
Abstract
As global temperatures rise, heat-related chronic health disorders are predicted to become more prevalent. We tested whether a single exposure to acute heat illness, using a preclinical mouse model of exertional heat stroke (EHS), can induce late-emerging health disorders that progress into chronic disease. Following EHS, mice were followed for 3 months; after two weeks of recovery, half were placed on a Western diet to determine if previous EHS exposure amplifies the negative consequences of an atherogenic diet. When compared to sham exercise controls, EHS-exposed mice exhibit accelerated diet-induced obesity, develop low level cardiac hypertrophy, develop accelerated diet-induced liver steatosis, severe hypoproteinemia and a loss of metabolic flexibility in the myocardium. The latter is characterized by a shift towards predominant glucose metabolism and glycolysis. These results demonstrate that a single exposure to severe exertional heat illness can induce long-lasting and unexpected health consequences in mammals and increased vulnerability to secondary metabolic stressors.

Similar content being viewed by others

Introduction
In the US in 2023, nearly 120,000 patient-health care contacts were recorded as heat-related emergencies, with 2300 heat-related deaths1. However, counting immediate deaths and injuries due to heat may be an underestimate of its overall negative health effects. Epidemiological studies have followed patient histories after exposure and reported significantly increased risks of cardiovascular, renal and liver disease over the next 12–14 years2,3,4. The underlying causes of these long-term, late-emerging consequences are poorly understood and poorly documented. The highest risk is seen in patients exposed to the most severe form of heat injury, i.e. heat stroke2,3, but significant risks and persistent symptoms appear to remain after exposure to less severe forms of heat illness such as heat exhaustion2,4,5. An outstanding problem concerns whether these delayed outcomes reflect a late emergence of pre-existing disease due to the stress of heat exposure; or whether heat illness itself is causative in chronic disorders that emerge later in life. To address this, we employed an established pre-clinical mouse model of exertional heat stroke (EHS) developed over the past 12 years. The mice were followed over three months of recovery, a time equivalent of > 9 human years based on conservative comparisons of mouse and human life spans6.
In previous studies, we demonstrated acute multi-organ injury within hours after EHS exposure in mice. The injuries resemble the acute clinical sequelae seen in humans7,8, including rhabdomyolysis, kidney injury, liver injury, gastro-intestinal injury and pro-inflammatory immune responses9,10. The mice generally recover function within a few days. However, in later experiments, using metabolomics, we intermittently evaluated myocardial metabolic health over two weeks of recovery11. To our surprise, myocardial metabolism recovered as expected over the first few days, but by 9–14 days female mice exhibited severe metabolic disorders in the heart, observations we later confirmed in males12. These changes were characterized by shifts toward glucose metabolism, blocks in the tricarboxylic acid cycle, lipid accumulation and altered purine metabolism11. When studied at 1 month, the female hearts appeared to largely recover13 but male hearts were not evaluated for longer than two weeks until the current results reported here.
In trying to link these prolonged and evolving disorders to underlying mechanisms, we began to define functional phenotypes within organ systems that could point to origins of later dysfunction. For example, female mice exposed to EHS recovered limb skeletal muscle function and body weight within ~1 week, but if re-exposed to EHS at two weeks of recovery, they exhibited sustained atrophy and neuromuscular weakness suggesting a loss of resilience to a second stress14. Immune blood cells harvested from female EHS mice after one month of recovery exhibited profound immunosuppression15 and skeletal muscle satellite stem cells from EHS mice had a reduced ability to proliferate when cultured in isolation15, also suggesting a loss of resilience to subsequent injury. We considered the possibility that these changes may reflect underlying maladaptive epigenetic mechanisms caused by the severe heat stress exposure. This kind of maladaptive change can make tissues or organ systems more vulnerable to secondary stressors throughout life16. This concept was supported by evidence of significant changes in global DNA methylation occurring across broad ranges of the genome, found one month after EHS in monocytes15, skeletal muscle17 and myocardium13 in females. However, it was extremely difficult to directly associate these specific DNA methylation changes with loss of function over time.
We hypothesized that this loss of resilience in various tissues may underlie the long-term and delayed health effects and vulnerabilities observed in human populations following heat injury, as discussed above. However, we speculated that it may require a second stressor for the effects to manifest themselves into chronic health disorders. An example of this kind of effect can be found in the long-term responses to other forms of environmental or social stressors, particularly experienced early in life, which become potent epigenetic stimuli for increased susceptibility to later obesity and heart disease8,18,19. In this study we pursued this idea by exposing mice to EHS and then, after two weeks of recovery, to a Western diet lasting 10 weeks. Western diets are known to be a form of metabolic stress that raises the probability of developing hypertension, heart disease, liver disease and metabolic syndrome in mice20 and humans21.
Our results demonstrate that a single exposure to EHS in both male and female mice results in a greater susceptibility to slowly developing obesity, liver steatosis and myocardial metabolic disorders that are consistent with developing heart disease. These late observations are stronger in males but are consistent with the concept that severe heat stress in mammals can contribute to long-lasting vulnerabilities to life-threatening chronic health disorders.
Results
Exertional heat stroke exposure increases susceptibility to obesity
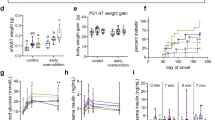
Adult male and female C57BL/6 mice were trained to exercise in a forced running wheel for three weeks. EHS exposure was induced by having the mice run in an incubator set to sex-specific environmental temperatures (Te); males = 34.5 °C; females = 37.5 °C. The protocol is outlined in Fig. 1a. The lower Te used for males was previously determined to compensate for their lower thermotolerance12,22. Results were compared against sham exercise controls (EXC), treated identically but with the absence of elevations in Te. Sixty-four mice were studied in 8 groups of 8, separated by sex, treatment, and diet (Fig. 1b). The specific Te used for each sex resulted in contrasting core temperature (Tc) profiles shown by typical examples in Fig. 1c. Overall, exercise duration was elevated in males using this protocol as compared to females (Fig. 1d), but males reached symptom limitation (i.e. unconsciousness) at a lower max core temperature (Tc,max) and were exposed to lower overall thermal loads, [i.e. time•(core temp.> 39.5 °C)]. Mice recovered consciousness and were upright within a few minutes after symptom limitation and exhibited normal mobility, drinking and grooming behavior within a few hours of exposure, with no outward signs of underlying health disturbances. No mice died or displayed apparent signs of morbidity at any stage of recovery in this study.

a Experimental Design. Mice were instrumented with telemetry transmitters in the abdominal cavity, allowed to recover and then were trained on voluntary running wheels in their cages. They were then exposed to exertional heat stroke (EHS) or a matched exercise control (EXC). The mice were returned to the vivarium on the same day and allowed to fully recover for two weeks prior to being placed on a standard diet (SD) or a Western diet (WD) for 10 weeks, after which they were euthanized. Cardiac ultrasound analyses were performed at the two-week recovery and near the end of the diet treatment. b Experimental groups of 8 for a total of 64 mice. All mice were contrasted against diet-matched exercise controls EXC. c Continuous core temperature measures in a typical male and a female mouse taken over the course of their EHS trials. d Grouped data for exercise duration in the heat, maximum core temperature and thermal load in both males and females (n = 16 per group, prior to diet treatment; note one mouse missing because of telemetry failure) (Statistical analyses sample T test and Wilcoxon (thermal area); all Benjamini-Hochberg (B-H) corrections < 0.2. e Total accumulated food intake measured in 1 week increments over the 10 wk diet intervention period (Two sample T tests; B-H corrections all < 0.05). f Body mass measured every 3 days over the course of the diet treatment (n = 8/group). Statistics: 2-way repeated measures MANOVA. Brackets and “Θ” at the far right of the graphs refer to repeated measures MANOVA significance, time X treatment; * symbolize within effects at specific time points or treatments. Means ± SD. (* or Θ) = P < 0.05; (** or ΘΘ < 0.01); (*** or ΘΘΘ <0.001); (**** or ΘΘΘΘ) <0.001. Panels (a, b): created with BioRender.com.
Two weeks after EHS, groups of mice were placed on either a Western diet (WD) or a standard vivarium diet (SD). The EHS-exposed mice exhibited greater total food intake over the 10-week diet phase, particularly on WD (Fig. 1e). All groups on WD also experienced greater weight gain during this period (Fig. 1f) but EHS mice on WD gained much more mass compared to EXC on WD. EHS mice on SD also gained significantly greater body mass over time compared to EXCs on SD (Fig. 1f), an effect more evident in males but significant in females. The data suggest that a single exposure to EHS in both male and female mice induces a long-lasting, whole body metabolic dysregulation, resulting in greater susceptibility to obesity.
Exertional heat stroke exposure affects myocardial health
Heart mass increased significantly in male mice on SD and WD after EHS when compared to diet-matched EXCs (Fig. 2a). When corrected for body size by expressing mass as a function of tibia length (Fig. 2a), male mice on both diets continued to show elevations in heart mass, whereas females did not. This suggests an ongoing cardiac hypertrophic response in males. Ventricular echocardiography revealed a significant elevation in posterior wall thickness and posterior wall velocity in female mice (but not males) at two weeks post EHS compared to time matched EXCs (Fig. 2b). This timepoint was previously identified using this mouse model as coincident with large metabolic abnormalities and local inflammation in female hearts after EHS11. However, after 10 wks on SD (12 wks post EHS) the female hearts exhibited a net loss of posterior wall thickness compared to their time matched controls (Fig. 2c), characterized by significantly narrower wall thickness in both diastole and systole. This suggests greater end-diastolic and end-systolic volumes, which would result in a thinner ventricular wall. In contrast, EHS males after 10 wks on SD exhibited elevations in %fractional shortening and posterior wall velocity but without changes in wall thickness (Fig. 2c). After 10 wks on WD, males showed a modest increase in anterior systolic wall thickness and females showed a modest increase in posterior systolic wall thickness (Fig. 2d).

a Cardiac mass and cardiac mass/tibia length in all groups at the end of the experiment (2 sample T or Wilcoxon (male WD) (B-H Q < 0.1). b Ventricular ultrasound measurements at the two-week recovery time point from exertional heat stroke (EHS) or exercise control (EXC) treatment. Measures include systolic and diastolic ventricular wall thickness, %fractional shortening and posterior wall velocity (n = 14–16/ group because all EHS or EXC mice received the same initial treatment up to this point (two female EXCs lost due to technical issues). c Ventricular ultrasound measures in standard diet (SD) mice, at the end of the study. d Ventricular ultrasound measures in Western diet (WD) mice at the end of the study. Means ± SD. a Statistical comparisons are two sample T test and Wilcoxon (male WD). b–d Statistical comparisons are two sample T test. * = P < 0.05,** < 0.01,*** < 0.001,**** < 0.001. All data sets tested for Benjamini-Hochberg B-H corrections and were < 0.18.
Untargeted metabolomics was performed on ventricular tissue from all mice at the end of the experiment. Traditional volcano plots (Fig. 3a, b and Supplemental Fig. S1, a, b) of metabolite responses, expressed as fold changes (FC) from diet- and sex-matched EXC data, are expressed on the X axis (log2FC) and as P values (-log10(P)) on the Y axis. Data that was considered significant is expressed in the shaded areas and colored symbols on the volcano plots when P < 0.05 and FC > ± 1.5. EHS mice on SD (Fig. 3a, b) exhibited many increased and decreased ventricular metabolites compared to matched EXCs. By far, the largest and most frequent changes were observed in males on SD (Fig. 3a). The results in mice on WD are shown in supplemental data Fig. S1. Mice on WD (both sexes) exhibited blunted metabolic responses to EHS when compared to EXC on WD. Data from Fig. 3 and Fig. S1 were re-evaluated using traditional forest plots which rank metabolic responses by P values (supplement Figs. S2 and S3). The specific fold changes and the corresponding Benjamini-Hochberg Q corrections for multiple testing (BH-Q) for each significant metabolite are listed for each metabolite that reached the P < 0.05 and FC > ± 1.5 criteria.

a, b Volcano plots showing significantly changed metabolites in each of the exertional heat stroke (EHS) groups compared to diet and sex-matched controls (EXC). The colored symbols display metabolites reaching a fold change (FC) of >±1.5 from their respective EXC and a significance level of P < 0.05. Red symbols in the red region were considered significantly increased, blue symbols in the blue region were considered significantly decreased. Benjamini Hochberg corrections for multiple sampling can be found in supplement Fig. S2. c a quantitative Venn diagram using Bioven software59 showing the number of biologically and statistically significant metabolites that overlap comparisons between EHS vs. EXC on SD (red), EHS vs. EXC on WD (blue), and EHS on WD vs. EXC on SD (yellow). In all cases metabolites selected had to meet the P < 0.05 and FC > ± 1.5 criteria.
In evaluating all of the data in all groups we found that the effects of WD on heart metabolomics were much more numerous and more significant than the effects of EHS. Powerful metabolic effects of WD on the heart have been previously reported23. We speculated that this would be a confounder for understanding the metabolomic changes specific to EHS because it would mask some changes that were influenced by both WD and EHS. We tested this by comparing the effects of EHS WD with EXC SD rather than with EXC WD. Overlapping metabolites are shown in the quantitative Venn diagram in Fig. 3c for all three comparisons of SD and WD. The number of significant changes (P < 0.05 and FC > ± 1.5) in EHS WD vs EXC SD were very large, mostly reflecting the responses to WD. Very few metabolites were completely unique in either of the other EHS groups, in either sex (blue and red spheres in Fig. 3c).
To provide a physiological context to the results, we sorted the metabolites into categories known to be important in myocardial metabolism and pathology24,25,26,27. We evaluated these specific metabolites in Figs. 4 and 5, not restricting the evaluations to limitations of thresholds for minimum fold changes or maximum P values. Therefore, for this secondary analysis, results for EHS WD were only compared to EXC SD.

Fold changes in specific metabolites in all EHS groups compared to their sex-matched EXC on SD. The metabolites chosen have previously been shown to have functional significance in myocardial metabolic disorders. a Changes in metabolites important in carbohydrate metabolism, glycolysis and alternative glycolytic pathways, including the pentose phosphate pathway (PPP), the polyol pathway (PolyP) and the hexosamine pathway (HP). b Changes in metabolites associated with both glycogenesis and glycogenolysis. Means ± SD or medians ±1 quartile for nonparametric data, n = 8 in each group. Benjamini Hochberg (B-H) corrected P values (Q) to account for errors due to multiple testing. * = P < 0.05,** < 0.01,*** < 0.001,**** < 0.001. Diagrammatic images created with BioRender.com.

Fold changes of specific metabolites in all EHS groups compared to their sex-matched EXC on SD. The metabolites chosen have functional significance to myocardial metabolic disorders. a Changes in metabolites identified as short chain fatty acids (SCFAs) or their respective -acyls that are generated in the gut or liver and are an important intermediary energy source. b Changes in metabolites identified as comprising the total “purine pool” in the heart that are a buffer against loss of intra-myocyte [ATP]. Means ± SD or medians ±1 quartile for nonparametric data n = 8 in each group. * = P < 0.05, **<0.01, ***<0.001, ****<0.001. Benjamini Hochberg corrected P (B.H.-Q) values are listed on the far right. Diagrammatic images created with BioRender.com.
Figure 4a summarizes the responses of myocardial carbohydrates involved in the early steps of glucose metabolism and glycolysis. Nearly all EHS-exposed animals, regardless of diet, shifted to much higher concentrations of simple carbohydrate metabolites compared to the median of their matched EXCs on SD. Nearly every metabolite essential for each step in glycolysis (right light blue panel) was increased in concentration in EHS mice. In addition, metabolites associated with parallel glycolytic pathways (e.g. pentose phosphate pathway, PPP; the hexosamine pathway, HP; and the polyol pathway, PolyP) were similarly elevated.
These results are supported by changes in metabolites related to glycogenesis and glycogenolysis (Fig. 4b) that are consistent with accelerated turnover of glycogen into maltose products and eventually into glucose. This suggests an underlying high turnover rate of glycogen, perhaps needed to meet the requirements for rapid glucose metabolism. Two regulatory molecules that can function to drive glycogenolysis were also elevated. Corticosterone, which stimulates glycogen synthesis and glucose uptake, was elevated in all EHS groups. In three of four EHS groups, 5-aminoimidazole-4-carboxamide-1-β-4-ribofuranoside (AICAR) was also increased. AICAR is an activator of AMP-kinase, which regulates many aspects of metabolism, but includes stimulation of glycogen synthesis28. In the presence of corticosterone it can stimulate glycogenolysis as well29. In male EHS groups on both diets, phosphocreatine (PCr) was elevated an average of ~2 fold compared to EXC SD; but this was not observed in females.
As shown in Fig. 5a, there were also consistent reductions in short-chain fatty acids (SCFAs) and their related acyl-carnitines and acyl-CoAs (≤ 5 C), (e.g. tylgyl carnitine and propionoyl carnitine). Some medium-chain acyl carnitines (e.g. valerylcarnitine (C5) and nonanylcarnitine (C9) were also significantly reduced in male SD EHS mice (P = 0.016 and P = 0.003, respectively). SCFAs are an important source of metabolic substrate in stressed hearts24.
Interestingly, in all EHS groups, there were significant reductions in nearly all metabolites that serve as an auxiliary “purine pool” for the myocyte (Fig. 5b). While ADP and AMP concentrations were not consistently altered, nearly all other purine metabolites were decreased, as well as some precursors necessary for purine synthesis (ribose, ribose 5-P and adenine).
Exertional heat stroke depletes vascular protein and accelerates diet-induced liver disease
An unexpected finding was a striking reduction in total plasma protein following EHS on both diets (Fig. 6a). Low plasma protein is considered a symptom and risk indicator for many chronic disease states, including liver cirrhosis30, heart failure31, acute kidney injury32 and chronic kidney disease33, all of which can be associated with severe heat illness. Alkaline phosphatase (ALP) was also depressed, similar to total plasma protein (Fig. 6a).

a Blood chemistry analyses of total plasma protein and alkaline phosphatase, an intracellular enzyme marker. Dotted areas are predicted values based on Otto et al.60 b Luminex® multiplex plasma protein analyses of soluble circulating adhesion molecules. Results in all exertional heat stroke (EHS) and exercise control (EXC) groups compared to a naïve strain-matched controls (blue hatched symbols), c Scoring of histological evidence of liver injury in terms of steatosis and ballooning, along with chemical analysis of plasma alanine aminotransferase (ALP), indicating damaged and/or fatty liver. n = 8 in most groups. In some biochemical analyses insufficient plasma sample was available for testing so the n =(4-8). No data samples were removed for any other reason. * = P < 0.05,** < 0.01, ***< 0.001, ****< 0.001. Statistical tests: Panel (a, c) two sample T or Wilcoxon with B-H Q corrections were below Q < 0.05 for both males and females. Panel b) ANOVA or Kruskal Wallis with post hoc tests were Dunnett T3 for multiple comparisons. Benjamini Hochberg corrected Q values were below 0.1 for all ANOVA P values.
Soluble vascular adhesion molecules are considered biomarkers of many types of cardiovascular diseases and inflammatory states34. These were evaluated in plasma and displayed in Fig. 6b. Because normal values in mice are not well established, we included our own subset of 5 male and 6 female adult C57BL/6 naïve control mice for comparison. In general, most adhesion molecules were suppressed in EHS-exposed mice, regardless of diet or sex. P-Selectin and E-Selectin were largely unchanged, though E-Selectin was depressed in EHS male mice on SD diet.
Livers were evaluated for histological characteristics of non-alcoholic fatty liver disease, based on the histological presence of “steatosis” and “ballooning.” As shown in Fig. 6c (right panel), WD caused an increased presence of both steatosis and ballooning, as expected. Using a scoring system from 1-5, both ballooning and steatosis were significantly amplified by EHS exposure in males on WD. Plasma alanine aminotransferase (ALT), a liver enzyme, commonly used to identify underlying liver disease, was modestly increased in female EHS mice on WD, but not males.
Discussion
The results demonstrate that when mice are exposed to a single exertional heat illness of sufficient intensity to simulate the acute pathophysiology of human heat stroke7,9, they can lose their long-term capacity to regulate body mass and food intake; they develop metabolic abnormalities in the myocardium consistent with early stages of heart disease; they exhibit signs of cardiac hypertrophy and appear more susceptible to diet-induced liver steatosis. These long-term effects are less predominant in females, possibly because of the inherent resistance of female mammals to many forms of heart disease35. Other outcomes include an unexpected and marked reduction in total plasma protein, ALP and endothelial-derived circulating adhesion molecules. These outcomes point to a complex and sustained multiorgan dysfunction following EHS.
The gain in body mass over the 10 wk diet intervention was most prominent in male mice but was also seen in females. The underlying cause is unknown but likely reflects some form of endocrine dysfunction since food intake was increased in EHS-exposed mice. A potential endocrine candidate may be elevated corticosterone, which was observed as a metabolite in myocardial tissue in EHS mice. Assuming this reflects circulating corticosterone, this could stimulate appetite and fat deposition while decreasing thermogenesis and metabolic rate36. Epigenetic responses to chronic stress (as reflected by high corticosterone) are known to contribute to the pathogenesis of obesity37.
Several metabolite shifts in ventricular tissue are notably similar to previous observations in rodent models of developing hypertrophy and also parallel data collected previously after only two weeks of recovery from EHS11,12. The most prominent feature, evident in all groups, was the elevation in nearly all simple sugars and glycolytic products, such as glucose, fructose and their phosphorylated intermediates (Fig. 4a). Shifts toward greater glucose metabolism and glycolysis, away from predominant fatty acid metabolism, is a characteristic of “hypertrophic growth and pathological remodeling” in the heart25 and has been referred to as evidence of a loss of “metabolic flexibility.”27 Elevations in ribose 5-P and fructose 6-P suggest activation of the pentose phosphate pathway, also known to be hyperactive in the hypertrophied heart25. Myocardial production of fructose can arise from the intracellular polyol pathway or through transport into myocytes from alternative GLUT transporters (e.g. GLUT5), expressed in the heart during metabolic stress38. Elevations in glucosamine-6P point to activation of the hexosamine biosynthetic pathway, another indicator of ongoing cardiac hypertrophic remodeling25,39.
The second most noticeable and reproducible metabolic alteration, 3 months after EHS, was the reduction in SCFAs and related low molecular weight fatty acid metabolites and acyls that largely arise from gut microbiota or the liver (Fig. 5a). These have proven to be important sources of energy in failing or stressed hearts, and a deficit is likely to contribute to the loss of metabolic flexibility24. The decrease in purine content within cardiac tissue (Fig. 5b) parallels development of pacing-induced heart failure in dogs, which is an observation reported to be directly proportional to losses of myocardial [ATP] with increasing progression of failure26. ATP was not a metabolite that was available in our metabolite library. Though relative [AMP] and [ADP] were not consistently affected at this recovery time point, in males, PCr was ~2 fold higher compared to SD EXC mice (Fig. 4b), with no accompanying change in creatinine. Taken together, we speculate that these data reflect a net elevation in PCr/ATP ratio, which is considered a prognostic indicator in the progression of heart failure40,41. We speculate that the elevation in PCr in this model could be compensatory for a weakened metabolic potential for rapid energy turnover.
One of the most puzzling findings was the loss of total plasma protein and the parallel losses seen in so many other plasma proteins. This is not a finding that has previously been associated with heat illness, but is common in many forms of systemic disease that involve the same organ systems that are affected by heat stress30,31,32,33. For example, 88% of human patients at hospital admission with heart failure have a secondary diagnosis of hypoproteinemia31. Hypoproteinemia must partially reflect reduced albumin since albumin makes up ~50% of the circulating plasma protein in mice and humans42. One mechanism could be an ongoing acute phase response in the liver due to inflammatory signaling. Since albumin is a ‘negative’ acute phase protein, its expected response would be to reduce plasma albumin concentrations43. Liver damage could also reduce the ability to maintain circulating albumin30 as does a condition called protein-losing gastroenteropathy44. Gastroenteropathy affects all plasma proteins due to damage to the gut mucosa and lymphatics, eventually causing a direct sink of proteins into the GI tract. GI injury is a susceptible organ system during severe heat stress9 and the time course of its complete recovery is not well understood.
The net loss of a variety of types circulating endothelial adhesion molecules (Fig. 6b) is also puzzling. In most forms of cardiovascular disease these biomarkers are increased, providing an indication of ongoing vascular inflammation and particularly atherosclerosis34. However, one known mechanism of reduced adhesion molecule expression is due to the suppressive effects of corticosteroids45,46 which were likely elevated in the vasculature in EHS mice, based on myocardial levels. Alternatively, the loss of these circulating adhesion molecules could also reflect the overall loss of plasma protein if it is due to leakage out of the vascular compartment.
Exactly how various forms of heat stress exposure can induce long term effects on whole body metabolism and specifically on heart metabolism is uncertain. Studies in heat stressed pigs have shown that, contrary to expectations, reductions in energy expenditure and loss of metabolic flexibility occur in the immediate recovery period following 5 days of continuous but moderate heat stress, with an apparent shift away from lipid metabolism47. Whether the nature of this effect can be similarly induced by intense, acute forms of heat (e.g. as seen in this study) is unknown, but the myocardial metabolomics we describe here are consistent with this outcome.
Like our emerging understanding of the delayed impact of concussion, long covid, post-traumatic stress and post-sepsis syndrome, surviving a severe heat injury in unacclimatized organisms appears to initiate a very complex and delayed pathological response that can elevate risk of developing chronic disease much later in time. If the relationship goes unnoticed, heat stress exposure may never be associated with the original environmental trigger. Recent surveys of heat stroke and heat injury victims after 6 and 12 months of recovery have reported that 47% continue to experience long-term physical complaints and mental health difficulties5. Yet, follow-up surveillance and long-term care for heat injury is not part of current practice guidelines in any relevant medical discipline. As areas of our world continue to warm and experience large swings of temperature with expected failures to electrical grids, the risk of both acute and chronic exposure to severe heat is likely to escalate48. Heat stroke may not be necessary to induce these kinds of effects, as recent studies in livestock show that even passive heat exposures of ~37°C (a common summer temperature in the U.S), sustained for 24 hours, induces structural and biochemical heart defects49. In addition, the close association between cardiovascular mortality across the US and the number of hot days (>32°C) within the same county suggest that these long-term outcomes may apply to general human populations over extensive times50. For example, “every additional extreme-heat day is associated with 0.12% higher monthly increase in cardiovascular mortality” in local populations50. Older, overweight individuals, or those with underlying health disorders are even more susceptible to heat stroke- induced heart disease3 and may require a much lower intensity of exposure to induce long-term effects. We suggest that the results of this study in young, healthy mice provide a cause-and-effect, logical framework and an experimental paradigm for defining severe heat illness as a relevant risk factor for future cardiovascular and metabolic disease.
Methods
Surgical and preparatory procedures
In this study, 64 male and female mice (C57BL/6 J), 8–10 weeks old, were obtained from The Jackson Laboratory (Bar Harbor, ME). All protocols and procedures used were reviewed and approved by the University of Florida Institutional Animal Care and Use Committee (IACUC #201910745). We complied with all relevant ethical regulations for animal use. Upon arrival, mice were randomly assigned into 8 groups (n = 8–10 in each) and were given access to a standard diet (SD) and water ad libitum while maintained on a 12 h dark/ 12 h light cycle at 22 ± 2 °C and 30%–60% humidity. One week later, all mice underwent isoflurane anesthesia for implantation of a sterile miniature temperature telemetry device (16.5 × 6.5 mm G2-E-Mitter; Starr Life Sciences, Oakmont, PA, USA). The mice were anesthetized with isoflurane using an Eagle Eye Model 150 anesthesia machine, Jacksonville, FL); i.e. 4% isoflurane, 0.4–0.6 L/min of O2 flow for rapid induction in an induction chamber, followed by a maintenance of 1.5%, 0.6 L/min using a nose cone. Adequate anesthetic depth was intermittently tested using a toe pinch-withdrawal test. Sterile veterinary eye lube was used to prevent corneal drying and the lower abdomen was shaved and disinfected. An initial dose of subcutaneous buprenorphine (0.1 mg/kg) was given at this time and the same dose given during 48 h of recovery, every 12 h. The surgical site was isolated with sterile adhesive bandages and a ∼1 cm midline incision made along the linea alba, beginning at 0.5 cm from the costal margin. The skin was separated from the muscle and a small incision made to penetrate the peritoneum. A miniature, sterile temperature telemetry device (16.5 × 6.5 mm G2 E-Mitter; Starr Life Sciences, Oakmont, PA) was placed into the intraperitoneal cavity in front of the caudal arteries and veins and dorsal to the digestive organs. Mice were then placed back in their clean cages with a mild heat source (Snugglesafe Microwavable Pad, Sussex, UK). Mice were monitored every 15 min during the first hour of recovery from anesthesia, then returned, singly housed, to the animal housing facility. A recovery period of 2 wks was provided after surgery and then voluntary running wheels were installed into each cage for the next three weeks until the EHS or control exercise protocols were performed51 (Fig. 1a–c). During the third week, the mice were brought to the laboratory on 4 occasions, 18–24 h apart, to practice using a forced running wheel (Lafayette Instrument Co., model 80840, Lafayette, IN), with speed incrementally increased using a preprogrammed protocol that was repeated in the EHS treatment9,51.
Exertional heat stroke and exercise sham protocols
Mice were brought to the laboratory and core temperature (Tc) was monitored overnight while mice remained at the light cycle, temperature and humidity of their vivarium. The next morning, between 0700 and 0800, the mice were randomized to either EHS or an identical exercise sham control protocol group (EXC). The EHS and EXC procedures were developed in previous studies9,51. The male and female mice were run while exposed to different environmental temperatures (Te) within the environmental chambers (Thermo-Forma 3940 Incubator: Thermo-Fisher, Waltham, MA). For male mice the Te was kept at 34.5 °C / ~ 40% relative humidity (RH) and 37.5 °C / ~ 40% RH for females. The lower temperature was used for males because of their markedly lower thermotolerance12,22 and because previous experiments showed that increasing the running time and speed in a lower Te in males resulted in similar metabolic myocardial dysfunction at two weeks of recovery that we previously reported in females11,12,22. Each EXC mouse ran identically in a second incubator for a duration and pattern that equaled the average duration and intensity of the EHS groups but with Te set to 22–23 °C. Mice were continuously monitored during the EHS and EXC protocols using infrared cameras. When the EHS mice reached “symptom limitation” the running wheels were immeditely stopped. Symptom limitation was defined as “apparent unconsciousness,” a form of central nervous system (CNS) dysfunction that resembles what is observed in ~80% of human heat stroke victims52. CNS dysfunction is a hallmark of heat stroke that distinguishes it from other, less serious forms of heat injury53. After completion, the mice were quickly removed from the running wheel and chamber to measure body mass and then were placed back into their home cages, which were equilibrated to room temperature. The cage was immediately returned to the environmental chamber at the specific temperature used for the EHS protocol for males and females. This was done to slow down cooling rate and to prevent the post-heat stroke hypothermic response common to rodents54. The mice were normally upright and breathing regularly within 15 min of symptom limitation and began grooming and displaying relatively normal resting behavior within the first hour. The mice had full access to food and water during recovery. No mice died or became morbid during or after the EHS protocol, prior to euthanasia and sample collection. Additional 1–2 mice above the target population of 8 mice per group were included in the study in order to make up for equipment failure or scheduling difficulties. The final group of 8 mice used for data collection was determined from the specific mice that were used for metabolomics analyses (randomly assigned from mice remaining at the end of the study).
Ultra-sound procedures of mouse heart
Fourteen days after EHS or EXC, mice were tested for heart function with an ultra-sound echocardiography system (LogiQe NextGen, SOUND Technologies, Carlsbad, CA). This was repeated at nine weeks after starting the diet interventions, discussed below. Mice underwent isoflurane anesthesia in the induction chamber (2% isoflurane at 0.5 L/min O2) and then were shaved from the neckline to mid-chest level; residual hair was removed with hair removal cream (Nair™, Church and Dwight Co., Ewing, NJ). Lubricating veterinary gel was applied to both eyes to prevent dryness. Anesthesia was continued throughout using a nose cone to maintain steady-state sedation, and the mouse was placed in a prone position on a heating pad with ECG leads. Electrode gel was applied to both the probe and the chest. The ultrasound probe was then lowered to the thorax and two-dimensional imaging was used to locate the parasternal short-axis view at the level of the papillary muscles. From this orientation, M-mode tracings of the left ventricle were obtained. Left ventricle fractional shortening and posterior wall velocity (PWV) were used to quantify systolic and diastolic dimensions and function. Measurement of ventricular wall thicknesses and time intervals were determined on 10-15 cardiac cycles and averaged for each mouse. Cardiac function was analyzed and confirmed by a coauthor blinded to the experimental groups.
Western diet and standard diet interventions
Two weeks after EHS or EXC, mice were randomized into either a Western diet (WD) group [Envigo,TD.88137 adjusted calories diet, 21% fat by weight (60% saturated fats); 48.5% carbohydrate by weight; 17.3% protein by weight)] or a standard diet (SD) group [Teklad 2919 global); 9% fat by weight (1.2% saturated fat), 44.9% carbohydrates; 19% protein by weight]55. Mice remained on their assigned diet for the remainder of the study (10 weeks). Both body weight and food weight were checked three times per week. Once per week, the food was discarded and replaced with 100 g of fresh WD or SD food. The mice continued to be singly housed during this time.
Tissue sample collection and evaluation
At wk 10 after the initiation of their specific diets (i.e., ~3 months after EHS exposure), mice were weighed and then underwent deep anesthesia for tissue collection. Note, this was performed approximately one week after the last ultrasound procedure. One day before tissue collection, mice were transported to the laboratory and maintained on a 12/12 light cycle to reduce the stress of transport prior to sample collection. On the day of tissue harvest, mice underwent rapid and deep isoflurane anesthesia9,51. After removing the hair over the upper abdomen and chest, a transthoracic cardiac stick was performed for a blood draw of 0.6-1 ml, which was immediately placed on ice. The thorax and abdominal cavity were then opened and euthanasia performed by removal of the heart from the great vessels. The heart ventricles, liver and forelimbs were collected and preserved or frozen for later analyses. Blood was centrifuged at 4 °C, 2000 rpm for 10 min. Basic blood biochemistry profile biomarkers were evaluated using an Abraxis Vetscan VS2 (Zoetis, Parsippany, NJ, US) and employing the Prep Profile II reagent rotor. Other plasma samples were analyzed using a multiplex MAGPIX Luminex Analyzer (Life Science; Hercules California, US 94547) and a 96 well reagent Milliplex MAP Mouse Cardiovascular Disease (CVD) bead assembly assay (MCD1MAG-77K) (MilliporeSigma, Rockville, MA; USA). Hearts were removed and rinsed in ice cold PBS, then gently blotted and weighed. While over ice, the heart was cut in half in a transverse cut, midway across the ventricles. The mid to apical part of the ventricles was flash-frozen and then stored at −80 °C for later metabolomics analyses.
The median lobe of the liver was preserved in 10% buffered formalin, then paraffin embedded and cut into 4 µm sections, followed by H&E staining. The sections were scored based on a previously developed scoring system56, with modifications applied to avoid ceiling effects of the measurement scales. Random sections from each mouse were divided into 3 equal areas of evaluation. Two trained and blinded scorers evaluated each of these images. The average score per sample was determined across the scorers and used for statistical testing. Two types of liver pathologies were evaluated: ballooning and steatosis. Steatosis is defined histologically extensive cytoplasmic vacuolization, whereas ballooning is a type of hepatocyte degeneration with marked swelling and enlargement of individual hepatocytes56. Both were graded from 0 to 5, with “0” = involvement of 0-5% of the image displaying steatosis or ballooning, 1 = 5–25%, 2 = 25–45%, 3 = 45–65%, 4 = 65–85% and 5 = > 85% of the image.
Metabolomics analyses
The frozen mid to apical ventricular samples (~50 mg) were shipped on dry ice to Metabolon, Inc. (Durham, NC, USA). As described by the Metabolon, Inc. protocols, protein homogenization and precipitation were performed in methanol and the sample vigorously shaken, followed by centrifugation. The extract was then divided into 5 fractions: two for reverse phase (RP) UPLC-MS/MS (ultrahigh performance liquid chromatography-tandem mass spectrometry). One was used for positive ion mode electrospray ionization (ESI) and one for negative ion mode ESI. Another fraction was used for hydrophilic UPLC-MS/MS with negative ion mode ESI. Samples were then extracted from their solvents, dried, and reconstituted in solvents compatible to elucidating negative, positive, basic, and acidic compounds. All samples were run through a dedicated C18 column (Waters UPLC BEH C18-2.1×100 mm, 1.7 μm). Then mass spectrometry was performed, and raw data files produced. Raw data files were compared to Metabolon’s library of compounds and metabolites and their relative concentrations evaluated by the authors as “batch-norm” imputed data. Eight hundred and seventy different metabolites of various types were detected for this study and reported for each sample.
Statistics and Reproducibility
A minimum required sample size for all groups was calculated using G*Power software57 and metabolomics data previously acquired in a mouse hearts from mice exposed to heat stroke12. An effect size of 1.33, SD = 0.6, power= 0.8 and α = 0.05 resulted in a minimum sample size of 8 in each group. For metabolomics, the batch normed EHS imputed data were normalized to their EXC, matched to a given diet and sex, so that results were expressed as a ± fold change from EXC. In a secondary analysis, the WD EHS data were also normalized to the EXC on SD data for all groups. This data is reported separately in the results. Fold change was expressed as fold from the median of the EXC metabolite concentrations because ~30% of the metabolite distributions were nonparametric.
All data were analyzed with SAS-JMP Pro 17, (Cary NC, USA), Real-statistics58 and/or GraphPad Prism 10.0 (Boston, MA, USA). Individual groups of data for each metabolite and group were tested for normality using the Shapiro-Wilk test and comparisons of variance using the Fishers F-test. Individual statistics used for each variable are described in figure legends. Groups being compared that deviated from normality were tested using nonparametric statistics: Mann-Whitney tests for two sample comparisons or Kruskal Wallis for multiple group comparisons. Parametric groups were tested using two sample T tests (or the Welch alternative for unequal variances) or ANOVA for multiple group comparisons. Post hoc analyses for ANOVA were limited to comparisons of just EHS groups against their matched EXC. The P-values for each appropriate test, along with a threshold change of ≥ 1.5 fold were used as cutoffs for volcano and forest plot analyses. Potential errors due to multiple testing were reported as either over or under a set threshold range, in the figure legends, or directly reported as Benjamini-Hochberg Q values for each outcome variable.
No outlier data were removed from any experiment. When sample size was reduced within a specific measurement, it represented a technical limitation due to insufficient plasma or tissue volume or equipment malfunction, scheduling, etc. Sample size, when different from n = 8 is explained in figure legends. All tests were performed using two sided tests at an α = 0.05. All tests were taken from distinct samples.
Data availability
All data used for the figures in this manuscript including the metabolomics raw data files are available on the data repository, Figshare, at the following URL https://doi.org/10.6084/m9.figshare.27910047.v1 Reprints and permissions information is available at www.nature.com/reprints.”
References
U.S. Environmental Protection Agenc. Climate Change Indicators in the United States. ww.epa.gov/system/files/documents/2024-09/climate_indicators_2024.pdf (2024).
Wang, J.-C. et al. The association between heat stroke and subsequent cardiovascular diseases. PLoS ONE 14, e0211386 (2019).
Tseng, M.-F. et al. Association between heat stroke and ischemic heart disease: A national longitudinal cohort study in Taiwan. Eur. J. Intern Med. 59, 97–103 (2019).
Wallace, R. F., Kriebel, D., Punnett, L., Wegman, D. H. & Amoroso, P. J. Prior heat illness hospitalization and risk of early death. Environ. Res. 104, 290–295 (2007).
Kruijt, N. et al. Exertional Heat Stroke and Rhabdomyolysis: A Medical Record Review and Patient Perspective on Management and Long-Term Symptoms. Sports Med. Open 9, 33 (2023).
Dutta, S. & Sengupta, P. Men and mice: Relating their ages. Life Sci. 152, 244–248 (2016).
Bouchama, A. et al. Classic and exertional heatstroke. Nat. Rev. Dis. Prim. 8, 8 (2022).
O’Connor, S. G. et al. Within-subject effects of environmental and social stressors on pre- and post-partum obesity-related biobehavioral responses in low-income Hispanic women: protocol of an intensive longitudinal study. BMC Public Health 19, 253 (2019).
King, M. A., Leon, L. R., Mustico, D. L., Haines, J. M. & Clanton, T. L. Biomarkers of multi-organ injury in a pre-clinical model of exertional heat stroke. J. Appl. Physiol. 118, 1207–1220 (2015).
King, M. A., Leon, L. R., Morse, D. A. & Clanton, T. L. Unique cytokine and chemokine responses to exertional heat stroke in mice. J. Appl. Physiol. 122, 296–306 (2017).
Laitano, O. et al. Delayed metabolic dysfunction in myocardium following exertional heat stroke in mice. J. Physiol. 598, 967–985 (2020).
Garcia, C. K. et al. Delayed metabolic disturbances in the myocardium after exertional heat stroke: contrasting effects of exertion and thermal load. J. Appl Physiol. 135, 1186–1198 (2023).
Murray, K. O., Brant, J. O., Kladde, M. P. & Clanton, T. L. Long-term epigenetic and metabolomic changes in the mouse ventricular myocardium after exertional heat stroke. Physiological Genomics 54, 486–500 (2022).
Alzahrani, J. M. et al. Neuromotor deficits and altered physiological responses to repeated exertional heat stroke exposures in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. https://doi.org/10.1152/ajpregu.00152.2022 (2022).
Murray, K. O. et al. Exertional heat stroke leads to concurrent long‐term epigenetic memory, immunosuppression and altered heat shock response in female mice. J. Physiol. 599, 119–141 (2021).
Murray, K. O., Clanton, T. L. & Horowitz, M. Epigenetic responses to heat: From adaptation to maladaptation. Exp. Physiol. 107, 1144–1158 (2022).
Murray, K. O. et al. Exertional heat stroke causes long-term skeletal muscle epigenetic reprogramming, altered gene expression, and impaired satellite cell function in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 326, R160–R175 (2024).
Tiffon, C. The Impact of Nutrition and Environmental Epigenetics on Human Health and Disease. Int J. Mol. Sci. 19, 3425 (2018).
Murphy, M., Cohn, D. & Loria, A. Developmental origins of cardiovascular disease: impact of early life stress in humans and rodents. Neurosci. Biobehav Rev. 74, 453–465 (2017).
Zheng, X. et al. Western diet augments metabolic and arterial dysfunction in a sex-specific manner in outbred, genetically diverse mice. Front. Nutr. 9, 1090023 (2023).
Odegaard, A. O., Koh, W. P., Yuan, J.-M., Gross, M. D. & Pereira, M. A. Western-Style Fast Food Intake and Cardiometabolic Risk in an Eastern Country. Circulation 126, 182–188 (2012).
Garcia, C. K. et al. Sex-dependent responses to exertional heat stroke in mice. J. Appl. Physiol. 125, 841–849 (2018).
Lee, J. et al. Myocardial metabolic alterations in mice with diet-induced atherosclerosis: linking sulfur amino acid and lipid metabolism. Sci. Rep. 7, 13597 (2017).
Palm, C. L., Nijholt, K. T., Bakker, B. M. & Westenbrink, B. D. Short-Chain Fatty Acids in the Metabolism of Heart Failure - Rethinking the Fat Stigma. Front Cardiovasc Med 9, 915102 (2022).
Tran, D. H. & Wang, Z. V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 8, e012673 (2019).
Shen, W. et al. Progressive loss of myocardial ATP due to a loss of total purines during the development of heart failure in dogs: a compensatory role for the parallel loss of creatine. Circulation 100, 2113–2118 (1999).
Vallerie, S. N. & Bornfeldt, K. E. Metabolic Flexibility and Dysfunction in Cardiovascular Cells. Arterioscler Thromb. Vasc. Biol. 35, e37–e42 (2015).
Longnus, S. L., Wambolt, R. B., Parsons, H. L., Brownsey, R. W. & Allard, M. F. 5-Aminoimidazole-4-carboxamide 1-beta -D-ribofuranoside (AICAR) stimulates myocardial glycogenolysis by allosteric mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 284, R936–R944 (2003).
Puthanveetil, P. et al. Cardiac glycogen accumulation after dexamethasone is regulated by AMPK. Am. J. Physiol.-Heart Circulatory Physiol. 295, H1753–H1762 (2008).
Carvalho, J. R. & Verdelho Machado, M. New Insights About Albumin and Liver Disease. Ann. Hepatol. 17, 547–560 (2018).
Karki, S., Gajjar, R., Bittar-Carlini, G., Jha, V. & Yadav, N. Association of Hypoalbuminemia With Clinical Outcomes in Patients Admitted With Acute Heart Failure. Curr. Probl. Cardiol. 48, 101916 (2023).
Wiedermann, C. J., Wiedermann, W. & Joannidis, M. Causal relationship between hypoalbuminemia and acute kidney injury. World J. Nephrol. 6, 176–187 (2017).
Haller, C. Hypoalbuminemia in renal failure: pathogenesis and therapeutic considerations. Kidney Blood Press Res 28, 307–310 (2005).
Blann, A. D. & Lip, G. Y. H. Cell Adhesion Molecules in Cardiovascular Disease and Its Risk Factors—What Can Soluble Levels Tell Us? J. Clin. Endocrinol. Metab. 85, 1745–1747 (2000).
Iorga, A. et al. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex. Differ. 8, 33 (2017).
Kuckuck, S. et al. Glucocorticoids, stress and eating: The mediating role of appetite‐regulating hormones. Obes. Rev. 24, e13539 (2023).
Xiao, Y., Liu, D., Cline, M. A. & Gilbert, E. R. Chronic stress, epigenetics, and adipose tissue metabolism in the obese state. Nutr. Metab. (Lond.) 17, 88 (2020).
Annandale, M. et al. Fructose Metabolism and Cardiac Metabolic Stress. Front Pharm. 12, 695486 (2021).
Kolwicz, S. C. & Tian, R. Glucose metabolism and cardiac hypertrophy. Cardiovasc. Res. 90, 194–201 (2011).
Samuel, T. J. et al. Myocardial ATP depletion detected noninvasively predicts sudden cardiac death risk in patients with heart failure. JCI Insight 7, e157557 (2022).
Wallis, J. et al. Supranormal myocardial creatine and phosphocreatine concentrations lead to cardiac hypertrophy and heart failure: insights from creatine transporter-overexpressing transgenic mice. Circulation 112, 3131–3139 (2005).
Zaias, J., Mineau, M., Cray, C., Yoon, D. & Altman, N. H. Reference Values for Serum Proteins of Common Laboratory Rodent Strains. J. Am. Assoc. Lab Anim. Sci. 48, 387–390 (2009).
Ehlting, C., Wolf, S. D. & Bode, J. G. Acute-phase protein synthesis: a key feature of innate immune functions of the liver. Biol. Chem. 402, 1129–1145 (2021).
Ozen, A. & Lenardo, M. J. Protein-Losing Enteropathy. N. Engl. J. Med. 389, 733–748 (2023).
Simoncini, T. et al. Estrogens and glucocorticoids inhibit endothelial vascular cell adhesion molecule-1 expression by different transcriptional mechanisms. Circ. Res 87, 19–25 (2000).
Wehling-Henricks, M., Lee, J. J. & Tidball, J. G. Prednisolone decreases cellular adhesion molecules required for inflammatory cell infiltration in dystrophin-deficient skeletal muscle. Neuromuscul. Disord. 14, 483–490 (2004).
Fausnacht, D. W. et al. Heat Stress Reduces Metabolic Rate While Increasing Respiratory Exchange Ratio in Growing Pigs. Anim. (Basel) 11, 215 (2021).
Romanello, M. et al. The 2023 report of the Lancet Countdown on health and climate change: the imperative for a health-centred response in a world facing irreversible harms. Lancet 402, 2346–2394 (2023).
Roths, M. et al. Environment-induced heat stress causes structural and biochemical changes in the heart. J. Therm. Biol. 113, 103492 (2023).
Khatana, S. A. M., Werner, R. M. & Groeneveld, P. W. Association of Extreme Heat and Cardiovascular Mortality in the United States: A County-Level Longitudinal Analysis From 2008 to 2017. Circulation 146, 249–261 (2022).
King, M. A., Alzahrani, J. M., Clanton, T. L. & Laitano, O. A Preclinical Model of Exertional Heat Stroke in Mice. J. Vis. Exp. https://doi.org/10.3791/62738 (2021).
Yezli, S. et al. Classic heat stroke in a desert climate: A systematic review of 2632 cases. J. Intern. Med. 294, 7–20 (2023).
DeGroot, D. W., O’Connor, F. G. & Roberts, W. O. Exertional heat stroke: an evidence based approach to clinical assessment and management. Exp. Physiol. 107, 1172–1183 (2022).
Leon, L. R., DuBose, D. A. & Mason, C. W. Heat stress induces a biphasic thermoregulatory response in mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 288, R197–R204 (2005).
Towler, D. A., Bidder, M., Latifi, T., Coleman, T. & Semenkovich, C. F. Diet-induced Diabetes Activates an Osteogenic Gene Regulatory Program in the Aortas of Low Density Lipoprotein Receptor-deficient Mice. J. Biol. Chem. 273, 30427–30434 (1998).
Ryu, J.-E. et al. Evaluation of Nonalcoholic Fatty Liver Disease in C57BL/6J Mice by Using MRI and Histopathologic Analyses. Comp. Med 65, 409–415 (2015).
Faul, F., Erdfelder, E., Buchner, A. & Lang, A.-G. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav. Res Methods 41, 1149–1160 (2009).
Zaiontz, C. Real Statistics Resource Pack software (Release 7.6) [Computer software]. https://www.real-statistics.com/ (2020).
Hulsen, T., de Vlieg, J. & Alkema, W. BioVenn – a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC Genomics 9, 488 (2008).
Otto, G. P. et al. Clinical Chemistry Reference Intervals for C57BL/6J, C57BL/6N, and C3HeB/FeJ Mice (Mus musculus). J. Am. Assoc. Lab Anim. Sci. 55, 375–386 (2016).
Acknowledgements
This research was supported by the U.S. Army Medical Research and Development Command W81XWH-19-2-0050 (T.L.C.), University of Florida Foundation BK Betty Stevens Professorship (UF Foundation F00294) (T.L.C.). JMA was supported by a fellowship from the Department of Exercise Physiology, College of Sport Sciences and Physical Activity, King Saud University, Kingdom of Saudi Arabia.
Author information
Authors and Affiliations
Contributions
J.M.A. performed the experiments, wrote the first draft and participated in all aspects of the project; A.J.S. and R.N.M. performed all of the ultrasound studies and associated data analyses, BJG and M.R. collected and analyzed animal experimental data; C.D., F.P.F. and D.A.M. performed biochemical and histological analyses. T.L.C. designed and oversaw all aspects of the project and was responsible for the final drafts of the manuscript. All participants contributed to evaluation of experimental outcomes and provided editorial input on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
We confirm that this work reflects a community of scholars that welcomes intellectual discussion and debate and that we worked within an organization (University of Florida, where the research work was performed) that is inclusive, does not restrict free speech, and is open to complaints of misconduct. The research group does not discriminate against persons based on race, religion, national origin, or sex. Animal welfare was considered for every aspect of the research work and met all local and U.S. regulations. The individuals who served as coauthors of this manuscript came from different countries, religions, sexes and ages, including undergraduate students, graduate students, technicians and senior faculty. Every aspect of the scientific work was transparent, without coercion or fraud.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Christopher Hine and Dario Ummarino. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alzahrani, J.M., Smuder, A.J., Gambino, B.J. et al. Mice develop obesity and lose myocardial metabolic flexibility months after exertional heat stroke. Commun Biol 8, 65 (2025). https://doi.org/10.1038/s42003-025-07484-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-07484-3
This article is cited by
-
Urban Land Use Transformation and Declining Cooling Services: A 40-year Assessment of Heat Stress Mitigation in a Hot and Arid City
Earth Systems and Environment (2025)
