
Abstract
Abnormal chromosome segregation (ACS) in preimplantation embryos causes miscarriages. For a normal pregnancy, it is necessary to reduce ACS occurrences in embryos. However, the causes of such abnormalities are unclear because no method to extract the segregated chromosomes from the blastomeres for detailed analysis. This study attempted to extract micronuclei derived from segregated chromosomes of mouse embryos. Some micronuclei in blastomeres were bound to the nucleus by DNA cross-links, some were bound to tubulin, and about half of the micronuclei had major satellite regions. By depolymerizing the cytoskeleton of blastomeres with cytochalasin B and colcemid, some micronuclei could be extracted from blastomeres of ACS embryos using a glass needle of a micromanipulator. DNA-sequencing results of each extracted micronucleus revealed that chromosomes in micronuclei were randomly selected, usually only one, and often contained a portion rather than the full length of the chromosome. This study allows a detailed analysis of micronuclei and facilitates the mechanism of the causes of ACS in embryos.
Similar content being viewed by others


Introduction
Chromosome aberrations in embryos are considered a possible cause of low birth rates; thus, to improve birth rates, research at infertility clinics focused on its detection and suppression1,2. A cause of chromosome aberrations in early embryos is abnormal chromosome segregation (ACS) and micronuclei formation from the abnormal chromosomes3,4,5. Research in the underlying causes of chromosome aberrations have been conducted primarily in cancer cells because of their availability. In such cells, micronuclei are a common outcome of cell division defects, including mitotic errors that missegregate intact chromosomes and errors in DNA replication or repair of chromosome fragments6,7. Zhang et al. reported that after mitosis, chromosomes from micronuclei can be reincorporated into daughter nuclei, potentially integrating mutations from the micronucleus into the genome8. However, unlike cancer cells, embryos constitute a limited sample for analysis, thus making studies of abnormal chromosomes in embryos extremely difficult9,10,11,12. If micronuclei of embryos are also reincorporated into blastomeres, as are cancer cells, embryos with ACS or micronuclei are highly likely to cause miscarriage after transfer into the recipient mother13,14,15.
Micronuclei vary in size; although large micronuclei can sometimes be observed with an ordinary microscope, many are difficult to find16,17. Basic research in animal embryos involves DNA staining and fluorescence microscopy using ultraviolet (UV) lamps18; however, human embryos should ideally be studied noninvasively19. In contrast, the biopsy technique collected a few blastomeres from an embryo, and next-generation sequencing (NGS) analysis can be used to determine the normality of the examined cells. However, this method not only does the biopsy and damages the embryo, but it also does a biopsy of only a portion of the embryo and does not necessarily reflect the normality of the entire embryo20. If collecting some blastomeres from embryos, it provides the average copy number of the karyotype, which could be 2n even when there is an error (i.e., one blastomere is 1n, and the other is 3n)15,21.
Clues for finding abnormal embryos include the presence of multiple nuclei large enough to be discernible in brightfield microscopy and the degree of irregular cell division by time-lapse observation16,17. However, in some previous studies, offspring have been born from zygotes with three or even four pronuclei22,23, making it difficult to accurately find abnormal embryos by these methods. In mouse studies, fluorescent staining of live embryos allows detecting even tiny micronuclei. Such studies showed that some ACS embryos can develop into normal offspring5,15. Thus, not only are ACS embryos difficult to find, but some can develop into normal offspring, making it difficult to strictly select them before transference to the mother. Therefore, to obtain healthy offspring without affecting the mother, it is necessary to clarify the mechanism of the incidence of ACS or micronuclei in embryos and reduce the number of ACS embryos rather than selecting only normal embryos.
Until now, results obtained from ACS and micronuclei have been limited by the need to stain the whole ACS embryo. NGS analysis of single blastomeres and micronuclei sequencing in embryos are possible10,24, but distinguishing between nuclei and micronuclei if both are contained in the same cell and cannot characterize micronuclei alone is difficult. Therefore, nothing has yet been determined as to whether specific chromosomes are selected for the micronucleus or randomly. It is also unknown whether the micronucleus contains the entire length of a chromosome or part of a chromosome, and if so, which part and whether it is part of more than one chromosome. If the details of the micronucleus could be examined, the ACS mechanism would be better understood, and it may eventually be possible to reduce the number of ACS embryos.
In this study, the morphology and structure of micronuclei of mouse ACS embryos at the two-cell stage were examined. To increase the occurrence of ACS embryos, embryos were produced by ICSI using freeze-dried (FD) spermatozoa25. The FD method allows the preservation of spermatozoa at room temperature, and many healthy offspring could be obtained from sperm after preservation for >1 year26. However, for an unknown reason, embryos fertilized with FD sperm have a high ACS incidence at the mitotic one-cell stage and produce some micronuclei at the two-cell stage27. A method of extracting micronuclei from blastomeres of two-cell embryos was established using a micromanipulator, and DNA-sequencing (DNA-seq) was attempted to analyze the extracted micronuclei.
Results
Classification of micronucleus at the two-cell stage
Initially, oocytes were fertilized with FD sperm by ICSI. Then, DNA damage in some zygotes was examined by γH2Ax immunostaining. Male pronuclei derived from FD sperm showed strong DNA damage compare to fresh sperm (Supplementary Fig. 1 and Supplementary Table 1). Some other zygotes were injected with H2B-mCherry mRNA. On the next day, two-cell stage embryos with visualized nuclei were rotated on inverted microscopy using a micromanipulator to observe the entire embryo in detail (Fig. 1a). First, to confirm that the staining method with H2B-mCherry did not affect chromosome segregation aberrations, 2-cell stage embryos stained with Hoechst or DAPI were compared with H2B-mCherry stained embryos (Supplementary Fig. 2 and Supplementary Table 2). The number of micronuclei did not differ between H2B-mCherry-RNA and DAPI staining but was partially lower than that obtained with Hoechst staining (Supplementary Table 2). However, when live embryos were simultaneously observed with brightfield and fluorescence, micronuclei stained with red H2B-mCherry could be better detected than with the low-concentration Hoechst stain, which is not toxic to embryos (Supplementary Fig. 2); a detailed examination of PFA fixed DAPI stained embryos showed similar results to our method. The results indicated that H2B-mCherry-RNA staining is more accurate in measuring micronuclei than Hoechst, although the possibility that this may affect the appearance of micronuclei at the 2-cell stage cannot be completely ruled out as RNA is injected into the 1-cell stage embryo.

a mRNA was injected into one-cell stage zygotes, and ACS embryos were classified by fluorescence observation at the two-cell stage. ES cells were also established from the classified ACS embryos. b The relationship between micronuclei and tubulin was observed by live-cell imaging. c The centromeres of micronuclei were observed by FISH. d A method was developed to extract micronuclei from two-cell stage embryos, and NGS analysis of the extracted micronuclei was performed by DNA-seq.
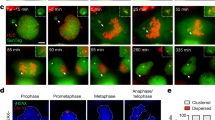
About half (45.3%) of the embryos had micronuclei and were determined to be ACS embryos Supplementary Table 3). ACS embryos were classified into three types based on the relationship between the micronucleus and nucleus, and perspective from which the micronucleus was intended to be extracted by micropipette (Fig. 2a). The most common ACS embryos, in which the micronucleus was in the cytoplasm of blastomeres disorganizedly and relatively distant from the nucleus (20.3%; Fig. 2b and Supplementary Table 3), are called “independent floating micronucleus“ (IFM) and “IFM embryos.” The next common embryos are those whose micronuclei were present in the same location in both blastomeres of the two-cell stage embryo (16.0%). This type of micronucleus is called “equal positioned micronuclei“ (EPM) or “EPM embryos.” The third type is the micronucleus located immediately adjacent to the nucleus (9.0%). This type of micronucleus is called “nuclear binding micronucleus” (NBM). The size and number of micronuclei differed in this type of ACS embryo. Some ACS embryos had a mixture of micronuclei classified into the above three categories. In such cases, micronuclei were classified by their most prominent features.

a Histones in two-cell stage embryos were observed by fluorescent microscopy, and micronuclei were classified into three categories based on their positional relationship to the nucleus. ACS Abnormal chromosome segregation, IFM Independent floating micronucleus, EPM Equal positioned micronuclei, NBM Nuclear binding micronuclei, NCS Normal chromosome segregation. Scale bar: 10 µm. b Rate of ACS embryos in each type of classification. c Blastocysts developed from each type of ACS or NCS embryo after 3 days of culture. Bar, 50 µm. d Developmental potential of each type of ACS or NCS embryos to the blastocyst. The error bar indicates standard error. e Fluorescence observation was performed at the two-cell stage to detect micronuclei (e-1 and e-2). Two-cell stage embryos were cultured individually to the blastocyst stage. Embryos that developed into blastocysts (e-3) were transferred to an ES cell establishment medium to observe outgrowth (e-4) and establishment of ES cells. Established ES cells were examined for their chromosome number (e-5) and the normality of the cell lines. Bar, 10 µm (e-1–e-3) and 100 µm (e-4).
Developmental potential of each ACS embryo type to the blastocyst and establishment of ES cell lines
To examine the ability of ACS embryos classified into the above three types to develop into blastocysts, ACS embryos in the late two-cell stage were cultured for 3 days. The rate of IFM embryos (21.5%) developing into blastocysts was slightly higher than EPM embryos (9.1%) and NBM embryos (13.0%), but all types were significantly lower than in normal chromosome segregation (NCS) embryos (59.2%) (Fig. 2c, d and Table 1). Considering even embryos with severe chromosomal abnormalities may develop to blastocysts, the health and further developmental potential of embryos cannot be determined by the blastocyst development rate alone. Although the birth rate can be investigated after embryo transfer, assessing the implantation and early fetus developmental potential of each ACS embryo in mice is not possible because > 10 embryos are transferred to a single recipient female. To examine the proliferation of the blastocysts derived from ACS embryos, the outgrowth rate, ES cell establishment rate, and normality of the karyotype of ES cell lines were examined (Fig. 2e and Supplementary Table 4). Seventy-six ACS-derived and 182 randomly selected NCS-derived blastocysts were cultured on feeder cells, resulting in 7 (9.2%) and 54 (29.7%) attached to feeder cells and showing outgrowths, respectively. Subsequently, all seven outgrowths from ACS embryos and 31 randomly selected outgrowths from NCS embryos were passaged, and all outgrowths were established as cell lines with ES cell-specific shapes, respectively. However, ACS embryo-derived ES cell lines had longer passage intervals than NCS embryo-derived ES cell lines because more cells were killed by passage processing. Because all embryos in this experiment are live cell imaged at the two-cell stage and cultured individually, it is possible to retrospectively examine the relationship between the type of micronuclei at the two-cell stage and ES cell establishment rates (Supplementary Fig. 3). As a result, no relationship was found between the type of micronuclei at the two-cell stage of ACS embryos and the establishment of ES cells (Table 1). The karyotype of ES cells was examined, and the percentage of cells showing an abnormal karyotype of ≤50% was considered an abnormal cell line (Supplementary Table 4, each cell line were examined 27–38 metaphase spreads). As a result, 4 of 6 (66.7%) of ACS embryo-derived ES cell lines examined were abnormal, whereas 2 of 9 (22.2%) of NCS embryo-derived ES cell lines were judged as abnormal.
Observation of microtubule dynamics by live cell imaging
In case tubulin cross-linking to the micronuclei could hinder micronucleus extraction from blastomeres, live cell imaging was performed up to 48 h after fertilization to examine tubulin dynamics in detail. At the two-cell stage, tubulin cross-linking between both cell nucleus disappearance in NCS embryos averaged 9.7 h (minimum 2.5 h, maximum 17.3 h, n = 9), whereas ACS embryos averaged 13.2 h (minimum 3.5 h, maximum 20 h, n = 8) (Fig. 3a–c and Supplementary Table 5). Several ACS embryos were observed in which tubulin remained around the micronuclei after the disappearance of tubulin cross-linking between both cell (Fig. 3b). Despite the small number of embryos examined, our results indicate that tubulin cross-linkage needs to be removed before extracting the micronuclei.

a–c Tubulin dynamics were observed from the one-cell to the four-cell stage by live cell imaging. Histones are stained by H2B-mCherry, and tubulin is stained by α-tubulin-EGFP. a NCS embryo. b ACS embryo. Tubulin was still observed after 19 h (b-3). b-4 shows a high magnification of the b-3 square. c ACS embryo. Tubulin disappeared within 6 h. c-4 shows a high magnification of the c-3 square. d, e Major satellite regions of micronuclei at two-cell embryos were observed by DNA-FISH. DNA is stained by DAPI (blue), and the major satellite is stained by DNA-FISH (green). d-3 or e-3 shows high magnification of the d-2 or d-3 square, respectively. The arrow shows a micronucleus with or without a major satellite. Whole Posi: The portion of micronuclei stained by DAPI and the portion stained by DNA-FISH almost overlap. Partial Posi: Only a portion of the DAPI-stained part of the micronucleus is stained by DNA-FISH. Bar, 10 µm.
Presence of major satellite regions in the micronucleus
The fact that tubulin was bound to micronuclei suggested that micronuclei may contain kinetochore. Because kinetochores are formed on centromeres, the major satellite regions in the vicinity of the centromere were examined by DNA-fluorescence in situ hybridization (FISH) to determine the presence or absence of the kinetochore. A total of 161 micronuclei were examined in 60 ACS embryos (average 2.7 micronuclei/embryo; Supplementary Table 6). Fifty-seven (35.4%) micronuclei had major satellite regions throughout the micronucleus, and 24 (14.9%) micronuclei were only partially present (Fig. 3d and Supplementary Table 7). Eighty (49.7%) micronuclei had no major satellite regions at all (Fig. 3e). Even in the same ACS embryos, the presence or absence of major satellite regions in micronuclei varied, and there was no clear difference between the three types of micronucleus (Supplementary Table 7).
Micronuclei extraction from ACS embryos
To analyze micronuclei in more detail, micronuclei were extracted from the blastomere of two-cell ACS embryos. Because many micronuclei bind with tubulin, not only cytochalasin B (CB) was used as an inhibitor of actin polymerization but also several inhibitors of tubulin polymerization were added before attempting to extract micronuclei by a small glass pipette using a micromanipulator. When demecolcine, nocodazole, or colcemid was added to the medium, micronuclei were most easily extracted when colcemid was added (Fig. 4a, b). In contrast, the extraction rate of micronucleus was slightly higher when middle (29–34 h after ICSI) two-cell stage embryos were used (78.9–81.3%) compared to early (24–28 h after ICSI) two-cell stage embryos (71.4–76.5%; Supplementary Table 8). Therefore, from this point on, CB and colcemid were added to the medium, and micronuclei were extracted from middle or later two-cell stage embryos.

a, b Micronuclei extraction from ACS embryos at the two-cell stage. The arrow shows a micronucleus. c Rate of micronuclei extracted embryos by type of ACS embryos. Blue, embryos from which one micronucleus could be extracted; red, embryos that attempted to extract one micronucleus but simultaneously extracted another micronucleus from another blastomere; black, embryos that could not be extracted because the nucleus was also sucked into the pipette when the micronucleus was taken out. d, e If the micronucleus is located close to the nucleus, when the micronucleus is pulled, the nucleus moves with it and is sucked into the pipette. f–i Embryos from which micronuclei could not be extracted were fixed and stained to examine the tubulin binding and presence or absence of major satellite regions. Circles or arrows indicate micronuclei. Numbers indicate micronucleus ID. When attempting to extract micronuclei, the nucleus also moved (f, g), so micronuclei extraction was stopped, the binding of tubulin to micronuclei (h), and the presence of major satellite regions was examined (i). The right section shows high magnification. j–n When micronucleus could be extracted, those micronuclei were injected into enucleated or intact oocytes and fixed and stained as above. j–l Micronuclei injected into enucleated oocytes. The arrow shows a micronucleus. m Micronucleus in enucleated oocytes derived from IFM embryos. m-2 and m-3 shows a high magnification of the square. n Two micronuclei in enucleated oocyte derived from EPM embryos. n-2 and n-3 shows a high magnification of the square. The arrow shows a micronucleus. Bar, 10 µm. o, p The relationships among DNA bridge, tubulin binding (o), and presence/absence of major satellite regions (p) in micronuclei that could not be extracted from embryo (o-1 or p-1) and micronuclei that could be extracted from embryo (o-2 or p-2) are summarized.
The rate of ACS embryos from which micronuclei could be extracted by ACS embryo type was 75.8%, 48.1%, and 6.7% for IFM, EPM, and NBM, respectively (Fig. 4c and Supplementary Table 9). With IFM embryos, it was easy to extract a single micronucleus (Movie S1); however, in EPM embryos, when extracting one micronucleus, the micronucleus of another blastomere often entered the glass pipette through the cell membrane at the same time (Movie S2). All remaining ACS embryos could not be extracted from the micronucleus because the micronucleus and nucleus are strongly binding together. If the micronucleus is aspirated into the pipette, the nucleus also enters the pipette (Fig. 4d, e and Movie S3).
Analysis of ACS embryos from which micronuclei could not be extracted
If the blastomere nucleus is about to enter the pipette together with the micronucleus (Fig. 4f, g), the extraction process is interrupted, and the embryos are fixed and stained for detailed analysis (Fig. 4h, i and Supplementary Fig. 4). First, histone H2B and tubulin were examined for 33 micronuclei in 16 ACS embryos. As a result, 19 (57.6%) of micronuclei showed a narrow histone or DNA bridge between micronuclei and nuclei (Fig. 4h), but no tubulin binding was observed (Table 2 and Supplementary Table 10). Eleven (33.3%) micronuclei were bound only to tubulin, not to the nucleus by DNA bridge. Neither DNA nor tubulin was bound to the remaining 3 (9.1%) micronuclei (Fig. 4o-1). The two micronuclei formed a DNA bridge between each other and also bound to tubulin.
DNA-FISH was performed on the same embryos to observe the presence or absence of a major satellite region in micronuclei (Fig. 4i). Although this second staining was difficult compared to the first one, major satellite regions could be examined in 11 of the 16 above embryos. Twenty-six micronuclei could be examined from these 11 embryos, and major satellite regions were found in nine of the 26 (34.6%) micronuclei (Fig. 4p-1).
Analysis of ACS embryos from which micronuclei could be extracted
To examine differences between micronuclei extracted from the embryo and those that are not, micronuclei must be examined after extraction. However, the extracted micronuclei are too small to handle, so we decided to inject the extracted micronuclei into oocytes for further examination. Sixty-three micronuclei that could be extracted from ACS embryos (Fig. 4j) were injected into the enucleated or intact oocytes (Fig. 4k, l) to examine for tubulin and major satellite regions on micronuclei (Table 2 and Supplementary Tables 11, 12, 13). All micronuclei could be extracted due to the absence of a DNA bridge to the nucleus, but 12 (19.0%) micronuclei (Fig. 4o, right) were found to have tubulin binding. When two micronuclei extracted simultaneously from EPM embryos were injected into one oocyte, micronuclei were connected by tubulin (Figs. 4n and Supplementary Fig. 5). DNA-FISH was performed on the same oocytes to observe the presence or absence of major satellite regions in micronuclei as above, and major satellite regions were found in seven of the 18 (38.9%) micronuclei (Fig. 4p, right). The relationships among DNA bridge, tubulin binding, and presence/absence of major satellite regions in micronuclei not extracting the embryo and micronuclei extracting the embryo are summarized in Fig. 4o, p. When micronuclei could not be extracted from ACS embryos, the DNA bridge between micronuclei and nuclei or tubulin binding to micronuclei was the main cause, and the presence or absence of major satellite regions did not seem to affect micronuclei extraction.
Analysis of extracted micronuclei by DNA-seq
Finally, DNA-seq analysis was performed on the 21 micronuclei extracted from ACS embryos, and 18 were successfully analyzed for DNA-seq (Fig. 5 and Supplementary Table 14). Eleven of 18 (61.1%) micronuclei contained a single chromosome, 5 (27.8%) contained two chromosomes, 1 (5.6%) contained four chromosomes, and the last one (ID 18) contained all chromosomes, except X and Y chromosomes (Fig. 5b). ID18 micronucleus was collected as it was identified as a usual micronucleus, but the number of chromosomes and the site of detection were quite different from the other micronuclei, so only the other micronuclei are summarized. Twenty-five chromosomes were found in the 17 micronuclei analyzed; excluding the same type, 14 different chromosomes were found in the micronuclei (Fig. 5d). There was a tendency for larger-sized chromosomes to be included in micronuclei, but the largest chromosome (chromosome 1) was not found in any micronuclei. The smaller chromosomes tended to be their full length contained within the micronucleus (chromosome No. 15 or 18), but a micronucleus containing the full length of chromosome 2 (the second largest chromosome) was also found. Supplementary Table 14 shows the ratio (%) of the length of the detected chromosome to its original length. Overall, 3 of 25 (12%) chromosomes contained their full length, 11 (44%) had more than half, and 11 (44.0%) had less than half the length of chromosome (Fig. 5c). When a portion of a chromosome was included in the micronucleus, the position on the chromosome varied from near the telomere side, centromere side, and intermediate site (Supplementary Figs. 6 and 7).

a Results of DNA-seq analysis of numbers mn2, mn9, and mn14 micronucleus. The mn2 contains the full length of chromosome 15, mn9 contains a part of chromosome 7, and mn14 contains a part of chromosomes 6 and 8. b The number of chromosomes in each micronucleus is shown in the pie chart. c The length of the chromosomes in each micronucleus is shown in the pie chart. Chromosomes < 50% of the full length were designated as <1/2, those with 50% to less than 100% were designated as >1/2, and those containing the full length were designated as Full. d Type and number of chromosomes found in micronuclei. The x-axis shows the type of chromosomes, and the y-axis shows the number of chromosomes found.
Detecting whether the micronucleus originates from sperm or oocyte
In this experiment, ICSI was performed with FD sperm derived from the C3H strain to determine if micronuclei were of sperm origin. Because oocytes are derived from BDF1 (C57BL/6N × DBA/2), polymorphism analysis of C3H can determine whether micronuclei are of sperm or oocyte origin. As a result, among the 18 successfully analyzed cases, sequence reads with C3H polymorphism from sperm were detected in 16 cases from the DNA of micronuclei (Supplementary Table 14). However, in one case, only chromosome 19 was included in the micronucleus, and its origin was from the oocyte (ID 4). Furthermore, four chromosomes were included in the micronucleus in one case, of which three were from the sperm origin and one was from the oocyte (ID 17). For samples containing all autosomes, relatively long chromosomes were derived from sperm; however, due to the low number of reads containing polymorphisms for relatively short chromosomes, it was impossible to determine their parental origin (ID 18). Overall, the chromosomes contained in micronuclei are significantly biased toward being derived from sperm.
Discussion
Here, we succeeded in extracting micronuclei from ACS embryos at the two-cell stage. To the authors’ knowledge, this is the first time micronuclei have been extracted using a micropipette and analyzed. Chromosomes in micronuclei were randomly selected and often contained a portion of the chromosome rather than the full length of the chromosome. In the future, this micronucleus extraction technique will be used as a new tool to analyze abnormally segregated chromosomes and remove them from blastomeres of ACS embryos.
This study found three types of micronuclei: IFM, in which micronuclei exist independently of the nucleus; EPM, in which two micronuclei exist at approximately the same location in both blastomeres; and NBM, in which micronuclei exist immediately adjacent to the nucleus (Fig. 2a). Previous studies investigated this question by fixing cells and examining them in detail10,21,28. Although cell types, observation methods, and classification purpose differ from this study, micronuclei classification results were similar. This may suggest that this pattern is common to micronuclei regardless of the cell type or the reason for ACS incidence. Unlike fixed cells, there were also live embryos, allowing us to classify micronuclei while they were alive and examine the characteristics and developmental potential of each classification.
The blastocyst rate of ACS embryos was lower than that of NCS embryos regardless of the type of micronuclei, but the outgrowth rate from resulting blastocysts and the ES cell establishment rate were comparable to NCS embryos (Table 1). Mashiko et al. reported that embryos determined to have ACS embryos but could develop into blastocysts had a similar offspring rate after transfer as NCS embryos because karyotypically normal cells survived and contributed to the development15. However, two-thirds of ES cell lines derived from ACS embryos had abnormal karyotype cells in more than half of the cells examined. This suggested that even if ACS embryos could develop into blastocysts, some blastomeres remained chromosomally abnormal, as reported previously29, and ES cell lines were also established from those partially abnormal blastocysts. Perhaps chromosomal aberrant cells in blastocysts are eliminated during fetus development. In contrast, establishing ES cell lines does not require such strict gene expression, and cells with chromosomal abnormalities can survive, which may have reduced the normal chromosome ratio of ES cell lines.
Micronuclei extraction was relatively easy once the method was established in IFM and EPM embryos (Fig. 4c). However, it was very difficult to extract micronuclei from NBM embryos, and most micronuclei that could not be extracted from embryos were bound to nuclei or tubulin. In contrast, micronuclei extracted from the embryo were not only not bound to the nucleus but showed no binding to tubulin. Regarding the presence or absence of major satellite regions in micronuclei, there was no significant difference between micronuclei extracted or not extracted from the embryo, suggesting that whether micronucleus extraction depends on its binding to the nucleus or tubulin and not on the presence of major satellite region.
In general, major satellite regions are located near the centromere, kinetochores are formed on the centromere, and tubulin binds to the kinetochores. In this study, however, tubulin binding was observed in many micronuclei even when major satellite regions were undetectable, and kinetochores were thought to be absent (Supplementary Table 10). If kinetochores are present but major satellite regions are absent, the DNA in the micronucleus is severely damaged. Studies of cancer cells have shown that micronuclei have extensive DNA damage due to impaired replication mechanisms and DNA repair30,31. In monkey embryos, Daughtry et al. reported that micronuclei in embryos also undergo extensive DNA damage similar to that in micronuclei of cancer cells, and some micronuclei may have a fragmentation between major satellite regions and centromeres, and denominated this phenomena chromosome breakage (or DNA breakpoints)24.
In this study, micronuclei were extracted from ACS embryos, and DNA-seq analysis was performed on them alone. The number of micronuclei analyzed is still small; it is clear that no chromosomes are prone to be micronuclei and that micronuclei often contain only a portion of a chromosomes rather than the full length (Fig. 5). Although more detailed studies are needed in the future, in micronuclei of embryos, there may not be a specific point within the chromosome that is prone to tearing but rather random tearing, which is different in somatic cells6.
The method developed in this study can not only analyze micronuclei derived from sperm but also remove micronuclei from embryos, which may contribute to investigating the causes of damage to sperm caused by FD treatment and the improvement of the production rate of offspring. However, the tendency of micronuclei to appear may vary depending on the type of sperm DNA damage. A previous study using X-irradiated spermatids reported the presence of kinetochores in 15% of the micronuclei32, different from this experiment using FD sperm (50.3% in total; Supplementary Table 7). While FD sperm are more resistant to strong radiation33 and high temperatures34, they are more damaged by drying than freezing process. Perhaps the incidence of micronuclei and the type/number of chromosomes in micronuclei are greatly influenced by the type of sperm damage. Our micronucleus analysis techniques may help to clarify the characteristics of damaged sperm in more detail.
This study established a new technique for extracting micronuclei and proved that micronuclei can be analyzed. Micronuclei are incorporated into the nucleus during the next cell division, introducing DNA damage in the micronucleus into the nucleus30. If micronuclei can be extracted from ACS embryos, it can avoid incorporating abnormal DNA into the nucleus. In the future, we would like to investigate how embryos from which micronuclei have been extracted develop. In contrast, micronuclei of NBM embryos look like a hernia in DNA35. It may be possible to reduce the abnormality by pushing the DNA into the nucleus before it is damaged in the micronucleus.
Herein, FD sperm were used to efficiently develop techniques to extract and analyze micronuclei. In the future, similar analysis will be performed for ACS occurring when fresh sperm is used for fertilization, and determining the cause of ACS in embryos may find ways to prevent ACS from occurring during infertility treatment, and treat chromosomal abnormalities.
Materials and methods
Animals
Eight- to 12-week-old ICR female and male, B6D2F1 (C57BL/6N × DBA/2) female and male, and C3H/He male mice were obtained from the Shizuoka Laboratory Animal Center (Hamamatsu, Japan). On the day of the experiment or after having finished all experiments, mice were euthanized by cervical dislocation. All experiments were conducted according to the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Committee of Laboratory Animal Experimentation of Yamanashi University (reference no. A4-10). All experiments were performed in accordance with these regulations and guidelines, which followed the ARRIVE guidelines. All mice were kept under specific pathogen-free conditions, with controlled temperature (25 °C), relative humidity (50%), and photoperiod (14L-10D). Mice were fed a commercial diet and provided distilled water ad libitum. We have complied with all relevant ethical regulations for animal use.
Media
HEPES-CZB medium36 and CZB37 were used for oocyte/embryo manipulation and incubation in 5% CO2 at 37 °C, respectively. The HTF medium38 was used for FD spermatozoa.
Preparation of FD spermatozoa
Both epididymides were collected from ICR, BDF1 or C3H male mice, and ducts were cut with a pair of sharp scissors. A few drops of the dense spermatozoa mass were placed into a centrifuge tube containing 2 ml HTF medium and incubated for 30 min at 37 °C in 5% CO2. The spermatozoa concentration was measured, and 50 µl aliquots of the spermatozoa suspension were dispensed into glass ampoules. The ampoules were flash-frozen in LN2 and FD using an FDU-2200 freeze dryer (EYELA, Tokyo, Japan). The cork of the freeze dryer was opened for at least 3 h until all samples were completely dry. After drying, ampoules were sealed by melting the ampoule necks using a gas burner under vacuum, as described previously39.
Oocyte preparation
Female mice were superovulated by injecting 5 IU equine chorionic gonadotropin and 5 IU human chorionic gonadotropin after 48 h. Cumulus-oocyte complexes (COCs) were collected from the oviducts of females 14 to 16 h later and moved to a Falcon dish containing HEPES-CZB medium. To disperse the cumulus, COCs were transferred into a 50 µl droplet of HEPES-CZB medium containing 0.1% bovine testicular hyaluronidase for 3 min. Cumulus-free oocytes were washed twice and moved to a 20 µl droplet of CZB for culture.
mRNA synthesis
α-Tubulin-EGFP and H2B-mCherry cloned into pcDNA3.1-poly(A) vector were used as a template for in vitro transcription40. The plasmids were linearized by ApaI at 37 °C for 3 h. Afterward, the plasmid was purified with phenol/chloroform and precipitated with ethanol. Purified DNA was dissolved in nuclease-free water as template DNA for subsequent in vitro transcription using the RiboMax Large-scale RNA Production System T7 kit (p1300; Promega). To improve translational efficiency, Ribo m7G Cap Analog (P1711; Promega) was added to the in vitro transcription mixture. The produced mRNA was treated with DNase I to eliminate the template DNA, purified with phenol/chloroform, and precipitated with ethanol. For further purification, the mRNA dissolved in RNase-free water was passed through the MicroSpin G25 column. The purified mRNA was stored at −80 °C until use.
ICSI
ICSI was performed as described previously36,41. Just before starting ICSI, the neck of an ampoule was punctured, and 50 µl sterile distilled water was immediately added and mixed with a pipette. For microinjection of spermatozoa, 1 to 2 µl spermatozoa suspension was moved directly to the injection chamber. The spermatozoa suspension was replaced every 30 min during the ICSI procedure. Several piezo pulses separated the spermatozoa head from the tail, and the head was injected into the oocyte. The oocytes that survived ICSI were incubated in a CZB medium at 37 °C with 5% CO2. Pronucleus formation was checked at 6 h after ICSI.
Microinjection of mRNA
During fluorescence observation of embryos, a zygote-labeled histone fluorescent tag was prepared. Histone H2B-mCherry mRNA or α-Tubulin-EGFP mRNA was diluted with nuclease-free water to 25–50 ng/μL before use. mRNA was injected into the cytoplasm of the zygote after ICSI, as described previously13,42. Briefly, microinjection was performed in HEPES-CZB on an inverted microscope with a micromanipulator (Narishige, Tokyo, Japan). The zona pellucida and cytosolic membrane were penetrated with a piezo drive (PRIME Tech, Tokyo, Japan). Ten minutes after microinjection, oocytes were washed and cultured in CZB. The mRNA injected oocytes were incubated at 37 °C under 5% CO2 in air for at least 2 h after injection to allow time for protein production.
Gamma-H2Ax assay
Gamma-H2Ax foci formation was used as marker of DNA double-strand breaks in male and female pronuclei43, and histone H3K9 me2 signals were used to distinguish between female and male pronuclei. After ICSI, all specimens were fixed with 4% paraformaldehyde for 10 h (PFA; Wako Pure Chemical, Osaka, Japan) containing 0.2% Triton X at RT for 20 min and stored in a refrigerator in phosphate-buffered saline (PBS) supplemented with 1% (w/v) bovine serum albumin (BSA, Sigma-Aldrich) until staining. Primary antibodies for zygote immunostaining included the antiphospho-H2Ax (Ser139) rabbit polyclonal antibody (1:500; Millipore-Merck, Darmstadt, Germany) and antihistone H3 (dimethyl K9) mouse monoclonal antibody (1:500; Abcam, Cambridge, UK). The secondary antibodies used were Aan lexa Fluor 488-labeled goat antimouse IgG (1:500; Molecular Probes, Eugene, OR, USA) and Alexa Fluor 568-labeled goat antirabbit IgG (1:500; Molecular Probes). DNA was stained with 4′6-diamidino-2-phenylindole (2 µg/mL; Molecular Probes). The brightness of each male pronucleus was measured using ImageJ and subtracted from that of the zygote cytoplasm.
Detection of ACS
After ICSI using ICR-derived FD sperm, the surviving ICR oocytes were injected with histone H2B-mCherry mRNA to visualize their nuclei or micronuclei. The next day, two-cell stage embryos were placed in a glass-bottomed chamber on a micromanipulator, and micronuclei in both blastomeres were examined in detail with a fluorescence microscope (Olympus IX73, Tokyo, Japan, or Keyence BZ-X710, Tokyo, Japan), while embryos were rotated with a glass pipette. First, we compared the incidence of ACS by Hoechst or DAPI staining of two-cell stage embryos not injected with H2B-mCherry mRNA to confirm that the H2B-mCherry mRNA injection into the zygote did not affect ACS incidence at 2-cell stage embryos. Images were acquired in 2–3 channels (brightfield, 368 nm blue, and 488 nm red fluorescence). For each embryo, 80 fluorescent images were acquired with a 20× objective lens, 1 μm apart in the z-axis for optical sectioning. Since this experiment confirmed that H2B-mCherry mRNA injection did not affect the incidence of ACS, all subsequent H2B-mCherry mRNA injections into zygote were performed to observe ACS. ACS embryos were categorized into three groups: IFM, in which micronuclei exist independently of the nucleus; EPM, in which two micronuclei exist at approximately the same location in both blastomeres; and NBM, in which micronuclei exist immediately adjacent to the nucleus.
In vitro development and establishment of ES cell lines
Two-cell stage embryos were prepared as described above except for the mouse strain, in which BDF1 sperm and oocytes were used. After determining whether embryos were ACS or NCS embryos, they were cultured individually for 3 days and examined for blastocyst development. When embryos developed into blastocysts, they were treated with acid Tyrode solution to remove the zonae pellucidae and used to establish ES cell lines, as described previously44,45, with slight modifications. Embryos were placed in 96-multiwell dishes precoated with mouse embryonic fibroblasts in 20% Knock-out Serum Replacement (Invitrogen, Carlsbad, CA, USA) and 0.1 mg/ml adrenocorticotropic hormone (fragments 1–24; American Peptide Company, Sunnyvale, CA, USA). Proliferating outgrowths were dissociated using trypsin and replated to fibroblasts until stable cell lines emerged.
Karyotyping of ES cell lines
To increase the metaphase stage of ES cells, 10 µl/ml demecolcine (045-18761; Wako, Japan) was added to the medium and cultured for 2 h. Cells were detached by trypsin, exposed to 0.075 M KCl solution for 20 min, fixed with Carnoy’s solution, and applied onto the glass slides. To count the number of chromosomes, the glass slides were stained with 4′,6-diamidino-2-phenylindole (DAPI) and observed under a microscope.
Observation of microtubule dynamics by live cell imaging
Histone H2B-mCherry (25–50 ng/µl) and α-tubulin-EGFP (25–50 ng/µl) mRNA injected two-cell embryos, as described above, were transferred to drops of CZB medium on a glass-bottomed dish and placed in the incubator of live cell imaging system as same as above. The embryos were cultured from the two-cell to the four-cell stage. For time-lapse observations, images were taken over 48 h at 15 min intervals. At each time point, 51 fluorescent images were taken 2 µm apart in the z-axis for optical sectioning.
Detection of major satellite regions by DNA-FISH
The preparation of the probe was described in a previous study46. Two-cell stage embryos were fixed and permeabilized in PBS containing 4% paraformaldehyde (PFA) and 0.2% Triton X-100 PBS for 40 min. After washing with 0.1% BSA/2× saline sodium citrate (SSC), embryos were incubated consecutively in 0.1% Tween-20/2× SSC containing 10%, 20%, and 50% formamide for 5, 5, and 30 min, respectively. Embryos were transferred to the probe solution against major satellite repeats and covered with mineral oil to avoid reducing the volume by evaporation in glass dishes. The glass dishes where embryos were in the probe solution were heated at 83 °C for 30 min. Denatured embryos and the probe were subjected to hybridization at 37 °C overnight on glass dishes. Embryos were washed in 0.1% BSA/2× SSC at 42 °C and 4× SSC/0.2% Tween-20 (4× SSCT) after blocking with 4% BSA/4× SSC for 10 min and washing with 4× SSCT. To detect digoxygenin (Dig)-labeled probe hybridized with major satellite repeats, embryos were incubated with fluorescein isothiocyanate-conjugated anti-Dig mouse antibody (200-092-156; 1:200; Jackson ImmunoResearch) diluted in 4× SSCT at 4 °C overnight. The antibody was detected using Alexa Fluor 488-conjugated goat antimouse IgG antibody (A11001; 1:100; Life Technologies) diluted in 4× SSCT for 45 min at room temperature. After washing with 4× SSCT, embryos were transferred to a Vectashield anti-bleaching solution containing DAPI (Dojindo). The images were acquired using an FV1200 confocal laser microscope (Olympus).
Micronuclei extraction from blastomeres
Histone H2B-mCherry mRNA injected two-cell embryos, as described above, were transferred into a droplet of H-CZB containing 5 µg/ml CB with or without 0.05 µg/ml demecolcine, 3 µg/ml nocodazole, or 0.1 µg/ml colcemid on the microscope stage for micronuclei extraction. The embryo and nucleus/micronucleus were observed in brightfield microscopy and fluorescence using the halogen and UV lamps of the microscope simultaneously, and micronuclei were removed with a pipette. When the inner diameter of the glass pipette was <7 µm, it was difficult to suck the micronucleus into the glass pipette because the micronucleus was very tiny and slightly glowing inside the cytoplasm of the blastomere in dim light. Too much cytoplasm was often sucked into a glass pipette until sucking the micronucleus, causing the blastomere to rupture. The optimal inner diameter of the glass pipette that facilitated micronuclei extraction from the blastomere was 7 to 10 µm. Using these pipettes, micronuclei were aspirated into the pipette before the faint fluorescence emitted by micronuclei faded. Micronuclei were isolated at early (24–28 h after ICSI) or middle (29–34 h after ICSI) two-cell stages. If the nucleus of the blastomere was pulled by the micronucleus when attempting to suck the micronucleus into the pipette, the micronucleus was determined to be an unextracted micronucleus, and the micronucleus extraction process was interrupted; otherwise, even the nucleus will be extracted with the micronucleus at the same time. In contrast, if the micronucleus was pulled to the micronucleus of the neighboring blastomere when it was sucked into the pipette, the extraction continued, and both micronuclei were removed simultaneously.
Observation of micronuclei that could not or could be extracted
Micronuclei that could not be extracted were stained to observe tubulin and major satellite regions, as described above. However, heat treatment during hybridization caused a complete loss of mCherry fluorescence, so the nuclei were stained again, this time with propidium iodide.
In contrast, when micronuclei could be extracted, they were injected into intact or enucleated oocytes, as described above. Enucleation was performed as described previously. In brief, oocytes were transferred to a droplet (~10 µl) of HEPES-CZB containing 5 µg/ml CB placed under oil in the operating chamber of a microscope stage. The oocyte was held with an oocyte-holding pipette, and its zona pellucida was perforated by applying several piezoelectric pulses with the tip of an enucleation pipette. The metaphase II chromosome spindle complex, distinguished as a translucent spot in the ooplasm, was drawn into the pipette with a small amount of accompanying ooplasm and gently pulled away from the oocyte until the stretched cytoplasmic bridge was pinched off. Injection of micronuclei into oocyte/enucleated oocytes was performed similarly to somatic cell nuclear transfer.
DNA-seq analysis of extracted micronuclei
In this experiment, FD sperm of the C3H strain were injected into BDF1 oocytes to determine the origin of micronuclei. Because BDF1 oocytes were produced by C57BL/6N × DBA/2, if a C3H/He-specific single nucleotide polymorphism (SNP) is found in the micronucleus, it is of sperm origin. The sample of a single micronucleus in 1 µl solution was placed in a PCR tube and immediately frozen in liquid nitrogen. Micronuclei samples were stored at −80 °C in a deep freezer until DNA amplification. DNA amplification was performed according to the manufacturer’s manual using the Single Cell GenomiPhi DNA Amplification Kit (Sigma-Aldrich, Inc., St. Louis, MO, USA). The amplified DNA was purified using AMPure XP (Beckman Coulter, Inc., Indianapolis, IN, USA), and library preparation was carried out according to the manufacturer’s instructions for the Nextera DNA Flex Library Prep (Illumina, San Diego, CA, USA). The resulting library was subjected to sequencing analysis using the Illumina miniSeq system.
The obtained fastq files were mapped to the mouse reference genome (GRCm38) with repeat sequences masked using RepeatMasker using bowtie247. Data on mouse strain SNPs48 were obtained from the “Mouse Genomes Project“ (https://www.sanger.ac.uk/data/mouse-genomes-project/) released by The Wellcome Sanger Institute. The percentage of chromosomes detected was determined by dividing the full chromosome length, centromeres, and telomeres was removed from the total length of the chromosome.
Statistics and reproducibility
All experiments were repeated at least thrice. These studies were performed independently twice or thrice, and similar results were obtained irrespective of the experimentalists. The embryo development and micronucleus extraction rates were evaluated using χ2 tests. The possibility of the micronuclei being of male (C3H) origin was assessed by binominal test. P < 0.05 was considered to represent a statistically significant difference. The code for the SNP identification and analysis used in this work is available on GitHub from https://github.com/tkohda/SNP_micronuclei.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Microarray and NGS data have been deposited in BioProject database (accession number: PRJDB17957). Source data can be obtained in Supplementary Data 1.
References
Berntsen, S. et al. A systematic review and meta-analysis on the association between ICSI and chromosome abnormalities. Hum. Reprod. Update 27, 801–847 (2021).
Belva, F. et al. Chromosomal abnormalities after ICSI in relation to semen parameters: results in 1114 fetuses and 1391 neonates from a single center. Hum. Reprod. 35, 2149–2162 (2020).
Budrewicz, J. & Chavez, S. L. Insights into embryonic chromosomal instability: mechanisms of DNA elimination during mammalian preimplantation development. Front. Cell Dev. Biol. 12, 1344092 (2024).
Mozdarani, H. & Nazari, E. Cytogenetic damage in preimplantation mouse embryos generated after paternal and parental gamma-irradiation and the influence of vitamin C. Reproduction 137, 35–43 (2009).
Yamagata, K., Suetsugu, R. & Wakayama, T. Assessment of chromosomal integrity using a novel live-cell imaging technique in mouse embryos produced by intracytoplasmic sperm injection. Hum. Reprod. 24, 2490–2499 (2009).
Klaasen, S. J. et al. Nuclear chromosome locations dictate segregation error frequencies. Nature 607, 604–609 (2022).
Terradas, M., Martin, M., Tusell, L. & Genesca, A. Genetic activities in micronuclei: is the DNA entrapped in micronuclei lost for the cell? Mutat. Res. 705, 60–67 (2010).
Zhang, C. Z. et al. Chromothripsis from DNA damage in micronuclei. Nature 522, 179–184 (2015).
Allais, A. & FitzHarris, G. Absence of a robust mitotic timer mechanism in early preimplantation mouse embryos leads to chromosome instability. Development 149, dev200391 (2022).
Brooks, K. E. et al. Molecular contribution to embryonic aneuploidy and karyotypic complexity in initial cleavage divisions of mammalian development. Development 149, dev198341 (2022).
Vazquez-Diez, C. & FitzHarris, G. Correlative live imaging and immunofluorescence for analysis of chromosome segregation in mouse preimplantation embryos. Methods Mol. Biol. 1769, 319–335 (2018).
Yao, T. et al. Live-cell imaging of nuclear-chromosomal dynamics in bovine in vitro fertilised embryos. Sci. Rep. 8, 7460 (2018).
Yamagata, K., Suetsugu, R. & Wakayama, T. Long-term, six-dimensional live-cell imaging for the mouse preimplantation embryo that does not affect full-term development. J. Reprod. Dev. 55, 343–350 (2009).
Hatano, Y., Mashiko, D., Tokoro, M., Yao, T. & Yamagata, K. Chromosome counting in the mouse zygote using low-invasive super-resolution live-cell imaging. Genes Cells 27, 214–228 (2022).
Mashiko, D. et al. Chromosome segregation error during early cleavage in mouse pre-implantation embryo does not necessarily cause developmental failure after blastocyst stage. Sci. Rep. 10, 854 (2020).
Daughtry, B. L. & Chavez, S. L. Time-lapse imaging for the detection of chromosomal abnormalities in primate preimplantation embryos. Methods Mol. Biol. 1769, 293–317 (2018).
Rodrigues, M. A. et al. The in vitro micronucleus assay using imaging flow cytometry and deep learning. NPJ Syst. Biol. Appl. 7, 20 (2021).
Yamagata, K. et al. Fluorescence cell imaging and manipulation using conventional halogen lamp microscopy. PLoS ONE 7, e31638 (2012).
Tan, T. C. Y. et al. Non-invasive, label-free optical analysis to detect aneuploidy within the inner cell mass of the preimplantation embryo. Hum. Reprod. 37, 14–29 (2021).
Rhon-Calderon, E. A. et al. Trophectoderm biopsy of blastocysts following IVF and embryo culture increases epigenetic dysregulation in a mouse model. Hum. Reprod. 39, 154–176 (2024).
Vazquez-Diez, C., Yamagata, K., Trivedi, S., Haverfield, J. & FitzHarris, G. Micronucleus formation causes perpetual unilateral chromosome inheritance in mouse embryos. Proc. Natl. Acad. Sci. USA 113, 626–631 (2016).
Bredbacka, P., Capalbo, A., Kananen, K., Picchetta, L. & Tomas, C. Healthy live birth following embryo transfer of a blastocyst of tetrapronuclear (4PN) origin: a case report. Hum. Reprod. 38, 1700–1704 (2023).
Yalcinkaya, E., Ozay, A., Ergin, E. G., Oztel, Z. & Ozornek, H. Live birth after transfer of a tripronuclear embryo: an intracytoplasmic sperm injection as a combination of microarray and time-lapse technology. Turk. J. Obstet. Gynecol. 13, 95–98 (2016).
Daughtry, B. L. et al. Single-cell sequencing of primate preimplantation embryos reveals chromosome elimination via cellular fragmentation and blastomere exclusion. Genome Res. 29, 367–382 (2019).
Wakayama, T. & Yanagimachi, R. Development of normal mice from oocytes injected with freeze-dried spermatozoa. Nat. Biotechnol. 16, 639–641 (1998).
Kamada, Y. et al. Assessing the tolerance to room temperature and viability of freeze-dried mice spermatozoa over long-term storage at room temperature under vacuum. Sci. Rep. 8, 10602 (2018).
Wakayama, S. et al. Healthy offspring from freeze-dried mouse spermatozoa held on the International Space Station for 9 months. Proc. Natl. Acad. Sci. USA 114, 5988–5993 (2017).
Gisselsson, D. et al. Abnormal nuclear shape in solid tumors reflects mitotic instability. Am. J. Pathol. 158, 199–206 (2001).
Rubio, C. et al. Impact of chromosomal abnormalities on preimplantation embryo development. Prenat. Diagn. 27, 748–756 (2007).
Sommer, S., Buraczewska, I. & Kruszewski, M. Micronucleus assay: the state of art, and future directions. Int. J. Mol. Sci. 21 (2020). https://doi.org/10.3390/ijms21041534
Terradas, M., Martin, M. & Genesca, A. Impaired nuclear functions in micronuclei results in genome instability and chromothripsis. Arch. Toxicol. 90, 2657–2667 (2016).
Collins, B. W., Howard, D. R. & Allen, J. W. Kinetochore-staining of spermatid micronuclei: studies of mice treated with X-radiation or acrylamide. Mutat. Res. 281, 287–294 (1992).
Wakayama, S. et al. Evaluating the long-term effect of space radiation on the reproductive normality of mammalian sperm preserved on the international space station. Sci. Adv. 7, eabg5554 (2021).
Wakayama, S. et al. Tolerance of the freeze-dried mouse sperm nucleus to temperatures ranging from -196 degrees C to 150 degrees C. Sci. Rep. 9, 5719 (2019).
Maciejowski, J. & Hatch, E. M. Nuclear membrane rupture and its consequences. Annu. Rev. Cell Dev. Biol. 36, 85–114 (2020).
Kimura, Y. & Yanagimachi, R. Intracytoplasmic sperm injection in the mouse. Biol. Reprod. 52, 709–720 (1995).
Chatot, C. L., Ziomek, C. A., Bavister, B. D., Lewis, J. L. & Torres, I. An improved culture medium supports development of random-bred 1-cell mouse embryos in vitro. J. Reprod. Fertil. 86, 679–688 (1989).
Quinn, P. Enhanced results in mouse and human embryo culture using a modified human tubal fluid medium lacking glucose and phosphate. J. Assist. Reprod. Genet. 12, 97–105 (1995).
Yang, L. L. et al. A novel, simplified method to prepare and preserve freeze-dried mouse sperm in plastic microtubes. J. Reprod. Dev. 69, 198–205 (2023).
Yamagata, K. et al. Noninvasive visualization of molecular events in the mammalian zygote. Genesis 43, 71–79 (2005).
Torikai, K. et al. Removal of sperm tail using trypsin and pre-activation of oocyte facilitates intracytoplasmic sperm injection in mice and rats. J. Reprod. Dev. 69, 48–52 (2023).
Ooga, M. et al. Parental competition for the regulators of chromatin dynamics in mouse zygotes. Commun. Biol. 5, 699 (2022).
Fernandez-Capetillo, O., Lee, A., Nussenzweig, M. & Nussenzweig, A. H2AX: the histone guardian of the genome. DNA Repair 3, 959–967 (2004).
Wakayama, T. et al. Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science 292, 740–743 (2001).
Wakayama, S. et al. Mice cloned by nuclear transfer from somatic and ntES cells derived from the same individuals. J. Reprod. Dev. 51, 765–772 (2005).
Ooga, M., Suzuki, M. G. & Aoki, F. Involvement of DOT1L in the remodeling of heterochromatin configuration during early preimplantation development in mice. Biol. Reprod. 89, 145 (2013).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Keane, T. M. et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 477, 289–294 (2011).
Acknowledgements
We thank Y.Kanda for assistance in preparing this manuscript. This work was partially funded by the Research Fellowships of Japan Society for the Promotion of Science for Young Scientists to D.I. (JP20J23364); to M.O. (17K15394); to S.W. (23K08843) to T.W. (23K18124 and 24K01779); the Naito Foundation and Takahashi Industrial and Economic Research Foundation (189) to S.W.; Asada Science Foundation and the Canon Foundation (M20-0008) to T.W. The authors would like to thank Editage for the English language review.
Author information
Authors and Affiliations
Contributions
I.S. and T.W. conceived and designed the study. I.S., H.S., Y.K., H.N., D.I., S.W., M.O., T.Kasai, T.Kohda, and T.W. performed experiments, analyzed the data, and interpreted the results. I.S. and T.W. wrote the manuscript. All authors read and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
All animal experiments were conducted according to the Guide for the Care and Use of Laboratory Animals and approved by the Institutional Committee of Laboratory Animal Experimentation of Yamanashi University (reference no. A4-10). All experiments were performed in accordance with these regulations and guidelines, which followed the ARRIVE guidelines.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Wee Wei Tee and Ophelia Bu. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shibasaki, I., Sugiyama, H., Kamada, Y. et al. Extracting and analyzing micronuclei from mouse two-cell embryos fertilized with freeze-dried spermatozoa. Commun Biol 8, 6 (2025). https://doi.org/10.1038/s42003-024-07358-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-024-07358-0
This article is cited by
-
Monosodium glutamate enhances freeze-dried mouse sperm quality during room-temperature storage
Scientific Reports (2025)
