/Bacteroides ratio is associated with more significant weight loss and a reduced risk of obesity after a high-fiber diet intervention26. Healthy overweight adults with higher Prevotella abundance lose more fat following the consumption of a whole-grain and fiber-rich ad libitum diet. This further supports the potential use of the gut microbiota composition as a biomarker for individualized obesity management. Research has also shown that individuals with a greater abundance of the Prevotella CAG 5226 from the Prevotella genus have better health outcomes27.
The Bacteroidota phylum primarily includes two genera: Bacteroides and Prevotella28. Prevotella is typically associated with a plant-based diet and is considered a “probiotic” bacteria that plays a beneficial role in the host. A decrease in Prevotella abundance has been associated with both human certain diseases and feed efficiency in cattle29,30,31,32,33. Prevotella possesses enzymes involved in the degradation of mucosal proteins, which may contribute to increased intestinal permeability. Prevotella has also been linked to carbohydrate and protein intake34,35. This is also evidenced by the high abundance of Prevotella in MSTN-KO cattle (Supplementary Information—Fig. S2).
The gut microbiota plays a critical role in providing the host with BCAAs and influencing host nutrient absorption and the immune system via the host epithelium36,37,38. Microbes produce lysine, an essential amino acid that the host can absorb and incorporate into host proteins39. An increase in the production of BCAAs (typical Bacillota/Bacteroidota ratio) by microbial communities has been associated with improved insulin sensitivity and protein synthesis40. BCAAs, including leucine, isoleucine, and valine, are essential amino acids that are crucial for muscle protein synthesis. Skeletal muscle, one of the largest organs in mammals41, stores protein as the primary form of energy, with BCAAs serving as the main energy source42. Higher doses of dietary BCAAs can stimulate protein synthesis43. Additionally, the Prevotella genus correlated positively with multiple amino acid metabolic pathways, including leucine biosynthesis, alanine and aspartate metabolism, and D-glutamate and D-glutamine metabolism34. Prevotella copri from the Prevotella genus and Bacteroides vulgatus from the Bacteroides genus are considered driving factors in the relationship between BCAA synthesis and insulin resistance. Prevotella copri can increase branched-chain amino acid levels in mice44. Parabacteroides merdae enhances the degradation of BCAAs and activates the mTORC1 pathway through BCAA regulation45. The Prevotella genus has enzymes involved in BCAA degradation, allowing them to utilize BCAAs as an energy source34. Prevotella_sp._CAG:732, Prevotella_sp._MSX73, and Prevotella_sp._MA2016 were the most critical bacteria for BCAA metabolism in the rumen of MSTN-KO cattle (Fig. 3C, Table 2). We observed an increase in the abundance of Prevotella copri and Parabacteroides merdae in the rumen of MSTN-KO cattle, indicating that MSTN-KO cattle not only increase BCAA synthesis but also promote BCAA degradation metabolism (Fig. 3A). In addition, in patients with type 2 diabetes, a significant decrease in Prevotella abundance is significantly associated with reduced traumatin and linolenic acid46. This finding is consistent with our results (Fig. 3D). The MSTN gene also affects the progression of diabetes47, and in terms of disease, the MSTN gene shows functional consistency with Prevotella.
BCAAs account for approximately 35% of the essential amino acids and approximately 21% of the total amino acids in muscle protein. They play various physiological roles, with the most significant being the promotion of protein synthesis in muscle, liver, heart, and adipose tissues, which are critical for maintaining the structure and functional integrity of organs in the body48. BCAAs also act as signaling molecules involved in the regulation of various physiological functions. Leucine, in particular, can stimulate protein synthesis, which is an energetically costly process associated with increased glucose demand by glucose-demanding tissues such as skeletal muscles, leading to enhanced insulin signaling, increased insulin sensitivity, and consequent insulin release49. BCAAs can promote muscle protein synthesis and increase muscle mass. Elite athletes typically have well-developed muscles, and the gut microbiota is positively correlated with branched-chain amino acid metabolism pathways44,50. β-Glucan inhibits intestinal flora BCAA biosynthesis by inhibiting Prevotella-mediated BCAA biosynthesis51. Low plasma BCAA levels may cause skeletal muscle to degrade since BCAAs are the primary energy source of skeletal muscle52. Supplementation with BCAAs has been proven to be effective in reducing muscle damage, accelerating the restoration of damaged muscles, and improving protein synthesis53,54. Different BCAAs have different mechanisms of action55. BCAAs rich in leucine can activate the mTOR signaling pathway in muscle cells, thereby promoting muscle protein synthesis through the activation of distinct intracellular pathways56. Isoleucine can promote muscle growth by upregulating the expression of important glucose transporters (GLUT1 and GLUT4) in muscles and increasing the expression of the glucose transporters SGLT1 and GLUT2 in the small intestine, promoting the development and health of skeletal muscle and the small intestine. BCAAs play a role in metabolism through SLC25A44, regulating energy homeostasis22. In this study, we observed an increase in the abundance of SLC family proteins in MSTN-KO cattle skeletal muscles, especially the SLC25 family members SLC25A6, SLC25A4, and SLC25A21 (Fig. 5D). In our previous study (https://doi.org/10.3390/ijms24098120)57, we investigated the changes in amino acid metabolism in the smooth muscle tissue of MSTN-KO cattle, which highlighted an improved efficiency of BCAA transport. This work focused on the molecular pathways and metabolic shifts in smooth muscle tissue, particularly in the context of MSTN gene editing. While that study provided valuable insights into the metabolic changes within muscle tissue, it primarily addressed the metabolic landscape from a tissue-specific perspective, with emphasis on transcriptomic and metabolomic alterations. In contrast, the current study explores the relationship between MSTN gene editing, rumen microbiota, and skeletal muscle, specifically how rumen microbial BCAA metabolism influences skeletal muscle BCAA levels. By integrating metagenomic analysis of rumen microbiota with the metabolic profiles of skeletal muscle, this study offers a broader view of the interplay between microbiota and host metabolism. Our findings demonstrate that MSTN gene editing not only impacts skeletal muscle development directly but also modulates the rumen microbiota’s role in BCAA metabolism, leading to a synergistic increase in skeletal muscle growth. Although nutrient metabolism can be identified through metagenomics, determining how organisms utilize and uptake nutrients remains challenging. For example, essential amino acids can exist as dipeptides, tripeptides, or oligopeptides of free amino acids, as well as part of complex protein breakdown originating from cells. We currently cannot ascertain the exact sources of amino acid fluctuations due to challenges in annotation and proper allocation of transport reactions58.
The intestinal flora plays two important roles in the initial steps of amino acid catabolism: deamination, which produces carboxylic acids and ammonia, and decarboxylation, which produces amines and carbon dioxide. Ammonia can inhibit mitochondrial oxygen consumption and reduce the metabolism of SCFAs by intestinal epithelial cells (IECs), potentially negatively impacting the host20. However, host IECs can regulate ammonia levels by converting ammonia to citrulline and glutamine; thus, the toxicity limits of proteolytic metabolism are not yet well understood. Acetic acid, as an important SCFA, shows a strong negative correlation with Prevotella (Fig. 3D). Additionally, host gut epithelial cells can also obtain nutrients from microbial metabolism. The increased muscle layer thickness in the rumen epithelium of MSTN-KO cattle may enhance nutrient absorption capacity (Fig. 4E). This physiological adaptation may be better suited to accommodate the diversity of rumen microbial metabolism. It is important to note that this study did not distinguish between microbial and host metabolites, however, given the significant association between the rumen microbiota and metabolic profiles, this study demonstrates a potentially important relationship for future studies. In recent years, many studies on rumen databases have significantly improved the annotation rates of rumen microorganisms59,60,61. Our next step is to further investigate the regulatory mechanisms of Prevotella and its relationship with other nutrient metabolism pathways through more gene-edited cattle. By expanding the rumen microbiome data of MSTN gene-edited cattle and integrating it with previously published rumen microbiome databases, we aim to improve rumen annotation rates and conduct more in-depth analyses.
In summary, MSTN-KO cattle exhibit increased levels of BCAAs in their skeletal muscles, leading to an increase in muscle mass. The increased levels of BCAAs are derived from the high abundance of the Prevotella genus in the rumen, particularly Prevotella_sp._CAG:732, Prevotella_sp._MSX73, and Prevotella_sp._MA2016. These species exhibit significant upregulation of genes related to BCAA synthesis pathways, leading to a significant alteration in BCAA biosynthetic processes in the rumen microbiota. A significant increase in the abundance of the Prevotella genus was positively correlated with traumatin, droperidol, and linoleic acid in the rumen but negatively correlated with LysoPS 20:4, LysoPE 17:1, and LysoPG 16:0. Additionally, myostatin gene knockout also enhances the expression of SLC family genes in muscle, accelerating the transport of BCAAs, which collectively contributes to increased muscle mass in MSTN-KO cattle (Fig. 6).

MSTN-KO cattle exhibit increased skeletal muscle mass due to elevated BCAA levels in muscle, derived from upregulated BCAA synthesis genes in rumen Prevotella species (Prevotella_sp._CAG:732, MSX73, and MA2016). Prevotella abundance correlates positively with traumatin, droperidol, and linoleic acid. Myostatin knockout also enhances solute carrier (SLC) gene expression in muscle, promoting BCAA transport, muscle fiber formation, and reduced intermuscular fat. The blue arrow represents an increase, and the red arrow represents a decrease.
Materials and methods
Animals
Myostatin-knockout (MSTN-KO) cattle were bred and genotyped in our laboratory62. To minimize the number of variables in the experiment, we sacrificed five male MSTN-KO cattle and three male wild-type (WT) cattle (approximately 2 years old, with an age difference of no more than 2 months) at the same time, and rumen fluid was collected for rumen metagenomic and metabolomic studies. The cattle on the farm were raised and managed by the same personnel under consistent conditions, and they were provided with the same diet, including free access to water. We have complied with all relevant ethical regulations for animal use. Ethical approval was received for the study from the Bioethics Committee of Inner Mongolia University (approval number: IMU-CATTLE-2023-058), and all procedures were conducted in accordance with their guidelines.
Physicochemical characteristics of muscle tissue
The physicochemical characterization (moisture, ash, fat, protein, and carbohydrate contents) of the muscle was performed according to standard procedures. Ash is the inorganic material left in the muscle after searing, and the amount of ash is calculated by searing and weighing. The percentage of meat moisture content was determined by the routine oven-drying method. The protein content was determined following the Kjeldahl method. Fat content was detected using the Soxhlet extraction method, and Soxhlet extraction instruments, ether, and a drying oven (Memmert, Germany) were used for fat content measurements. Gross energy was determined by automatic adiabatic oxygen bomb calorimetry (Parr 1281, Automatic Energy Analyzer, Moline, IL, USA). Amino acid quantification was carried out by an amino acid analyzer (HITACHI L-8900, Japan).
Hematoxylin–eosin (HE) staining
The muscle and rumen samples were cut into approximately 0.5 cm × 0.5 cm specimens using a sterile surgical blade. The samples were then fixed in 4% paraformaldehyde. Following fixation, the specimens were embedded in paraffin blocks and cut into 2–4 μm sections using a Leica SM 2000 R microtome. The sections were dried overnight at 37 °C.
After dewaxing, the muscle and rumen samples paraffin sections were subjected to HE staining. Subsequently, the sections were dehydrated using increasing concentrations of ethanol and made transparent using xylene.
Rumen fluid sampling
The rumen fluid samples (50–100 mL) were collected from MSTN-KO and WT cattle within 2 h after slaughter. The ruminal contents were manually squeezed to obtain a rumen fluid sample. The rumen fluid sample also contained small particles. The bottle containing the rumen fluid sample was immediately immersed in an ice box to prevent microbial contamination. Ruminal fluid samples collected from each cattle were divided into triplicates, transferred to the laboratory, and stored at −80 °C until DNA extraction of microorganisms and metabolite extraction from the rumen.
Metagenomic sequencing and analysis
We extracted DNA from rumen fluid samples using the E.Z.N.A. Stool DNA Kit (D4015-02, Omega, USA) and stored at −80 °C until use in the PCR. A DNA library was constructed using the TruSeq Nano DNA LT Library Preparation Kit (FC-121-4001, Illumina, USA), and metagenome sequencing was performed using HiSeq4000 (Illumina, U.S.A.) strategies with a PE150 sequencing mode.
To obtain valid reads for further analysis, we processed the raw sequencing reads. First, sequencing adapters were removed from sequencing reads using Cutadapt v1.963. Second, low-quality reads (quality scores < 20), short reads (<100 bp), and reads containing more than 5% “N” were trimmed using the sliding-window algorithm method in Fqtrim v0.9.464. Third, the reads were aligned to the host genome using Bowtie2 v2.2.065 to remove the host genome (Bos_taurus_UMD_3.1)66,67,68 contamination. Once quality-filtered reads were obtained, they were de novo assembled to construct the metagenome for each sample by IDBA-UD v1.1.169. Contigs longer than 500 bp were retained for subsequent clustering analysis. The assembly quality was evaluated using the QUAST program. MetaGeneMark (v3.26)70 was employed to predict coding sequences (CDS) from contigs ≥500 bp. CDS sequences shorter than 100 nt were filtered out based on the prediction results. The remaining CDS sequences were clustered using CD-HIT (v4.6.1)71 with identity ≥95% and coverage ≥90% to generate representative unigenes. Clean data from each sample were aligned to the gene sequences using Bowtie2, and the number of reads mapped to each gene was counted. Genes with reads mapped ≤2 across all samples were filtered out, resulting in the final set of unigenes for further analysis. The abundance of each gene in each sample was calculated based on the number of mapped reads and the length of the gene (Supplementary Information—Table S3).
Species annotation was performed by aligning the unigenes to the NCBI NR database (v20230717) using DIAMOND (v0.9.14) with the BLASTp algorithm (e-value ≤ 1e−5). For each unigene, the alignment results with e-values ≤ 10 times the minimum e-value were selected for species classification. Species annotation was carried out using a method similar to the lowest common ancestor (LCA) algorithm in MEGAN software, incorporating the NCBI taxonomic classification system. This allowed us to obtain species annotation information across various taxonomic levels (SuperKingdom, Phylum, Class, Order, Family, Genus, Species). The abundance information of unigenes was combined with species classification to derive abundance at each taxonomic level, calculated as the sum of the abundances of unigenes annotated to that taxonomic level. We then analyzed differences in microbial abundance across different taxonomic levels. Functional analyses were conducted using the GO (gene ontology, http://geneontology.org/), KEGG (Kyoto Encyclopedia of Genes and Genomes, http://www.kegg.jp/kegg/download), CAZy (v20220806, http://www.cazy.org/), and eggNOG (v4.5) databases72. We used DIAMOND software to align unigene sequences with the protein sequences in the KEGG pathway, CAZy, and eggNOG database (blastp, evalue ≤ 1e−5), and selected the highest scoring KEGG, CAZy, and eggNOG alignment result as the annotation for each unigene. By integrating the hierarchical structure of the KEGG pathway, CAZy, and eggNOG database, we were able to statistically analyze the information at various levels within the KEGG, CAZy, and eggNOG framework. R v4.2.1 was utilized for principal component analysis (PCA), richness, and diversity. LEfSe analysis was performed using OmicStudio tools (https://www.omicstudio.cn/tool/). Pathway analysis of differentially abundant metabolites was conducted using MetaboAnalyst 5.0 (https://www.metaboanalyst.ca/)73.
Untargeted metabolome analysis
The rumen fluid samples were thawed on ice, and 20 μL of each sample was extracted with 120 μL of precooled 50% methanol, vortexed, and then simmered. The samples were then kept overnight at −20 °C. The supernatants were then put into fresh 96-well plates after 20 min of spinning at 4,000 × g. Before the LC‒MS measurements, the materials were kept at −80 °C. All samples were acquired by the LC‒MS system following the manufacturer’s instructions (quality control: a mixture of 10 μL per sample). All chromatographic separations were performed using an ultra-performance liquid chromatography (UPLC) system (SCIEX, UK). An ACQUITY UPLC T3 column (100 mm × 2.1 mm, 1.8 µm, Waters, UK) was used for reversed-phase separation at 35 °C.
The LC‒MS raw data files were converted into mzXML format and then processed by the XCMS74, CAMERA75, and metaX76 toolboxes implemented with R v4.2.1. Each ion was identified by comprehensive information on retention time and m/z data. The HMDB and KEGG databases were used to annotate metabolites using exact m/z data of the samples.
Transcriptome analysis
Total RNA was isolated and purified using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s instructions. The RNA concentration and purity of each sample were quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop, Wilmington, DE, USA). RNA integrity was assessed using a Bioanalyzer 2100 (Agilent, CA, USA), with a RIN value greater than 7.0, and was further verified by denaturing agarose gel electrophoresis. Poly(A) RNA was isolated from 1 μg of total RNA using Dynabeads Oligo(dT)25-61005 (Thermo Fisher, CA, USA) through two rounds of purification.
The purified Poly(A) RNA was fragmented into small pieces using the Magnesium RNA Fragmentation Module (NEB, cat. E6150, USA) under 94 °C for 5–7 min. These fragmented RNA pieces were then reverse transcribed into cDNA using SuperScript™ II Reverse Transcriptase (Invitrogen, cat. 1896649, USA). Following this, second-strand cDNA was synthesized with U-labeling using E. coli DNA polymerase I (NEB, cat. M0209, USA), RNase H (NEB, cat. M0297, USA), and dUTP solution (Thermo Fisher, cat. R0133, USA). A single A-base was then added to the blunt ends of the DNA fragments to prepare them for ligation to indexed adapters. Each adapter contained a T-base overhang to facilitate the ligation to the A-tailed DNA fragments. Single- or dual-indexed adapters were ligated to the fragments, followed by size selection using AMPureXP beads. After the U-labeled second-strand DNA was treated with the heat-labile UDG enzyme (NEB, cat. M0280, USA), the ligated products were amplified by PCR under the following conditions: initial denaturation at 95 °C for 3 min; 8 cycles of denaturation at 98 °C for 15 s, annealing at 60 °C for 15 s, and extension at 72 °C for 30 s; and a final extension at 72 °C for 5 min. The final cDNA library had an average insert size of 300 ± 50 bp. Finally, paired-end sequencing (PE150, 2 × 150 bp) was performed on an Illumina Novaseq™ 6000 according to the manufacturer’s recommended protocol.
The raw sequencing data were subjected to quality control, including the removal of reads containing adaptor contamination using Cutadapt software (v1.9). After removing low-quality and undetermined bases, we used HISAT2 software (v2-2.0.4)77 to map the reads to the genome. The mapped reads of each sample were assembled using StringTie (v1.3.4 d)78 with default parameters. Then, all the transcriptomes from all the samples were merged to reconstruct a comprehensive transcriptome using gffcompare software. After the final transcriptome was generated, StringTie and Ballgown were used to estimate the expression levels of all the transcripts, and the expression levels of the mRNAs were determined by calculating the FPKM.
To identify differentially expressed mRNAs, the R package DESeq279 was used to select transcripts with a fold change >2 or <0.5 and a P value < 0.05. GO enrichment and KEGG enrichment analyses were performed on the differentially expressed mRNAs using specialized tools80,81.
Statistics and reproducibility
All data were presented in the form of mean ± standard deviation (SD). Statistical analysis was performed using GraphPad Prism 8.3.0. An unpaired t-test was used to analyze the differences between the two groups. The significance threshold was set at P < 0.05. *P < 0.05, **P < 0.01, ***P < 0.001. For all experiments, biological replicates were used, with each replicate representing a separate animal. For the metagenomic study, five MSTN-KO cattle and three WT cattle were included, and for the muscle transcriptomic study, three cattle were included per group.
Data availability
All data supporting the findings of this study are available within the paper and as Supplementary Information. Source data for figures can be found in Supplementary Data 1. The metagenome sequence data of the rumen have been deposited in the Sequence Read Archive (SRA) under the accession number PRJNA1165944. The transcriptome sequence data has been deposited in the SRA under the accession number PRJNA1181640.
References
Ticinesi, A. et al. Gut microbiota, muscle mass and function in aging: a focus on physical frailty and sarcopenia. Nutrients 11, 1633 (2019).
Giron, M., Thomas, M., Dardevet, D., Chassard, C. & Savary-Auzeloux, I. Gut microbes and muscle function: can probiotics make our muscles stronger? J. Cachexia Sarcopenia Muscle 13, 1460–1476 (2022).
Chen, L. H. et al. Probiotic supplementation attenuates age-related sarcopenia via the gut-muscle axis in SAMP8 mice. J. Cachexia Sarcopenia Muscle 13, 515–531 (2022).
Bäckhed, F., Manchester, J. K., Semenkovich, C. F. & Gordon, J. I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl Acad. Sci. USA 104, 979–984 (2007).
Bischoff, S. C. et al. Towards a multidisciplinary approach to understand and manage obesity and related diseases. Clin. Nutr. 36, 917–938 (2017).
Canfora, E. E., Jocken, J. W. & Blaak, E. E. Short-chain fatty acids in control of body weight and insulin sensitivity. Nat. Rev. Endocrinol. 11, 577–591 (2015).
Shen, H., Lu, Z., Xu, Z., Chen, Z. & Shen, Z. Associations among dietary non-fiber carbohydrate, ruminal microbiota and epithelium G-protein-coupled receptor, and histone deacetylase regulations in goats. Microbiome 5, 123 (2017).
Hernández, M. A. G., Canfora, E. E., Jocken, J. W. E. & Blaak, E. E. The Short-Chain Fatty Acid Acetate in Body Weight Control and Insulin Sensitivity. Nutrients 11, 1943 (2019).
Tang, G. et al. Butyrate ameliorates skeletal muscle atrophy in diabetic nephropathy by enhancing gut barrier function and FFA2-mediated PI3K/Akt/mTOR signals. Br. J. Pharm. 179, 159–178 (2022).
Parada Venegas, D. et al. Short chain fatty acids (SCFAs)-mediated gut epithelial and immune regulation and its relevance for inflammatory bowel diseases. Front. Immunol. 10, 277 (2019).
Balestrino, M. & Adriano, E. Beyond sports: efficacy and safety of creatine supplementation in pathological or paraphysiological conditions of brain and muscle. Med. Res. Rev. 39, 2427–2459 (2019).
Fu, S. K. et al. Effect of daily oral lactobacillus plantarum PS128 on exercise capacity recovery after a half-marathon. Nutrients 13, 4023 (2021).
Ma, N. & Ma, X. Dietary amino acids and the gut-microbiome-immune axis: physiological metabolism and therapeutic prospects. Compr. Rev. Food Sci. Food Saf. 18, 221–242 (2019).
Martínez-Álvaro, M. et al. Bovine host genome acts on rumen microbiome function linked to methane emissions. Commun. Biol. 5, 350 (2022).
Zhang, A. et al. Cadmium exposure modulates the gut-liver axis in an Alzheimer’s disease mouse model. Commun. Biol. 4, 1398 (2021).
Wang, W. et al. Tauroursodeoxycholic acid inhibits intestinal inflammation and barrier disruption in mice with non-alcoholic fatty liver disease. Br. J. Pharm. 175, 469–484 (2018).
Ferrucci, L. & Fabbri, E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat. Rev. Cardiol. 15, 505–522 (2018).
Van Hul, M. & Cani, P. D. The gut microbiota in obesity and weight management: microbes as friends or foe? Nat. Rev. Endocrinol. 19, 258–271 (2023).
Fang, H. et al. Effects of dietary inclusion of shrimp paste on growth performance, digestive enzymes activities, antioxidant and immunological status and intestinal morphology of hybrid snakehead (Channa maculata ♀ × Channa argus ♂). Front. Physiol. 10, 1027 (2019).
Oliphant, K. & Allen-Vercoe, E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome 7, 91 (2019).
Peng, H., Wang, Y. & Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 39, 6747–6756 (2020).
Yoneshiro, T. et al. BCAA catabolism in brown fat controls energy homeostasis through SLC25A44. Nature 572, 614–619 (2019).
Macke, E., Callens, M., De Meester, L. & Decaestecker, E. Host-genotype dependent gut microbiota drives zooplankton tolerance to toxic cyanobacteria. Nat. Commun. 8, 1608 (2017).
Abulizi, N. et al. Gut mucosal proteins and bacteriome are shaped by the saturation index of dietary lipids. Nutrients 11, 418 (2019).
Wang, Y. N. et al. Effects of probiotics and prebiotics on intestinal microbiota in mice with acute colitis based on 16S rRNA gene sequencing. Chin. Med. J. (Engl.) 132, 1833–1842 (2019).
Hjorth, M. F. et al. Prevotella-to-Bacteroides ratio predicts body weight and fat loss success on 24-week diets varying in macronutrient composition and dietary fiber: results from a post-hoc analysis. Int. J. Obes. 43, 149–157 (2019).
Jacky, D. et al. Effects of OsomeFood Clean Label plant-based meals on the gut microbiome. BMC Microbiol. 23, 88 (2023).
Tett, A., Pasolli, E., Masetti, G., Ercolini, D. & Segata, N. Prevotella diversity, niches and interactions with the human host. Nat. Rev. Microbiol. 19, 585–599 (2021).
Gu, N. et al. Prevotella copri transplantation promotes neurorehabilitation in a mouse model of traumatic brain injury. J. Neuroinflamm. 21, 147 (2024).
Wang, D. D. et al. The gut microbiome modulates the protective association between a Mediterranean diet and cardiometabolic disease risk. Nat. Med. 27, 333–343 (2021).
Brooke, C. G., Najafi, N., Dykier, K. C. & Hess, M. Prevotella copri, a potential indicator for high feed efficiency in western steers. Anim. Sci. J. 90, 696–701 (2019).
Xue, M. Y., Sun, H. Z., Wu, X. H., Liu, J. X. & Guan, L. L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 8, 64 (2020).
Jiang, B. et al. Multi-omics reveals the mechanism of rumen microbiome and its metabolome together with host metabolome participating in the regulation of milk production traits in dairy buffaloes. Front. Microbiol. 15, 1301292 (2024).
Petersen, L. M. et al. Community characteristics of the gut microbiomes of competitive cyclists. Microbiome 5, 98 (2017).
Morton, E. R. et al. Variation rural African gut microbiota is strongly correlated with colonization by entamoeba and subsistence. PLoS Genet. 11, e1005658 (2015).
Metges, C. C. Contribution of microbial amino acids to amino acid homeostasis of the host. J. Nutr. 130, 1857s–1864s (2000).
Hou, J., Xiang, J., Li, D., Liu, X. & Pan, W. Gut microbial response to host metabolic phenotypes. Front. Nutr. 9, 1019430 (2022).
Eckburg, P. B. et al. Diversity of the human intestinal microbial flora. Science 308, 1635–1638 (2005).
Metges, C. C., Petzke, K. J. & Hennig, U. Gas chromatography/combustion/isotope ratio mass spectrometric comparison of N-acetyl- and N-pivaloyl amino acid esters to measure 15N isotopic abundances in physiological samples: a pilot study on amino acid synthesis in the upper gastro-intestinal tract of minipigs. J. Mass Spectrom. 31, 367–376 (1996).
Lynch, C. J. & Adams, S. H. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat. Rev. Endocrinol. 10, 723–736 (2014).
Holstein, I. et al. Post-transcriptional regulation of MRTF-A by miRNAs during myogenic differentiation of myoblasts. Nucleic Acids Res. 48, 8927–8942 (2020).
Harper, A. E., Miller, R. H. & Block, K. P. Branched-chain amino acid metabolism. Annu. Rev. Nutr. 4, 409–454 (1984).
Kamei, Y., Hatazawa, Y., Uchitomi, R., Yoshimura, R. & Miura, S. Regulation of skeletal muscle function by amino acids. Nutrients 12, 261–271 (2020).
Liu, X. et al. Prevotella copri alleviates sarcopenia via attenuating muscle mass loss and function decline. J. Cachexia Sarcopenia Muscle 14, 2275–2288 (2023).
Qiao, S. et al. Gut Parabacteroides merdae protects against cardiovascular damage by enhancing branched-chain amino acid catabolism. Nat. Metab. 4, 1271–1286 (2022).
Li, W., Li, L., Yang, F., Hu, Q. & Xiong, D. Correlation between gut bacteria Phascolarctobacterium and exogenous metabolite α-linolenic acid in T2DM: a case-control study. Ann. Transl. Med. 10, 1056 (2022).
Bitar, M. S. et al. Hydrogen sulfide donor NaHS improves metabolism and reduces muscle atrophy in type 2 diabetes: implication for understanding sarcopenic pathophysiology. Oxid. Med. Cell Longev. https://doi.org/10.1155/2018/6825452 (2018).
Neinast, M. D. et al. Quantitative analysis of the whole-body metabolic fate of branched-chain amino acids. Cell Metab. 29, 417–429.e414 (2019).
Xiao, F. et al. Leucine deprivation increases hepatic insulin sensitivity via GCN2/mTOR/S6K1 and AMPK pathways. Diabetes 60, 746–756 (2011).
Lee, J. et al. BCAA supplementation in mice with diet-induced obesity alters the metabolome without impairing glucose homeostasis. Endocrinology 162, bqab062 (2021).
Nie, Q. et al. Bioactive dietary fibers selectively promote gut microbiota to exert antidiabetic effects. J. Agric. Food Chem. 69, 7000–7015 (2021).
Jindal, A. & Jagdish, R. K. Sarcopenia: ammonia metabolism and hepatic encephalopathy. Clin. Mol. Hepatol. 25, 270–279 (2019).
Roberson, P. A. et al. Protein supplementation throughout 10 weeks of progressive run training is not beneficial for time trial improvement. Front. Nutr. 5, 97 (2018).
Kato, H. et al. Leucine-enriched essential amino acids attenuate muscle soreness and improve muscle protein synthesis after eccentric contractions in rats. Amino Acids 47, 1193–1201 (2015).
Yu, D. et al. The adverse metabolic effects of branched-chain amino acids are mediated by isoleucine and valine. Cell Metab. 33, 905–922.e906 (2021).
Drummond, M. J., Dreyer, H. C., Fry, C. S., Glynn, E. L. & Rasmussen, B. B. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J. Appl Physiol. (1985) 106, 1374–1384 (2009).
Gu, M. et al. Combined transcriptome and metabolome analysis of smooth muscle of myostatin knockout cattle. Int. J. Mol. Sci. 24, 8120 (2023).
Burkovski, A. & Krämer, R. Bacterial amino acid transport proteins: occurrence, functions, and significance for biotechnological applications. Appl. Microbiol. Biotechnol. 58, 265–274 (2002).
Smith, R. H., Glendinning, L., Walker, A. W. & Watson, M. Investigating the impact of database choice on the accuracy of metagenomic read classification for the rumen microbiome. Anim. Microbiome 4, 57 (2022).
Blanco-Míguez, A. et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 41, 1633–1644 (2023).
Nguyen, T. T. M. et al. Comparative analysis of functional diversity of rumen microbiome in bison and beef heifers. Appl. Environ. Microbiol. 89, e0132023 (2023).
Zhao, Y. et al. Growth traits and sperm proteomics analyses of myostatin gene-edited Chinese yellow cattle. Life (Basel) 12, 627 (2022).
Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12 (2011).
Pertea, G. fqtrim: v0.9.4 release. (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Zimin, A. V. et al. Mis-assembled “segmental duplications” in two versions of the Bos taurus genome. PLoS ONE 7, e42680 (2012).
Zhou, J. et al. Discovering copy number variation in dual-purpose XinJiang brown cattle. Front. Genet. 12, 747431 (2021).
Merchant, S., Wood, D. E. & Salzberg, S. L. Unexpected cross-species contamination in genome sequencing projects. PeerJ 2, e675 (2014).
Peng, Y., Leung, H. C., Yiu, S. M. & Chin, F. Y. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428 (2012).
Zhu, W., Lomsadze, A. & Borodovsky, M. Ab initio gene identification in metagenomic sequences. Nucleic Acids Res. 38, e132 (2010).
Li, W. & Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006).
Jensen, L. J. et al. eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 36, D250–D254 (2008).
Pang, Z. et al. MetaboAnalyst 5.0: narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 49, W388–w396 (2021).
Smith, C. A., Want, E. J., O’Maille, G., Abagyan, R. & Siuzdak, G. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 78, 779–787 (2006).
Kuhl, C., Tautenhahn, R., Böttcher, C., Larson, T. R. & Neumann, S. CAMERA: an integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 84, 283–289 (2012).
Wen, B., Mei, Z., Zeng, C. & Liu, S. metaX: a flexible and comprehensive software for processing metabolomics data. BMC Bioinforma. 18, 183 (2017).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Young, M. D., Wakefield, M. J., Smyth, G. K. & Oshlack, A. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11, R14 (2010).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484 (2008).
Acknowledgements
This study was supported by National Natural Science Foundation of China (32360837, 32341052), Scientific and Technological Innovation 2030 (2023ZD0404803), Inner Mongolia Open Competition Projects (2022JBGS0025), Inner Mongolia Science and Technology Leading Team (2022LJRC0006), Inner Mongolia Science and Technology Major Projects (2021ZD0009, 2021ZD0008, 2022ZD0008, 2023KJHZ0028), Inner Mongolia Young Talents Projects (NJYT23138), Inner Mongolia Natural Science Foundation (2023MS03004), Central Government Guides Development (2022ZY0212), National Agricultural Science and Technology Project (NK2022130203), Collaborative Innovation among Universities in Hohhot (XTCX2023-06), Ministry of Education Engineering Centre Project (JYBGCSYS2022), and Xinjiang Uygur Science and Technology Major Project (2023A02011-1).
Author information
Authors and Affiliations
Contributions
L.Y. and G.L. conceived and designed the experiment. C.H. drafted the paper. C.H. performed bioinformatics analyses. C.H., Z.H., L.B., and J.L. performed the experiments and interpreted the result data. X.L., Y.Z., C.B., G.S., L.Y., and G.L. revised the paper. L.Y. and G.L. contributed resources and funding. All authors agree to be accountable for all aspects of work, ensuring integrity and accuracy.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All the authors have agreed on the publication.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Sabina La Rosa and David Favero. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hai, C., Hao, Z., Bu, L. et al. Increased rumen Prevotella enhances BCAA synthesis, leading to synergistically increased skeletal muscle in myostatin-knockout cattle. Commun Biol 7, 1575 (2024). https://doi.org/10.1038/s42003-024-07252-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-07252-9
