
Abstract
Ubiquitin conjugating E2 enzymes are a set of ~40 proteins that play a central role in the ubiquitination cascade. They transfer ubiquitin from an E1 enzyme to substrates with the help of an E3 enzyme. The members of the E2 family share structural similarity in their conserved UBC fold. This complicates an assessment of the specificity of E2-E3 interactions. We identified a nanobody that binds to the ‘backside’ region of Ube2G2, an E2 involved in ER protein quality control. This binding does not affect ubiquitin loading but shows varying degrees of inhibition on E3-mediated ubiquitination, in the order HRD1 > CHIP >> TRC8. A naturally occurring segment that binds Ube2G2’s backside, referred to as G2BR (Ube2G2 Binding Region), shows a similar inhibitory effect depending on the identity of the interacting E3. The G2BR in the Ube2G2-cognate E3 Gp78 enhances Ube2G2’s activity, but its deletion results in a similar inhibition upon addition of the nanobody. Occupation of a single binding site on an E2 can thus affect its interactions with different E3s.
Similar content being viewed by others


Introduction
E2 conjugating enzymes act as intermediaries in ubiquitination, a post-translational modification that determines the fate of its target protein. This can range from proteasomal degradation to altering its interaction network or cellular location1,2,3. Ubiquitination involves the conjugation of ubiquitin (Ub), a 76-residue protein, to a target via a cascade of E1, E2, and E3 enzymes, typically on lysine residues1,2,3. Ub itself has seven lysine residues, enabling the formation of chains of diverse topologies with different functional outcomes, depending on linkage types2,3. The E1 or Ub-activating enzyme loads a Ub onto its active site cysteine in an ATP-dependent manner to yield Ub attached to the E1 in a thioester bond. Ub is then transferred to an E2 enzyme, and with the help of an E3 ligase to the substrate’s lysine(s)2,3. If the E3 is of the RING (Really Interesting New Gene) or U-box domain family, the E2 directly transfers Ub to the substrate. For HECT (Homologous to the E6-AP Carboxyl Terminus) or RBR (RING in Between RING) family E3s, Ub is first transferred to the E3 in thioester linkage and then to the substrate. While E3 ligases generally control substrate specificity and Ub linkage types, RING family E3s rely on E2 enzymes to determine Ub linkage types (e.g., K48, K63, K11) and facilitate Ub transfer, with E3s acting as scaffolds to bring E2 and the substrate together1,2,3.
Humans have approximately 2 E1s, ~40 E2s and >600 E3s4,5,6. The central question of how a few dozen E2s discriminate between hundreds of E3s, and possibly thousands of substrates remains unresolved. The structural similarities among E2s, all of which share a conserved UBC domain and differ by terminal extensions or insertions, adds to the puzzle1,4,7. E2s may manage complex protein-protein interactions by conformational changes upon binding diverse partners, although pinpointing specific sites of interaction on these small 15–20 kDa E2 proteins is challenging. The backside region of E2s, thus named because of its position opposite the catalytic site, lacks direct interactions with E1 or E3 proteins (Fig. 1)1. Nonetheless, this backside region plays a crucial role in allosteric, non-covalent regulatory effects. These can lead to inhibition8,9 or activation10,11,12,13, depending on the particular E2 and its interacting partners1,8,9,10. Here we use nanobodies to explore whether backside binding can lead to both activating and inhibitory outcomes on one and the same E2. Nanobodies are of course distinct from cellular E2-interacting proteins, but have the advantage of binding to a specific, defined epitope.

Structures of E2 conjugating enzymes involved in ubiquitination are shown with their respective PDB IDs. Ube2G2 is depicted with the conserved E1, E3, backside region, acidic loop, and active site cysteine along with both termini. Note the conserved fold among most of the members of the E2 family. *Indicates the two catalytically inactive E2s.
Nanobodies (or VHHs) are recombinantly expressed antibody fragments derived from the variable domain of heavy-chain-only antibodies present in camelids14,15. They retain the specificity and affinity of conventional antibodies, but offer advantages such as high stability, ease of bacterial production, and efficient tissue penetration due to their small size ( ~ 15 kDa)14,15. Nanobodies can stabilize proteins in specific conformations, as best studied for GPCRs16. We have previously identified a nanobody that targets Ube2J1, which allosterically regulates the activity of Ube2J1 near a site that is phosphorylated upon cellular activation17.
Ube2G2 is an E2 also involved in protein quality control at the endoplasmic reticulum (ER). It catalyzes the transfer of Ub in K48-Ub linkage and targets misfolded proteins for degradation18. Ube2G2 functions in the ER-associated degradation (ERAD) pathway together with E3 ligases that include Gp78/AMFR (Glycoprotein 78/ Autocrine motility factor receptor)19,20,21, HRD1 (3-Hydroxy-3-methylglutaryl reductase degradation)22, TRC8/RNF139 (Translocation in renal carcinoma chromosome 8 gene/Ring finger protein 139)23,24, and MARCH623,25. Each of these E3s focuses on distinct substrates26. Ube2G2 is also essential for degrading Class I HLA molecules in cells infected with human cytomegalovirus (CMV) as a means of evading recognition by cytotoxic T cells27. Although specific examples of Ube2G2’s role in ERAD and the discovery of the E3 TRC8 were obtained by dissecting the mechanism of action of the CMV-encoded proteins US2 and US11, which target Class I HLA molecules for proteasomal degradation27, the nuances of Ube2G2’s interactions with different E3s remain unclear. Ube2G2 is tethered to the ER through interaction with ancient ubiquitous protein 1 (AUP1), a protein also involved in ERAD and in the regulation of lipid metabolism28,29,30. This interaction involves a G2BR (Ube2G2 Binding Region) segment that binds to Ube2G2’s backside. A G2BR is also present in the E3 Gp78, thought to enhance Ube2G2’s activity through allosteric effects31,32, although inhibitory consequences of the G2BR-Ube2G2 interaction have also been reported28.
Using a nanobody that binds to the backside of Ube2G2, we report that occupation of the backside region does not significantly alter the ability of Ube2G2 to engage in thioester formation, but differentially reduces its E2-dependent ubiquitination of substrates through E3 ligase interaction (Table 1). The ‘CUE-G2BR’ of AUP1 and the isolated G2BR segments of AUP1 and Gp78 all show a similar inhibitory trend (Table 1). This nanobody-based approach shows how a particular structural feature on Ube2G2 can affect its conformation and function.
Results
Identification of a nanobody that recognizes Ube2G2 by screening using phage display
To generate a nanobody against human Ube2G2, we immunized an alpaca with purified recombinant Ube2G2 and constructed a nanobody phage display library. This library was then screened by panning on immobilized Ube2G2. Following two rounds of panning, we identified a nanobody, VHH12G2 (Fig. 2A). VHH12G2 was obtained by bacterial periplasmic expression and purified to homogeneity (yield: ~ 6-10 mg/L). Upon mixing of VHH12G2 with Ube2G2, we confirmed their ability to interact in solution by size exclusion chromatography (SEC; Fig. 2B) At a 2:1 molar ratio of VHH12G2:Ube2G2, the VHH12G2 complex elutes at a larger Stokes radius than does free Ube2G2, with both proteins present in the complex in equal amounts, as verified by SDS-PAGE (Fig. 2B). We determined the binding affinity of VHH12G2 for Ube2G2 by microscale thermophoresis and obtained a value of 1.3 ± 0.49 μM (Fig. 2C). VHH12G2 fails to recognize Ube2G2 in immunoblot. The epitope recognized by VHH12G2 is thus conformationally sensitive. VHH12G2 does not cross-react with other E2s we tested, including E2s with acidic loops (Ube2G1, Ube2R1), a distantly related E2 (Ube2H), an E2 involved in ERAD (Ube2J1), or with an E2 that possess only the core UBC domain (Ube2D2), as examined by size exclusion chromatography (Supplementary Fig. 1). We conclude that VHH12G2 is specific and binds to Ube2G2 in a conformationally sensitive manner.

A Amino acid sequence of VHH12G2 with complementarity determining regions (CDRs) highlighted according to the IMGT database56. B Size exclusion chromatography (SEC) profiles that illustrate binding of Ube2G2 to VHH12G2 with milli absorbance units (mAU) plotted against fraction numbers. The elution profiles for Ube2G2 (green), VHH12G2 (blue) and a 2:1 molar ratio mixture of VHH12G2: Ube2G2 (red) are shown. Protein fractions from the indicated peaks were analyzed by SDS-PAGE (shown on the right). Note the co-elution of VHH12G2 and Ube2G2, distinguishable by a subtle difference in migration, in the peak fraction (red trace). C Microscale thermophoresis of the affinity (Kd) measurement of VHH12G2 for Ube2G2 tagged with Alexa Fluor 647. The binding data, averaged from three independent experiments, are presented with the estimated binding constant indicated. The raw relative fluorescence data for all MST runs is displayed on the right.
VHH12G2 binds to the backside of Ube2G2
To identify the epitope on Ube2G2 recognized by VHH12G2, we used NMR and acquired transverse relaxation optimized (TROSY) version of 15N-1H heteronuclear single quantum coherence (HSQC) spectra of 15N labeled Ube2G2, in the presence and absence of an excess of VHH12G2. Peaks for each residue were assigned based on the published NMR structure (PDB: 2KLY33). We estimated the lower limits of the combined chemical shift changes per residue (or their low bounds in case of ambiguous assignments) upon VHH binding and plotted these shifts for Ube2G2. Based on the magnitude of the chemical shifts observed, interaction of VHH12G2 with Ube2G2 likely involves residues G23 to G27, with significant effects seen throughout the protein, especially near the N- and C- termini (residues K14 to A56, S115 to A136, and K153 to L165) (Fig. 3A). Consistent with this interpretation, we observed varying degrees of reduced binding of VHH12G2 to N- or C-terminally tagged Ube2G2 based on a minimal shift in SEC profiles of Ube2G2-bound VHH12G2 and a corresponding reduction from the expected 1:1 elution ratio of VHH12G2:Ube2G2 as judged by SDS PAGE (Fig. 3D, E).

A NMR 15N-TROSY-HSQC spectrum of 15N labeled Ube2G2 with VHH12G2 (red) overlaid on the spectrum of 15N labeled (Black) Ube2G2 alone. The estimated combined chemical shifts for individual Ube2G2 residues are shown in ppm (parts per million) values for VHH12G2 in the middle panel. Residues that show detectable chemical shifts relative to unliganded Ube2G2 are mapped onto the NMR structure of Ube2G2 (PDB: 2KLY) to the right, with coloring schemes indicating the magnitude (red>orange) of chemical shifts. B NMR 15N-TROSY-HSQC spectrum of Ube2G2 with the weakly binding VHH02G2 (blue); description otherwise similar to (A). C NMR 15N-TROSY-HSQC spectrum of Ube2G2 with a negative control VHHSMT3 (green) overlaid on the spectrum of 15N Ube2G2 spectrum (black). The 15N-TROSY-HSQC spectrum of Ube2G2 alone (black) at the bottom. D–E Size exclusion chromatography (SEC) profiles of VHH12G2 in the presence of (D) C-terminally tagged Ube2G2, and (E) N-terminally tagged Ube2G2. All binding studies were carried out with a 2-fold molar excess of VHH12G2 over Ube2G2. SDS-PAGE analysis of samples corresponding to the VHH-E2 complex peaks (red) is shown in the middle panel. SDS PAGE of various ratios of E2:VHH12G2 is shown to the right to indicate the expected degree of Coomassie staining for both proteins. Abbreviations: SBP: Streptavidin binding protein, SMT3: suppressor of MIF2 mutations, yeast SUMO protein.
To better identify Ube2G2 residues recognized by VHH12G2, we performed mutagenesis and changed the 6 residues with the most pronounced chemical shifts on Ube2G2 to Ala ( > 0.2 ppm from Fig. 3A; G23,I24,V25,G27,F34,G52). These mutants include Ube2G2F34A; a single residue mutant with the most pronounced chemical shift, Ube2G2I24A,V25A, Ube2G2Triple (I24A,V25A,F34A); where all non-Gly residues were mutated to Ala, and Ube2G2AllAla; where all 6 residues were mutated to Ala (Supplementary Fig. 2). The elution peaks of the Ube2G2:VHH12G2 complex migrate with decreasing apparent molecular weight with increasing number of mutations introduced, reflecting different dynamics in the association between Ube2G2 mutants and VHH12G2 (Supplementary Fig. 2B)34. The Ube2G2AllAla mutant runs at a higher apparent molecular weight on SDS PAGE compared to Ube2G2WT (close to VHH12G2, making it difficult to separate them even on a 20% SDS PAGE), and elutes at a larger Stokes’ radius as analyzed by SEC (Supplementary Fig. 2A,E). Only the Ube2G2AllAla mutant has very poor binding to VHH12G2 as judged by SEC (Supplementary Fig. 2A). Ube2G2AllAla is the only mutant incapable of forming a thioester with ubiquitin (Supplementary Fig. 2C,D). This result is consistent with the proposed mode of binding of VHH12G2 but could also be explained by a failure to properly fold or a conformational change of the mutant.
While screening the immune library against a different E2, we identified another nanobody, VHH02G2. VHH02G2 does not bind to Ube2G2 as shown by SEC (Supplementary Fig. 3A), but it interacts weakly with Ube2G2 as seen by NMR spectroscopy (Fig. 3B). Residues near the acidic loop region appear to be involved (Fig. 3B). This weak interaction supports the flexible nature of the acidic loop region, consistent with previous reports33,35. These weak interactions are not a generic feature of VHHs, since a nanobody that recognizes yeast SUMO, VHHSMT3, does not show interactions with Ube2G2 as inferred from the NMR spectrum, nor does it bind to Ube2G2 by SEC (Fig. 3C, Supplementary Fig. 5A). We could therefore use these VHHs as controls.
We used alpha fold 3.0 prediction software36 to see whether it would accurately predict the region on Ube2G2 to which VHH12G2 binds. The top-scoring model, with the best confidence metrics, showed an interaction of VHH12G2 with Ube2G2’s backside (Supplementary Fig. 4A). However, VHH02G2 (weak binding) and VHHSMT3 (non-interacting) failed to yield satisfactory binding details, likely due to forced fit interactions (Supplementary Fig. 4B, C). While the individual predicted protein folds had good confidence scores, the ‘interface predicted template modeling’ (ipTM) scores were very low (best score is above 0.8; values for predicted VHH-E2 interactions were between 0.17 – 0.24), indicating a great degree of uncertainty. Thus, such predictions which are helpful for visualization should be validated with biochemical experiments.
Binding of VHH12G2 reduces the ubiquitination activity of Ube2G2 in the presence of E3 proteins
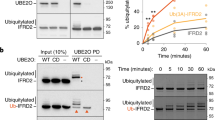
The ‘backside’ region to which VHH12G2 binds is at a considerable distance from Ube2G2’s active site cysteine (Cys89). We therefore did not expect to see a significant change in Ube2G2’s ability to form its thioester-linked Ub derivative. Indeed, when we measured Ube2G2~Ub thioester formation in the presence of Uba1 (E1), Ub, and Ube2G2, we saw no significant differences in Ub loading in the presence of a 3-fold molar excess VHH12G2 over Ube2G2 as analyzed by SDS-PAGE under non-reducing conditions (Fig. 4B). When performing SDS-PAGE under reducing conditions (with addition of DTT), only iso-peptide Lys-linked ubiquitinated Ube2G2 species persist. Formation of Ub-modified Ube2G2 (Ube2G2Ub) was reduced only very slightly. However, there is no loss of Ube2G2’s Ub-loading activity in the presence of VHH12G2.

A Schematic representation of the ubiquitination cascade. The E1 enzyme activates ubiquitin (Ub) in the presence of ATP and transfers it to the active site cysteine (-SH) of an E2 enzyme (Ub loading). Subsequently, an E3 enzyme with a RING/U-box domain facilitates the transfer of Ub directly from the E2 to the substrates. B Thioester Ub loading assays of Ube2G2 in the presence and absence of VHH12G2, analyzed by SDS-PAGE under non-reducing (left, -DTT) and reducing (right, +DTT) conditions. Reaction mixtures contained human E1 (Uba1), human Ube2G2, human Ub, and VHH12G2 added at a 3-fold molar excess over Ube2G2. Samples were collected at the indicated time points. Thioester Ub complexes detected under non-reducing conditions are labeled as Ube2G2Ub1/2/X based on the number of Ubs attached. SDS-PAGE under reducing conditions destroys any Ub thioesters and shows Lys-linked Ube2G2-Ub adducts. C–E Immunoblots of ubiquitination assays performed with the indicated E3 domains: (C) RING domain of human HRD1 (GST-HRD1), (D) RING domain of human TRC8 (GST-TRC8), and (E) full-length human CHIP (U-box type E3). Immunoblots with anti-Ubiquitin (top), anti-HA (middle, to detect HA tagged VHH), and anti-Ube2G2 (bottom) are shown for (C–E), respectively. Identifiable ubiquitinated species are indicated next to the blots where possible. Ub/Ub2 denotes unanchored ubiquitin.
E3 enzymes facilitate the ubiquitination of substrates and impose processivity on the formation of Ub chains4. Ube2G2, which plays a key role in protein quality control via the ERAD pathway, interacts with several E3s to generate K48-linked poly-Ub chains37. These E3s mainly belong to the RING family, which transfers Ub from E2~Ub to substrates38,39. Because the relevant E3s are membrane-anchored through one or more transmembrane segments, we expressed their cytoplasmically oriented catalytic portions in E.coli. To determine if VHH12G2 interferes with the interaction of Ube2G2 with its partner E3s, we used recombinantly expressed GST-tagged cytoplasmic RING domains from ER-resident HRD1 (aa236-444) and TRC8 (aa513-664) in ubiquitination assays. We also included the cytoplasmic ERAD-associated E3 CHIP (carboxyl terminus of Hsc70-interacting protein)40,41, a U-Box domain-containing E3. All ubiquitination assays with E3s were analyzed under reducing conditions. While we observe some ubiquitination of Ube2G2 upon inclusion of the E3 enzymes (Fig. 4C-E), the predominant ubiquitinated species appear to be other proteins, likely contaminants from the E1 and E3 purification process (Supplementary Fig. 6A).
For HRD1, the addition of excess VHH12G2 over Ube2G2 reduced the overall amount of ubiquitinated species, suggesting the involvement of Ube2G2’s backside in its interaction with HRD1 (Fig. 4C). For TRC8, VHH12G2 had a lesser impact, with only a mild reduction in the formation of higher molecular ubiquitinated species above 250 kDa (Fig. 4D). The presence of free Ub ( ~ 10 kDa) remaining even at the later time points also indicated reduced ubiquitination. For CHIP, we likewise observed reduced higher molecular weight ubiquitinated species over time (Fig. 4E).
Since VHH02G2 weakly interacts with Ube2G2’s acidic loop near the active site Cys89, we investigated whether its presence allows VHH02G2’s ubiquitination by proximity. However, the inclusion of VHH02G2 did not result in its ubiquitination in the presence or absence of HRD1 (Supplementary Fig. 3B,C). Ube2G2 therefore does not promote ubiquitination of a weakly binding VHH despite its interaction near the active site and the presence of a catalytically active E3. As a further specificity control, VHHSMT3 showed no effect on any of the E3-mediated ubiquitination assays (Supplementary Fig. 5B-J). All purified proteins used in this study are shown in Supplementary Fig. 6, and SDS PAGE gels corresponding to the ubiquitination reactions shown in main figures are presented in Supplementary Fig. 7.
The G2BR segment binds to the ‘backside’ of Ube2G2 and displaces VHH12G2 showing concomitant reduction in E3-mediated activity
A unique region found in ER-resident AUP1 and in the E3 Gp78 is referred to as the G2BR. G2BR binds to Ube2G2’s backside region, as shown by X-ray crystallography (PDB: 3H8K35 and PDB:7LEW29) (Fig. 5A), and NMR32. AUP1 has multiple roles, including the regulation of lipid droplet formation and exerting protein quality control through the ERAD pathway30. It recruits Ube2G2 from the cytoplasm to the ER via its G2BR segment to enable HRD1-mediated ubiquitination28,29. The effect of G2BR binding on Ube2G2’s activity varies, depending on experimental conditions and whether the G2BR is part of an E3 with a RING domain, like Gp7829,32. The backside region has been shown to be inhibitory for other E2s8,13,42, which has made it a target for the design of inhibitors9. Since VHH12G2 also shows an inhibitory effect on E2-E3 enzyme activity, we examined the effect of the soluble portion of AUP1, which participates in ERAD (the CUE-G2BR segment), as well as that of the isolated G2BR segments from AUP1 and Gp78.

A Predicted binding region of VHH12G2 based on NMR data and alpha fold 3.0 (left). X-ray crystal structures of G2BR regions from Gp78 (middle) and AUP1 (right) are shown for comparison29. B SEC profile of a 2-fold molar excess of CUE-G2BRAUP1 over Ube2G2 added to a mixture of prebound 2:1 molar ratio of VHH12G2:Ube2G2 (dotted red line). Profiles of CUE-G2BRAUP1 (pink), Ube2G2 (green), and VHH12G2 (blue) are also shown. The purified portion of AUP1 (aa292-410, CUE-G2BR) is depicted in a cartoon on the right (boxed). Fractions corresponding to the mixture of CUE-G2BRAUP1 + (Ube2G2 + VHH12G2) are shown below the cartoon in an SDS PAGE gel. C SEC profile of a 2-fold molar excess of Gp78 over Ube2G2 added to a mixture of VHH12G2:Ube2G2 similar to (B). Profiles of GST-Gp78 (black), Ube2G2 (green), and VHH12G2 (blue) are shown. The purified GST tagged Gp78 region (aa309-643, RING – CUE -G2BR) is shown in the boxed cartoon on the right. D–F Immunoblots with anti-ubiquitin antibody of ubiquitination assays performed in the presence and absence of a 3-fold molar excess of VHH12G2, a 2-fold molar excess of CUE-G2BRAUP1, or both over Ube2G2 with (D) GST-HRD1 RING, (E) 6xHis-CHIP, and (F) GST-TRC8 at indicated time points. TM Transmembrane, AT Acyltransferase, CUE Coupling of Ubiquitin to ER degradation, RING Really Interesting New Gene, G2BR Ube2G2 Binding Region.
We compared the NMR data for the G2BR peptide when bound to Ube2G2 from reference32, with our NMR data for the Ube2G2:VHH12G2 complex (Supplementary Fig. 8A,B). While the overall binding profile is similar, there are subtle differences, clarifying the different mode of engagement of Ube2G2 by the G2BR peptide vs VHH12G2. The G2BR segments bind to Ube2G2 with low nM affinity (4-10 nM29), while VHH12G2 binds with ~1 μM affinity (Fig. 2C). The magnitude of chemical shifts in ppm values are much larger for G2BR binding, consistent with it being a strong binder.
Since G2BR binds with higher affinity on Ube2G2’s backside binding region than VHH12G2, we tested the stability of the VHH12G2 complex in the presence of the AUP1 C-terminal region (aa292-410, hereafter referred to as CUE-G2BRAUP1), and the cytoplasmic Gp78 (N-terminal GST-tagged, 309-643, RING–CUE-G2BR, referred to as GST-gp78). The affinity of GST-Gp78 for Ube2G2 is ~5 nM21. We added a 2-fold molar excess of VHH12G2 over Ube2G2, followed by a similar 2-fold molar excess of CUE-G2BRAUP1 or GST-Gp78, and subjected the mixture to SEC. Both CUE-G2BRAUP1 and GST-Gp78 displaced VHH12G2 from Ube2G2 (Fig. 5B, C). This displacement is most likely due to the lower affinity of VHH12G2 for Ube2G2 but could also be due to differences in the conformational changes induced by binding of either VHH12G2 or G2BR.
To test if the CUE-G2BRAUP1 portion of AUP1 involved in ERAD also results in a similar reduction of activity with E3s, we added a 3-fold molar excess of CUE-G2BRAUP1over Ube2G2, or VHH12G2, or both (Fig. 5D-F). For HRD1, the presence of CUE-G2BRAUP1 resulted in a greater reduction in ubiquitination compared to VHH12G2 (Fig. 5D). For CHIP, VHH12G2 showed a greater reduction than CUE-G2BRAUP1(Fig. 5E). The impact of CUE-G2BRAUP1 on TRC8 was negligible (Fig. 5F). Since CUE-G2BRAUP1 displaces VHH12G2, it overrides the effect of the VHH when added together. Thus, the CUE-G2BR segment in AUP1 similarly reduces Ube2G2’s activity with HRD1 and CHIP, but not that of TRC8.
The CUE domain interacts with poly-Ub chains and aids in Ub chain assembly43,44. Therefore, some of the reduction in activity observed for CUE-G2BRAUP1 might be attributed to the CUE domain. To test this, we purified the G2BR regions by appending them to C-terminus of a GFP construct with a Sortase A motif (LPETG) placed between GFP and a streptavidin binding protein (SBP) tag: GFP-(Lys-Pro-Glu-Thr-Gly)-SBP-His6-G2BRAUP1/Gp78. Since the G2BR region is a short peptide ( ~ 3 kDa), it is difficult to purify without attached tags. We employed Sortase A 5 M enzyme to cleave and isolate the SBP-His6-G2BR constructs (Fig. 6A), which bound to Ube2G2, as confirmed by SEC (Fig. 6B). Substituting CUE-G2BRAUP1 in the previous assays to the SBP-His6-G2BR constructs of either AUP1 or Gp78 showed similar trends (Fig. 6C). The G2BR segment was therefore the major contributor to the reduction in activity seen in Fig. 5D-F. The SBP-His6 segment alone showed no binding to Ube2G2 (Fig. 6D), nor did it impact the ubiquitination assay with E3s (Fig. 6E).

A Constructs of G2BR appended to the C terminal end of a streptavidin binding protein tag (SBP) and 6xHis (His6) tag. SDS PAGE gels of the purified constructs are shown to the right. B SEC binding profiles of a mixture of 2-fold molar excess of SBP-His6-G2BR variations with Ube2G2 (red) along with unbound Ube2G2 (green). SDS PAGE gels of fractions corresponding to the mixture are shown below. Positions of Ube2G2 and SBP-His6-G2BR variations are indicated next to the gels on the right. C Anti-ubiquitin immunoblots of ubiquitination assays performed in the presence and absence of a 3-fold molar excess of VHH12G2, or SBP-His6-G2BRAUP1, or SBP-His6-G2BRgp78 with E3s: GST-HRD1 (left), His6-CHIP (middle), and GST-TRC8 (right). Identified species of ubiquitinated Ube2G2 and free unanchored Ub chains (Ub, Ub2) are indicated on the right. D SEC profiles of a mixture of 2-fold molar excess of SBP-His6 over Ube2G2 (red) along with unbound Ube2G2 (green). SDS PAGE gel fractions of the mixture are shown below. E Anti-ubiquitin immunoblots of ubiquitination assays performed similarly in the presence and absence of a 3-fold molar excess of SBP-His6 with E3s: GST-HRD1 (left), His6-CHIP (middle), and GST-TRC8 (right).
Thus, we have shown that backside binding VHH12G2, CUE-G2BRAUP1, and the isolated G2BR regions from AUP1 and Gp78 (SBP-His6 tagged) all reduce Ube2G2’s activity with E3s to varying degrees.
VHH12G2 shows release of Gp78 from Ube2G2 prior to transfer of Ub3 chains to substrate
Gp78 is a Ube2G2-specific E3 involved in ERAD and functions in maintaining protein quality control19. As one of the best-studied E2-E3 interactions, how Gp78 interacts with Ube2G2 is well understood31,32,35. At the ER, Gp78’s G2BR binds to Ube2G2, thus enhancing Ube2G2’s interaction with the RING domain through an allosteric change in the complex31. This enhanced RING binding causes a secondary allosteric change in Ube2G2 that decreases its affinity for G2BR, ultimately leading to the dissociation of the E2-E3 complex31.
Although VHH12G2 is displaced from Ube2G2 in the presence of GST-Gp78 (Fig. 5C), we examined the impact of adding VHH12G2 to the ubiquitination assay. Surprisingly, we saw an increased presence of unanchored Ub3 chains in the presence of an excess of VHH12G2 over Ube2G2 (Fig. 7A). VHHSMT3 or VHH02G2 failed to do so (Fig. 7B, Supplementary Fig. 3D). The release of unanchored Ub3 chains was also seen when using isolated SBP-His6-G2BRAUP/Gp78 segments (Fig. 7C). Ub3 chains can be seen (but faintly) in the presence of only Gp78, but the significant increase seen upon inclusion of VHH12G2 suggests that this is due to backside binding.

A–C Anti-ubiquitin Immunoblots of ubiquitination assays with E3 GST-Gp78 at indicated time points in the presence or absence of a 3-fold molar excess of (A) VHH12G2, (B) unrelated VHHSMT3, and (C) VHH12G2 or G2BR segments appended to SBP-His6 tags over Ube2G2. D Schematic of the Gp78 mechanism of action on Ube2G2 from reference31, along with the proposed explanation for the effect when VHH12G2 or isolated G2BR binds.
The Ub3 species observed in reducing conditions (i.e. upon addition of DTT, Fig. 7A-C) can only originate from thio-ester linked Ub chains on the active site of Ube2G2. The consistent observation of prominent Ub3 bands in the presence of backside binders to Ube2G2 (VHH12G2 and SBP-His6-G2BRAUP1/Gp78), suggests that at some point, there should exist a fraction of Ube2G2-Ub3 chains occupied by the backside binders (Fig. 7D). En bloc transfer of pre-formed Ub chains from Ube2G2 to substrates with Gp78 have been shown in previous studies20,21,32,37. In our case, without an E3, we only see formation of unanchored Ub2 chains by Ube2G2 (Fig. 4B), suggesting that the interactions with Gp78 are responsible for the formation of these Ube2G2-Ub3 species. The formation of larger Ub chains cannot be excluded and may escape detection under the assay conditions used. All of this could point to an additional intermediary step in the existing model of Ube2G2-Gp78 interaction31, which involves the detachment of Gp78 from Ube2G2, allowing a backside binder to sequester Ube2G2-Ub3 species as illustrated in Fig. 7D.
Gp78 without G2BR has reduced E3 activity; VHH12G2 and SBP-His6-G2BRAUP1 further reduce Gp78ΔG2BRc activity
The presence of G2BR in Gp78 increases its activity compared to its G2BR-lacking variant, suggesting that G2BR’s backside binding enhances its ubiquitination activity31. This effect was attributed to the allosteric changes that result from the initial G2BR binding, as explained in the previous section31. This G2BR confers specificity of Gp78 to Ube2G2, as Gp78 does not interact effectively with other E2s18,20,32. We used Gp78ΔG2BRc (GST-Gp78 aa309-573, RING–CUE), and confirmed that deletion of G2BR abrogates binding of Gp78 to Ube2G2 (Fig. 8A). We also saw a similar reduced ubiquitination activity of Ube2G2 with Gp78ΔG2BRc compared to Gp78 (Fig. 8B). Gp78ΔG2BRc retains E3 functionality through its RING domain and shows activity comparable to Ube2G2 interacting with other E3s like TRC8 or CHIP (Fig. 4D, E). The ability of Gp78 to productively engage Ube2G2 is highlighted by the near-complete depletion of free Ub within 30 min, in contrast to the persistence of free Ub even after 2 hours with other E3s used in this study (HRD1, TRC8, CHIP). Addition of an excess of VHH12G2 over Ube2G2 in the Gp78ΔG2BRc ubiquitination assay resulted in further decreased ubiquitination, which was not seen with VHHSMT3 (Fig. 8B). This suggests that the inhibitory effects of backside binding by VHH12G2 persist after G2BR removal (Fig. 8B).

A Cartoon representing the region of Gp78ΔG2BRc (aa309-573) appended to GST for purification (top). SEC profiles show a mixture of a 2-fold molar excess of Ube2G2 over GST-Gp78ΔG2BRc (red) along with unbound Ube2G2 (green). The corresponding fractions of the mixture are shown below in an SDS PAGE gel. B Anti-ubiquitin immunoblot of ubiquitination assays performed with GST-Gp78, and GST- Gp78ΔG2BRc in the presence and absence of a 3-fold molar excess of VHH12G2, or unrelated VHHSMT3 over Ube2G2 for the indicated time points. C Anti-ubiquitin immunoblot of ubiquitination assays performed similar to (B) using isolated G2BR constructs appended to SBP-His6 tags. D Anti-ubiquitin immunoblot of ubiquitination assays with GST-Gp78 in the presence and absence of a 3-fold molar excess of CUE-G2BRAUP1, SBP-His6-G2BRAUP1 or Gp78 over Ube2G2 at indicated time points. E SEC profile of the ubiquitination assay sample at the 30-minute time point with a 3-fold molar excess of CUE-G2BRAUP1 (red) indicated by an arrow in D. Profiles of CUE-G2BRAUP1 (black) and Ube2G2 (green) are also shown. SDS PAGE analysis of the ubiquitination assay SEC fraction (red) is displayed to the right, with the major eluted species indicated at the bottom.
The addition of a synthetic G2BR peptide in trans was previously shown to enhance E3 activity in vitro with Gp78ΔG2BR32. In a similar type of assay, we added SBP-His6-G2BRAUP1/Gp78 in excess over Ube2G2 with Gp78ΔG2BRc (Fig. 8C). While we did not see a significant increase in E3 activity, inclusion of SBP-His6-G2BRAUP1 showed a slight decrease comparable to that seen with VHH12G2 (Fig. 8C). However, this effect was not seen with SBP-His6-G2BRGp78 (Fig. 8C). This may be an indication of the subtle differences in interaction between the two G2BRs of AUP1 and Gp78 on Gp78’s activity without its internal G2BR segment.
The CUE domain of AUP1 sequesters Ub and reduces ubiquitination activity of Gp78
Both ER resident AUP1 and Gp78 contain CUE-G2BR domains. Sequestration of Ube2G2 by AUP1 prevents its degradation and facilitates Ube2G2’s participation in HRD-mediated ERAD28,29. The interaction between Gp78 and Ube2G2 is terminated upon execution of Gp78’s E3 ubiquitination activity31. To identify if AUP1-bound Ube2G2 can function with Gp78, Ube2G2’s activity with excess CUE-G2BRAUP1 was assessed in the presence of Gp78. Figure 8D shows a significant reduction in Ube2G2’s activity with GST-Gp78 when CUE-G2BRAUP1 is used, an effect not seen with isolated G2BRs. As the CUE domain is the primary difference, we analyzed the reaction mixture at 30 min time point by SEC to analyze possible interactions. CUE-G2BRAUP1 not only co-elutes with Ube2G2 (Fig. 8E) as seen also from Fig. 5B, but also with free Ub. Excess CUE-G2BRAUP1 co-elutes with free Ub, suggesting that the observed inhibition may stem from sequestration of Ub by the CUE domain, thereby reducing its availability for Gp78’s activity. This likely mirrors the CUE domain’s dual role in cells: sequestration of Ub and ensuring the proper orientation of poly Ub chains for enhanced processivity with E3s20,43,44.
We also investigated whether Ube2G2 remains bound to the isolated G2BRs present in excess with Gp78ΔG2BRc as shown in Fig. 8D, given the hypothesis that Gp78’s RING domains might trigger an allosteric effect and detach the G2BRs from the E2. We saw co-elution of Ube2G2 with G2BRs, indicating that they remain bound. However, the fraction of Ube2G2 that is unbound and available for Gp78’s activity cannot be assessed unambiguously (Supplementary Fig. 9). We might therefore be observing the combined effect of bound and unbound G2BRs on activity.
Discussion
Here we showed that engagement of the ‘backside’ of Ube2G2 by either VHH12G2, by CUE-G2BRAUP1 or by isolated G2BR segments generally results in a reduction of Ube2G2’s activity when assayed with various E3s, in declining order: HRD1 > CHIP > > TRC8, with the exception of the G2BR-containing E3 Gp78 (Table 1). We confirmed the activating effect of G2BR in cis within E3 Gp7820. However, deletion of the G2BR from Gp78 (rendering it more like a RING E3) still resulted in inhibition when the nanobody occupied Ube2G2’s backside. These results underscore how the occupancy of an interaction site on an E2 can influence interactions with different E3s, suggesting that specific E2 conformations are optimized for interaction with particular E3s.
While reports indicate that G2BR peptide added in trans can show increased Ube2G2 activity in vitro with RING domains of HRD1, TRC8 and Gp78ΔG2BR29,32, we did not observe such stimulation in our assays. This discrepancy could stem from differences in our ubiquitination assay setup, particularly our inclusion of a pre-incubation step with Ube2G2 and various backside binding proteins, which ensures that the results reflect the behavior of Ube2G2-bound species. The assays reported in32 were done with the GST-tagged RING domains of E3s immobilized on GST beads, and only bead-bound materials were analyzed. Our findings fit reports that show G2BR enhances Gp78 activity in cis20. Addition of G2BR segments in trans followed by an assessment of the totality of ubiquitinated species resulted in a reduction in total ubiquitinated species (Fig. 8C).
VHH12G2 and the G2BRs from AUP1 and Gp78 bind to the backside of Ube2G2 but interact differently. The contact residues on Ube2G2 involved in the binding of both G2BRs include T17, P20, V25, A26, G27, M29, E31, E38, L40, I41, M42, E45, E50, F51, G52, V53, K156, V159, Q160, L163, G164, L16529, with partial overlap from VHH12G2 at residues G23, I24, V25, G27, F34, and G52 (chemical shifts >0.2 ppm; shared contact residues in bold italic). While allosteric shifts after G2BR binding occur near the active site residues 96-108 in Ube2G2’s crystal structure32, our NMR data indicate minimal chemical shifts in these regions (Fig. 3A). This, together with comparisons of the NMR profiles (Supplementary Fig. 8A,B), suggests that the conformational change imposed by binding of G2BR and VHH12G2 must differ. Our NMR data show significant shifts across the protein, excluding the active site, hinting at a specific Ube2G2 conformation that might hinder ubiquitination in concert with HRD1 or CHIP. This is noteworthy, as TRC8 and Gp78 do not show significant inhibition in this configuration. If backside binding is inhibitory, it may not matter for the ERAD pathways that involve TRC8 or Gp78.
Backside binding on E2s can either enhance or inhibit activity, depending on the E2 and its interacting partner(s). In activating scenarios, the backside typically interacts weakly with Ub, enhancing the processivity of poly-Ub chain formation, as seen with Ube2D2 (UbcH5b)45,46, Ube2D3 (UbcH5c)10, Ube2B (Rad6b)47, the SUMO-conjugating E2 Ube2I11, as well as in the case of Ube2G2-Gp78, where cis binding increases processivity31,32,35. For the Ube2D family, non-covalent weak interactions with Ub on the E2 backside (Kd ~200-300 μM) increases affinity and processivity with E3s by formation of self-assembled oligomers of Ube2D~Ub molecules45,46. For Ube2B, interaction with Ub on Ube2B’s backside ( ~ 600 μM) is required for poly-ubiquitin chain formation in the absence of an E347. The E3 Rad18 has a similar R6BD (Rad6 binding domain) region that binds to Ube2B’s backside (Kd ~62 μM), with an orientation perpendicular to that of G2BR’s binding to Ube2G2. Rad18’s R6BD binding to Ube2B competes with the weak interaction with Ub on its backside, directing Ube2B to mono-ubiquitinate its substrate, PCNA47. Conversely, examples of inhibition include the backside binding of Ube2R2 by a small molecule CC06518, inhibition of Ube2E3 by Ub’s K48 interaction on the backside42, and various E2s being inhibited by targeted Ub variants9. Our findings generally indicate an inhibitory effect of backside binding, except for Gp78’s G2BR in cis. We hypothesize that the observed inhibition results from an allosteric change on Ube2G2 upon engagement of the backside, disrupting Ube2G2’s weak interactions with Ub48, a phenomenon also reported for other E2s10,46,47. Despite the addition of a molar excess of backside binders relative to Ube2G2, we can rule out molecular crowding as the cause of inhibition, because assays done under similar conditions with the non-interacting VHHSMT3 (Supplementary Fig. 5 B-J) or SBP-His6 alone (Fig. 6E, Supplementary Fig. 6D) show no inhibition. Binding of VHH12G2 may sterically hinder E3 RING engagement. We mapped the residues chemically shifted upon binding of VHH12G2 to Ube2G2 onto the published X-ray structure of the G2BR-RING bound to Ube2G2 (PDB: 4LAD). This shows that the residues most significantly shifted upon binding of VHH12G2 do not fully coincide with the E3 binding region (Supplementary Fig. 8C). Our data show that ubiquitination in the presence of the RING domain of the TRC8 E3 is not significantly inhibited by the presence of VHH12G2. Steric hindrance with E3 binding site therefore is unlikely to be the reason for the reduction in activity.
NMR data suggest that Ube2G2 does not bind to the U-box domain and does not show enhanced ubiquitination in the presence of CHIP49. However, our assays showed a time-dependent increase in CHIP-dependent ubiquitination with Ube2G2, especially for higher molecular weight species (Fig. 4E). In vitro, CHIP can thus interact and function with Ube2G2, although at a lower rate compared to TRC8 or HRD1 (Fig. 4C-E). Since CHIP is also involved in ERAD, it may interact with Ube2G2 in cells and not just in vitro.
In the ERAD pathway, TRC8 in partnership with Ube2G2 plays a particularly important role in the degradation of Class I HLA molecules27,50. Although Ube2G2 shows activity with TRC8 in vitro, backside binding shows only minimal inhibition of activity (Fig. 4D). The association between Ube2G2 and TRC8 may therefore not rely on backside binding, or an alternative mode of interaction may exist between these two proteins.
AUP1 and Gp78 are proteins integral to ERAD and target different types of misfolded proteins within the ER. AUP1 collaborates with HRD1 to manage luminal misfolded proteins, forming a large complex that facilitates substrate recognition and translocation across the ER28,29,30. Conversely, Gp78 primarily addresses misfolded membrane proteins on the cytoplasmic side of the ER19,20,21. Distinctions between HRD1 and Gp78 within the ERAD context include their substrate specificity, molecular partnerships, and possibly their mode of action, though they may also function sequentially within the same pathway51. Local Ub concentrations may influence certain E3-mediated ubiquitination reactions52. Our observation that CUE-G2BRAUP1 sequesters free Ub, resulting in a reduced pool of Ub for Gp78’s activity (Fig. 8E) suggests that local Ub concentration could explain the independent or sequential ERAD pathways adopted by HRD1 and Gp7852.
The pan-ubiquitin blots in this study likely detect contaminating proteins rather than specific substrates. While HERP is Ube2G2’s main substrate of Gp7837, G2BR’s displacement of VHH12G2 prevents study of reduced ubiquitination. Substrates for Ube2G2 with E3s HRD1, TRC8, or CHIP amenable to purification have not been reported. The sparse representation of these ubiquitinated products poses a challenge for detection by mass spectroscopy. The development of artificial substrates targeted to E3s by other means may resolve these limitations53. Analysis of specific substrates in vitro or in cellular systems is beyond the scope of this study.
AUP1’s G2BR binds to Ube2G2, resulting in a notable inhibition of HRD1’s ubiquitination activity in vitro (Fig. 5D). In the cellular environment, AUP1 binds to Ube2G2 to prevent its degradation28,29. This binding has also been implicated in the activation of Ube2G2 to serve ERAD in cells29. The discrepancy between results obtained in vitro and in cells suggests that compartmentalization in a membrane environment, or alternatively, the presence of additional proteins within the HRD1-SEL1 complex, may relieve the inhibition seen in vitro. Overexpression of GFP-tagged AUP1 lacking the G2BR in HeLa cells [19] showed increased ubiquitination in the fraction immunoprecipitated with an anti-GFP antibody, which implies that the G2BR negatively regulates ubiquitination. Our findings corroborate this and show that G2BR binding to the ‘backside’ of Ube2G2 restricts its ubiquitination activity. Thus, regulation of ubiquitination in vivo remains a challenging puzzle.
Materials and methods
Nanobody library preparation and bacteriophage screening
An alpaca was immunized with 100 μg of purified recombinant Ube2G2 in four rounds of immunization at biweekly intervals. Following this, total RNA was extracted (RNeasy mini kit, Qiagen, 74104) from lymphocytes and converted to cDNA by reverse transcription (SuperScript III First-Strand Synthesis System, Thermo Fisher,18080051). VHHs were amplified from cDNAs and cloned into a phagemid vector to create a Ube2G2-targeted nanobody phagemid library in TG1 E. coli strain (Agilent, 200123). Phage display library was produced by infecting TG1 library with helper phage M13KO7 (NEB, N0315S) and grown O/N in 2YT media (18 g/L tryptone, 10 g/L yeast extract, 5 g/L NaCl) with Ampicillin (100 μg/mL), and Kanamycin (50μg/mL) at 30 °C, 250 rpm. Phages were precipitated from the supernatant the next day by adding 1/6th volume of 2.5 M NaCl, 20% w/v PEG 6000 at 4 °C for 1 h. Precipitated phages were centrifuged at 10,000xg for 20 min at 4 °C and phage pellets were resuspended in TBS (50 mM Tris-HCl, pH 7.5, 150 mM NaCl). The phage was re-precipitated similarly to reduce bacterial pellet contamination. Phage-based screening was done using streptavidin binding protein (SBP) tagged Ube2G2 (Ube2G2-SBP-6xHis) immobilized on streptavidin-coated magnetic beads (Dynabeads™ MyOne™ Streptavidin T1, Thermo Fisher, 65601). Purified phage displaying VHHs were pre-cleared against SBP-6xHis tag by allowing them to bind to 40 μg GFP-SBP-6xHis on 100 μL beads. After pre-clearing, unbound phages were allowed to bind to 40 μg Ube2G2 on beads for 30 min on a rotator. The beads were washed thrice in TBST (TBS + 0.1% Tween 20) and eluted with 0.2 M Glycine pH 2.2 for 10 min and later neutralized to pH 7.5 using 1 M Tris-HCl pH 9.1. Eluted phages were infected on log phase ER2738 E. coli strain (NEB, E4104) for propagation. A second round of screening was similarly done on the phage display library obtained from the first round, with 2 μg Ube2G2 on beads. After the two rounds of screening, highly enriched VHH sequences were selected and cloned into pHEN6 periplasmic vector with a C-terminal HA-6xHis tag for subsequent experiments. VHH02G2 sequence: QVQLVETGGGLVQPGESLTLSCTVSGSDFSDWAMTWFRQTPENQRELVATISNGAGSVDYYADSVKGRFIISRDNAKNTLYLQMNNLKTEDTGVYICNQRGYFGQGTQVTVSS. VHHSMT3 sequence: QLQLVETGGGLVQPGGSLRLSCVASGFAFSDSVITWVRQAPGKGPEWVAIIGYTGKARSYANSVKGRFTIDRDNDKNTVYLQMNNLTPDDTALYYCVRGDVPNGDGLERGQGTQVTVSS.
Protein purifications
Nanobody sequences cloned into the pHEN6 vector were transformed into WK6 E. coli cells and grown in TB (Terrific Broth, RPI) to 0.4-0.8 OD at 600 nm at 37 °C, 250 rpm. Nanobody expression was induced by adding 1 mM IPTG (Chem-Impex, 00194) followed by incubation at 30 °C, 250 rpm for 5–6 h. Cells were pelleted at 5000xg for 10 min and 30 mL TES buffer (0.2 M Tris pH 8.0, 0.5 M sucrose, 0.65 mM EDTA, protease inhibitor tablet) (cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail, Sigma, 11836170001) was added per 1 L pellet and incubated for 1 h at 4 °C. Osmotic shock was induced by adding 150 mL ice-cold water for 30 min to release the periplasmic fraction, after which cells were pelleted at 5000xg for 10 min. Supernatant containing VHH was purified using Ni-NTA affinity chromatography (Ni-NTA agarose beads, Qiagen, 30210) followed by size exclusion chromatography (SEC) using Hiload Superdex 75 16/600 column (Cytiva, 28989333) in ÄKTA pure 25 L FPLC.
All cytoplasmic purifications were done similarly. BL21 (DE3) cells were transformed with expression plasmids in the following vectors. pET SUMO vector: 6xHis-SUMO-Ube2G2 (Ube2G2 Uniprot: P60604). pET28 vector: 6xHis-Sortase A 5 M (Addgene: 51140), 6xHis-CHIP (1-303, Uniprot: Q9UNE7), His6-Ube2R1 (Uniprot: P49427), 6xHis-Ube2J1 (Uniprot: Q9Y385). Modified pET28 vector with a C-terminal sortag motif (‘LPETG’54) followed by SBP tag55 and 6xHis tag (--LPETG-SBP-6xHis): Ube2G2-SBP-6xHis, Uba1-SBP-6xHis (Uba1 Uniprot: P22314), GFP-SBP-6xHis (GFP Uniprot: B7UCZ6), Ube2H-SBP-6xHis (Uniprot: P62256), Ube2G1-SBP-6xHis (Uniprot: P62253), Ube2D2-SBP-6xHis (Uniprot: P62837). pGEX vector: GST-HRD1 (aa236-444, Addgene: 185349), GST-TRC8 (aa513-664, Addgene: 185352), GST-Gp78c (aa309-643, Addgene: 11429), Gp78ΔG2BRc (modified from GST-Gp78, aa309-573), GST-AUP1 (aa292-410, Addgene: 185331). G2BR segments were appended to the end of GFP-SBP-6xHis constructs after adding a ‘GGSS’ linker. Sequences of G2BRAUP1: SSWARQESLQERKQALYEYARRRFTER, G2BRGp78: SADERQRMLVQRKDELLQQARKRFLNKS. Ube2G2 mutants were made on 6xHis-SUMO-Ube2G2 construct and purified similarly.
Transformed E. coli were grown at 37 °C, 250 rpm in TB until OD600 nm reached 0.4-0.8. Expression was induced with 1 mM IPTG and growth at 30 °C, 250 rpm for 5–6 h. Cells were lysed using a homogenizer (Avastin Emulsiflex C3) in lysis buffer (50 mM Tris HCl pH 7.5, 150 mM NaCl, protease inhibitor tablet, DNAse) (DNase I, Sigma, 10104159001). Lysate was cleared by centrifugation at 20,000 rpm for 20 min at 4 °C using JA20 rotor on Beckman coulter JE centrifuge. The cleared lysate was further purified on Ni-NTA beads or GSTrap™ HP column (Cytiva, 17528101). For Ni-NTA beads, columns were washed with TBS containing 10 mM Imidazole and eluted in 250 mM Imidazole in TBS. For GST-tagged proteins, cleared lysate was diluted in TBS + 1 mM DTT and filtered using 0.22 μm before injection into the FPLC column. Column washes were done using TBS. Bound proteins were eluted using 20 mM L-reduced glutathione in TBS with 1 mM DTT. To obtain untagged Ube2G2, purified 6xHis-SUMO-Ube2G2 was cleaved using SUMO protease (sigma SAE0067) following manufacturer’s instructions. Cleaved materials were further purified using Ni-NTA resin and SEC. GST-AUP1 was cleaved to obtain the CUE-G2BRAUP1 using PreScission protease (Cytiva, 27084301) and further purified on a GSTrap column and by SEC.
To obtain SBP-6xHis-G2BR segments, Sortase A 5M-mediated cleavage was performed using (Gly)3 nucleophile (Chem Impex, 04555) on purified GFP-LPETG-SBP-6xHis-G2BR constructs in TBS with 10 mM CaCl2 for 60 min at 25 °C54. Concentrations used for the Sortase reactions were 2.5 μM Sortase A 5 M, 50 μM GFP-SBP-6xHis-G2BR, and 2.5 mM (Gly)3. After cleavage, the reaction mixture was passed through a Ni-NTA column to remove uncleaved components, (Gly)3 and 6xHis tagged Sortase A 5 M. The isolated G2BR segments were further purified using SEC.
Protein concentrations were determined using NanoDrop One (Thermo Scientific, ND-ONE-W) and corrected for absorbance using their extinction coefficients.
SEC binding experiments
All binding experiments were carried out in 250 μL in TBS. Proteins were added at the indicated molar ratios (2-fold molar excess of binding partner over Ube2G2, unless otherwise stated) and incubated at 25 °C for 30 min on a rotator. 200 μL of sample was then applied to a Superdex 75 10/300 (Cytiva, 17517401) column on FPLC at a flow rate of 0.5 mL/min and 0.5 ml fractions were collected. For CUE-G2BRAUP1/GST-gp78c binding to Ube2G2:VHH12G2, 2:1 molar ratio of VHH:Ube2G2 was incubated first for 30 min, followed by addition of CUE-G2BRAUP1 or GST-gp78c, and further incubated for 10 min prior to SEC.
SEC binding experiments following ubiquitination assays were performed similarly. Reactions were performed in 250 μL, and after 30 min of reaction at 37 °C, a 200 μL aliquot was applied to Superdex 75 10/300 column for SEC. All SEC experiments were performed at 4 °C. SEC profiles were plotted using GraphPad Prism. Maximum peak values per profile were normalized to 100 milli absorbance units (mAU) and overlaid for comparisons.
Microscale thermophoresis
Ube2G2 protein was C terminally tagged with Alexa Fluor 647 (AF647) using Sortase A 5 M mediated conjugation54. A sortase A motif ‘LPETG’ was included in the Ube2G2-LPETG-SBP-6xHis construct. A reaction mixture containing 1.6 μM 6xHis-Sortase A 5 M, 31 μM Ube2G2-LPETG-SBP-6xHis, 300 μM (Gly)3 -AF647 in TBS with 10 mM CaCl2 was incubated at 25 °C for 30 min at 300 rpm. After incubation, the mixture was applied to Ni-NTA agarose to remove (Gly)3 -AF647, unreacted 6xHis tagged Ube2G2 and Sortase A 5 M. The flow through was collected and further purified by SEC to remove any remaining (Gly)3 -AF647 nucleophile. Purity of the conjugated protein was confirmed by SDS PAGE and by its absorbance at 280 and 650 nm (Supplementary Fig. 6B). Microscale thermophoresis was performed using Monolith NT.115pico Instrument (Nano Temper) at the Center for Macromolecular Interactions at Harvard Medical School. Binding experiments were replicated in 3 trials and fitting was done using Kd model from MO Affinity Analysis software. The sample measurements were taken using distinct aliquots of proteins for each trial.
(Gly)3 -AF647 was synthesized by conjugating AF647 (Alexa Fluor™ 647 C2 Maleimide, Thermo Fisher, A20347) to (Gly)3 peptide (synthesized using standard solid-phase peptide synthesis protocols) using a thio-Michael addition reaction. The final product was purified using reverse-phase HPLC and its identity was confirmed using liquid chromatography-mass spectrometry (Waters UPLC-MS Acquity).
NMR analysis of VHH binding to Ube2G2
15N labeled 6xHis-SUMO-Ube2G2 in BL21 (DE3) cells were grown in M9 minimal media (42.3 mM Na2HPO4, 22 mM KH2PO4, 8.6 mM NaCl, 2 mM MgSO4, 0.1 mM CaCl2, 0.5% w/v glucose, 0.0002% w/v Thiamine hydrochloride, 1 g/L 15NH4Cl) until OD600 nm reached 0.4. Protein expression was induced using 1 mM IPTG and the culture was grown at 30 °C, 250 rpm for 20 h. 6xHis-SUMO-Ube2G2 protein was purified using Ni-NTA affinity chromatography and Ube2G2 was isolated using SUMO protease and further purified in NMR buffer (20 mM NaPi, 50 mM NaCl, 0.5 mM EDTA, 1 mM TCEP, pH 7.0) by SEC as described under ‘protein purifications’. Nanobodies – VHH12G2, VHH02G2, and VHH1SMT3 were also purified in the same NMR buffer by SEC, as described under ‘protein purifications’. For NMR experiments, 15N labeled Ube2G2 was used at 0.5 mM concentration with 10% D2O in the presence or absence of a 110% molar equivalent of VHHs. 15N-TROSY HSQC data was collected on a Bruker Advance II 600 MHz spectrometer equipped with a Prodigy cryogenic probe at 25 °C. The number of complex points acquired were 512 in the direct 1H dimension and 128 in the indirect 15N dimension, and the number of scans used was 8. Data processing was done using the nmrPipe software, data analysis, transferring of peak assignments and spectral plot generations were performed using the CARA software. The combined chemical shift change values were calculated as sqrt((DeltaHcs)^2 + (DeltaNcs/5.0)^2).
In vitro ubiquitination assays
For all in vitro ubiquitination assays, backside binding proteins or control proteins (VHH12G2, VHH02G2, VHHSMT3, CUE-G2BRAUP1, SBP-His6-G2BRAUP1/Gp78, SBP-His6) were preincubated at a 3-fold molar excess over Ube2G2 in TBS (50 mM Tris-HCl pH 7.5, 150 mM NaCl) for 30 min at 25 °C on a rotator before the ubiquitination reaction. For Ube2G2:VHH12G2:CUE-G2BRAUP1 reactions, a 3:1 ratio of VHH12G2:Ube2G2 was incubated first for 30 min, followed by addition of 2-fold molar excess of CUE-G2BRAUP1 over Ube2G2 and further incubation for 10 min. After complex formation, purified recombinant E1 (Uba1-SBP-6xHis), Ubiquitin (R&D, U-100H-10M) and respective E3s were added, and the reaction was started by adding MgCl2 (5 mM) and ATP (2 mM). Reactions were performed at 37 °C with gentle agitation (300 rpm). At various time points 50 μL aliquots were taken, and the reaction was quenched with 15 μL of 5x laemmli buffer (350 mM Tris-HCl pH 6.8, 30% glycerol, 10% w/v SDS, 600 mM DTT, 0.01% w/v bromophenol blue) in reducing ( + DTT) or non-reducing (-DTT) conditions. Samples were boiled at 95 °C for 5 min before loading on an SDS PAGE gel. Final concentration of components used for the assay were: 0.1 μM E1, 4 μM Ube2G2, 12 μM of Ube2G2 backside binding proteins or control proteins, 10 μM ubiquitin, 8 μM or 12 μM CUE-G2BRAUP1 (as indicated in figure legends), 4 μM GST-HRD1, 4 μM GST-TRC8, 4 μM 6xHis-CHIP, 1 μM GST-gp78, and 1μM Gp78ΔG2BRc. After the final time point, 50 μL of the reaction was similarly quenched and 30 μL was loaded on a 13% SDS PAGE gel and stained by Coomassie (InstantBlue® Coomassie Protein Stain, Abcam, ab119211). All E3 ubiquitination assays were analyzed under reducing conditions. All ubiquitination experiments were repeated thrice, a representative blot of which is displayed in the figures. The assays were performed on different days with different aliquots of purified proteins.
Immunoblots
For ubiquitination assays, 10 μL of the quenched reaction was run on 13% SDS PAGE gels and transferred to a PVDF membrane using the Trans-Blot turbo transfer system (Biorad, 1704150). The membrane was blocked with 5%BSA (w/v) in TBST (TBS + 0.1% Tween 20) for 1 h at 25 °C with shaking, followed by addition of primary antibodies diluted in blocking buffer; 1:1000 Direct-Blot™ HRP anti-Ubiquitin Antibody (Biolegend 646303, Clone P4D1), or 1:10,000 of HRP conjugated anti-HA monoclonal antibody (Invitrogen 26183-HRP), or 1:1000 of anti-Ube2G2 rabbit monoclonal antibody (Cell signaling technology, 63182), for 1 h at RT. Membranes were washed in TBST thrice. HRP conjugated antibodies were visualized using Pierce™ ECL Western Blotting Substrate (Thermo Fisher, 32106) and imaged using ChemiDoc MP (Biorad, 12003154). For anti-Ube2G2 blots, membranes were incubated with goat anti-Rabbit-HRP (Abclonal, AS014) at 1:10,000 dilution for 30 min, washed in TBST thrice, developed similarly and imaged.
PDB images and alpha fold
Alpha fold 3.0 predictions were done by uploading the sequences of Ube2G2, and VHHs using the online version of the software. Images of proteins from protein data bank (PDB) and AlphaFold output were rendered using ChimeraX 1.2.4 software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
All unedited and uncropped gel images are included in Supplementary Fig. 10. All other data can be obtained from the corresponding author, Pavana Suresh, pavana.suresh@childrens.harvard.edu.
References
Stewart, M. D., Ritterhoff, T., Klevit, R. E. & Brzovic, P. S. E2 enzymes: more than just middle men. Cell Res. 26, 423–440 (2016).
Komander, D. & Rape, M. The ubiquitin code. Annu. Rev. Biochem 81, 203–229 (2012).
Swatek, K. N. & Komander, D. Ubiquitin modifications. Cell Res. 26, 399–422 (2016).
Gundogdu, M. & Walden, H. Structural basis of generic versus specific E2-RING E3 interactions in protein ubiquitination. Protein Sci. 28, 1758–1770 (2019).
Schulman, B. A. & Harper, J. W. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat. Rev. Mol. Cell Biol. 10, 319–331 (2009).
Ye, Y. & Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 (2009).
Wenzel, D. M., Stoll, K. E. & Klevit, R. E. E2s: structurally economical and functionally replete. Biochem J. 433, 31–42 (2011).
Ceccarelli, D. F. et al. An allosteric inhibitor of the human Cdc34 ubiquitin-conjugating enzyme. Cell 145, 1075–1087 (2011).
Garg, P. et al. Structural and Functional Analysis of Ubiquitin-based Inhibitors That Target the Backsides of E2 Enzymes. J. Mol. Biol. 432, 952–966 (2020).
Brzovic, P. S., Lissounov, A., Christensen, D. E., Hoyt, D. W. & Klevit, R. E. A UbcH5/ubiquitin noncovalent complex is required for processive BRCA1-directed ubiquitination. Mol. Cell 21, 873–880 (2006).
Knipscheer, P., van Dijk, W. J., Olsen, J. V., Mann, M. & Sixma, T. K. Noncovalent interaction between Ubc9 and SUMO promotes SUMO chain formation. EMBO J. 26, 2797–2807 (2007).
Lewis, M. J., Saltibus, L. F., Hau, D. D., Xiao, W. & Spyracopoulos, L. Structural basis for non-covalent interaction between ubiquitin and the ubiquitin conjugating enzyme variant human MMS2. J. Biomol. NMR 34, 89–100 (2006).
Kumar, P. et al. Role of a non-canonical surface of Rad6 in ubiquitin conjugating activity. Nucleic Acids Res. 43, 9039–9050 (2015).
Muyldermans, S. Nanobodies: natural single-domain antibodies. Annu Rev. Biochem 82, 775–797 (2013).
Ingram, J. R., Schmidt, F. I. & Ploegh, H. L. Exploiting Nanobodies’ Singular Traits. Annu Rev. Immunol. 36, 695–715 (2018).
Steyaert, J. & Kobilka, B. K. Nanobody stabilization of G protein-coupled receptor conformational states. Curr. Opin. Struct. Biol. 21, 567–572 (2011).
Ling, J. et al. A nanobody that recognizes a 14-residue peptide epitope in the E2 ubiquitin-conjugating enzyme UBC6e modulates its activity. Mol. Immunol. 114, 513–523 (2019).
Fang, S. et al. The tumor autocrine motility factor receptor, gp78, is a ubiquitin protein ligase implicated in degradation from the endoplasmic reticulum. Proc. Natl Acad. Sci. USA 98, 14422–14427 (2001).
Chen, B. et al. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc. Natl Acad. Sci. USA 103, 341–346 (2006).
Liu, W., Shang, Y. & Li, W. gp78 elongates of polyubiquitin chains from the distal end through the cooperation of its G2BR and CUE domains. Sci. Rep. 4, 7138 (2014).
Li, W. et al. Mechanistic insights into active site-associated polyubiquitination by the ubiquitin-conjugating enzyme Ube2g2. Proc. Natl Acad. Sci. USA 106, 3722–3727 (2009).
Kikkert, M. et al. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 279, 3525–3534 (2004).
Stefanovic-Barrett S. et al. MARCH6 and TRC8 facilitate the quality control of cytosolic and tail-anchored proteins. EMBO Rep. 19, e45603 https://doi.org/10.15252/embr.201745603 (2018).
Jo, Y., Hartman, I. Z. & DeBose-Boyd, R. A. Ancient ubiquitous protein-1 mediates sterol-induced ubiquitination of 3-hydroxy-3-methylglutaryl CoA reductase in lipid droplet-associated endoplasmic reticulum membranes. Mol. Biol. Cell 24, 169–183 (2013).
Hassink, G. et al. TEB4 is a C4HC3 RING finger-containing ubiquitin ligase of the endoplasmic reticulum. Biochem J. 388, 647–655 (2005).
Stevenson, J., Huang, E. Y. & Olzmann, J. A. Endoplasmic Reticulum-Associated Degradation and Lipid Homeostasis. Annu Rev. Nutr. 36, 511–542 (2016).
van de Weijer, M. L. et al. Multiple E2 ubiquitin-conjugating enzymes regulate human cytomegalovirus US2-mediated immunoreceptor downregulation. J. Cell Sci. 130, 2883–2892 (2017).
Klemm, E. J., Spooner, E. & Ploegh, H. L. Dual role of ancient ubiquitous protein 1 (AUP1) in lipid droplet accumulation and endoplasmic reticulum (ER) protein quality control. J. Biol. Chem. 286, 37602–37614 (2011).
Smith, C. E. et al. A structurally conserved site in AUP1 binds the E2 enzyme UBE2G2 and is essential for ER-associated degradation. PLoS Biol. 19, e3001474 (2021).
Spandl, J., Lohmann, D., Kuerschner, L., Moessinger, C. & Thiele, C. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J. Biol. Chem. 286, 5599–5606 (2011).
Das, R. et al. Allosteric regulation of E2:E3 interactions promote a processive ubiquitination machine. EMBO J. 32, 2504–2516 (2013).
Das, R. et al. Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol. Cell 34, 674–685 (2009).
Ju, T., Bocik, W., Majumdar, A. & Tolman, J. R. Solution structure and dynamics of human ubiquitin conjugating enzyme Ube2g2. Proteins 78, 1291–1301 (2010).
Mayer, C. L. et al. Size-exclusion chromatography can identify faster-associating protein complexes and evaluate design strategies. BMC Res Notes 2, 135 (2009).
Chakrabarti, K. S., Li, J., Das, R. & Byrd, R. A. Conformational Dynamics and Allostery in E2:E3 Interactions Drive Ubiquitination: gp78 and Ube2g2. Structure 25, 794–805 e5 (2017).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature https://doi.org/10.1038/s41586-024-07487-w (2024).
Li, W., Tu, D., Brunger, A. T. & Ye, Y. A ubiquitin ligase transfers preformed polyubiquitin chains from a conjugating enzyme to a substrate. Nature 446, 333–337 (2007).
Tiwari, S. & Weissman, A. M. Endoplasmic reticulum (ER)-associated degradation of T cell receptor subunits. Involvement of ER-associated ubiquitin-conjugating enzymes (E2s). J. Biol. Chem. 276, 16193–16200 (2001).
David, Y., Ziv, T., Admon, A. & Navon, A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J. Biol. Chem. 285, 8595–8604 (2010).
Jiang, J. et al. CHIP is a U-box-dependent E3 ubiquitin ligase: identification of Hsc70 as a target for ubiquitylation. J. Biol. Chem. 276, 42938–42944 (2001).
Matsumura, Y., Sakai, J. & Skach, W. R. Endoplasmic reticulum protein quality control is determined by cooperative interactions between Hsp/c70 protein and the CHIP E3 ligase. J. Biol. Chem. 288, 31069–31079 (2013).
Nguyen, L. et al. The ubiquitin-conjugating enzyme, UbcM2, is restricted to monoubiquitylation by a two-fold mechanism that involves backside residues of E2 and Lys48 of ubiquitin. Biochemistry 53, 4004–4014 (2014).
Bagola, K. et al. Ubiquitin binding by a CUE domain regulates ubiquitin chain formation by ERAD E3 ligases. Mol. Cell 50, 528–539 (2013).
von Delbruck, M. et al. The CUE Domain of Cue1 Aligns Growing Ubiquitin Chains with Ubc7 for Rapid Elongation. Mol. Cell 62, 918–928 (2016).
Buetow, L. et al. Activation of a primed RING E3-E2-ubiquitin complex by non-covalent ubiquitin. Mol. Cell 58, 297–310 (2015).
Sakata, E. et al. Crystal structure of UbcH5b~ubiquitin intermediate: insight into the formation of the self-assembled E2~Ub conjugates. Structure 18, 138–147 (2010).
Hibbert, R. G., Huang, A., Boelens, R. & Sixma, T. K. E3 ligase Rad18 promotes monoubiquitination rather than ubiquitin chain formation by E2 enzyme Rad6. Proc. Natl Acad. Sci. USA 108, 5590–5595 (2011).
Bocik, W. E., Sircar, A., Gray, J. J. & Tolman, J. R. Mechanism of polyubiquitin chain recognition by the human ubiquitin conjugating enzyme Ube2g2. J. Biol. Chem. 286, 3981–3991 (2011).
Soss, S. E., Yue, Y., Dhe-Paganon, S. & Chazin, W. J. E2 conjugating enzyme selectivity and requirements for function of the E3 ubiquitin ligase CHIP. J. Biol. Chem. 286, 21277–21286 (2011).
Stagg, H. R. et al. The TRC8 E3 ligase ubiquitinates MHC class I molecules before dislocation from the ER. J. Cell Biol. 186, 685–692 (2009).
Zhang, T., Xu, Y., Liu, Y. & Ye, Y. gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell 26, 4438–4450 (2015).
Yau, R. & Rape, M. The increasing complexity of the ubiquitin code. Nat. Cell Biol. 18, 579–586 (2016).
Lips, C. et al. Who with whom: functional coordination of E2 enzymes by RING E3 ligases during poly-ubiquitylation. EMBO J. 39, e104863 (2020).
Popp M. W., Antos J. M., Ploegh H. L. Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Protein Sci. 15, 1–15. https://doi.org/10.1002/0471140864.ps1503s56 (2009).
Keefe, A. D., Wilson, D. S., Seelig, B. & Szostak, J. W. One-step purification of recombinant proteins using a nanomolar-affinity streptavidin-binding peptide, the SBP-Tag. Protein Expr. Purif. 23, 440–446 (2001).
Giudicelli, V., Brochet, X. & Lefranc, M. P. IMGT/V-QUEST: IMGT standardized analysis of the immunoglobulin (IG) and T cell receptor (TR) nucleotide sequences. Cold Spring Harb. Protoc. 2011, 695–715 (2011).
Acknowledgements
This research was supported by NIH grant 5DP1AI150593-05. The authors thank the Center for Macromolecular Interactions at Harvard Medical School for microscale thermophoresis measurements. We thank the Bio-molecular NMR facility & Dana-Farber Cancer Institute NMR core for the NMR measurements. We thank Sushruth Muralidharan for helpful discussions.
Author information
Authors and Affiliations
Contributions
H.P. and P.S. contributed to conception and design of experiments. P.S., C.W., N.B., Z.J.S, N.P., and I.S. performed experiments. P.S. and H.P. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics
Animal study using Alpaca (Vicugna Pacos) was approved by IACUC University of Massachusetts Amherst. The study was conducted in accordance with the local legislation and institutional requirements. All methods reported are in accordance with ARRIVE guidelines.
Peer review
Peer review information
Communications Biology thanks Albert Lee and the other, anonymous, reviewers for their contribution to the peer review of this work. Primary Handling Editors: Christina Karlsson Rosenthal. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Suresh, P., Wijne, C., Sun, ZY.J. et al. A nanobody that binds to the backside of the ubiquitin conjugating enzyme Ube2G2 differentially affects interactions with its partner E3 Ligases. Commun Biol 8, 614 (2025). https://doi.org/10.1038/s42003-025-08038-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08038-3
