
Abstract
This study aims to evaluate differences between Infinium MethylationEPIC (EPICv1) and Infinium MethylationEPICv2 (EPICv2) arrays in estimating DNAm age with eleven DNAm clocks using buffy coat, peripheral blood mononuclear cell (PBMC), and saliva from 16 healthy middle-aged individuals. DNAm ages were estimated using six principal component-based (PC) clocks (PCHorvath1, PCHorvath2, PCHannum, PCPhenoAge, PCGrimAge, and PCDNAmTL) and five non-PC clocks (DunedinPACE, DNAmFit, YingCausAge, YingAdaptAge, and YingDamAge) across all biological samples. Agreement between arrays was assessed using Spearman correlation, Bland-Altman plots, and Wilcoxon Signed-Rank test. The 16 individuals with median age of 48 [43.5;53.8] years, were predominantly female, Chinese and non-smokers. High correlations (ρ > 0.8) were observed between EPICv1 and EPICv2 except for DunedinPACE, YingDamAge and YingAdaptAge. PC-based clocks showed lower systematic bias (MAPE:0.118-8.98%) compared to non-PC-based clocks (MAPE:5.31-21.2%). Saliva samples demonstrated greatest variability between arrays. EPICv2 introduces systematic biases especially in non-PC-based clocks and between different biological samples.
Similar content being viewed by others


Introduction
Deoxyribonucleic acid (DNA) methylation (DNAm) involves the addition of a methyl group to the cytosine base of cytosine-phosphate-guanine (CpG) sites in DNA and plays a crucial role in gene regulation1,2. Global DNA hypomethylation and local CpG island hypermethylation patterns are associated with ageing and replicative senescence3,4. DNAm clocks are algorithms developed to estimate DNAm age based on DNAm levels at sites distributed across the genome. Biological age refers to an individual’s age defined by the level of age-dependent biological changes, such as molecular and cellular damage accumulation5. DNAm clocks were developed using DNAm levels measured with the Illumina HumanMethylation27K (HM27K), Infinium HumanMethylation450K (HM450K) and Infinium MethylationEPICv1 (EPICv1) BeadChip arrays. Human Infinium BeadChip assays have progressed through several generations, including HM27K, which focused on analysing promoter CpG methylation6. HM450, its successor, expanded the number of CpG sites to include gene body CpG methylation, highlighting the role of gene body methylation in regulating gene expression6. EPICv1 further broadened the scope by focusing significantly on cis-regulatory elements, acknowledging the importance of tissue-specific methylation signatures in these enhancers6. EPICv1 has since been extensively utilised to profile DNA cytosine modifications due to their cost-effectiveness, precise quantification, and user-friendly data analysis, particularly beneficial for large cohort studies6.
The recently launched Illumina InfiniumMethylationEPICv2 BeadChip (EPICv2) introduces several significant advancements, including use of lower input DNA and improve coverage in enhancer, super-enhancers and CTCF binding regions, over its predecessor, EPICv12,6. The EPICv2 retains 83% of the CpG sites from EPICv1 and 81% from HM4506. The removal and addition of certain CpG sites and signal background deviation in EPICv2 may introduce bias in DNAm age estimation due to the absence of certain CpG sites and deviation in methylation levels6. In addition, methylation levels differ between biological samples, introducing differing DNAm age predictions7,8,9,10. For instance, there are differences in DNAmGrimAge2 between blood and saliva samples from mothers (aged 39-42 years) who were free of major medical conditions and disabilities11. Previous studies examining DNAm age estimation across EPICv2 and EPICv1 were limited to cell line models6 and whole blood samples from 5-year-old children12. These sample choices constrain interpretation, as cell lines may not reflect DNAm patterns of human biological samples, and paediatric samples deviate from the adult blood samples typically used to train DNAm clocks12. Moreover, these studies evaluated only a subset of available DNAm clocks, notably omitting fourth-generation clocks that offer biological interpretability, such as YingCausAge, YingAdaptAge, and YingDamAge6,12.
The first aim of this study is to analyse the number of missing CpG sites in eleven human DNAm clocks across EPICv1 and EPICv2 arrays and to assess the enrichment of these missing CpG sites within specific genomic features and CpG contexts. The second aim is to evaluate the agreement of DNAm age estimations using the EPICv2 array compared to the EPICv1 array utilising buffy coat, peripheral blood mononuclear cells (PBMC) and saliva obtained from healthy middle-aged individuals.
Results
Participant characteristics
Participant characteristics are given in Table 1. The median age of the participants was 48 [43.5; 53.8] years, with 9 of 16 participants being female and 14 of 16 participants being Chinese. Thirteen of 16 participants self-reported as non-smokers. Most participants (11/16) had self-reported alcohol consumption less than once per week. The median of Body Mass Index (BMI), waist-to-hip ratio, brachial systolic pressure and brachial diastolic pressure of the participants were 22.2 [21.1; 23.3] kg/m², 0.79 [0.76; 0.82], 115 [109;126] mmHg and 74.8 [69.4;79.3] mmHg.
DNAm clock CpG coverage in EPICv1 and EPICv2 arrays
The number of CpG sites included in the 11 DNAm clocks and the CpG coverage using EPICv1 and EPICv 2 is shown in Table 2. The number of CpG sites utilised in the 11 DNAm clocks ranges between 71 and 1331. EPICv1 includes all CpG sites for most DNAm clocks except Horvath1, Hannum, DNAmFitAge and YingCausAge with 5.38%, 8.45%, 1.44%, 28.2% missing CpG sites respectively. Less than 10% the CpG sites are missing in EPICv2 in most DNAm clocks including Horvath1, Hannum, PhenoAge, Horvath2, YingAdaptAge and YingDamAge. However, for GrimAge, DNAmTL, DNAmFitAge, DunedinPACE and YingCausAge, 15.6% to 24.6% CpG sites are missing in EPICv2. The CpG loci identifiers of the missing CpG sites of each DNAm clock in EPICv1 and EPICv2 are given in Table S1A and Table S1B respectively. For genomic feature, the missing CpG sites of most DNAm clocks are enriched in the gene body and the intergenic region (Table S2). For CpG context, the missing CpG sites of most DNAm clocks are enriched in CpG islands and open sea (Table S3).
DNAm age correlation in EPICv1 and EPICv2 with the same biological sample types
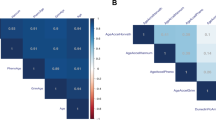
The Spearman correlations between DNAm clocks assessed using EPICv1 and EPICv2 arrays showed consistently high correlations across all biological samples in all PC clocks, YingCausAge and DNAmFitAge (ρ > 0.844) (Fig. 1). PC-based DNAm clocks showed higher correlation between EPICv1 and EPICv2 than non-PC-based DNAm clocks. YingDamAge and YingAdaptAge exhibited high Spearman correlations between EPICv1 and EPICv2 in buffy coat and PBMC samples (ρ > 0.815). YingDamAge and YingAdaptAge had moderate correlations between EPICv1 and EPICv2 in saliva sample (ρ =0.629 and ρ =0.553 respectively). DunedinPACE had high correlation between EPICv1 and EPICv2 in buffy coat and saliva samples (ρ > 0.774), and moderate correlations in PBMC samples (ρ =0.512).

Each cell corresponds to correlation coefficient (ρ), coloured in blue or red for low Spearman correlation (ρ < 0.7) and high Spearman correlation (ρ > 0.7) respectively.
Agreement and difference of DNAm clocks between EPICv1 and EPICv2 across different biological sample types
The agreement of DNAm ages between EPICv1 and EPICv2 was evaluated using Bland-Altman plots, as shown in Fig. 2. The mean difference (SD), limit of agreement and MAPE are summarised in Table S4. Across all biological samples, mean differences were positive for PCHorvath1 (range: 1.03 to 4.03 years), PCHorvath2 (range: 0.66 to 2.77 years), PCHannum (range: 0.27 to 1.20 years), PCDNAmTL (range: 0.002 to 0.009 kb), and DunedinPACE (range: 0.045 to 0.094). Mean differences were negative for PCPhenoAge (range: -1.96 to -0.82 years), PCGrimAge (range: -0.72 to -0.34 years), DNAmFitAge (range: -4.35 to -2.34 years), YingCausAge (range: -8.17 to -3.48 years), and YingDamAge (range: -6.27 to -3.60 years). For YingAdaptAge, the mean difference was negative in buffy coat (-4.55 years) and saliva (-4.67 years), but positive in PBMC (1.62 years). The smallest absolute mean difference between EPICv1 and EPICv2 was observed for PCHannum in buffy coat (mean difference ± SD: 0.27 ± 0.80) and the largest absolute mean difference between EPICv1 and EPICv2 was observed for YingCausAge in PBMC (mean difference ± SD: -8.17 ± 3.12 years). Lower MAPE between EPICv1 and EPICv2 were observed in PC-based DNAm clocks (MAPE: buffy coat: 0.118 to 4.28%; PBMC: 0.176 to 3.57%; saliva: 0.278 to 8.98%) compared to non-PC-based DNAm clocks (MAPE: buffy coat: 8.27 to 15.0%; PBMC: 5.31 to 21.2%; saliva: 6.52 to 14.3%). The 95% limits of agreement across DNAm clocks were consistently widest in the saliva samples compared to both buffy coat and PBMC samples across all DNAm clocks.

Black line indicates the mean difference, with the red box marked up the 95% of the limits of agreement, and grey line indicates the zero of the y-axes, across three different biological samples on each clock.
Differences in DNAm ages between the EPICv1 and EPICv2 across 11 DNAm clocks in buffy coat, PBMC, and saliva were assessed using Wilcoxon Signed-Rank test (Fig. 3& Figure S1). Across all biological samples, significant differences in DNAm ages between EPICv1 and EPICv2 were observed for PCHorvath1, PCHorvath2, PCPhenoAge and DNAmFitAge (Fig. 3). In both PBMC and buffy coat, significant differences were observed for DunedinPACE, YingDamAge and YingCausAge. In buffy coat, significant differences were observed for PCGrimAge and YingAdaptAge. No significant difference was observed in any biological samples for PCHannum and PCDNAmTL. The smallest absolute median difference between EPICv1 and EPICv2 was observed in PCHannum (0.306 years) and the largest absolute median difference was observed in YingCausAge (8.43 years). The results of Wilcoxon Signed-Rank test are summarised in Table 3. Individual-level comparisons of DNAm age estimates between EPICv1 and EPICv2 revealed consistent increase or decrease of DNAm ages across all biological samples (Figure S1). Cell compositions estimated using DNAm data across all biological samples also showed high spearman correlation and high agreement between EPICv1 and EPICv2 in most cell compositions except for Neutrophils and Eosinophils in PBMC (Figure S1 and S2). EpiDISH did not estimate the Eosinophils in buffy coat and Eosinophils, CD8T and B cells in Saliva.

Significance test was done by Wilcoxon signed rank test, where *, ** and *** indicates statistically significance after Bonferroni correction. *** indicates p-values < 0.0000909. ** indicates p-values < 0.000909. * indicates p-adj < 0.00455. Note: PCDNAmTL has a unit of kb and DunedinPACE has no unit as it is defined as pace of ageing of an individual ageing relative to the normative rate of one year of ageing per year of calendar time.
Discussion
DNAm clocks were highly correlated between EPICv1 and EPICv2, except for DunedinPACE in PBMC samples and YingDamAge and YingAdaptAge in saliva samples. However, statistically significant differences were observed in DNAm age derived from EPICv1 and EPICv2. Saliva samples displayed the highest variability of difference between EPICv1 and EPICv2 for DNAm ages as compared to buffy coat and PBMC samples. Cell compositions estimated using DNAm data across all biological samples also showed high spearman correlation and high agreement.
The high correlation and high agreement of DNAm ages between EPICv1 and EPICv2 are attributed to the preservation of most CpG sites from existing DNAm clocks in EPICv2 and the high correlation between the DNAm levels at shared CpG sites of EPICv1 and EPICv2 observed in four human cell lines (GM12878, LNCaP, K562, and HCT116)6. In most clocks, statistically significant differences in DNAm ages between EPICv1 and EPICv2 were found. The consistent direction of within-person DNAm age differences between EPICv1 and EPICv2 indicates that the observed discrepancies are due to systematic differences introduced by arrays used, rather than random inter-individual variability. There is no distinct enrichment of missing CpG sites within genomic regions and CpG contexts in EPICv2 compared to EPICv1, thus this systematic offset could be attributed to the greater number of missing CpG sites used to measure various DNAm clocks in EPICv2 compared to EPICv1. Furthermore, the missing CpG sites imputation such as GLMNET could not fully compensate this discrepancy and might introduce noise for CpG sites that are completely missing from EPICv213,14,15. Previous studies have found that each clock contains CpG sites with unreliable methylation beta values, which can affect the reproducibility of DNAm ages16. The possibility of cell composition effect should be minimally as the biological sample used for both arrays are identical and the cell compositions predicted by EpiDISH exhibited a high spearman correlation and agreement between the arrays. Therefore, the systematic differences observed between EPICv1 and EPICv2 suggest that direct comparisons of DNAm ages derived from different arrays should be interpreted with caution, especially for small clinical trials and population studies with small effect size17,18. Researchers and clinicians should account for these cross-array biases8,9 and development of better imputation or cross-array calibration algorithms would be essential to enhance the reliability of existing DNAm clocks in EPICv219.
PC-based clocks exhibit lower mean differences and smaller variability of differences between EPICv1 and EPICv2 than non-PC-based clocks across biological samples. This is consistent with previous study, which have shown that principal component analysis (PCA) enhances the reliability and reproducibility of DNAm age estimation20. This is achieved by aggregating information from many age-related CpGs to improve the signal quality and reduce the effect of inherent technical noise associated with individual CpGs20. Future construction of DNAm clocks should consider the incorporation of PCA to enhance the reliability and reproducibility of the DNAm clocks.
Saliva samples displayed the highest variability in DNAm ages differences between EPICv1 and EPICv2. This can be attributed to the heterogenous cell types in saliva, including epithelial cells and leukocytes, potentially leading to greater variations in DNAm ages20,21,22. Blood-based tissues, buffy coat and PBMC, tend to have higher quality DNA with less variability of stability, purity, and quantity compared to saliva, contributing to less variability in DNAm ages estimation23,24. However, there is only a saliva specific cell-type deconvolution algorithm for children and not adults25. Modifying the array probes can impact cell composition, as indicated by the moderate correlations observed in some cell composition predictions. Therefore, cell composition adjustment in saliva and development of cell-type deconvolution algorithms specific to saliva and EPICv2 array would be essential for saliva to be used in cohort studies or commercial epigenetic tests24.
The Illumina EPICv2 array offers cost effective DNAm profiling for large cohort studies. However, this platform provides limited coverage of the whole methylome (937,690 out of 28 million CpGs, 3.5%)6,26. To address these limitations, whole-genome bisulphite sequencing (WGBS) has emerged as the gold standard for comprehensive methylation analysis, enabling the identification of complete mechanistic processes underlying DNAm levels across 28 million CpGs27. Traditional bulk DNAm measurements, whether from arrays or WGBS, represent the average DNAm levels in populations of cells, potentially diluting the tissue-specific and cell type-specific ageing signatures27. Single-cell WGBS allows high-resolution examination of DNAm patterns, revealing whether age-related DNAm changes are uniform across cells or emerge from population-level variations27. This insight can help explain the variability in DNAm age estimations between tissues27. Therefore, single-cell WGBS would be essential to understand the mechanism of epigenetic regulation in ageing and enable the construction of a DNAm clock with CpGs causally linked to ageing processes with minimal discrepancies in DNAm age estimations between biological samples27.
This study is the first comprehensive analysis of DNAm age difference between EPICv1 and EPICv2 in three commonly used biological samples including buffy coat, PBMC and saliva. The small sample size, ethnicity consisting of mostly Chinese and age range of 40-60 years may limit the generalisability of the findings and the robustness of imputation.
This study underscores the necessity of platform-specific considerations in DNAm age estimation. The observed systematic offsets between EPICv1 and EPICv2, particularly in non-PC-based clocks and high variability of differences in saliva samples, suggest that future research should incorporate PC analysis in the construction of DNAm clocks and a method to adjust for these biases. The small effect sizes changes or difference observed in DNAm age measurements from EPICv2 should be interpreted cautiously, as they may reflect systematic platform differences rather than meaningful age-related changes, given that these clocks were not originally trained on EPICv2.
Methods
Study design
This study is a cross-sectional analysis nested within the randomised clinical trial Does Alpha-ketoglutarate supplementation lower BiologicaL agE in middle-aged adults (ABLE) study28, which evaluates the effect of Calcium Alpha-Ketoglutarate (Ca-AKG) on biomarkers of aging in individuals 40-60 year of age. A total of 16 screening participants were included in this sub-study and before randomization. All participants provided written informed consent. The study has been approved by NUS IRB (NUS-IRB-2021-946) and registered at clinicaltrial.gov (NCT05706389).
Participants characteristics
Demographic and lifestyle characteristics, including age, gender, race, education level, type of housing, smoking status and alcohol consumption status were collected from participants through participant survey after biological samples collection. Race is classified as Chinese and Caucasian. Education levels are classified as undergraduate and graduate. Housing types are classified as private housing and public housing. Smoking status is defined as non-smoker and ex-smoker. Alcohol consumption is self-reported and detailed in terms of the number of servings and the frequency of intake per week, in the past year. Alcohol consumption is categorised as never, less than once per week, and more than once per week within the past year. Height and weight were measured using a Seca 213 Stadiometer (Germany) and a Seca 813 digital floor scale with high capacity (Germany), respectively. The body mass index (BMI) was subsequently calculated as weight/height² (kg/m²). Waist and hip circumferences were measured using a Seca 201 Ergonomic circumference measuring tape, Germany, and the waist-to-hip ratio was subsequently calculated.
Biological samples collection and preparation
All blood and saliva were collected after a minimum of 8 hours of overnight fasting, in the morning, between 8.30 and 9.30 am28. A total of 8 mL of venous blood was collected, followed by collecting 4 mL of saliva through passive drooling into a saliva collection tube (Isohelix, Kent, UK) pre-filled with a guanidine-free DNA stabilisation buffer. Whole blood and saliva samples were transported at a temperature of 4 °C and processed within 1 hour after the sample collection. The saliva collection tube was transferred and stored in a -80 °C freezer.
The Anticoagulant Citrate Dextrose Solution, Solution A (ACD-A) blood tubes were gently mixed by inversion after collection, and then centrifuge at 2500 rpm for 15 min at 23 °C with brake off (acceleration: 9, deceleration: 0). After aliquoting the top layer of plasma, the buffy coat was drawn and stored at –80 °C freezer. Whole blood was diluted with phosphate-buffered saline (PBS), layered over 15 mL of Ficoll media, and centrifuge at 400 g for 30 min at 23 °C with brake off (acceleration: 9, deceleration: 0). The PBMC layer was extracted, washed twice with PBS. The cell pellet was thoroughly resuspended after each wash. The PBMC cell count was performed using the Countess II FL Automated Cell Counter and stored at –80 °C freezer. The biological samples stored at –80 °C freezer were delivered with dry ice for DNA extraction.
DNA for biological samples were extracted using the Qiagen QIAamp DNA Mini Kits and further processed in a single batch to minimize the batch effect. Quality of DNA samples were evaluated using the Agilent Tape Station (Agilent, Santa Clara, CA). Quantification of DNA from study participant were performed using the Qubit™ fluorometer (Thermo Fisher Scientific, Waltham, MA). The EPICv1 and EPICv2 were used for high-throughput measurement of DNA methylation on a genome-wide scale. For each methylation chip experiment, approximately 1μg of genomic DNA from each study participant and from each biological specimen was bisulphite treated using the Zymo EZ DNA Methylation Kit (Zymo, Irvine, CA) to convert non-methylated cytosine nucleotides to uracil for subsequent methylation profiling. The bisulfite treated DNA were denatured into single strands and hybridized to the Infinium EPIC BeadChip via allele-specific annealing to either the methylation-specific probe or the non-methylation probe. Hybridization to the chip was followed by single-base extension with labelled di-deoxynucleotides. Hybridized BeadChips were subsequently stained, washed, and scanned to determine intensities of methylated and unmethylated bead types using Illumina’s iScan system.
DNAm analysis
Identical DNAm samples of buffy coat, PBMC and saliva biological samples were processed in parallel on the EPICv1 and EPICv2 Beadchips (Illumina Inc., San Diego, CA). Each sample was prepared from a single DNA extraction and analysed on both array versions to ensure direct comparability of DNAm measurements. DNAm age was predicted using methylation data from EPICv1 and EPICv2 BeadChips across different biological samples (buffy coat, PBMC, and saliva). Noob normalisation was performed to correct for biases associated with type I and type II CpG sites29. Eleven human DNAm clocks were calculated, 6 principal component-based DNAm clocks (PC clocks) consisting of Horvath1 DNAm age(PCHorvath1)30, Horvath2 DNAm age (PCHorvath2)31, Hannum DNAm age (PCHannum)32, DNAm PhenoAge (PCPhenoAge)33, DNAm GrimAge (PCGrimAge)34, and DNAmTL (PCDNAmTL) (an estimator for telomere length in kilobases)35 and non-PC clocks including DunedinPACE (pace of ageing)36, DNAmFit37, YingCausAge38, YingAdaptAge38 and YingDamAge38. The PC clocks were calculated, in accordance with established protocols20. The units of measurement for the DNAm clocks utilised in this study are in years, with the exception of two clocks PCDNAmTL (unit in kilobase, kb)35 and DunedinPACE (no unit). DunedinPACE is defined as pace of ageing of an individual ageing relative to the normative rate of one year of ageing per year of calendar time36. Cell compositions were predicted using EpiDISH for all biological samples including saliva39. Note that, although EpiDISH was not specifically trained on saliva data, its estimates for saliva serve as approximations. Regularised generalised linear regression model (GLMNET) was used to impute missing CpG sites used in the calculation of DNAm clocks and cell compositions in EPICv240. This imputation approach leverages the substantial overlap of CpG sites between EPICv1 and EPICv2 arrays, using these shared sites as predictors to estimate methylation values for missing sites. The GLMNET algorithm employs elastic net regularization, which combines L1 and L2 penalties to handle high-dimensional data while preventing overfitting.
Assessment of missing CpG sites and enrichment
This study assessed CpG site coverage for 11 human DNAm clocks across the EPICv1 and EPICv2 arrays, quantifying missing CpG sites and evaluating their enrichment within specific genomic regions and CpG contexts using the Bioconductor packages including IlluminaHumanMethylation 450kanno.ilmn12.hg19, EPICanno.ilm10b4.hg19 and EPICv2anno.20a1.hg38 and clock coefficients from Biolearn41. The CpG loci identifiers, unique alphanumeric codes assigned by Illumina to specific CpG sites in the genome, were compiled for the missing sites. Genomic regions refer to distinct elements within the genome with specific biological functions or structural characteristics. These genomic regions include 1st Exon (first exon), 5’ UTR (5’ Untranslated Region), Body (coding region), IGR (intergenic region), TSS1500 (region within 1500 base pairs upstream of the transcription start site), TSS200 (region within 200 base pairs upstream of the transcription start site) and 3’ UTR (3’ Untranslated Region)42. CpG contexts refer to the occurrence and position of CpG within the genome. These CpG contexts consist of island, opensea, shelf and shore42.
Statistical and reproducibility
Descriptive statistics were performed and data presented as median and interquartile interval (IQR) or number and percentage (%). Analyses involved comparing DNAm age predictions between EPICv1 and EPICv2 using Spearman correlation and Bland-Altman plots to assess agreement between EPICv1 and EPICv2. A Spearman correlation coefficient (ρ) greater than 0.7 indicates good agreement, ρ value between 0.5 and 0.7 was defined as moderate agreement, and ρ value below 0.5 was defined as poor agreement between the two methods43. The mean difference of EPICv2 and EPICv1 was defined as the average of the measurements of EPICv2 -EPICv1. The standard deviation of difference was calculated as well. The mean difference in Bland-Altman plots was interpreted as DNAm ages in years for all DNAm clocks except PCDNAmTL and DunedinPACE. For PCDNAmTL, the mean difference was interpreted as telomere length in kilobases (kb). For the DunedinPACE clock, the mean difference was interpreted as the mean difference in the pace of ageing. To standardise the difference across various DNAm clocks, the mean absolute percentage error (MAPE) was calculated to compare across DNAm clocks44. Box plots were used to visualize the difference of DNAm ages between EPICv1 and EPICv2 and significance differences were tested by Wilcoxon Signed-Rank test. Bonferroni multiple testing correction was applied by number of DNAm clocks (n = 11). All statistical analyses were performed using R (version 4.4.1) or Python (3.12.0). All tests are two-sided with significance at p < 0.05, unless otherwise stated. Each comparison group consisted of 16 samples. Specifically, 16 buffy coat, 16 saliva, and 16 PBMC samples were analysed at both EPICV1 and EPICV2. All datasets used in this study are provided and links are provided for software used.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The data supporting the findings of this study are available from Prof. Andrea B. Maier upon appropriate approval. Please note that access to the data may be subject to certain restrictions to comply with ethical guidelines or institutional policies. Data used for figures and Supplementary Figs. are provided in Supplementary Data 1-4.
Code availability
All original R code (version 4.4.1) used to generate the CpG missing sites of each clocks is available on GitHub: https://github.com/TayJianHua/EPICv2-vs-EPICv1-DNAm-comparison. Clocks coefficients and clock calculation codes are available in: https://bio-learn.github.io/clocks.html.
References
Angeloni, A. & Bogdanovic, O. Sequence determinants, function, and evolution of CpG islands. Biochem Soc. Trans. 49, 1109–1119 (2021).
Noguera-Castells, A., García-Prieto, C. A., Álvarez-Errico, D. & Esteller, M. Validation of the new EPIC DNA methylation microarray (900K EPIC v2) for high-throughput profiling of the human DNA methylome. Epigenetics 18, 2185742 (2023).
Johnson, A. A. et al. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 15, 483–494 (2012).
Booth, L. N. & Brunet, A. The aging epigenome. Mol. Cell 62, 728–744 (2016).
Moqri, M. et al. Biomarkers of aging for the identification and evaluation of longevity interventions. Cell 186, 3758–3775 (2023).
Kaur, D. et al. Comprehensive evaluation of the Infinium human MethylationEPIC v2 BeadChip. Epigenetics Commun. 3, 6 (2023).
Murata, Y. et al. Evaluation of the usefulness of saliva for DNA methylation analysis in cohort studies. Neuropsychopharmacol. Rep. 39, 301–305 (2019).
Fernandez-Jimenez, N. et al. Comparison of Illumina 450K and EPIC arrays in placental DNA methylation. Epigenetics 14, 1177–1182 (2019).
Solomon, O. et al. Comparison of DNA methylation measured by Illumina 450K and EPIC BeadChips in blood of newborns and 14-year-old children. Epigenetics 13, 655–664 (2018).
Cheung, K., Burgers, M. J., Young, D. A., Cockell, S. & Reynard, L. N. Correlation of Infinium HumanMethylation450K and MethylationEPIC BeadChip arrays in cartilage. Epigenetics 15, 594–603 (2020).
Lu, A. T. et al. DNA methylation GrimAge version 2. Aging (Albany NY) 14, 9484–9549 (2022).
Lussier, A. A. et al. Technical variability across the 450K, EPICv1, and EPICv2 DNA methylation arrays: lessons learned for clinical and longitudinal studies. Clin. Epigenetics 16, 166 (2024).
Giuili, E. et al. Comprehensive evaluation of the implementation of episignatures for diagnosis of neurodevelopmental disorders (NDDs). Hum. Genet. 142, 1721–1735 (2023).
Li, J. et al. Comparison of the effects of imputation methods for missing data in predictive modelling of cohort study datasets. BMC Med. Res. Methodol. 24, 41 (2024).
Lena, P. D., Sala, C., Prodi, A. & Nardini, C. Methylation data imputation performances under different representations and missingness patterns. BMC Bioinforma. 21, 268 (2020).
Sugden, K. et al. Patterns of Reliability: Assessing the Reproducibility and Integrity of DNA Methylation Measurement. Patterns (N. Y) 1, 100014 (2020).
Koncevičius, K. et al. Epigenetic age oscillates during the day. Aging Cell 23, e14170 (2024).
McEwen, L. M. et al. Systematic evaluation of DNA methylation age estimation with common preprocessing methods and the Infinium MethylationEPIC BeadChip array. Clin. Epigenetics 10, 123 (2018).
Zhang, G. et al. Across-Platform Imputation of DNA Methylation Levels Incorporating Nonlocal Information Using Penalized Functional Regression. Genet Epidemiol. 40, 333–340 (2016).
Higgins-Chen, A. T. et al. A computational solution for bolstering reliability of epigenetic clocks: Implications for clinical trials and longitudinal tracking. Nat. Aging 2, 644–661 (2022).
Dunnet, M. J. et al. Leukocyte-specific DNA methylation biomarkers and their implication for pathological epigenetic analysis. Epigenetics Commun. 2, 5 (2022).
Zheng, S. C. et al. A novel cell-type deconvolution algorithm reveals substantial contamination by immune cells in saliva, buccal and cervix. Epigenomics 10, 925–940 (2018).
Nishitani, S., Parets, S. E., Haas, B. W. & Smith, A. K. DNA methylation analysis from saliva samples for epidemiological studies. Epigenetics 13, 352–362 (2018).
Wolf, S. E. et al. Cross-tissue comparison of telomere length and quality metrics of DNA among individuals aged 8 to 70 years. PLoS One 19, e0290918 (2024).
Middleton, L. Y. M. et al. Saliva cell type DNA methylation reference panel for epidemiological studies in children. Epigenetics 17, 161–177 (2022).
Libertini, E. et al. Saturation analysis for whole-genome bisulfite sequencing data. Nat. Biotechnol. 34, 691–693 (2016).
Bell, C. G. et al. DNA methylation aging clocks: challenges and recommendations. Genome Biol. 20, 249 (2019).
Sandalova, E. et al. Alpha-ketoglutarate supplementation and BiologicaL agE in middle-aged adults (ABLE)-intervention study protocol. Geroscience 45, 2897–2907 (2023).
Teschendorff, A. E. et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196 (2013).
Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 14, R115 (2013).
Horvath, S. et al. Epigenetic clock for skin and blood cells applied to Hutchinson Gilford Progeria Syndrome and ex vivo studies. Aging (Albany NY) 10, 1758–1775 (2018).
Hannum, G. et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367 (2013).
Levine, M. E. et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging (Albany NY) 10, 573–591 (2018).
Lu, A. T. et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging (Albany NY) 11, 303–327 (2019).
Lu, A. T. et al. DNA methylation-based estimator of telomere length. Aging (Albany NY) 11, 5895–5923 (2019).
Belsky, D. W. et al. DunedinPACE, a DNA methylation biomarker of the pace of aging. Elife 11, e73420 (2022).
McGreevy, K. M. et al. DNAmFitAge: biological age indicator incorporating physical fitness. Aging (Albany NY) 15, 3904–3938 (2023).
Ying, K. et al. Causality-enriched epigenetic age uncouples damage and adaptation. Nat. Aging 4, 231–246 (2024).
Teschendorff, A. E., Breeze, C. E., Zheng, S. C. & Beck, S. A comparison of reference-based algorithms for correcting cell-type heterogeneity in Epigenome-Wide Association Studies. BMC Bioinforma. 18, 105 (2017).
Wong, W. W., Griesman, J. & Feng, Z. Z. Imputing genotypes using regularized generalized linear regression models. Stat. Appl Genet Mol. Biol. 13, 519–529 (2014).
Ying, K. et al. A Unified Framework for Systematic Curation and Evaluation of Aging Biomarkers. bioRxiv, 2023.2012.2002.569722 (2024). https://doi.org/10.1101/2023.12.02.569722
Tian, Y. et al. ChAMP: updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 33, 3982–3984 (2017).
Mukaka, M. M. Statistics corner: A guide to appropriate use of correlation coefficient in medical research. Malawi Med J. 24, 69–71 (2012).
Bland, J. M. & Altman, D. G. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1, 307–310 (1986).
Acknowledgements
This study was funded by a National University Health System (NUHS) internal grant (A-000-2469-00-00), NUS Yong Loo Lin School of Medicine grant, and a generous gift from the Lien Foundation (A-000-6347-00-00).
Author information
Authors and Affiliations
Contributions
J.T. and Y.C. performed majority of the statistical analysis and prepared the manuscript. Z.L. and L.G. were responsible for data and sample collection. R.D. processed the DNA samples, measured DNA methylation and advised on the analysis. R.B., J.G., and S.H. performed imputation of EPICv2 data and the calculation of DNAm ages. W.W., E.S., R.D., B.K.K., S.H. and A.B.M. provided guidance and reviewed the manuscript. A.B.M supervised the entire project and preparation of the manuscript.
Corresponding author
Ethics declarations
Competing interests
B.K.K served on the Scientific Advisory Board for Ponce De Leon Health. S.H. and R.B. are co-founder of the non-profit Epigenetic Clock Development Foundation. J.G. is the informatics scientist of the non-profit Epigenetic Clock Development Foundation. Other authors have no conflict of interest to disclose.
Peer review
Peer review information
Communications Biology thanks Kristine Haftorn and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Rosie Bunton-Stasyshyn & Christina Karlsson Rosenthal.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Tay, J.H., Chew, Y.E., Wang, W. et al. DNAm age differences between infinium methylationEPICv1 vs EPICv2 in buffy coat, PBMC, and saliva samples. Commun Biol 8, 654 (2025). https://doi.org/10.1038/s42003-025-08021-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08021-y
