
Abstract
Histone deacetylase 8 (HDAC8) is a well-known epigenetic regulator for cancer therapy. However, developing targeted inhibitors for HDAC8 is challenging due to a limited understanding of its structural dynamics, which is crucial for ligand interaction. Here, we employed an integrated approach, including native mass spectrometry (native MS), hydrogen-deuterium exchange mass spectrometry (HDX-MS), and molecular dynamics (MD) simulation, to investigate the inhibition mechanism and dynamic regulation of human HDAC8 (hHDAC8) by selective inhibitor PCI-34051, compared with the pan-inhibitor SAHA. Our results revealed that PCI-34051 engages with an expanded set of residues and conforms more aptly to the binding channel of hHDAC8, stabilizing the flexible loops surrounding the binding channel. Moreover, this dynamic stabilization effect is not limited to the binding regions, but also extends to distant regions (such as L2, α5, and α1 + α2), with L3 serving as a critical structural bridge. Overall, these results show the structural and dynamic regulations of hHDAC8 by PCI-34051, which induces a lower energy state for the protein-ligand system compared to SAHA, thus showing better inhibitory effects. In addition, it also suggests that certain regions, specifically loops L2 and L3, within the hHDAC8 protein could be key regions for targeted intervention.
Similar content being viewed by others
Introduction
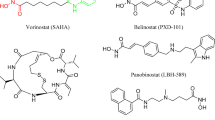
HDAC8 is a Zn2+-dependent class I HDAC and plays a pivotal role in epigenetic regulation by catalyzing the removal of acetyl groups from acetyl-lysines (Fig. 1A)1,2. This enzyme is a well-recognized target for cancers, correlated with diseases such as childhood neuroblastoma, T-cell lymphoma3, Cornelia de Lange syndrome4, and parasitic and viral infections5,6. Although several pan-HDAC inhibitors, such as suberoylanilide hydroxamic acid (SAHA/Vorinostat, Fig. 1B), have gained U.S. Food and Drug Administration approval for the clinical treatment of cancers and other diseases, they often exhibit limited selectivity across various HDAC isoforms due to the strong affinity of the hydroxamate group (zinc binding group, ZBG) to Zn2+ ion at the shared catalytic pocket, thus leading to undesirable side effects7,8. Furthermore, off-target effects of pan-inhibitors caused high toxicity in vivo9,10. Therefore, developing isoform-selective inhibitors has become an inevitable trend for the future of drug development, although the high structural similarity in the active sites across different HDAC isoforms presents a significant challenge in designing isoform-selective inhibitors8,11,12,13. By far, only a handful of human HDAC8 (hHDAC8) selective inhibitors have been reported, including “linkerless” inhibitors14, NCC-14915, and PCI-34051 (Fig. 1C)16. Modification in the linker region has been one of the strategies for developing selective inhibitors17, for example, compared to SAHA, PCI-34051 features a larger linker group that interacts specifically with the hHDAC8 catalytic pocket, thereby contributing to its selectivity. Encouragingly, despite targeting the common catalytic pocket, PCI-34051 exhibits a preference for hHDAC8, demonstrating a more potent inhibitory effect compared to SAHA, with IC50 values of 410 nM for SAHA and 10 nM for PCI-3405116, and Kd values of for 1200 nm for SAHA and 75.1 nm for PCI-3405113,18. Moreover, PCI-34051 has shown reduced toxicity and has recently advanced into clinical research, marking a significant step forward in the development of more selective and safer hHDAC8 inhibitors16,19,20.

A The catalytic process of hHDAC8. B The chemical structure of SAHA. C The chemical structure of PCI-34051. D The loops and helices’ name of hHDAC8 (PDB: 3F07). L1: 30–36; L2-A: 97–107; L2-B: 84–92; L3: 136–156; L4: 176–182; L5: 202–226; L6: 263–280; L7: 301–307; L8: 341–358; α1: 21–29; α2: 37–48; α3: 64–70; α4: 73–83; α5: 108–126; α6: 157–169; α7: 183–190; α8: 237–254; α9: 281–294; α10: 308–323; α11: 358–373.
Beyond the static structures, the protein conformational dynamics are also crucial in drug design due to their role in regulating protein-ligand interactions21,22. In hHDAC8, a series of flexible loops (Fig. 1D), including L1 (residues 30–36), L2-A (97–107), L2-B (84–92), L3 (136–156), L4 (176–182), L5 (202–226), L6 (263–280), L7 (301–307), and L8 (341–358), bridge its core secondary structures to form the catalytic pocket (also known as the binding channel), which contains a zinc ion23,24. These loops enhance the entrance flexibility of the binding channel, facilitating interactions with various inhibitors that induce unique conformational adjustments at this entry point24. NMR spectroscopy has revealed that inhibitor binding to hHDAC8 can affect distant regions such as α1 + α2, mediated by L125. Moreover, molecular dynamics (MD) simulations have shown that SAHA can modulate the conformation of L1 and L226,27. Additionally, X-ray crystallography has captured the stabilization effects on L2 induced by inhibitor binding28. Despite these advances, the current methods still leave gaps in our understanding of the global structural changes of hHDAC8 before and after inhibitor binding, particularly the overall dynamic changes upon selective inhibitor binding.
Romier et al. explored the interaction between hHDAC8 and its selective inhibitor PCI-34051 using molecular docking and relatively short 100 ns MD simulations13. These simulations revealed that PCI-34051 adopts an L-shaped conformation within the binding pocket, interacting with the catalytic tyrosine (Y306) and the L1 and L6 loops of hHDAC8. In contrast, in other HDAC isozymes, a steric L1−L6 lock prevents the binding of L-shaped inhibitors with rigid linker regions. This investigation highlighted the difference in the binding pocket of hHDAC8 and other subtypes of HDACs; however, the short duration of the simulations might not adequately reflect the full dynamics of hHDAC8. Thus, the dynamic properties of hHDAC8 before and after binding are still ambiguous. Although molecular docking and MD simulations are foundational in silico tools for drug discovery, integrating experimental techniques and extended MD simulations offers a more comprehensive understanding of the dynamic and structural interactions of hHDAC8 with selective inhibitors, thereby better informing the design of new inhibitors.
Structural mass spectrometry (MS) methods, such as hydrogen-deuterium exchange MS (HDX-MS), native MS, and crosslinking MS, have shown great potential in characterizing the structures, dynamics, and conformational changes of native proteins or complexes rapidly and sensitively29,30,31. HDX-MS reports solvent accessibility and hydrogen bond (HB) stability through monitoring the deuterium exchange on protein backbone amide hydrogens30,32,33,34. Since deuterium labeling is initiated in D2O solution, it preserves protein dynamic information in an aqueous state35,36,37. Therefore, HDX-MS can obtain the dynamic information that is lost in X-ray diffraction and cryo-electron microscopy, and overcome the molecular weight limitation in NMR38,39,40,41. Consequently, it has been used for probing protein-ligand interactions42, protein dynamics43, and even allosteric regulation44. Furthermore, the protection factor (PF), defined as kclose/kopen, can be calculated using the equation kint/kobs-1 (where kobs is the observed exchange rate)45. Therefore, PF is negatively correlated with the local Gibbs free energy (ΔG) 45. Our previous work, as well as Phillips’s work, successfully used the PF extracted from HDX-MS data to assess local stability changes within the allosteric regulation of glycogen phosphorylases46,47. Apart from HDX-MS, native MS also provides valuable higher-order structure information such as binding stoichiometry and subunit arrangements48. The ability to maintain noncovalent interactions of protein-ligand complexes makes it highly complementary to biophysical methods and suitable for studying protein-ligand interactions49, metal-containing proteins50, and even large protein complexes51. Additionally, some unique dynamic motions in proteins can be monitored by native MS52,53.
Here, we conducted an integrative study employing native MS, HDX-MS, and MD simulation to explore the interactions and selectivity mechanism of two hHDAC8 inhibitors: the pan-inhibitor SAHA and the selective hHDAC8 inhibitor PCI-34051, within a dynamic context. Native MS revealed a more stable binding of PCI-34051 to hHDAC8 and unveiled a significant structural impact by this inhibitor compared to SAHA. To further investigate the structural basis of these phenomena, HDX-MS was employed to provide higher-resolution insights into inhibitor binding. The analyses demonstrated that PCI-34051 induces a lower energy state in hHDAC8 than SAHA, highlighting broader binding and more distant conformational changes within hHDAC8-inhibitor complexes. MD simulations were then conducted to elucidate the residual interactions of these dynamic events. They pinpointed L3 as a pivotal structural bridge connecting the binding site to peripheral secondary structures (L2-A and L2-B, α1, α2, α4, and α5). These simulations also supported that PCI-34051 engages more extensively with residues and fits more suitably into the malleable binding channel of hHDAC8. Our integrative approach provides a detailed overview of the binding mechanism of selective hHDAC8 inhibitors and the protein dynamic alterations upon binding, thereby offering essential insights for the development of targeted inhibitors.
Results
Native MS of hHDAC8-inhibitor complexes
The hHDAC8 sample was firstly checked for its molecular weight and activity. Denatured LC-MS shows the molecular weight of the hHDAC8 sample to be 42,823.5 Da, which aligns with its theoretical weight (42,822.2 Da) with a ppm of 30.4 (Fig. S1). Titration of Zn²+ to the hHDAC8 sample was performed and a 1:1.4 ratio (hHDAC8:Zn2+) was found to reach the maximum activity of hHDAC8, excess Zn2+ leads to a decrease in activity (Fig. S2A)54. The ZnCl2-activated hHDAC8 was subsequently used in all following experiments. Subsequent inhibition assays show that both SAHA and PCI-34051 can significantly inhibit hHDAC8 activity at a ratio of 1:5 (Fig. S2B). Further native MS experiment displays that hHDAC8 was likely bound with multiple metal ions. By gradually increasing the sample cone (SC) voltage from 40 V (Fig. S3A) to 80 V (Fig. S3B), and to 150 V (Fig. S3C), the results revealed that apart from Zn2+ binding, two extra K+ ions and one Na+ ion bound to hHDAC8. The binding of two K+ ions has been previously reported in hHDAC8 crystal structures23,24,28,55,56,57.
The binding stoichiometry, structural changes, and dynamics upon inhibitor binding were then evaluated for SAHA and PCI-34051. We first examined these samples using the Q-ToF instrument (Waters Synapt G2-Si). Both SAHA and PCI-34051 bind to hHDAC8 at a stoichiometry of 1:1 (molar ratio) (Fig. 2A–C). Specifically, as shown in Fig. 2B, a mass shift of ~262.4 Da was observed for the SAHA group, which is roughly consistent with the presence of one SAHA molecule (Theo. M.W. 264.3250 Da). However, a mass shift of only 235.6 Da was found in the PCI-34051 group (Theo. M.W. 296.3260 Da) (Fig. 2C). To elucidate the unexpected mass shift, we further examined the samples using the higher-resolution Orbitrap instrument (Orbitrap Fusion Lumos Tribrid). To better eliminate nonspecific adducts, we increased the in-source dissociation voltage to 20 eV. Using the hHDAC8 + Zn2+ + K+ + Na+ state as the reference peak, the SAHA group exhibited a mass shift (264.47 Da) corresponding to the addition of a single SAHA molecule (Figs. 2D, E); however, the PCI-34051 group displayed a mass shift (296.22 Da) corresponding to the incorporation of a single PCI-34051 molecule, accompanied by the concurrent loss of one K+ and one Na+ (Fig. 2F). To our best knowledge, this marks the first observation that the binding of an hHDAC8 inhibitor in solution results in the dissociation of specific metal ions. This discovery indicates that the inhibitor’s interaction extends beyond the orthosteric site, eliciting a structural reorganization that transcends the immediate binding region. The differential binding patterns of SAHA and PCI-34051 suggest distinct impacts on the structure and dynamics of hHDAC8, with PCI-34051 demonstrating a more profound structural alteration.

A–C Native mass spectra of unbound hHDAC8, hHDAC8-SAHA, and hHDAC8-PCI-34051 on Q-ToF instrument. The peak of unbound hHDAC8 is highlighted by orange circles. The peaks corresponding to inhibitors-bound hHDAC8 are highlighted by orange circles with a black dot inside. D–F The deconvoluted native mass spectra of hHDAC8, hHDAC8-SAHA, and hHDAC8-PCI-34051 on Orbitrap instrument.
HDX-MS experimental optimization and PF analysis
To acquire higher-resolution structural and dynamic insights, HDX-MS experimental conditions were evaluated and optimized, including stability test (Fig. S4), quench conditions (Table S1), and digestion time (Table S2). The protein:inhibitor incubation ratios were then evaluated by native MS for rough ranges and later by HDX-MS for optimum values, aiming to detect significant binding events in HDX-MS while avoiding nonspecific binding. Native MS experiments reveal that the hHDAC8:SAHA ratio should be in between 1:10 and 1:20 (Fig. S5B, C), while a ratio between 1:5 and 1:10 for hHDAC8:PCI-34051 (Fig. S5D, E). Subsequent optimization using HDX-MS further reveal that a ratio of 1:15 for hHDAC8:SAHA and a ratio of 1:8 for hHDAC8:PCI-34051 resulted in significant HDX changes (Fig. S6). The formal protein-inhibitors binding experiments yielded a final sequence coverage of 96.4% with a redundancy of 3.02 for the SAHA group, and a sequence coverage of 96.4% with a redundancy of 2.88 for the PCI-34051 group (Fig. S7). A detailed summary of HDX-MS data is provided in Table S3.
PF values were then computed based on the HDX-MS data and mapped onto the hHDAC8 structure (PDB code: 3F07). PF is inversely proportional to ΔG, meaning that lower PF values correspond to higher residue dynamics, and vice versa45. Therefore, ΔPF reflect the changes in the local dynamics of regions before and after inhibitor binding. Overall, the PF values generated from HDX-MS data exhibit a strong alignment with the B-factor of the crystal structure (Fig. S8A, B). For instance, as shown in Fig. S8C, D, the PF values of sequence regions including the N-terminal and L2 are lower than other regions, which indicate relatively high flexibility in these regions. To further compare the structural and dynamic alterations upon inhibitor binding, the ΔPF values for both SAHA and PCI-34051 groups were calculated, respectively (Figs. 3A, B). We found residues 75–95 (α4 and L2-B) exhibited significant ΔPF changes upon both SAHA and PCI-34051 binding. The structural stabilizing effect of these regions are in line with previous research: In the study by Dowling et al., the L2 loop of unbound hHDAC8 was found to be highly dynamic, rendering its structural information unresolved in crystal structures, while upon inhibitor binding, the structure of L2 (residues 84–107) became resolvable, suggesting that inhibitor binding indeed contributes to stabilizing the L2 region28. Additionally, residues 152–157 (part of L3), residues 108–110 (L2-A tail), and residues 108–125 (α5) display distinctive ΔPF increases only in the PCI-34051 group, implying the stronger stabilizing effect of PCI-34051 on the system (Figs. 3C, D). These findings align with the native MS observation of metal losses due to PCI-34051 binding. The PF increase in the 152–157 region can be attributed to the binding effects, similar stabilizing effects have been previously reported in the interactions between F152 of L3 and other inhibitors (e.g., “linkerless” inhibitors)13,57. However, the stabilization of residues 108–110 and α5 (residues 111–125) upon inhibitor binding has not been previously reported. When observing the crystal structure of hHDAC8, hydrophobic interactions are found between L3 and α5 (Fig. 3D). Meanwhile, Y154 on L3 links with Y75 on α4 through H-bond, suggesting that the inhibitor’s interactions with region L3 may contribute to the dynamic alterations in α5 and α4.

A, B The change of ln(PF) from hHDAC8 binding with SAHA and PCI-34051. C, D The change of ln(PF) from hHDAC8 binding with SAHA and PCI-34051 mapped onto crystal structure (PDB: 3F07). The gray sphere refers to the Zn2+ ion and the purple sphere refers to the K+ ion. Only residues exhibiting significant PF changes are highlighted within the structures.
HDX-MS differences upon inhibitor binding
To gain more detailed insights into the binding site and structural alterations, a comprehensive analysis of peptide-level HDX differences and kinetic plots was conducted (Fig. 4A, B). Overall, all peptides with significant deuterium uptake changes (Fig. S9) in either the SAHA or PCI-34051 group exhibited a decreasing deuterium incorporation compared to the unbound state (Fig. 4C, D and Supplementary Data 1 and 2). The deuterium uptake spectra of these peptides show no bimodal or multimodal uptake distributions (Supplementary Data 3), indicating that the structural changes most likely happen on a single conformer58. According to their locations in the crystal structures (Fig. 4A, B), these peptides were categorized into three groups. Firstly, the regions forming the binding channel of hHDAC8 are shown in pink (peptide 28–44 (α1-L1-α2), 132–152 (part of L3), 152–157 (part of L3), 201–216 (L5), 268–277 (L6) and 300–311 (L7)). Secondly, the regions in purple-blue represent areas situated > 20 Å away from the binding channel with significant decreases in deuterium uptake (e.g., peptide 71–95 (α4 + L2-B)) suggesting that the binding of inhibitors leads to distant conformational changes on hHDAC8. Finally, L2-A (peptide 100–110, 96–106), located between the binding regions and the distant conformational changes regions, also shows an HDX decrease. It was previously identified as the binding region in some literature24,28, but considered to be a distant conformational change in others26,27. Furthermore, the HDX kinetic plots were categorized into four types as defined by Wilson’s group39: type I peptides show no significant changes, type II peptides exhibit “transient” changes (show significant changes in early or middle timepoints), type III peptides display “permanent” alterations within all timepoints (show significant changes in all timepoints), while type IV peptides show a mixture of type II and type III.

A, B HDX difference plots for the SAHA group and PCI-34051. The classification of regions is highlighted on the crystal structure (PDB code: 3F07). Pink refers to Binding regions. Purple blue refers to distant conformational change regions. Yellow refers to L2-A. C, D The representative peptides show significant HDX changes of SAHA and PCI-34051.
In the SAHA group, 4 of the 6 significant HDX-decreasing peptides (Fig. 4C, labeled in pink) are located in the binding regions, including peptides 28–44, 132–152, 268–277, and 300–311. Among them, peptides 132–152 (L3), 268–277 (L6), and 300–311 (L7) exhibit type II patterns upon SAHA binding (Fig. 4C), indicating a relatively weak association or high turnover in these regions39. Only peptides 28–44 (α1-L1-α2) show a type IV pattern, indicating a relatively strong association or low turnover compared to the other peptides. In contrast, the reduction in deuterium uptake is more pronounced in the PCI-34051 group (Fig. 4B) than in the SAHA group (Fig. 4A). In total, 12 HDX-decreasing peptides were observed. Six of them are located at the binding region and they are all in loop structure (Fig. 4D, labeled in pink). Among which, peptides 152–157 (part of L3), and 201–216 (L5) are unique binding ones that only occur in PCI-34051 group, while 132–152 (L3) shows a type III pattern, peptides 28–44 (L1), 152–157 (L3) and 300–311 (L7) exhibit type IV patterns (Fig. 4D). These patterns indicate strong association or low turnover. Additionally, peptides 201–216 (L5) and 268–277 (L6) show a type II pattern, but the HDX difference for 268-–277 (L6) is also larger than that observed in the SAHA group. These observations are consistent with native MS data and the Kd values of each inhibitor reported in the literature13,18, suggesting a tighter interaction with PCI-34051. The occurrence of binding peptides 152–157 and 201–216 in the PCI-34051 group indicates stronger and broader ligand-protein interactions induced by PCI-34051. Due to the high structural similarity between Schistosoma mansoni HDAC8 (smHDAC8) and hHDAC8, smHDAC8 has been frequently used as a model protein for obtaining hHDAC8 crystal structures with and without ligand binding5,13. In Romier’s study, they reported that H141 and H142 in L3 of smHDAC8 (corresponding to H142 and H143 of hHDAC8), F216 in L5 (F208 of hHDAC8), and Y341 in L7 (Y306 of hHDAC8) of smHDAC8 form key interactions with PCI-340513. Additionally, K20 in L1 (K31 of hHDAC8) and P291 in L6 (P273 of hHDAC8) create a sub-pocket for PCI-34051 binding. These sites are all located within the HDX-decreasing regions identified above, confirming our findings that they are critical binding regions. Additionally, when observing the structures of smHDAC8 (PDB: 4BZ5), the smHDAC8-SAHA complex (PDB: 4BZ6)5, and the smHDAC8-PCI-34051 complex (PDB: 6HQY)13, it is interesting to find that K20 (K31 in hHDAC8) in L1 and Y341 (Y306 in hHDAC8) in L7 underwent significant changes in residue orientation (Fig. S10). Understanding how molecules recognize and interact with one another is fundamentally important in diverse biological processes such as protein-ligand interactions. The induced-fit model builds upon the lock-and-key hypothesis as well as conformational selection are the most recognized models59,60,61,62. From our HDX-MS data (Supplementary Data 3), the deuterium uptake spectra of the binding channel loops show no bimodal or multimodal uptake distributions, suggesting that the conformational changes in the binding channel loops are most likely being induced-fit. Similar ligand-induced active site stabilization was also observed in Konermann’s study of enzyme conformational dynamics during ligand binding using HDX-MS42.
Apart from the binding regions, the distant conformational changes also differ between the two inhibitor groups (Fig. 4A, B). Specifically, in the SAHA group, the α4 + L2-B region (peptide 71–95) shows a type II pattern (Fig. 4C); while upon PCI-34051 binding, α4 + L2-B (peptide 71–95), α3 (peptide 67–74), α5 (peptide 111–125), as well as a loop on the surface of hHDAC8 (peptide 48–66), display a type II or a type IV pattern (Fig. 4D), suggesting more intensive regulatory structural effects induced by PCI-34051. This is consistent with our PF analysis as well as literature reports28. These results strongly support the idea that inhibitor binding induces substantial structural stabilization in this region, and such effects are mostly induced through the structural communication network from binding sites to distant sites63,64,65. These distant regions identified in our experiments are spatially located at the periphery of binding regions (Fig. 4C, D), also suggesting a transfer effect from the binding channel to the distant regions. According to the crystal structure, the binding loop L3 interacts with adjacent secondary structures (α1, α4, α5, and L2-A) through H-bonds (Fig. 5A), including N136-Y24 (α1), K146-H71 (α4), Y154-Y75 (α4), D157-T119 (α5), N136-T119 (α5), A149-Y96 (L2-A), F152-C102 (L2-A), and Y154-P103 (L2-A). Meanwhile, the binding loop L1 interacts with α2 and α1 through H-bonds of I34-R37, K36-D29. Therefore, the inhibitors’ binding to L3 and L1 is likely to trigger dynamic fluctuations in the peripheral regions (such as α1, α2, α4, α5, and L-2A) through these interactions. Additionally, intra-loop H-bonds are observed within L1, L2-A, L2-B, and L3 (Fig. 5A), which means the conformational change of one residue may influence the whole loop. Taken together, we therefore observed the trend of HDX decreasing that communicated from binding regions to distant regions.

A Depiction of H-bonds within the structural domain encompassing L1, L2, L3, α1, α2, α4, and α5, as determined from the crystal structure (PDB ID: 3F07). Intrahelical hydrogen bonds have been omitted for clarity. The illustration does not include non-bonded main chain or side chain interactions. B Analysis of the perturbations in residue interactions upon PCI-34051 binding, as revealed by molecular dynamics (MD) simulations. Enhanced interactions between residues are indicated by red lines, whereas diminished interactions are denoted by blue dashes. The intensity of the color gradient corresponds directly to the magnitude of the interaction change. L3 is highlighted in blue on this structure.
MD simulation confirms the binding regions and distant conformational changes
To further investigate how the structural stabilization effects in binding regions impact the distant regions in the dynamic circumstance, we performed 1 μs MD simulations for unbound hHDAC8, hHDAC8-SAHA, and hHDAC8-PCI-34051 complexes. MD trajectories reveal that L2-A becomes more stable and shifts closer to the inhibitor binding sites upon inhibitor binding (Fig. S11). To quantify these changes, we calculated the Root Mean Square Fluctuation (RMSF) for each residue across the three groups and compared differences between the inhibitor-bound and unbound states. It shows that L2-A indeed undergoes significant conformational stabilization, with a more pronounced effect observed in hHDAC8-PCI-34051 (Fig. S12). However, contact probability calculations reveal that L2-A exhibits no contact with the inhibitors (Fig. S13 and Table S4). These results highlights the distant regulation role of L2-A and meanwhile fills the uncertain role of L2-A in HDX-MS results. Although the Y100-D101 of L2-A was reported to be interacted with the substrate in Vannini’s early investigation66. Both Hansen and Wu observed conformational stabilization of L2-A without interaction with the inhibitor26,27, which aligns with our findings. They attribute this stabilization to the mediation of either L1 or the F152 (L3) & F208 (L5). To obtain a thorough interaction map of the secondary structures, the residual interaction network (combining salt-bridges, hydrogen bonds, and hydrophobic interactions) within hHDAC8 in the presence or absence of PCI-34051 was analyzed. Upon PCI-34051 binding, enhanced residual interactions were found within the binding regions (L1 and L3) and their neighboring regions (Fig. 5B). Specifically, the interactions of L1-a5, L1-L2-A, L3-α1, L3-α5, and L3-L2-A were enhanced, and these neighboring regions further interact with L2-B, α4, and α5. Therefore, a clear route can be extracted from the PCI-34051 group, which starts from the binding site (Zn) towards L2-B, α4, and α5 regions, where L3 connects to most secondary structures through an increased number of hydrogen bonds. Meanwhile, along these routes, the reduced H-bonds were also observed within inner hHDAC8. Nevertheless, these changes underline the residual interaction rearrangements across these secondary structures where L3 functions as a critical mediating bridge, elucidating how binding events precipitate widespread structural influences.
In addition to these regions, the contact probability, RMSF of local regions, and the changes in the inhibitor binding free energy (ΔGbinding) have also been examined to explore the structural-to-affinity relationship. Residues with a contact probability >0.2 are considered as significant contacted residues. In SAHA group, these residues are located at L1 (K33, I34), L3 (H143, F152), L4 (D178, L179, H180), L6 (D267), L7 (G304, G305, Y306) (Table S4 and Fig. S13A). These residues in PCI-34051 group are located at L1 (K33, P35), L3 (F152), L4 (D176, D178, L179, H180, H181), L6 (D267, M274), L7 (G304, G305, Y306, L308), β5 (a β-sheet linked with L5, T197, S199, L200, H201), β6 (a β-sheet adjacent to β5, S226) (Table S4 and Fig. S13B). Specifically, F152 of L3 show flexible conformation before inhibitors binding (Fig. S11A–C). But after binding, it aligns towards the inhibitors due to π-π stacking towards the inhibitors’ cap regions in both SAHA (Fig. S11D–F) and PCI-34051 groups (Fig. S11G–I), further emphasizing binding of the inhibitors can affect the conformation of L3. Nevertheless, PCI-34051 induced a more intensive stabilization effect towards L3 than SAHA according to PF analysis (Fig. 3). In fact, owing to the aromatic rings in the linker and cap regions of PCI-34051, π–π stacking interactions between PCI-34051 and aromatic residues of hHDAC8 such as F152, F208, and Y306 can also be found in Romier’s previous study13, which correlates with our study. Additionally, as observed in HDX-MS our experiments, broader contacts between PCI-34051 and the binding pocket of hHDAC8 can be also seen in MD simulations (Fig. S13). For the protein dynamics, L5 shows clear movement with high RMSF in unbound hHDAC8 (Fig. S12A); however, PCI-34051 binding markedly reduces the flexibility in this region (Fig. S12B). This may due to the fact that PCI-34051 binds directly with β5, adjacent to L5, thereby stabilizing the region. Furthermore, L6 shows increased RMSF upon inhibitor binding, indicating destabilization in this region. This effect is likely induced by steric hindrance from the inhibitor, with PCI-34051 causing a more pronounced impact compared to SAHA, suggesting a stronger conformational displacement in this region by PCI-34051. The dynamic changes in these regions is consistent with the high plasticity of the hHDAC8 binding channel as reported in the literature13,23,24. To further assess the differences in binding affinity of these two ligands, we calculated the ΔGbinding through MM/PBSA (Table 1). The results show that PCI-34051 has a lower ΔGbinding (−38.05 kJ/mol) compared to that of SAHA (−33.04 kJ/mol), which is consistent with its lower experimental Kd value compared to SAHA13,18.
Overall, our integrative data together with the findings in the literature13,23,24 point out that the high plasticity of hHDAC8’s binding channel is key to accommodate PCI-34051. The lower ΔGbinding as well as Kd of PCI-34051 suggest that PCI-34051 fits the hHDAC8 pocket more adequately than SAHA, leading to a lower energy state for the system. This binding effect is transmitted through L3 to its surrounding structures, such as L2-A, L5, and even the periphery of hHDAC8.
Discussion
By integrating data from native MS, HDX-MS, MD simulations, and insights from relevant literature, we have obtained a comprehensive understanding of the structural and dynamic processes in hHDAC8 upon inhibitor binding. Following coordination with the zinc ion, PCI-34051 interacts with residues in the binding channel, which most likely causes an induced-fit conformational change of the binding channel loops. Subsequently, the system experiences an energy-decreasing event, characterized by significant structural changes resulting in the loss of K+ and Na+ ions as well as distant stabilizing regulation (evidenced by decreases in HDX or RMSF) in regions such as L2-A, α5, and even α1 + α2. During the distant structural regulation, L3 serves as a structural bridge, linking the binding regions and distant regions through the residual communication network. In comparison to PCI-34051, SAHA, with a flexible fatty chain as the linker group, interacts with fewer residues and forms a weak interaction with hHDAC8, thus stabilizing the energy state less intensively.
In elucidating the selective mechanism of PCI-34051, we integrated findings from Romier’s study, which demonstrated that the exposure of Y306 forms a sub-pocket receptive to PCI-3405113. We further infer that the dynamics of hHDAC8’s binding channel also contribute to the selectivity. Specifically, the plastic binding channel of hHDAC8, unlike those of other HDAC subtypes, can adapt to accommodate PCI-34051’s rigid and bulky linker group. This adaptation is evidenced by the extensive binding interface resulting from PCI-34051 interactions, as revealed through our HDX-MS results and dynamic analyses. Conversely, SAHA can access the binding channels of various HDAC subtypes due to its thinner linker group (Fig. 1B).
Combining the understanding of the structural and dynamic characteristics of hHDAC8, we explored how these features could guide inhibitor development. Firstly, the insertion of an aromatic ring into the inhibitor’s linker enhances its affinity to hHDAC8, which is supported by the quantitative structure-activity relationship studies67,68,69 and Romier’s study13. Secondly, considering the plasticity of hHDAC8’s binding channel surface across class I HDACs, increasing the size and rigidity of the inhibitor’s linker region may enhance selectivity. Thirdly, in addition to hHDAC8, other class I HDACs like HDAC1-3, functioning within protein-protein complexes, may also exhibit stabilization effects at distant conformational change regions identified in our experiments, potentially affecting their stability and functions. Indeed, literature reports have suggested the development of potential allosteric inhibitors targeting distal structural elements (L2-A region) of class I HDACs70. Lastly, HDAC covalent inhibitors and selective hHDAC8 degraders have been developed with improved inhibition effects and fewer side effects71,72. These novel avenues also provide potential druggable target sites, awaiting further structural studies.
In summary, enhancing inhibition with improved selectivity and reduced toxicity remains a key challenge in developing selective hHDAC8 inhibitors. In this study, we used native MS, HDX-MS, and MD simulations to elucidate the binding mechanisms and dynamic effects of hHDAC8 with selective inhibitor PCI-34051 and the pan-inhibitor SAHA. Our findings revealed differing mechanisms between PCI-34051 and SAHA, with PCI-34051 inducing a lower energy state in hHDAC8. Furthermore, we investigated hHDAC8’s dynamic properties, characterized by its dynamic binding channel surface and distant structural regulatory effects upon inhibitor binding. This comprehensive approach provides valuable insights into the structural and dynamic alterations in hHDAC8, informing the design of selective and potentially allosteric inhibitors. Overall, our study highlights the efficacy of an integrative structural approach in understanding the structure and dynamics of protein-ligand interactions, guiding therapeutic development.
Methods
Materials
The hHDAC8 sample was expressed by Atagenix (Wuhan, China) with a C-terminal 6×His-tag, and stored in 50 mM Tris-HCl (pH 8.0), 150 mM KCl, and 1 mM DTT. SAHA was purchased from Sigma-Aldrich (St. Louis, USA). PCI-34051 was purchased from Selleck (Houston, USA). Ammonium acetate, ammonia, cesium iodide, dimethyl sulfoxide (DMSO), guanidine hydrochloride, potassium monophosphate, potassium dihydrogen phosphate, tris(2-carboxyethyl)phosphine hydrochloride (TCEP) were purchased from Sigma-Aldrich (St. Louis, USA). Acetonitrile (LC-MS grade) and formic acid (MS grade) were purchased from Fisher Scientific (Waltham, USA). Deuterium oxide was purchased from Cambridge Isotope Laboratories (Tewksbury, USA). HDAC assay kit was purchased from Active Motif (Carlsbad, USA). Borosilicate glass capillary (1.0 mm o.d./0.58 mm i.d.) was purchased from Sutter (Novato, USA). Ultra-centrifugal filters with 10 kD molecular weight (MW) cut-off were purchased from Millipore (Burlington, USA).
Activity test of hHDAC8
The hHDAC8 sample was first titrated with the ZnCl2 to test the enzyme activity’s dependence on Zn2+. hHDAC8 was mixed with ZnCl2 at molar ratios of 1:0, 1:1, 1:1.4, 1:1.8, and 1:2.2 respectively, at 4 °C for 1 h, with a final protein concentration of 7.5 μM. Subsequently, hHDAC8 was mixed with the BOC-(Ac)Lys-AMC substrate at final concentrations of 3 μM and 100 μM, respectively, in a total reaction volume of 50 μL. The reaction plate was incubated at 37 °C for 1 h, followed by addition of 50 μL developing solution and left to stood for 10–15 min. Fluorescence intensity was read with an excitation wavelength of 360 nm and an emission wavelength of 460 nm.
For inhibitor effect testing, hHDAC8 was pre-activated using the optimal ZnCl2 incubation ratio determined earlier. The protein-to-substrate ratio remained the same as described above. In the inhibitor groups, inhibitors including SAHA and PCI-34051 were added to a final concentration of 15 μM for each inhibitor group, respectively. The reactions were carried out under identical conditions as described for the ZnCl2 activity assay.
Native mass spectrometry
The ZnCl2-activated hHDAC8 was buffer-exchanged into 200 mM ammonium acetate buffer using 10 kD MW cut-off ultra-centrifugal filters. For inhibitor ratio optimization, SAHA was reacted at a molar ratio of 1:10 and 1:20, and PCI-34051 was reacted at a molar ratio of 1:5 and 1:10 at 37 °C for 30 min. The mass spectrum of the 1:10 group of each inhibitor was chosen for observing the binding phenomenon. hHDAC8 containing 1% DMSO was taken as the blank group. Native MS experiments were first conducted on a Synapt G2-Si Q-ToF mass spectrometer (Waters, USA). The samples were then sprayed from house-prepared borosilicate capillaries to the nano-electrospray source. The capillary voltage was 1.4–1.5 kV, the sampling cone voltage was varied from 40 to 150 V, the source offset was 80 V, the source temperature was 80 °C, the cone gas flow was 50 L/Hr, and the purge gas flow was 600 mL/h. These samples were then injected into Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific, USA) with a spray voltage of 1.4–1.7 kV, in source dissociation voltage of 20 eV, and Orbitrap resolution of 30000. The native MS spectra acquired from the Orbitrap instrument were deconvoluted using UniDec software73.
Hydrogen-deuterium exchange mass spectrometry
hHDAC8 was prepared at a preliminary concentration of 20 μM. For inhibitor ratio optimization, SAHA was reacted at a molar ratio of 1:10, 1:15, and 1:20, PCI-34051 was reacted at a molar ratio of 1:5, 1:8, and 1:10 at 37 °C for 30 min. The ratio was finally determined to be 1:15 and 1:8 for SAHA and PCI-34051, respectively, to achieve a maximum amount of protein-drug complex but avoid nonspecific binding. HDX reaction was initiated at 20 °C, and 3 μL of 20 μM protein or protein-inhibitor samples were diluted into 57 μL of phosphate buffer (composition see Table S1) in H2O (pH 7.4, non-labeling reference) or phosphate buffer in D2O (pD 7.4, labeling experiment). At five labeling time points 10 s, 1, 10, 60, and 240 min, aliquots were quenched with a quench buffer containing 0.1 M K2HPO4, 1 M Gnd-HCl, and 0.5 M TCEP in H2O (1:1 v/v) at pH 2.5 and 0 °C for 2 min.
The quenched samples were then injected into the HDX ACQUITY system (Waters, USA). The sample first flowed through a pepsin digestion column (Enzymate BEH Pepsin Column, 300 Å, 5 µm, 2.1 mm × 30 mm) at a rate of 100 μL/min at 15 °C. The peptide products flowed into a trap column (BEH C18 Vanguard, 130 Å, 1.7 μm, 2.1 mm × 5 mm) for desalting and enrichment. After 4 min trapping, the samples were then injected into an analytic column (BEH C18, 130 Å, 1.7 μm, 1.0 mm × 100 mm). The LC gradient was run at 50 μL/min from 2 to 35% mobile phase B in 6 min with a 1 min hold at 35%, followed by a flush up to 85% in 0.5 min. A clean blank of 0.1% formic acid (aqueous) was run after every sample injection with the same procedure as the sample. Mobile phase A was 0.1% formic acid (aqueous) and mobile phase B was 0.1% formic acid (acetonitrile).
Data acquisition was performed on the Synapt G2-Si instrument in positive polarity and resolution mode. The capillary voltage was set to 3 kV, the sampling cone voltage was set to 30 V, and the trap collision voltage was set to 4 V (low energy) and 20–35 V ramping (elevated energy). Mass spectra were acquired at the m/z range of 50–2000 using MSE acquisition mode. Source and desolvation temperatures were set to 80 and 250 °C, respectively. MS was calibrated using sodium formate, and lock mass was corrected using Leu-enkephalin. Each labeling sample was performed in triplicate.
Peptides were assigned using the ProteinLynx Global Server (PLGS, Waters, USA) software package. DynamX 3.0 software (Waters, USA) was used to calculate and compare the deuterium uptake of each assigned peptide. Deuteros software74 was used to calculate the confidence interval of significant HDX difference. PyHDX45 was used to generate residue-level PF from HDX difference data. A summary table of the HDX-MS data can be found in the Supplementary Information (Table S3). Protein structures were visualized with PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.).
Modeling, molecular dynamics simulations and data analyze of SAHA and PCI-34051 with hHDAC8
The crystal structure of hHDAC8 (PDB code: 1T69) was used as the starting conformation for our MD simulations of hHDAC8 in the apo and SAHA-bound states. The loop-missing structure was constructed using the structure predicted by AlphaFold2. The two bound potassium ions in the binding sites (denotated as K+1 and K+2) were modelled based on the crystal structure of schistosoma mansoni hHDAC8 (smHDAC8) complexed with PCI inhibitor (PDB code: 6HQY). The PCI-bound hHDAC8 complex was also obtained by structural alignment between smHDAC8 and hHDAC8. H142 and H181 were protonated according to pKa values predicted by ProPka375. Force field parameters for the inhibitors were generated using the CHARMM General Force Field (CGenFF). Both SAHA and PCI were negatively charged (with −1e). The CHARMM36m force field was used for the protein76.
The models of the protein or the inhibitor-bound complex were then placed into a periodic cubic box with sides of 8.3 nm solvated with TIP3P water molecules containing Na+ and Cl- ions at 0.15 M, resulting in ∼54,000 atoms in total for each system. The systems were then energy minimized and equilibrated in a stepwise manner using 1 ns NVT simulations and a following NPT simulation. Finally, 1000 ns were simulated for each trajectory. Neighbor searching was performed every 20 steps. As the crystal structures of HDAC8 has been deposited, the the brute-force MD simulation timescale is not needed in our simulation. The PME algorithm was used for electrostatic interactions with a cut-off of 1.2 nm. A reciprocal grid of 72 × 72 × 72 cells was used with 4th order B-spline interpolation. A single cut-off of 1.272 nm was used for Van der Waals interactions. Temperature coupling was done with the V-rescale algorithm. Pressure coupling was done with the Parrinello-Rahman algorithm. The hydrogen mass repartitioning technique77 was used allowing simulations to be performed with an integration time step of 4 fs. All simulations were performed using a GPU-accelerated version of Gromacs 2021.3 in three replicates78.
Protein structures were visualized using PyMOL. The root-mean-square deviation and RMSF per residue of the trajectories were analyzed using Gromacs gmx tools. The ion density map was analyzed using GROmaρs79. The protein-ligand interactions were analyzed using getcontacts (https://getcontacts.github.io/). The long-range communication was analyzed using the Pyintergraph tool80. To compare the binding affinities of PCI and SAHA with hHDAC8, we used the molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) method (https://github.com/Valdes-Tresanco-MS/gmx_MMPBSA) to estimate binding free energies. Confirmed the protonation state of the structure and input the trajectory files xtc, tpr, and index files, set the polar or non-polar environment according to the default parameters, and ran gmx apbs to produce the results.
Statistics and reproducibility
All analyses related to enzyme activity tests were performed using one-way analysis of variance (ANOVA) with the GraphPad Prism 8 software package (GraphPad, San Diego, CA). The statistical significance of HDX-MS data was assessed at a 99% confidence level using a two-tailed t-test with 99% confidence limits and 2 degrees of freedom, conducted within the Deuteros software package. The methods for calculating contact probability, RMSF, and residual interactions are described in the “Modeling, Molecular Dynamics Simulations, and Data Analysis of SAHA and PCI-34051 with hHDAC8” section. All experiments were conducted in three replicate measurements. The sample sizes (n = 3) in each figure are indicated in the respective figure legend. MD trajectories were run in three replicates.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The native MS and HDX-MS raw data was deposited on MassIVE database (http://massive.ucsd.edu) under accession number MSV000096946 and MSV000096388. The lists of HDX states for the peptides are provided in Supplementary data 4 and 5. The list of residual RMSF values is included in the Supplementary data 6. The residual PF values are lists in Supplementary data 7 and 8. The HDX differences for each peptide were listed in Supplementary data 9 and 10. The changes of residual interaction list are provided in Supplementary data 11. The source data of Fig. S2 are provided in Supplementary data 12 and 13. The crystal structures from other publications referenced in this paper are accessible under the PDB ID 1T69, 3F07, 4BZ5, 4BZ6, and 6HQY.
Code availability
The initial coordinate and simulation input files and a coordinate file of the final output are provided in a GitHub repository and can be accessed via the following URL: https://github.com/YuX-Luo/HDAC8_inhibitors_coordination_files.
References
Falkenberg, K. J. & Johnstone, R. W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 13, 673–691 (2014).
Micelli, C. & Rastelli, G. Histone deacetylases: structural determinants of inhibitor selectivity. Drug Discov. Today 20, 718–735 (2015).
Oehme, I., Deubzer, H. E., Lodrini, M., Milde, T. & Witt, O. Targeting of HDAC8 and investigational inhibitors in neuroblastoma. Expert Opin. Inv. Drug 18, 1605–1617 (2009).
Kaiser, F. J. et al. Loss-of-function HDAC8 mutations cause a phenotypic spectrum of Cornelia de Lange syndrome-like features, ocular hypertelorism, large fontanelle and X-linked inheritance. Hum. Mol. Genet 23, 2888–2900 (2014).
Marek, M. et al. Structural basis for the inhibition of histone deacetylase 8 (HDAC8), a key epigenetic player in the blood fluke schistosoma mansoni. PLoS Pathog. 9, e1003645 (2013).
Yamauchi, Y. et al. Histone deacetylase 8 is required for centrosome cohesion and influenza A virus entry. PLoS Pathog. 7, e1002316 (2011).
Fan, W. et al. Histone deacetylase inhibitor based prodrugs. Eur. J. Med Chem. 203, 112628 (2020).
Luo, Y. & Li, H. Structure-based inhibitor discovery of class i histone deacetylases (HDACs). Int. J. Mol. Sci. 21, 8828 (2020).
Chakrabarti, A. et al. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Future Med. Chem. 8, 1609–1634 (2016).
Zhang, L. H., Chen, Y. M., Jiang, Q. X., Song, W. G. & Zhang, L. Therapeutic potential of selective histone deacetylase 3 inhibition. Eur. J. Med. Chem. 162, 534–542 (2019).
Millard, C. J., Watson, P. J., Fairall, L. & Schwabe, J. W. R. Targeting class i histone deacetylases in a “complex” environment. Trends Pharm. Sci. 38, 363–377 (2017).
Gupta, P., Reid, R. C., Iyer, A., Sweet, M. J. & Fairlie, D. P. Towards isozyme-selective HDAC inhibitors for interrogating disease. Curr. Top. Med. Chem. 12, 1479–1499 (2012).
Marek, M. et al. Characterization of histone deacetylase 8 (HDAC8) selective inhibition reveals specific active site structural and functional determinants. J. Med. Chem. 61, 10000–10016 (2018).
Krennhrubec, K., Marshall, B. L., Hedglin, M., Verdin, E. & Ulrich, S. M. Design and evaluation of ‘Linkerless’ hydroxamic acids as selective HDAC8 inhibitors. Bioorg. Med. Chem. Lett. 17, 2874–2878 (2007).
Suzuki, T. et al. Rapid discovery of highly potent and selective inhibitors of histone deacetylase 8 using click chemistry to generate candidate libraries. J. Med. Chem. 55, 9562–9575 (2012).
Balasubramanian, S. et al. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 22, 1026–1034 (2008).
Hailu, G. S. et al. Lysine Deacetylase inhibitors in parasites: past, present, and future perspectives. J. Med. Chem. 60, 4780–4804 (2017).
Singh, R. et al. Kinetic and thermodynamic rationale for suberoylanilide hydroxamic acid being a preferential human histone deacetylase 8 inhibitor as compared to the structurally similar ligand, trichostatin A. Biochemistry 52, 8139–8149 (2013).
Wang, F. et al. Combined anti-PD-1, HDAC inhibitor and anti-VEGF for MSS/pMMR colorectal cancer: a randomized phase 2 trial. Nat. Med. 30, 1035–1043 (2024).
Rettig, I. et al. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 6, e1657 (2015).
Amaral, M. et al. Protein conformational flexibility modulates kinetics and thermodynamics of drug binding. Nat. Commun. 8, 2276 (2017).
Boehr, D. D., Nussinov, R. & Wright, P. E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 5, 789–796 (2009).
Vannini, A. et al. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc. Natl. Acad. Sci. USA 101, 15064–15069 (2004).
Somoza, J. R. et al. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 12, 1325–1334 (2004).
Werbeck, N. D. et al. A distal regulatory region of a class I human histone deacetylase. Nat. Commun. 11, 3814 (2020).
Kunze, M. B. A. et al. Loop interactions and dynamics tune the enzymatic activity of the human histone deacetylase 8. J. Am. Chem. Soc. 135, 17862–17868 (2013).
Zhou, J., Huang, Y., Cheng, C., Wang, K. & Wu, R. Intrinsic dynamics of the binding rail and its allosteric effect in the class I histone deacetylases. J. Chem. Inf. Model. 57, 2309–2320 (2017).
Dowling, D. P., Gantt, S. L., Gattis, S. G., Fierke, C. A. & Christianson, D. W. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry 47, 13554–13563 (2008).
Zhang, M. M. et al. An Integrated approach for determining a protein-protein binding interface in solution and an evaluation of hydrogen-deuterium exchange kinetics for adjudicating candidate docking models. Anal. Chem. 91, 15709–15717 (2019).
Huang, R. Y. C. & Chen, G. D. Higher order structure characterization of protein therapeutics by hydrogen/deuterium exchange mass spectrometry. Anal. Bioanal. Chem. 406, 6541–6558 (2014).
Liu, T., Limpikirati, P. & Vachet, R. W. Synergistic structural information from covalent labeling and hydrogen-deuterium exchange mass spectrometry for protein-ligand interactions. Anal. Chem. 91, 15248–15254 (2019).
Masson, G. R. et al. Recommendations for performing, interpreting and reporting hydrogen deuterium exchange mass spectrometry (HDX-MS) experiments. Nat. Methods 16, 595–602 (2019).
Trabjerg, E., Nazari, Z. E. & Rand, K. D. Conformational analysis of complex protein states by hydrogen/deuterium exchange mass spectrometry (HDX-MS): challenges and emerging solutions. Trac-Trend Anal. Chem. 106, 125–138 (2018).
Masson, G. R., Jenkins, M. L. & Burke, J. E. An overview of hydrogen deuterium exchange mass spectrometry (HDX-MS) in drug discovery. Expert Opin. Drug Discov. 12, 981–994 (2017).
Kahsai, A. W., Rajagopal, S., Sun, J. & Xiao, K. Monitoring protein conformational changes and dynamics using stable-isotope labeling and mass spectrometry. Nat. Protoc. 9, 1301–1319 (2014).
Zhang, Z. Complete extraction of protein dynamics information in hydrogen/deuterium exchange mass spectrometry data. Anal. Chem. 92, 6486–6494 (2020).
Hodge, E. A., Benhaim, M. A. & Lee, K. K. Bridging protein structure, dynamics, and function using hydrogen/deuterium-exchange mass spectrometry. Protein Sci. 29, 843–855 (2020).
Niu, B. et al. Protein footprinting and X-ray crystallography reveal the interaction of PD-L1 and a macrocyclic peptide. Biochemistry 59, 541–551 (2020).
Narang, D., James, D. A., Balmer, M. T. & Wilson, D. J. Protein footprinting, conformational dynamics, and core interface-adjacent neutralization “hotspots” in the SARS-CoV-2 spike protein receptor binding domain/human ACE2 interaction. J. Am. Soc. Mass Spectrom. 32, 1593–1600 (2021).
Liyanage, O. T., Quintero, A. V., Hatvany, J. B. & Gallagher, E. S. Distinguishing carbohydrate isomers with rapid hydrogen/ deuterium exchange-mass spectrometry. J. Am. Soc. Mass Spectrom. 32, 152–156 (2021).
Liu, X. R., Zhang, M. M. & Gross, M. L. Mass spectrometry-based protein footprinting for higher-order structure analysis: fundamentals and applications. Chem. Rev. 120, 4355–4454 (2020).
Fast, C. S., Vahidi, S. & Konermann, L. Changes in enzyme structural dynamics studied by hydrogen exchange-mass spectrometry: ligand binding effects or catalytically relevant motions? Anal. Chem. 89, 13326–13333 (2017).
Zhang, J., Balsbaugh, J. L., Gao, S., Ahn, N. G. & Klinman, J. P. Hydrogen deuterium exchange defines catalytically linked regions of protein flexibility in the catechol O-methyltransferase reaction. Proc. Natl. Acad. Sci. USA 117, 10797–10805 (2020).
Ducharme, J., Polic, V., Thibodeaux, C. J. & Auclair, K. Combining small-molecule bioconjugation and hydrogen-deuterium exchange mass spectrometry (HDX-MS) to expose allostery: the case of human cytochrome P450 3A4. ACS Chem. Biol. 16, 882–890 (2021).
Smit, J. H. et al. Probing universal protein dynamics using hydrogen-deuterium exchange mass spectrometry-derived residue-level gibbs free energy. Anal. Chem. 93, 12840–12847 (2021).
Kish M. et al. Allosteric regulation of glycogen phosphorylase by order/disorder transition of the 250’ and 280s Loops. Biochemistry 62, 1360–1368 (2023).
Huang J. et al. Insights into phosphorylation-induced protein allostery and conformational dynamics of glycogen phosphorylase via integrative structural mass spectrometry and in silico modeling. ACS Chem. Biol.17, 1951–1962 (2022).
Li, H., Nguyen, H. H., Ogorzalek Loo, R. R., Campuzano, I. D. G. & Loo, J. A. An integrated native mass spectrometry and top-down proteomics method that connects sequence to structure and function of macromolecular complexes. Nat. Chem. 10, 139–148 (2018).
Li, H., Wongkongkathep, P., Van Orden, S. L., Ogorzalek Loo, R. R. & Loo, J. A. Revealing ligand binding sites and quantifying subunit variants of noncovalent protein complexes in a single native top-down FTICR MS experiment. J. Am. Soc. Mass Spectrom. 25, 2060–2068 (2014).
Li, H. et al. Structural characterization of native proteins and protein complexes by electron ionization dissociation-mass spectrometry. Anal. Chem. 89, 2731–2738 (2017).
Yin, Z., Huang, J., Miao, H., Hu, O. & Li, H. High-pressure electrospray ionization yields supercharged protein complexes from native solutions while preserving noncovalent interactions. Anal. Chem. 92, 12312–12321 (2020).
Natalello, A., Santambrogio, C. & Grandori, R. Are charge-state distributions a reliable tool describing molecular ensembles of intrinsically disordered proteins by native MS? J. Am. Soc. Mass Spectrom. 28, 21–28 (2017).
Santambrogio, C., Natalello, A., Brocca, S., Ponzini, E. & Grandori, R. Conformational characterization and classification of intrinsically disordered proteins by native mass spectrometry and charge-state distribution analysis. Proteomics 19, e1800060 (2019).
Hu, E. et al. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 275, 15254–15264 (2000).
Whitehead, L. et al. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Biorg Med. Chem. 19, 4626–4634 (2011).
Dowling, D. P., Gattis, S. G., Fierke, C. A. & Christianson, D. W. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry 49, 5048–5056 (2010).
Tabackman, A. A., Frankson, R., Marsan, E. S., Perry, K. & Cole, K. E. Structure of ‘linkerless’ hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. J. Struct. Biol. 195, 373–378 (2016).
James, E. I., Murphree, T. A., Vorauer, C., Engen, J. R. & Guttman, M. Advances in hydrogen/deuterium exchange mass spectrometry and the pursuit of challenging biological systems. Chem. Rev. 122, 7562–7623 (2022).
Fischer, E. Einfluss der configuration auf die wirkung der enzyme. Ber. Dtsch Chem. Ges. 27, 2985–2993 (1894).
Koshland, D. E. Application of a theory of enzyme specificity to protein synthesis. Proc. Natl. Acad. Sci. USA 44, 98–104 (1958).
MONOD, J., WYMAN, J. & CHANGEUX, J. P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 12, 88–118 (1965).
Holyoak, T. Molecular recognition: lock-and-key, induced fit, and conformational selection. In Encyclopedia of Biophysics (ed Roberts G. C. K.) 1584–1588 (Springer Berlin Heidelberg, 2013).
Zhang, Y. et al. Intrinsic dynamics is evolutionarily optimized to enable allosteric behavior. Curr. Opin. Struct. Biol. 62, 14–21 (2020).
Motlagh, H. N., Wrabl, J. O., Li, J. & Hilser, V. J. The ensemble nature of allostery. Nature 508, 331–339 (2014).
Goodey, N. M. & Benkovic, S. J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 4, 474–482 (2008).
Vannini, A. et al. Substrate binding to histone deacetylases as shown by the crystal structure of the HDAC8-substrate complex. Embo Rep. 8, 879–884 (2007).
Banerjee, S., Adhikari, N., Amin, S. A. & Jha, T. Histone deacetylase 8 (HDAC8) and its inhibitors with selectivity to other isoforms: an overview. Eur. J. Med. Chem. 164, 214–240 (2019).
Manal, M. et al. Novel HDAC8 inhibitors: a multi-computational approach. SAR QSAR Environ. Res. 28, 707–733 (2017).
Ortore, G., Di Colo, F. & Martinelli, A. Docking of hydroxamic acids into HDAC1 and HDAC8: a rationalization of activity trends and selectivities. J. Chem. Inf. Model. 49, 2774–2785 (2009).
McClure, J. J. et al. Development of allosteric hydrazide-containing class i histone deacetylase inhibitors for use in acute myeloid leukemia. J. Med. Chem. 59, 9942–9959 (2016).
Raouf, Y. S. et al. Discovery of YSR734: a covalent HDAC inhibitor with cellular activity in acute myeloid leukemia and duchenne muscular dystrophy. J. Med. Chem. 66, 16658–16679 (2023).
Huang, J. et al. Structure-based discovery of selective histone deacetylase 8 degraders with potent anticancer activity. J. Med. Chem. 66, 1186–1209 (2022).
Marty, M. et al. Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal. Chem. 87, 4370–4376 (2015).
Lau, A. M. C., Ahdash, Z., Martens, C. & Politis, A. Deuteros: software for rapid analysis and visualization of data from differential hydrogen deuterium exchange-mass spectrometry. Bioinformatics 35, 3171–3173 (2019).
Olsson, M. H. M., Sondergaard, C. R., Rostkowski, M. & Jensen, J. H. PROPKA3: consistent treatment of internal and surface residues in empirical pK(a) predictions. J. Chem. Theory Comput. 7, 525–537 (2011).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Hopkins, C. W., Le Grand, S., Walker, R. C. & Roitberg, A. E. Long-time-step molecular dynamics through hydrogen mass repartitioning. J. Chem. Theory Comput. 11, 1864–1874 (2015).
Abraham, M. J. et al. GROMACS: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1-2, 19–25 (2015).
Briones, R., Kutzner, BiauC., de Groot, C. & Aponte-Santamaria, B. L. C. GROma rho s: a GROMACS-based toolset to analyze density maps derived from molecular dynamics simulations. Biophys. J. 116, 4–11 (2019).
Sora, V. et al. PyInteraph2 and PyInKnife2 to analyze networks in protein structural ensembles. J. Chem. Inf. Model. 63, 4237–4245 (2023).
Acknowledgements
H.L. would like to thank the Pearl River Talent Recruitment Program for support. H.L. thanks Prof. Yong Wang from Zhejiang University for the help and instructions on the MD simulations and helpful discussion. Y.L. would like to thank Xi Chen for the instruction of the HDX-MS system. X.C. thanks Dr. Smit for the help and instructions on the use of PyHDX software. This work was supported in part by the NSFC (No. 81872836, 91953102), Natural Science Foundation of Guangdong Province (No. 2022A1515010965, 2019A1515011265), Guangdong Provincial Key Laboratory of Construction Foundation (No. 2023B1212060022).
Author information
Authors and Affiliations
Contributions
Yuxiang Luo: investigation, data analysis, writing–original draft preparation, conceptualization. Zhaoyue Yan: investigation, data analysis review & editing. Xiakun Chu: investigation, data analysis, writing–review & editing. Ying Zhang: investigation. Yufan Qiu: investigation. Huilin Li: investigation, conceptualization, writing–review & editing, project administration, funding acquisition, supervision.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Jonathan Phillips and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Antonio Calabrese and Laura Rodríguez Pérez. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, Y., Yan, Z., Chu, X. et al. Binding mechanism and distant regulation of histone deacetylase 8 by PCI-34051. Commun Biol 8, 221 (2025). https://doi.org/10.1038/s42003-025-07649-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-07649-0
This article is cited by
-
Decoding HDACs and its inhibitors-artificial intelligence assisted smart software based super computational modelling technology in targeting cancer and neurological disorders of the brain
Network Modeling Analysis in Health Informatics and Bioinformatics (2025)
