
Abstract
Global elimination of hepatitis C virus (HCV) will require an effective cross-genotype vaccine. The HCV E2 envelope glycoprotein is the main target of neutralizing antibodies but also contains epitopes that elicit non-neutralizing antibodies which may provide protection through Fc effector functions rather than direct neutralization. We determined cryo-EM structures of a broadly neutralizing antibody, a moderately neutralizing antibody, and a non-neutralizing antibody bound to E2 to resolutions of 3.8, 3.3, and 3.7 Å, respectively. Whereas the broadly neutralizing antibody targeted the front layer of E2 and the non-neutralizing antibody targeted the back layer, the moderately neutralizing antibody straddled both front and back layers, and thereby defined a new neutralizing epitope on E2. The small size of complexes between conventional (monovalent) Fabs and E2 (~110 kDa) presented a challenge for cryo-EM. Accordingly, we engineered bivalent versions of E2-specific Fabs that doubled the size of Fab–E2 complexes and conferred highly identifiable shapes to the complexes that facilitated particle selection and orientation for image processing. This study validates bivalent Fabs as new fiducial markers for cryo-EM analysis of small proteins such as HCV E2 and identifies a new target epitope for vaccine development.
Similar content being viewed by others


Introduction
Hepatitis C virus (HCV) is a major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma, infecting 1–2% of the world population and causing an estimated 240,000 deaths in 2022 alone1. Remarkable advances have been made in the treatment of patients with persistent HCV infection using direct-acting antiviral (DAA) therapies2. However, an effective prophylactic HCV vaccine is still much needed because of the high cost of DAA treatment and the increase in new infections and poor awareness of carriers, especially in developing countries3.
The feasibility of developing a prophylactic HCV vaccine is supported by clinical observations that ~25% of individuals who become infected with HCV are able to clear the infection spontaneously4. Spontaneous clearance of HCV infection is associated with the early appearance of broadly neutralizing antibodies that bind to HCV envelope glycoproteins and neutralize multiple HCV variants5,6. The HCV E2 envelope glycoprotein is the main target of neutralizing antibodies and mediates viral entry into host cells by interacting with multiple cell surface receptors, including CD81, claudin, occludin, and scavenger receptor-B17. A schematic representation of E2 is shown in Fig. 1a. The central β-sandwich of E2 is flanked by front (residues 424–459) and back (residues 597–645) layers8,9. The CD81 binding loop comprises residues 519–535.

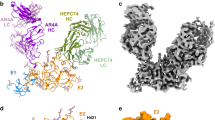
a Schematic representation of HCV E2 (residues 384–746) colored by structural components, with variable regions (VRs) in yellow, central β-sandwich in red, front layer in blue, CD81 binding loop in cyan, back layer in purple, and stalk and transmembrane region in green. E2 N-linked glycans are indicated. Conserved disulfide bonds are drawn as red dashed lines. b (left) Final reconstructed cryo-EM map of the Fab HC84.26.5D–E2 complex using cryoSPARC77. (center) Overall structure of the Fab HC84.26.5D–E2 complex (ribbon diagram). E2 is green; L chain is blue; H chain is yellow. (right) EM map of the interface between E2 and HC84.26.5D. The side chains of interacting residues are shown in stick representation with carbon atoms in green (E2), cyan (L), or yellow (H), nitrogen atoms in blue, and oxygen atoms in red. c (left) Final cryo-EM map of the Fab HC84.26.5D–E2–Fab CBH7 complex. (center) Structure of the Fab HC84.26.5D–E2–Fab CBH7 complex. E2 is green; VL of HC84.26.5D is blue; VH of HC84.26.5D is yellow; L chain of CBH7 is magenta; H chain of CBH7 is orange. (right) EM map of the interface between E2 and CBH7 showing interacting residues. d (left) Final cryo-EM map of the Fab CBH4B–E2 complex. (center) Structure of the Fab CBH4B–E2 complex. E2 is green, L chain is brown; H chain is cyan. (right) EM map of the interface between E2 and CBH4B showing interacting residues.
Neutralizing antibodies protect against infection by HCV and other enveloped viruses by inhibiting virus attachment, entry, fusion, and/or egress from host cells. Much less is known about the functional significance of non-neutralizing antibodies, although there is abundant evidence that non-neutralizing antibodies can also be protective against diverse viruses, including alphaviruses10,11, HIV-112, Lassa fever virus13, West Nile virus14, Crimean-Congo hemorrhagic fever virus15, and SARS-CoV-216. However, the mechanism of protection involves effector functions mediated through Fc receptors, not direct neutralization. For example, non-neutralizing antibodies can protect humanized mice against HIV-1 infection in an entirely Fc receptor-dependent manner12. Non-neutralizing antibodies might offer similar protection against HCV, although this possibility has not been tested in vivo. Conversely, it has also been proposed that non-neutralizing antibodies may negatively modulate neutralizing antibodies, thereby exacerbating HCV infection17,18,19. Here we determined cryo-EM structures of a broadly neutralizing antibody (HC84.26.5D), a moderately neutralizing antibody (CBH7), and a non-neutralizing (CBH4B) antibody bound to HCV E2, and compared these structures with X-ray structures of previously reported antibody–E2 complexes8,9,20,21,22,23,24,25,26 and with models predicted by the deep learning method AlphaFold27.
HC84.26 is a broadly neutralizing antibody isolated from an individual with chronic HCV infection28. It recognizes a conformational epitope on the E2 envelope glycoprotein of HCV, designated as antigenic domain D, and neutralizes genotypes 1a, 2a, 4a, 5a, and 6a. HC84.26 inhibits E2 binding to the viral receptor CD8128. Affinity maturation of HC84.26 was carried out to isolate variants with improved binding and neutralization activities29. One of these affinity-matured variants, HC84.26.5D, showed increased neutralization of diverse HCV isolates in vitro and was able to prevent acute HCV infection in humanized mice.
CBH4B is a non-neutralizing antibody isolated from a chronically infected blood donor that recognizes a conformational epitope on HCV E2, designated as antigenic domain A, which comprises a cluster of highly immunogenic determinants that account for a substantial portion of the antibody response to E230,31,32,33,34. Antibodies to antigenic domain A, which may act as an immunological decoy, have the potential to interfere with virus-neutralizing antibodies17,18,19. CBH4B does not block binding of E2 to CD81.
CBH7 is a moderately neutralizing antibody that recognizes a conformational epitope on E2, designated as antigenic domain C30,35. It neutralizes genotypes 1 and 2 but not genotypes 3, 4, and 533,36,37. Neutralization is mediated by blocking E2 binding to CD81. CBH7 has bidirectional competition with the non-neutralizing antigenic domain A antibody CBH4B31. Although CBH4B blocks CBH7 binding to E2 by nearly 40%, the neutralization potency of CBH7 against genotype 1a is not inhibited by CBH4B35.
We used single-particle cryo-EM to determine structures of antibodies HC84.26.5D, CBH4B, and CBH7 bound to the full extracellular domain of E2 from HCV strain 1b09. The relatively small size of a complex between a conventional (i.e., monovalent) Fab fragment of these antibodies and E2 (~110 kDa) presents a challenge for cryo-EM because small targets often lack recognizable shape features that can facilitate initial image alignment at low resolution. Structure determination is further complicated by the intrinsic flexibility of the elbow regions linking the variable (V) and constant (C) domains of Fabs, which may limit their ability to orient particles accurately38. To overcome these obstacles, we engineered dimeric bivalent versions of Fab HC84.26.5D and Fab CBH4B that doubled the effective size of their complexes with E2 and conferred a highly recognizable shape to the complexes to facilitate image processing. We previously employed this method to determine the cryo-EM structure of a ~100 kDa complex between LAG3 and a therapeutic antibody targeting this immune checkpoint receptor39. Its application to Fab–E2 complexes, reported here, establishes the general utility of bivalent Fabs as new fiducial markers for cryo-EM analysis, particularly for small proteins. In addition, the CBH7–E2 structure permitted identification of a new neutralizing epitope on E2, thereby advancing our knowledge of the antibody response to HCV and defining a new target epitope for rational vaccine design.
Results
Cryo-EM structure determination of Fab–E2 complexes using bivalent Fabs
Conventional Fabs are monovalent monomers. We recently reported the crystal structure of a bivalent dimeric form of Fab HC84.26.5D created by deleting a single residue, VHSer113 (Kabat numbering), in the elbow region linking the VH and CH1 domains40. Three-dimensional domain swapping of VL and VH domains with the corresponding V domains of a symmetry-related Fab molecule in the crystal resulted in formation of a doughnut-shaped Fab dimer with a rectangular hole in which the two VL/VH modules are separated by ~25 Å. In agreement with the crystal structure, a low resolution (7 Å) cryo-EM reconstruction of the Fab HC84.26.5D–E2 complex showed that the dimeric Fab bound two E2 molecules (~220 kDa)40. However, in the present study, at a global resolution of 3.8 Å, the Fab HC84.26.5D–E2 complex formed a superassembly of ~440 kDa composed of four E2 molecules bound to a dimer of Fab dimers. Formation of this dimer of dimers is facilitated by contacts between the CL domains of two domain-swapped dimers arranged in parallel. This arrangement is stabilized by hydrogen bonds between backbone atoms of CLLeu153 from each dimer (Supplementary Fig. 1). Based on local resolution analysis and side-chain features in the Fab–E2 interfaces, a resolution of 3.8 Å was achieved for two locally refined cryo-EM maps of the Fab HC84.26.5D–E2 complex as determined from gold standard FSC curves (Table 1) (Supplementary Fig. 2) (Fig. 1b).
To define the epitope recognized by antibody CBH7, we solved the cryo-EM structure of a ternary complex of monomeric Fab CBH7, dimeric Fab HC84.26.5D, and E2 to 3.3 Å resolution using dimeric Fab HC84.26.5D as a fiducial marker (Table 2) (Supplementary Fig. 3) (Fig. 1c). In contrast to Fab HC84.26.5D, deletion of VHSer113 alone was insufficient to convert monomeric Fab CBH4B to a dimer, which instead required deletion of four elbow region residues, VHSer112–CHSer115. We determined the structure of the Fab CBH4B–E2 complex using dimeric Fab CBH4B as a fiducial marker and locally refined the Fab–E2 interface to 3.7 Å resolution based on gold standard FSC curves (Table 3) (Supplementary Fig. 4) (Fig. 1d).
To examine rotameric outliers in the Fab HC84.26.5D–E2, Fab CBH7–E2, and Fab CBH4B–E2 structural models (Tables 1–3), we analyzed rotamers for contact residues from E2 and the CDR loops (Supplementary Table 1). We focused on the locally refined models used for interface evaluation. No rotameric outliers were observed among the contact residues in any of the three structures. To examine the Ramachandran statistics, we analyzed dihedral angles for interfacial residues in the Fab CBH7–E2 and Fab CBH4B–E2 complexes (Supplementary Table 1). In the Fab CBH7–E2 and Fab CBH4B–E2 interfaces, only two (non-glycine) and one residue, respectively, fall within the allowed region, while the remaining residues are in the favored region of the Ramachandran plot.
We previously reported the affinity of monomeric versus dimeric forms of Fab HC84.26.5D for E240. As measured by biolayer interferometry (BLI), the affinity of monomeric Fab HC84.26.5D for E2 (KD = 3.8 nM) was essentially identical to that of dimeric Fab HC84.26.5D (4.9 nM). We performed similar BLI affinity measurements for Fab CBH4B (Supplementary Fig. 5). The affinity of monomeric Fab CBH4B for E2 (KD = 19 nM) is comparable to that of dimeric Fab CBH4B (KD = 27 nM). Thus, dimerization of Fab HC84.46.5D and Fab CBH4B did not have a major impact on affinity for E2.
Although dimeric Fab HC84.26.5D and Fab CBH4B display similar overall architectures in the HC84.26.5D–E2 and CBH4B–E2 complexes, the disposition of V and C domains is different in the two doughnut-like structures, with central holes of different shapes and dimensions: a diamond-shaped hole of 38 × 46 Å for HC84.26.5D versus a rectangular-shaped hole of 46 × 47 Å for CBH4B (Supplementary Fig. 6a, b). Of note, the shape of dimeric Fab HC84.26.5D bound to E2 in the cryo-EM structure is significantly different from that of unbound dimeric Fab HC84.26.5D in the crystal structure (Supplementary Fig. 6c)40, indicating flexibility. To understand the domain movements underpinning dimerization of Fab CBH4B and Fab HC84.26.5D, we compared dimeric Fab CBH4B and Fab HC84.26.5D with monomeric Fab CBH7, whose crystal structure we determined to 1.6 Å resolution (Table 4). As calculated using the program DynDom (https://dyndom.cmp.uea.ac.uk/dyndom)41, the angle of rotation around the switch peptide of the CBH4B H chain (residues 110–113), which links VH and CH1, is 76° with respect to CBH7 (Fig. 2a). The angle of rotation around the switch peptide of the CBH4B L chain (residues 105–110), which links VL and CL, is 109° with respect to CBH7. Likewise, the angles of rotation around the switch peptides of the Fab HC84.26.5D H (residues 110–113) and L chains (106–109) are 112° and 68° with respect to the corresponding H and L chains of CBH7 (Fig. 2a). These rotations result in more extended conformations of the H and L chains of dimeric CBH4B and HC84.26.5D compared to the H and L chains of monomeric CBH7. This is reflected in reductions in total buried surface area between VH and CH1 from 1670 Å2 for CBH7 to 643 Å2 for CBH4B (745 Å2 for HC84.26.5D) and in total buried surface area between VL and CL from 1724 Å2 for CBH7 to 621 Å2 for CBH4B (742 Å2 for HC84.26.5D), as analyzed by PDBePISA (https://www.ebi.ac.uk/pdbe/pisa/)42.

a (left) Comparison of H chain conformations in unbound monomeric Fab CBH7 (magenta), E2-bound HC84.26.5D (yellow), and E2-bound CBH4B (green). The switch peptide linking VH and CH1 is blue. H chains were superposed through their VH domains. Angles of rotation around the switch peptide with respect to Fab CBH7 are indicated. (right) Comparison of L chain conformations in unbound monomeric Fab CBH7 (magenta), E2-bound HC84.26.5D (yellow), and E2-bound CBH4B (green). The switch peptide linking VL and CL is blue. L chains were superposed through their VL domains. Angles of rotation around the switch peptide with respect to Fab CBH7 are indicated. b (top) Crystal structure of monomeric Fab CBH7 with elbow region between VH and CH1 domains framed in black. The elbow angle is 133°. The elbow region between VL and CL domains is framed in red. (middle) Close-up view of standard ball-and-socket joint between VH and CH1 in Fab CBH7 formed by VH residues Val11, Thr110, and Ser112 (socket) and CH1 residues Phe145 and Pro146 (ball). (bottom) Close-up view of residues forming the elbow region between VL and CL. c (top) Cryo-EM structure of Fab HC84.26.5D in the complex with E2 (not shown) with elbow region between VH and CH1 framed in black. The elbow angle is 202°. The elbow region between VL and CL domains is framed in red. (middle) Close-up view of residues forming ball-and-socket joint in Fab CBH7 but rearranged in the Fab HC84.26.5D–E2 complex. (bottom) Close-up view of residues forming the elbow region between VL and CL. d (top) Cryo-EM structure of Fab CBH4B in the complex with E2 (not shown) with elbow region between VH and CH1 framed in black. The elbow angle is 215°. The elbow region between VL and CL domains is framed in red. (middle) Close-up view of residues forming ball-and-socket joint in Fab CBH7 but rearranged in the Fab CBH4B–E2 complex. (bottom) Close-up view of residues forming the elbow region between VL and CL.
The angles of rotation around the switch peptides of the H and L chains of dimeric Fab CBH4B with respect to monomeric Fab CBH7 (76° and 109°, respectively) differ significantly from the corresponding angles of rotation for dimeric Fab HC84.26.5D (112° and 68°, respectively) (Fig. 2a). The different geometries of the two domain-swapped Fab dimers are most likely a consequence of the different elbow region deletions: VHSer112–CHSer115 in Fab CBH4B versus only VHSer113 in Fab HC84.26.5D. Antibody molecules contain a highly conserved ball-and-socket joint between VH and CH1 that, in monomeric Fab CBH7, comprises VH residues Val11, Thr110, and Ser112, which form the socket, and CH1 residues Phe145 and Pro146, which form the ball (Fig. 2b)43. Although these five residues are present in dimeric Fab CBH4B and Fab HC84.26.5D, they do not form a canonical ball-and-socket joint. Conformational differences in the elbow region between VL and CL (residues 106–110) are shown in Fig. 2b.
Structure of the Fab HC84.26.5D–E2 complex
Superposition of two locally refined complexes in the Fab HC84.26.5D–E2 superassembly gave a root-mean-square difference (RMSD) of 0.4 Å for 818 α-carbon atoms, indicating close similarity. Therefore, the following description of Fab–E2 interactions applies to both refined complexes. E2 and HC84.26.5D adopt similar overall folds in the cryo-EM and crystal structures20,40, with RMSD of 1.5 Å for superposition of 215 α-carbons of E2 (Supplementary Fig. 7a) and RMSD of 0.9 Å for superposition of 196 α-carbons of the Fab VLVH module (Supplementary Fig. 7b). We were able to model the complete interface between E2 and HC84.26.5D, including all six complementarity-determining regions (CDRs) of the antibody. By contrast, in the crystal structure of Fab HC84.26.5D40, no density feature was detected for VHCDR2 residues Ser25–His29, implying flexibility. In the crystal structure of E2 from HCV strain 1b0920, E2 contains eight disulfide bonds formed by 16 highly conserved cysteine residues that impart to E2 its globular conformation. We could model all eight disulfides in the cryo-EM structure of E2 from this strain (Fig. 1b–d), in agreement with the crystal structure20. We were also able to trace structural features for at least two N-acetylglucosamine residues at six of 10 potential N-glycosylation sites on the neutralizing face of E2, with additional sugar moieties visible at three of these sites (Supplementary Fig. 8).
Neutralizing epitope recognized by HC84.26.5D
Most broadly neutralizing antibodies targeting E2, including HC84.26.5D, contain an H chain encoded by the VH1-69 gene segment44. Previous structural studies of VH1-69-encoded neutralizing antibodies bound to E2 revealed that they are characterized by a hydrophobic tip of their germline-encoded VHCDR2 loop and a long somatically-generated VHCDR3 loop (15–22 residues)8,20,21,22,23,24,26,44. HC84.26.5D has a considerably shorter VHCDR3 (10 residues), which lacks the intraloop disulfide that stabilizes VHCDR3 in several other VH1-69 broadly neutralizing antibodies. In the HC84.26.5D–E2 complex, HC84.26.5D contacts antigenic site 412 (AS412; residues Gln412–Arg424), front layer, CD81 binding loop, central β-sandwich, and back layer of E2 (Supplementary Table 2) (Fig. 3a, b). HC84.26.5D buries 1124 Å2 of solvent-accessible surface on E2, compared to 633–1204 Å2 for other front-layer-specific antibodies. Moreover, most E2 residues that contact HC84.26.5D are 90–100% conserved among all HCV genotypes, in agreement with the broad neutralizing capacity of this antibody. VH dominates the interactions of HC84.26.5D with E2, accounting for 71% (868 Å2) of the surface area on E2 buried at the interface, compared to 563–1192 Å2 for other front-layer-specific broadly neutralizing antibodies. Superposition of the HC84.26.5D–E2 complex onto complexes involving other VH1-69-encoded neutralizing antibodies (AR3X, HEPC3, RM2-01, and 1382_01_H05)20,21,22,23,24,26 revealed that the antibodies engage E2 with different angles of approach (Fig. 3a) and with footprints that map to different but overlapping regions of the antigenic surface (Fig. 3b).

a Surface representations of HC84.26.5D–E2, AR3X–E2 (PDB code 6URH), HEPC3–E2 (6MEI), 1382_01_H05–E2 (7RFC), and RM2-01–E2 (7JTF) complexes20,23,24,26. All structures are superposed on E2 of the HC84.26.5D–E2 complex to illustrate differences in the angle of approach of VH1-69 neutralizing antibodies with respect to E2. The Fab constant domains are not shown for clarity. E2 surfaces are colored by structural components: hypervariable regions, yellow; front layer, cyan; β-sandwich, orange; CD81 binding loop, blue; post-VR3, gray; AS412 region, pink; back layer, green. H and L chains are shown in dark gray and white, respectively. b Epitopes of the front layer-specific neutralizing antibodies are colored on the E2 surface based on their structural components, with interacting residues labeled. Footprints for H chain CDRs are displayed separately.
The surface area on E2 buried by VHCDR3 of HC84.26.5D (258 Å2) is considerably less than the surfaces buried by VHCDR3 of AR3X (486 Å2), HEPC3 (580 Å2), and 1382_01_H05 (532 Å2), but comparable to the surface buried by VHCDR3 of macaque RM2-01 (257 Å2) (Fig. 4a). Therefore, VHCDR3 of HC84.26.5D plays a less prominent role in E2 recognition than VHCDR3 of most other front layer-specific neutralizing antibodies. This is attributable to the particular angle of approach of HC84.26.5D rather than to its short VHCDR3. Indeed, HC84.26.5D VHCDR1 and VHCDR2, which bury 220 Å2 (28%) and 315 Å2 (40%), respectively, of E2 surface, make contributions similar to VHCDR3 258 Å2 (32%) (Fig. 4a). Whereas other VH1-69 antibodies contact E2 almost entirely via VH19, VL of HC84.26.5D accounts for 29% (330 Å2) of E2 surface buried at the interface. The surface area on E2 buried by HC84.26.5D (1122 Å2) is greater than the surfaces buried by HEPC3 (972 Å2), RM2-01 (658 Å2), and 1382_01_H05 (1014 Å2), but less than that by AR3X (1181 Å2). The larger surface buried by AR3X is attributable to an unusual 14 amino acid insertion in VHCDR226.

a Comparison of surface areas buried by CDRs of HC84.26.5D, AR3X, HEPC3, 1382_01_H05–E2, and RM2-0120,23,24,26. b Surface representations of the HC84.26.5D–E2 and other front layer-binding antibody–E2 structures. E2 surfaces are colored by structural components: hypervariable regions, yellow; front layer, cyan; β-sandwich, orange; CD81 binding loop, blue; post-VR3, gray; back layer, green. The locations of helices α1, α2, and η2 and of Cys429 are marked. The VHCDR1 (red), VHCDR2 (blue), and VHCDR3 (green) loops are mapped onto the E2 surface. c Interactions of VHCDR1–3 and VLCDR1–3 loops of HC84.26.5D with E2. E2 surfaces are colored by structural components: hypervariable regions, yellow; front layer, cyan; β-sandwich, orange; CD81 binding loop, blue; back layer, green. CDR1 (red), CDR2 (blue), and CDR3 (green) loops of VL and VH are shown. Interacting residues are drawn in stick representation and labeled. Hydrogen bonds are indicated by black dashed lines.
In the HC84.26.5D–E2 structure, the VHCDR1 is loop oriented towards the C-terminal portion of the highly conserved AS412 epitope (residues 412–423), with which it makes two hydrogen bonds (VHCDR1 Thr28 Oγ1–O His421 E2 and VHCDR1 His29 N–O His421 E2) and multiple hydrophobic contacts with Trp420 and Ile422 (Supplementary Table 2) (Fig. 4b). The tip of VHCDR2 of HC84.26.5D (I52-P52a-D53-F54; Kabat numbering) conforms to the hydrophobic motif (I/V52-P52a-X53-F54) characteristic of other VH1-69-encoded broadly neutralizing antibodies8,20,21,22,23,24,26. This tip interacts extensively with a highly conserved hydrophobic groove between the front layer and the CD81 binding loop, with which it makes two hydrogen bonds and a π–cation interaction with Trp529 via VHCDR2 Arg55 (Supplementary Table 2) (Fig. 4b). In addition, VHCDR2 contacts back layer residue Tyr613, in agreement with previous epitope mapping28. VHCDR3 packs against a hydrophobic surface on E2 formed by two 310 helices in the front layer, helix α1 (residues 438–442) and a short helix η2 (residues 447–449), and one 310 helix in the back layer, helix α2 (residues 614–617). Further linking VHCDR3 to the front layer are two hydrogen with helix α1 residues Leu441 and Phe442 (VHCDR3 Ser98 Oγ–O Leu441 E2 and VHCDR3 Gly100 N–O Phe442 E2), which also contact VHCDR2 (Supplementary Table 2) (Fig. 4b). In agreement with the cryo-EM structure, a previous mutagenesis study identified Leu441 and Phe442 as key residues for HC84.26.5D binding and HCV neutralization by this broadly neutralizing antibody28. Of note, neutralization of genotype 2b, where Phe442 is mutated to Leu442, was reduced 10-fold compared to other HCV genotypes.
Comparison of HC84.26.5D with other E2 front layer-specific neutralizing antibodies
Human neutralizing antibodies encoded by VH1-69 and rhesus neutralizing antibodies encoded by VH1.36, an ortholog of the human VH1-69 gene45, make similar overall footprints on E2 but differ in the position of their CDR loops on the antigen surface, as shown by a comparison of E2 complexes with HC84.26.5D, AR3X, HEPC3, 1382_01_H05, and RM2-01 (Figs. 3b and 4c)20,23,24,26. The binding mode of HC84.26.5D, in which hydrophobic tip of VHCDR2 interacts with the E2 front layer near Cys429 and helix α1, most closely resembles the binding modes of human neutralizing antibody 1382_01_H0524 and rhesus neutralizing antibody RM2-0123 (Fig. 4c). By contrast, VHCDR2 of AR3X and HEPC3 interacts with a hydrophobic pocket formed by front layer helices α1 and η2 and back layer helix α2 (Fig. 4c)20,26. This pocket is occupied by VHCDR3 of HC84.26.5D, whose short length (10 residues) compared to VHCDR3 of 1382_01_H05 and RM2-01 (17 and 15 residues, respectively) allows an angle of approach that orients VHCDR3 towards helix η2. In addition, VHCDR1 of HC84.26.5D interacts with the AS412 epitope and back layer residue Pro612, whereas VHCDR1 of 1382_01_H05 and RM2-01 contacts Trp529 at the tip of the CD81 binding loop. Interactions with E2 may stabilize VHCDR1 of HC84.26.5D, which is disordered in the crystal structure of the unbound antibody40.
Structural analyses of HCV E2 bound to different broadly neutralizing antibodies have shown that the E2 front layer can adopt at least two distinct conformations, designated A and B22. In the A conformation (e.g., in complexes with AR3X and HEPC3), large sections of the E2 back layer are obscured by the front layer, whereas in the B conformation (e.g., in complexes with 212.1.1 and HC1AM), back layer residues are more accessible for direct interaction with broadly neutralizing antibodies. E2 in complex with HC84.26.5D clearly adopts the A conformation in which front layer helix α1 has undergone a substantial movement relative to its position in the B conformation (Supplementary Fig. 9). The flexibility of the E2 front layer is further underscored by examination of the recent cryo-EM structure of the E1/E2 homodimeric complex46, which does not include bound antibodies. As illustrated in Supplementary Fig. 9, residues 426–459 of the E2 front layer adopt a conformation in the E1/E2 homodimeric complex that is distinct from the A and B conformations of E2 in antibody-bound structures22.
Structure of the CBH7–E2 complex identifies a new neutralizing epitope
CBH7 is a neutralizing antibody that targets antigenic domain C and blocks E2 binding to CD8130,35. To examine recognition of domain C by this antibody, we determined the cryo-EM structure of E2 bound to CBH7 to 3.3 Å resolution (Table 2) (Fig. 5a). Similar to the HC84.26.5D–E2 structure, we were able to model the entire E2 molecule in the CBH7–E2 complex, except hypervariable domain 1 (HVR1). In the CBH4B–E2 complex, antigenic site AS412 (residues Gln412–Arg424), part of the front layer (residues Thr425–Cys452), and most of the CD81 binding loop (residues Gly523–Val536) were disordered. Superposition of E2 in the CBH4B–E2 and HC84.26.5D–E2 structures gave an RMSD of 1.1 Å for 217 α-carbon atoms, while superposition of E2 in the CBH7–E2 and HC84.26.5D–E2 structures gave an RMSD of 1.1 Å for 166 α-carbon atoms. Thus, E2 adopts a similar fold in complexes with all three antibodies. The E2 front layer in the CBH7–E2 complex is in the A conformation, but is disordered in the CBH4B–E2 complex.

a Overall structure of the CBH7–E2 complex. E2, blue; H chain, dark gray, L chain, salmon. CH1/CL domains are not shown. VHCDR1, VHCDR2, and VHCDR3 are colored red, green, and blue, respectively. (top inset) β-strand 8 of E2 runs anti-parallel to VHCDR2 and is stabilized by four hydrogen bonds denoted by black dashed lines. Interacting residues are shown in stick representation. (middle inset) Interactions of VHCDR1 and VHCDR3 with E2. (bottom inset) Interactions of VHCDRs with hydrophobic surface on E2. b Superposition of E2 of the CBH7–E2 and CD81–E2 (7MWX)48 complexes to illustrate the retracted conformation of the AS412 epitope in CBH7-bound E2. Shifting AS412 towards the CD81 binding loop creates steric clashes with CD81. E2 is blue and magenta in the CBH7–E2 and CB81–E2 complexes, respectively; CD81 is green. c Overall structure of the CBH4B–E2 complex. E2, blue; H chain, pink; L chain, green. VHCDR1, VHCDR2, and VHCDR3 are colored red, green, and blue, respectively. (top inset) Hydrogen bond interactions of VHCDRs with the E2 back layer. (bottom inset) Interactions of VHCDRs with hydrophobic surface on E2. d Binding modes of CBH7 and CBH4B on E2 showing β-hairpin formed by β-strands 7 and 8 that separates antigenic domain A from antigenic domain C.
CBH7 buries 1192 Å2 of solvent-accessible surface on E2 that straddles both neutralizing and non-neutralizing faces, with 13 antibody residues contacting 21 E2 residues. The CBH7–E2 interface exhibits moderate shape complementarity, based on a shape correlation statistic (Sc)47 of 0.60 (Sc = 1.0 for interfaces with geometrically perfect fits) compared to 0.54 for the HC84.26.5D–E2 interface. The relatively short VHCDR3 (13 residues) of CBH7 plays a less dominant role in E2 recognition than the longer VHCDR3 of most other VH1-69 HCV antibodies, accounting for 25% (316 Å2) of total Fab buried surface area at the interface with E2 compared to 42% (462 Å2), 49% (497 Å2), 54% (416 Å2), 53% (556 Å2), and 39% (302 Å2) for 1198_05_G10 (20 residues), AR3B (19 residues), AR3C (18 residues), HEPC3 (17 residues), and 212.1.1 (16 residues), respectively. Similar to the HC84.26.5D–E2 complex, VL of CBH7 contributes significantly to contacts with E2 and accounts for 30% (377 Å2) of total Fab buried surface for CBH7 compared to 27% (332 Å2) for HC84.26.5D. Other VH1-69 antibodies contact E2 almost exclusively through VH20.
Structural studies have shown that the CD81 binding site of E2 comprises the AS412 epitope (residues 412–423), the front layer (residues 424–459), the CD81 binding loop (residues 519–535), and residues 616–617 of the back layer48. The CD81 binding loop transitions from a retracted to an extended conformation upon CD81–E2 complex formation. All the determined neutralizing antibody–E2 structures, including HC84.26.5D–E2 and CBH7–E2, have the CD81 binding loop in the retracted position8,20,21,22,23,24,25,26. Binding of CBH7 to E2 alters the conformation of the AS412 epitope from that found in the CD81–E2 complex, shifting it towards the CD81 binding loop and creating steric clashes with CD81 (Fig. 5b). Repositioning of the AS412 epitope is driven by hydrophobic contacts between VHCDR1 Tyr32 and AS412 Trp420 combined with a side-chain–side-chain hydrogen bond between VHCDR1 Asn31 and AS412 His421 (Supplementary Table 3) (Fig. 5a). Residues 56–59 of VHCDR2 form a β-strand that runs anti-parallel to β-strand 8 on the back layer of E2 (residues 637–644) in the CBH7–E2 complex. This anti-parallel arrangement is stabilized by four main-chain–main-chain hydrogen bonds (VHCDR2 Glu57 N–O Arg639 E2, VHCDR2 Glu57 O–N Arg639 E2, VHCDR2 Tyr59 N–O Glu637 E2, and VHCDR2 Tyr59 O–N Glu637 E2) and a salt bridge (VHCDR2 Glu57 Oε1–Nη1 Arg639 E2) (Supplementary Table 3) (Fig. 5a). VHCDR2 Phe54, located at the hydrophobic tip of the VHCDR2 loop characteristic of VH1-69-encoded neutralizing antibodies, interacts with a highly conserved hydrophobic patch formed by residues Tyr507, Pro513, and Val515 of the E2 β-sandwich core (Fig. 5a). VHCDR3 Tyr99 mediates additional hydrophobic interactions with this patch, while VHCDR3 Gly96, Tyr97, and Ile98 contact AS412 Trp420 and His421.
The epitope recognized by CBH7 is distinct from the epitopes recognized by HC84.26.5D and CBH4B, as illustrated in Fig. 6. It is also distinct from the epitopes described in previous structural studies of antibody–E28,9,20,21,22,23,24,25,26 or antibody–E1E249,50 complexes. The CBH7–E2 structure, therefore, defines a new neutralizing epitope on E2. Figure 6 shows the footprint on E2 of CBH7 and its relation to the footprints of HC84.26.5D and CBH4B. Whereas HC84.26.5D targets the E2 front layer (Fig. 3a) and CBH4B targets the back layer (Fig. 5c), CBH7 straddles both front and back layers, such that the CBH7 epitope is clearly distinct from the HC84.26.D and CBH4B epitopes, despite some overlap. HC84.26.5D is representative of neutralizing antibodies that target the E2 front layer, as is evident by comparing the footprints on E2 of HC84.26.5D, AR3X, HEPC3, RM2-01, and 1382_01_H05 (Fig. 3b). CBH4B is representative of non-neutralizing antibodies, such as 2A129, that target the E2 back layer (Fig. 7a) (see below).

Footprints of HC84.26.5D, CBH7, and CBH4B on E2 are colored according to epitope, with E2 depicted as a gray surface. Residues that interact exclusively with HC84.26.5D are cyan; residues that interact exclusively with CBH7 are pink; residues that interact exclusively with CBH4B are green. Residues interacting with both HC84.26.5D and CBH7 are yellow; residues interacting with both CBH4B and CBH7 are red.

a Crystal structures of 2A12–E2 (4WEB), E1–E2 (6WO5), and HEPC46–E2 (6MEJ) complexes and cryo-EM structure of CBH7–E2 complex. E1 is a non-neutralizing antibody, not the E1 envelope glycoprotein. The structures are superposed on E2 of the CBH4B–E2 complex to illustrate differences in the orientation of the non-neutralizing antibodies with respect to E2. b Axes of angles of approach of E1 (salmon), 2A12 (green), HEPC46 (maroon), and CBH4B (cyan) over E2 (blue). c Electrostatic surface potential map of E2 in the CBH4B–E2 and 2A12–E2 complexes shows that the VHCDR loops of 2A12 only interact with the back layer, whereas the VHCDR loops of CBH4B also contact pVR3. The back layer and pVR3 regions are outlined with black dashes. The electropositive patch on the back layer of E2 is labeled. VHCDR1, VHCDR2, and VHCDR3 are colored red, green, and blue, respectively. d Surface representation of E2 (magenta) in the HEPC46–E2 and E1–E2 complexes shows positioning of VHCDR loops. In E1, VHCDR1 contacts the CD81 binding loop (light blue), and VHCDR3 interacts with β-sandwich turn T542–N548 (orange), while in HEPC46, all VHCDRs anchor this β-sandwich turn. VHCDR1, VHCDR2, and VHCDR3 are colored red, green, and blue, respectively. e The tips of the VHCDR2 loops of E1 (salmon) and HEPC46 (maroon) interact with the same hydrophobic patch on E2 formed by β-sandwich residues P513–V516.
To better understand the newly defined CBH7 epitope, we used molecular dynamics (MD) simulations of glycosylated unbound E2 to assess the effects of selected E2 variants on epitope accessibility and mobility. We calculated epitope accessible surface area (ASA) and mobility (root-mean-square fluctuation; RMSF) for E2 with deleted HVR1 (ΔHVR1), E2 with deleted N-glycans 1, 4, and 6 (ΔN1/N4/N6), and a triple mutant representing E2 polymorphisms (I538V/Q546L/T563V), in comparison with wild-type 1b09 E2 (Supplementary Table 4). Each of the E2 variants has been previously shown to affect broadly neutralizing antibody neutralization, leading to increased antibody sensitivity (ΔHVR1, ΔN1/N4/N6)51,52 or increased antibody resistance (I538V/Q546L/T563V)53, although effects varied depending on the HCV isolate context and antibody. Average epitope surface accessibility increased for the E2 variants in comparison with the wild-type 1b09 for both probe sizes, with probes reflecting a water molecule (1.4 Å radius) or antibody CDR loops for a heavy or light chain V domain (10 Å radius). The largest epitope accessibility increases were observed for the E2 polymorphism triple mutant for both probe sizes, which increased the average accessible surface area by ~10–15%. Predicted epitope mobility was affected most for the ΔHVR1 mutant, which resulted in a ~30% reduction in average epitope residue mobility (1.39 Å RMSF, versus 1.99 Å average RMSF for wild-type). While these predicted epitope effects are intriguing, experimental studies to assess CBH7 binding and neutralization effects of these variants would be needed, as CBH7 was not tested with those variants previously51,52,53, and possible comparative MD simulations with longer time scales (to better predict larger-scale E2 motions) and full E1E2 context would additionally be useful.
Comparison of the crystal structure of unbound Fab CBH7 with the cryo-EM structure of Fab CBH7 bound to E2 showed small yet relevant differences in CDR loop conformation (Supplementary Fig. 10a). The RMSD for the superposition of V domains, based on 217 Cα atoms, is 0.8 Å, indicating that the overall topology of the bound and unbound states is conserved, with the exception of minor rotameric differences in the CDR loops. For example, upon complex formation, the side chains of VLCDR3 Phe94 and Leu95 reorient towards a hydrophobic groove on E2 formed by turns 510–513 and 544–546 of the central β-sandwich and the back layer 310 helix (residues 633–635). Notably, the inherent flexibility of the elbow region connecting the V and C domains permits changes in the rotational angles of the switch peptides between E2-bound and unbound CBH7, as calculated using DynDom41 (Supplementary Fig. 10b). In E2-bound CBH7, the rotational angles of the switch peptides around the H chain (residues 112–116) and the L chain (residues 105–106) are altered by 27° and 32°, respectively, relative to unbound CBH7 (Supplementary Fig. 10b). These rotations facilitate the movement of the V domains toward the antigen, resulting in elbow angle shifts from 133° to 166°, while maintaining the local conformations of the CDRs (Supplementary Fig. 10a, c, d).
Structure of the CBH4B–E2 complex
CBH4B is a non-neutralizing antibody targeting antigenic domain A of E2 that does not block E2 binding to CD8130,31,32,33,34. To examine recognition of domain A by this antibody, we determined the cryo-EM structure of E2 bound to CBH4B to 3.7 Å resolution (Table 3). CBH4B binds to the non-neutralizing face of E2 at a location distant from the CD81 binding site comprising the back layer (residues 597–646) and post-variable 3 region (pVR3; residues 581–596) (Fig. 5c). The CBH4B–E2 structure agrees with previous epitope mapping34. Surprisingly, antigenic site AS412 and the front layer, which are well defined in the CBH7–E2 and HC84.26.5D–E2 complexes, are partly disordered in the CBH4B–E2 complex. It may be that CBH4B binding to the back layer of E2 destabilizes the front layer. Alternatively, the front layer may be intrinsically flexible and require binding of antibodies like CBH7 or HC84.26.5D that target the neutralizing face of E2 for stabilization. Indeed, all the determined antibody–E28,20,21,22,23,24,25,26 and antibody–E1E249,50 structures in which the front layer and AS412 epitope are clearly defined involve antibodies bound to the neutralizing face.
The CBH4B–E2 interface exhibits moderate shape complementarity (Sc = 0.58)47, similar to the CBH7–E2 and HC84.26.5D–E2 interfaces (0.60 and 0.54, respectively). CBH4B buries 813 Å2 of solvent-accessible surface on E2, with VL contributing only 120 Å2 (15%). VHCDR2, which contacts pVR3, plays a dominant role in E2 recognition, accounting for 51% (416 Å2) of total Fab buried surface with E2 compared to only 30% (240 Å2) for the relatively long VHCDR3 (16 residues), which contacts the back layer. CBH4B forms a dense network of five hydrogen bonds with back layer residues Arg630, Tyr632, and Arg639: VHCDR3 Asp97 Oδ2–Nη1 Arg630 E2, VHCDR3 Asp97 Oδ2–Nη2 Arg630 E2, VHCDR3 Val100 N–O Arg630 E2, VHCDR3 Val100 O–N Tyr632 E2, and VHCDR1 Tyr32 Oη–Nη1 Arg639 E2 (Supplementary Table 5) (Fig. 5c). CBH4B contacts β-strand 7 on the back layer of E2 (residues 625–632), whereas CBH7 contacts β-strand 8 (residues 637–644). The β-hairpin formed by these two strands separates antigenic domain A (CBH4B) from antigenic domain C (CBH7) (Fig. 5d). In agreement with the CBH4B–E2 and CBH7–E2 structures, CBH4B does not interfere with HCV neutralization by CBH735. VHCDR2 Phe54, located at the tip of the VHCDR2 loop, interacts with a hydrophobic groove on the back layer of E2 formed by Leu603, Cys607, Val629, Ala642, and Cys644 (Fig. 5c). Collectively, these residues are only moderately conserved across HCV genotypes: 89% for Leu603, 100% for Cys607, 70% for Ile626, 56% for Phe627, 74% for Cys629, and 54% for Leu640.
Previous studies have used non-neutralizing antibodies directed against the central β-sandwich (HEPC46 and E1) or back layer (2A12) of E2 to facilitate crystallization of E2 bound to front layer-specific neutralizing antibodies8,9,20,22. The H chains of HEPC46 and 2A12 are encoded by the human IGHV1-18 and mouse IGHV14-3 genes, respectively, while the H chain of E1, like the H chains of CBH4B and CBH7, is encoded by the human VH1-69 gene. Although both CBH4B and 2A12 bind to the back layer, their different angles of approach result in different sets of interactions, such that CBH4B contacts pVR3 as well as the back layer, whereas 2A12 contacts only the back layer (Fig. 7a–c). Likewise, CBH7, E1, and HEPC46 bind distinct, but overlapping, epitopes on E2. E1 engages the CD81 binding loop via VHCDR1 and β-sandwich turn 542–548 via VHCDR321. This β-sandwich turn is anchored by all three VHCDR loops of HEPC46 (Fig. 7d). Interestingly, the tips of the VHCDR2 loops of E1 (Val54) and HEPC46 (Tyr53) interact with the same hydrophobic patch on E2 formed by residues 513–516 of the central β-sandwich as the tip of the VHCDR3 loop (Tyr99) of CBH7 (Fig. 7d).
Surprisingly, superposition of the CBH4B–E2 structure onto the existing E1E2 structures49,50 revealed a partial overlap of CBH4B with E1 (Supplementary Fig. 11a), even though this antibody binds HCV virions30,31,32,33,34. Moreover, CBH4B bound two different recombinant forms of E1E2, one soluble and the other membrane-bound, albeit with 240-fold lower affinity compared to E2 alone for soluble E1E2 (Kd = 19 nM versus 0.08 nM) and 40-fold lower affinity compared to E2 alone for membrane-bound E1E2 (Kd = 3.1 nM versus 0.08 nM) (Supplementary Fig. 11b). Such affinity reductions were not observed for other E2-specific antibodies tested, including HC84.26.5D. One interpretation of these results is that binding of CBH4B to E1E2 requires a significant rearrangement of E1 and E2 subunits from the arrangement seen in current E1E2 structures46,49,50, and that this rearrangement is associated with an energetic cost that lowers affinity. If so, this would imply that the E1E2 heterodimer can adopt multiple conformations, as demonstrated for E2 alone22.
To further examine E2 recognition by CBH4B, we calculated the mean Shannon sequence entropy across residues of the CBH4B epitope on E2, weighted by residue buried surface area, and compared it against the respective values for HC84.26.5D and CBH7. This analysis revealed that while all three epitopes have high residue conservation (>80% weighted mean), the CBH4B epitope is slightly more conserved than the HC84.26.5D and CBH7 epitopes, with a weighted mean sequence entropy of 13 and a weighted mean residue conservation of 86%. The HC84.26.5D epitope yielded weighted mean sequence entropy and conservation values of 20 and 82%, respectively, while for the CBH7 epitope these values were 17 and 84%, respectively. For comparison, the HEPC46 epitope is considerably more variable than the CBH4B, HC84.26.5D, or CBH7 epitopes, with mean sequence entropy and conservation values of 51 and 55%, respectively. As exemplified by CBH4B, targeting a conserved epitope is necessary but not sufficient for broad neutralization; non-neutralizing antibodies have been observed targeting conserved sites on other viruses, such as HIV gp41 and the SARS-CoV-2 fusion peptide54, and such antibodies can play a role in viral control outside of direct neutralization.
AlphaFold predictions of antibody–E2 complexes
We used the AlphaFold v2.3 deep learning modeling tool to predict the antigen-binding mode for antibodies HC84.26.5D, CBH7, and CBH4B, and compared the models to the experimentally determined cryo-EM structures. The AlphaFold models of the HC84.26.5D–E2 (Supplementary Fig. 12), CBH7–E2 (Supplementary Fig. 13), and CBH4B–E2 (Supplementary Fig. 14) complexes superimposed very well to the respective cryo-EM structures, and the AlphaFold predicted antigenic site is nearly identical. The models have relatively high AlphaFold confidence scores, 0.88, 0.91, and 0.85, for HC84.26.5D–E2, CBH7–E2, and CBH4B–E2, respectively, which is indicative of AlphaFold antibody–antigen modeling success based on previous benchmarking55, and all three modeled complexes have medium accuracy based on CAPRI criteria56 and ligand RMSDs of <4.0 Å (Supplementary Figs. 12–14).
Superposition of E2 from the AlphaFold models and cryo-EM for the HC84.26.5D–E2, CBH7–E2, and CBH4B–E2 complexes gives RMSDs of 1.12 Å, 1.41 Å, and 1.24 Å for 215, 202, and 169 Cα atoms, respectively. The predicted AlphaFold models for E2 in all the three complexes (Supplementary Figs. 12b, 13b, and 14b) recapitulate the α/β folding and overall architecture of the cryo-EM structures. However, structural alignments between cryo-EM and AlphaFold models of the CBH7–E2 complex revealed significant differences in the conformation of the E2 front layer (Thr425–Cys459) (Supplementary Fig. 13e). The predicted structure of the front layer in CBH7–E2 adopts a unique conformation of peptide downstream of helix α1 (Phe442–His445), where there is a turn at residues His434–Gly436 toward helix α2 (Arg614–His617), while in the cryo-EM structure the front layer is extended toward the CD81 binding loop (Thr519–Val536). This conformational variation in the front layer may be attributed to HC84.26.5D bound to the front layer of E2 in the cryo-EM HC84.26.5D–E2–CBH7 ternary complex structure, and no HC84.26.5D present in the CBH7–E2 complex AlphaFold model. In the HC84.26.5D–E2 and CBH7–E2 complexes, AlphaFold models have AS412 (Gln412–Asn423) in a β-hairpin conformation turned at residues Thr416–Ser419, as observed in E2412–424 peptide bound to HCV157, AP3358, 19B3, or 22D1159 (Supplementary Figs. 12c and 13e). In contrast, AS412 in the cryo-EM structures adopts extended coils with partially open hairpin conformations, containing a turn (HC84.26.5D–E2) that is also observed in E2412–424 bound with HC33.160 and 3/1161. In the cryo-EM HC84.26.5D–E2 structure, the N-terminal region of AS412 (Gln412–Asn415) forms a turn and moves toward the C-terminus of a proximal β-sandwich (Val516–Thr518). Superposition of VL/VH domains for the HC84.26.5D–E2, CBH7–E2, and CBH4B–E2 complexes gives RMSDs of 0.86 Å, 0.73 Å, and 1.00 Å for 199, 212, and 217 Cα atoms, respectively. Thus, the AlphaFold model for VL/VH is similar to the cryo-EM structures, and residues in the predicted structured region have high confidence scores (average pLDDT scores of VL/VH regions are above 92) that correlate with model accuracy. Buried surface area and antigen–antibody interaction analysis predict models of CDR loops of comparable quality to the cryo-EM structures, except the VHCDR1 loop (pLDDT = 85.6) in the HC84.26.5D–E2 AlphaFold model (Supplementary Fig. 12e), where the tip of VHCDR1 moves toward the β-hairpin turn (Thr416–Ser419). The extended conformation of AS412 in cryo-EM exposed the back layer, allowing VHHis29 to interact with Pro612 of the back layer and π–π stacking interaction with His412.
Discussion
We determined cryo-EM structures of a broadly neutralizing antibody (HC84.26.5D), a moderately neutralizing antibody (CBH7), and a non-neutralizing (CBH4B) antibody bound to HCV E2, and compared these structures with crystal structures of other antibody–E2 complexes8,9,20,21,22,23,24,25,26 and with models predicted by AlphaFold27. Whereas HC84.26.5D targeted the E2 front layer and CBH4B targeted the back layer, CBH7 straddled both front and back layers. The epitope recognized by CBH7, designated as antigenic domain C35, is clearly distinct from the epitopes recognized by HC84.26.5D and CBH4B (antigenic domains D and A, respectively)28,31, despite some overlap (Fig. 6). Some residues that are part of the HC84.26.5D epitope, 612 and 613, are located in the conventional usage of the back layer location but are clearly available on the surface by the binding of this antibody. This is consistent with a previous report of other broadly neutralizing antibody structures involving back-layer residues22. The CBH7 epitope, unlike the HC84.26.5D and CBH4B epitopes, was not defined in previous structural studies of antibody–E28,9,20,21,22,23,24,25,26 or antibody–E1E249,50 complexes and therefore extends our knowledge of antibody recognition of E2 to a new neutralizing epitope.
Although neutralizing antibodies have been the focus of most studies of antiviral immune responses, non-neutralizing antibodies have been shown to be protective against a number of enveloped viruses, including alphaviruses10,11, West Nile virus14, HIV-112, Crimean-Congo hemorrhagic fever virus15, Lassa fever virus13, and SARS-CoV-216. However, whereas neutralizing antibodies inhibit virus attachment and entry, non-neutralizing antibodies act through Fc effector functions. For example, antibody-mediated protection against Mayaro virus, an arthritogenic alphavirus, did not require direct neutralization as myeloid cells expressing Fcγ receptors could mediate viral clearance11. Similarly, activation of cellular effector functions by non-neutralizing antibodies was found to be critical for protection against Lassa fever virus13. In another study, non-neutralizing antibodies elicited by mRNA vaccination protected against lethal SARS-CoV-2 infection via Fc-mediated effector functions16. Whether non-neutralizing anti-E2 antibodies such as CBH4B can protect against HCV infection in vivo has not been investigated. On the other hand, there is some evidence that non-neutralizing antibodies may exacerbate HCV infection by negatively modulating neutralizing antibodies17,18,19. In addition, non-neutralizing antibodies such as CBH4B fall within antigenic domain A30,31,32,33,34, which is highly immunogenic and may serve as a decoy to deflect the immune response from less immunogenic regions such as antigenic domains B and D that mediate virus neutralization.
Structure determination of small proteins (<70 kDa) by cryo-EM represents a considerable challenge, even with recent technical advances such as Volta phase plates. Whereas large proteins usually have sufficient shape features for image alignment, small proteins often lack recognizable features to facilitate particle selection and orientation for image processing. Conventional (i.e., monomeric) Fabs (~50 kDa) can serve as fiducial markers by increasing particle size and assisting in orientation62,63,64. However, the inherent flexibility of the elbow regions connecting V and C domains may reduce the ability of Fabs to orient particles accurately38. One approach to overcoming this problem is engineering the elbow region of the H chain to lock the Fab in a defined conformation65. In another approach, target-binding camelid antibodies (nanobodies) were rigidly attached to two scaffolds: (1) the Fab of an antibody specific for the nanobody and (2) a nanobody-binding protein A fragment fused to maltose-binding protein66. These fiducial markers of ~120 kDa, called Legobodies, were used to determine cryo-EM structures of proteins of ~25 kDa.
We previously engineered a dimeric bivalent Fab for use as a fiducial marker to determine the cryo-EM structure of the immune checkpoint receptor LAG3 (~50 kDa)39. This dimeric Fab doubled the effective size of its complex with LAG3 and conferred a highly identifiable doughnut-like shape to the complex. The application of this method to Fab–E2 complexes demonstrates the general utility of bivalent Fabs as fiducial markers for cryo-EM analysis of small proteins. In addition, we have developed robust design principles for constructing bivalent Fabs for different antibodies39,40. Dimerization of Fabs can be reliably induced by deleting one (VHSer113), two (VH112Ser and VH113Ser or CH114Ala and CH115Ser), three (VH112Ser–CH114Ala), or four (VH112Ser–CH115Ser) residues in the elbow region linking the VH and CH1 domains, depending on the antibody. Thus, deletion of a single residue (VHSer113) sufficed for dimerization of Fab HC84.26.5D, whereas dimerization of Fab CBH4B required deletion of four residues (VH112Ser–CH114Ala). Importantly, all elbow region residues involved in deletions to create dimeric Fabs are either invariant (CH114Ala and CH115Ser) or highly conserved (VH112 and VH113) in antibody sequences and are located at the opposite end of the VH domain from the antigen-binding CDR loops.
Legobodies and other nanobody-based fiducial markers must be isolated on a case-by-case basis from phage or yeast display libraries or from immunized camelids67,68, which can present challenges for broad application of nanobodies to structure determination. By comparison, thousands of conventional antibodies targeting thousands of proteins are already available, particularly against targets of biomedical interest such as viral glycoproteins. For example, the ABCD (AntiBodies Chemically Defined) database (https://web.expasy.org/abcd/) contains 26,410 sequenced antibodies recognizing 4292 different targets (January 2024).
The success of AlphaFold27 in modeling the CBH7 and CBH4B interactions with E2 provides additional support for its utility in predicting some unseen antibody–antigen complex structures. While AlphaFold’s training set includes structures from the PDB released through September 2021, and thus would possibly include the previously described HC84.26.5D–E2434–446 peptide complex29 as well as a number of front layer-specific antibody–E2 structures, no structures from that potential set appear to reflect the binding modes or key elements of the CBH7–E2 or CBH4B–E2 complexes (e.g., key CDR loop interactions with E2). However, recent AlphaFold benchmarking55 and results from a recent community CASP/CAPRI assessment56 demonstrate that there is room for improvement, and a sizable proportion of antibody–antigen complexes are unable to be modeled correctly by AlphaFold2. The recently released AlphaFold3 method reportedly improves success for antibody–antigen modeling versus AlphaFold2, although its capability to generate high-accuracy models still appears to be limited69. Nevertheless, AlphaFold model confidence scores are indicators of accuracy for this class of interfaces55, as seen in the high scores for the well-modeled complexes in this study, thus enabling researchers to prioritize likely accurate models versus likely incorrect ones for subsequent experimental validation and hypothesis testing, including for antibody complexes with HCV E2 and E1E2.
Methods
Protein expression and purification
To produce monovalent or bivalent Fab fragments, codon-optimized genes encoding the VH and CH1 domains of the H chain and the VL and CL domains of the L chain of HC84.26.5D, CBH7, and CBH4B were synthesized chemically (GeneScript) and cloned into the mammalian expression vector pcDNA 3.4-TOPO. For secretion, we used signal sequences from the corresponding germline VH and VL genes in the IMGT database (http://www.imgt.org/). A streptavidin tag (WSHPQFEK) was attached to the C-terminus of the CH1 domains for affinity purification. The gene encoding the VHCH1 region of bivalent Fab HC84.26.5D contained a deletion of one amino acid, VHSer113 (Kabat numbering), in the elbow region between VH and CH1 compared to the wild-type monovalent Fab40. The gene encoding the VHCH1 region of bivalent CBH4B contained a deletion of four amino acids (VHSer112–CHSer115) in the elbow. Fab CBH7 was produced only in monovalent form. Both bivalent and monovalent Fabs were expressed by co-transfecting equimolar amounts of H and L chain plasmids into Expi293 with Expifectamine (Thermo Fisher). After 72 h incubation, cultures were harvested by centrifugation, and supernatants were dialyzed against 100 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 1 mM EDTA. Recombinant Fabs were purified from culture supernatants using consecutive streptavidin affinity (IBA Lifesciences) and Superdex 200 (GE Healthcare) columns. A codon-optimized gene encoding the full-length E2 ectodomain from HCV strain 1b09 (residues 384–643) was synthesized chemically (GeneScript) and cloned into pcDNA 3.4-TOPO with an N-terminal immunoglobulin κ signal sequence for secretion and a C-terminal His6 tag for affinity purification. Soluble E2 was purified from culture supernatants of transfected Expi293 cells using sequential HisTrap Ni2+-NTA (GE Healthcare) and Superdex 200 columns as described70. Dimeric Fab HC84.26.5D–E2, Fab CBH4B–E2, and Fab HC84.26.5D–E2–Fab CBH7 complexes were purified for cryo-EM with a Superdex 200 column from equimolar mixtures of Fab and E2 proteins.
Crystallization, data collection, and structure determination of Fab CBH7
Crystals of monomeric Fab CBH7 (8 mg/ml) were grown at 20 °C by vapor diffusion in sitting drops in 100 mM sodium acetate (pH 4.5) and 25% (w/v) polyethylene glycol 3350. For data collection, crystals were cryoprotected with 20% (w/v) glycerol, flash-cooled, and stored in liquid nitrogen. X-ray diffraction data were collected at beamline 23-ID-B of the Advanced Photon Source, Argonne National Laboratory. Diffraction data were indexed, integrated, and scaled using the program HKL300071. Initial phases were obtained by molecular replacement with the program Phaser72 using E2-specific Fab HEPC3 (PDB accession code 6MED)20 as a search model. Manual model building and corrections were performed using Coot73 with iterative cycles of refinement using REFMAC574. Water molecules were added manually at 1σ contour level for 2FO−FC and at 3σ contour level for FO−FC in the electron density maps. The final model was validated with PROCHECK75 and MolProbity76. Data collection, processing, and refinement statistics are summarized in Table 4.
Cryo-EM sample preparation and data acquisition
For each complex (Fab HC84.26.5D–E2, Fab CBH4B–E2, and Fab HC84.26.5D–E2–Fab CBH7), 3 μl of protein was applied to the carbon side of Quantifoil R1.2/1.3 (300 mesh) glow-discharged (PELCO easiGlow) grids. The grids were blotted for 2.5–5.5 s with a wait time of 5–10 s and vitrified by plunging into liquid ethane using an FEI Vitrobot Mark IV set at 4 °C and 100% humidity. The grids were transferred into grid boxes and stored in liquid nitrogen for cryo-EM imaging. Grids were screened for quality using an in-house Thermo Fisher Scientific (TFS) 200 kV Talos Arctica transmission electron microscope (TEM) equipped with a Falcon 3EC direct electron detector (DED). High-quality grids were imaged using a TFS 300 kV Titan Krios microscope equipped with a Gatan K3 DED in counting mode at the National Cryo-Electron Microscopy Facility (NCEF) in Frederick, MD. Automated data collection was performed at a magnification of 81,000X over a defocus range of −1.0 to −2.5 μm with a total dose of 50.0 e−/Å2 fractionated over 40 raw frames. For the Fab HC84.26–E2 complex, 9653 movies were collected with pixel size, dose rate, and exposure time of 1.12 Å/pixel, 16.54 e−/pixel/sec, and 3.8 s, respectively. For the Fab CBH4B–E2 complex, 7168 movies were collected with pixel size, dose rate, and exposure time of 1.17 Å/pixel, 18.04 e−/pixel/sec, and 3.5 s, respectively. For the Fab HC84.26.5D–E2–Fab CBH7 complex, 7058 movies were collected with pixel size, dose rate, and exposure time of 1.12 Å/pixel, 17.4 e−/pixel/sec, and 3.6 s, respectively. Data for all the complexes were processed with cryoSPARC v477.
Single-particle cryo-EM image processing
For the Fab HC84.26.5D–E2 complex, data were motion corrected using patch motion correction, and CTF information was estimated using patch CTF estimation. Low-quality micrographs were eliminated based on relative ice thickness, total full frame motion, and CTF fit resolution worse than 6 Å. To generate initial templates, 500 micrographs were used for blob picking with a blob size of 100–200 Å diameter. 2D class averages were generated after extracting the particles with a 320 pixel box and a binning factor of 2 (a pixel size of 2.24 Å). Unique 2D class averages with prominent protein features were used as templates for further particle picking from all high-quality micrographs. Template picking yielded 2,422,548 particles extracted with a 320 pixel box and a binning factor of 2 (a pixel size of 2.24 Å). These particles were subjected to iterative rounds of 2D classification to eliminate contaminants and damaged particles. After multiple rounds of 2D classification, 695,972 cleaned particles were extracted at full resolution and used for ab initio reconstruction (K = 5) with no symmetry (C1) and C2 symmetry independently, and an initial alignment resolution of 8 Å. In C1 and C2 symmetry, the best resolved classes with 180,188 and 357,752 particles, respectively, were subjected to heterogeneous refinement followed by non-uniform and CTF refinement to yield cryo-EM maps at resolutions of 3.8 Å and 3.5 Å, respectively, based on the gold standard Fourier shell correlation (GSFSC) of with a criterion of 0.143. In the C2 symmetry map, certain loops and side chains were missing, which reappeared after relaxing the symmetry from C2 to C1, resulting in a resolution of 3.7 Å determined by GSFSC curve with a threshold of 0.143. The final reconstruction of the Fab HC84.26.5D–E2 complex identified as a superassembly with four E2 molecules bound to four domain-swapped Fab HC84.26.5D molecules (Supplementary Fig. 1). To delineate Fab HC84.26.5D–E2 interactions, the best resolvable interface analyzed by local resolution estimation was subjected to local refinement using a soft mask (dilation radius = 4, soft padding width = 2) covering one E2 and one Fab HC84.26.5D molecule on two independent 3D reconstructions. The final maps for two reconstructions estimated at 3.8 and 3.7 Å based on GSFSC curves of 0.143 criterion after post-processing had B factors of −128 and −119 Å2. Data processing details are summarized in Supplementary Fig. 2.
For the Fab CBH4B–E2 and Fab HC84.26.5D–E2–Fab CBH7 complexes, the same methods were followed for template picking and particle cleaning as described above. Template picking yield 4,606,895 (Fab CBH4B–E2) and 3,265,955 (Fab HC84.26.5D–E2–Fab CBH7) particles. 2D and 3D classification were carried out to select consistent particle classes. Finally, 200,763 (Fab CBH4B–E2) and 165,691 (Fab HC84.26.5D–E2–Fab CBH7) particles were selected for 3D refinements. Reported resolution maps at 3.7 Å (Fab CBH4B–E2) and 3.3 Å (Fab HC84.26.5D–E2–Fab CBH7) were generated after post-processing with B factors of −137 Å2 and −79 Å2, respectively, based on GSFSC curve of 0.143 criterion. The final reconstruction map of the Fab CBH4B–E2 complex showed density features for only one E2 molecule (Fig. 1d). The Fab CBH4B–E2 interface was locally refined by generating a soft mask (dilation radius = 4, soft padding width = 2) covering E2, VL, and VHCH1. The final reconstruction map estimated at 3.4 Å resolution based on GSFSC curve of 0.143 criterion after post-processing had a B factor of −126 Å2. Data processing details are summarized in Supplementary Figs. 3 and 4.
Model building and refinement
Crystal structures of dimeric Fab HC84.26.5D (PDB code 6X9X)40, HCV E2 (6MEI)20, and Fab CBH7 (8TFE) were used as initial models and docked into the EM maps of the Fab HC84.26.5D–E2, Fab CBH4B–E2, and Fab HC84.26.5D–E2–Fab CBH7 complexes using UCSF Chimera (https://www.cgl.ucsf.edu/chimera/). The models were manually fitted in Coot73 and subjected to iterative real-space refinement using PHENIX with secondary structure and geometry restraints. The MolProbity server76 was used to identify geometrical outliers and problematic regions in the model, which were manually corrected in Coot73. Molecular dynamics flexible fitting (MDFF) was performed to improve the overall refinement statistics and minimize clashes. Parameters for cryo-EM data collection and modeling statistics are summarized in Tables 1–3. Contact residues were identified with the CONTACT program78 and were identified as residues containing an atom 4.0 Å or less from a residue of the binding partner. Structural alignments and superposition were performed with PyMOL (http://www.pymol.org/pymol). Figures were created using PyMOL, UCSF Chimera (https://www.cgl.ucsf.edu/chimera/), and UCSF ChimeraX (https://www.cgl.ucsf.edu/chimerax/). Figures were labeled with Adobe Illustrator and Microsoft PowerPoint.
Biolayer interferometry
The binding affinity of Fab fragments for soluble HCV E2 was assessed using an Octet RED96 system (Pall ForteBio) with Ni2+-NTA biosensors. Biosensors were first loaded with 2 µg/ml of purified His6-tagged E2 for 600 s, followed by stabilization using a cross-linker mixture containing 0.1 M 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide and 0.025 M N-hydroxysuccinimide for 60 s. The reaction was subsequently quenched with 1 M ethanolamine (pH 8.0) for 60 s. Fab binding kinetics were determined by monitoring association for 300 s and dissociation for 300 s across a two-fold serial dilution series of each Fab. Kinetic parameters, including the equilibrium dissociation constant (KD), were derived by globally fitting association and dissociation curves using Octet Data Analysis 10.0 software.
Epitope conservation and entropy analysis
The 1b09 E2 sequence was aligned to a set of pre-aligned HCV reference E2 sequences downloaded from the Los Alamos National Laboratory (LANL) HCV database79 (N = 187 sequences) using the MAFFT multiple sequence alignment program80. This was used to calculate residue conservation and Shannon sequence entropy for each 1b09 E2 position. Entropy values, calculated in units of nats, were scaled to a 1–100 range based on a maximum entropy of 2.4 nats, which corresponds approximately to the highest observed values across the E2 positions. Weighted averages of epitope residue conservation and entropy values were calculated using residue-level interface buried surface area values calculated by PDBePISA (https://www.ebi.ac.uk/pdbe/pisa/)42. Each residue’s conservation or entropy value was weighted by its fractional contribution to the total interface buried surface area.
Molecular dynamics simulations
MD simulations were performed using NAMD 3.081 and the CHARMM36m force field82, with HCV strain 1b09 E2 (residues 384–643) from the CBH7-bound structure used as input for simulations. AlphaFold227 in the ColabFold framework83 was used to model structurally unresolved E2 residues, including HVR1, using the CBH7-bound cryo-EM E2 coordinates from this study as a ColabFold custom input template. N-glycans with the mannose (Man3GlcNAc2) glycoform, which was noted previously to be a common component of mammalian expressed E2 N-glycans70, were modeled onto all E2 N-glycosylation sites using a previously described glycan building and refinement protocol in Rosetta84. E2 variant input structures were generated through modifications of the wild-type 1b09 E2 input coordinates to remove residues, remove N-glycans, or mutate residues. MD simulation inputs were prepared with CHARMM-GUI85, solvated in a periodic octahedral box with TIP3P water molecules and 0.15 M NaCl ions placed with Monte Carlo sampling, and hydrogen mass repartitioning was applied. Following initial minimization and equilibration, all systems were run for 500 ns using 4 fs timesteps at 310 °K, constraining hydrogen-containing bonds with the SHAKE/RATTLE algorithm. This was performed for six independent replicates for each E2 (wild-type or variant) input. Residue surface accessibility calculations were performed with 600 representative structures from each E2 (100 structures sampled evenly from each E2 replicate trajectory), using Rosetta (ref. 86) for residue surface accessibility calculations, as used previously with glycosylated E2 models70.
Structure prediction
Structural modeling of antibody–E2 complexes was performed using locally installed AlphaFold227 (v2.3), with default parameters. Individual chain structural templates available in the PDB as of the running date (May 2023) were permitted, and only variable domains were modeled for antibodies. Twenty-five models were generated per complex, and the complexes were ranked by AlphaFold2 multimer confidence score, which is a combination of pTM and interface pTM (ipTM) confidence scores (0.2 × ipTM + 0.8 × ipTM)87. The top-ranked model for each complex was selected for analysis.
Data availability
X-ray crystallographic data have been deposited in the Protein Data Bank (PDB) under accession code 8TFE. Cryo-EM data have been deposited in the PDB under accession codes 8TGV, 8TGZ, 8U9Y, 8THZ, 8TZY, and 8TXQ and the Electron Microscopy Data Bank (EMDB) under accession codes 41245, 41247, 42041, 41275, 41774, and 41703. Molecular dynamics simulation files have been deposited in Zenodo (https://zenodo.org/records/15353525). Numerical source data for Supplementary Fig. 5 are in Supplementary Data 1.
References
Global Hepatitis Report https://www.globalhep.org/sites/default/files/content/resources/files/2024-04/2024%20Global%20Hepatitis%20Report%20-%20WHO.pdf (2024).
Feeney, E. R. & Chung, R. T. Antiviral treatment of hepatitis C. BMJ 348, g3308 (2014).
Rosen, H. R. “HepC, where art thou?”: What are the remaining (fundable) questions in hepatitis C virus research? Hepatology 65, 341–349 (2017).
Micallef, J. M., Kaldor, J. M. & Dore, G. J. Spontaneous viral clearance following acute hepatitis C infection: a systematic review of longitudinal studies. J. Viral Hepat. 13, 34–41 (2006).
Osburn, W. O. et al. Clearance of hepatitis C infection is associated with the early appearance of broad neutralizing antibody responses. Hepatology 59, 2140–2151 (2014).
Bailey, J. R. et al. Broadly neutralizing antibodies with few somatic mutations and hepatitis C virus clearance. JCI Insight 2, e92872 (2017).
Zeisel, M. B., Fofana, I., Fafi-Kremer, S. & Baumert, T. F. Hepatitis C virus entry into hepatocytes: molecular mechanisms and targets for antiviral therapies. J. Hepatol. 54, 566–576 (2011).
Kong, L. et al. Hepatitis C virus E2 envelope glycoprotein core structure. Science 342, 1090–1094 (2013).
Khan, A. G. et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 509, 381–384 (2014).
Schmaljohn, A. L., Johnson, E. D., Dalrymple, J. M. & Cole, G. A. Non-neutralizing monoclonal antibodies can prevent lethal alphavirus encephalitis. Nature 297, 70–72 (1982).
Earnest, J. T. et al. The mechanistic basis of protection by non-neutralizing anti-alphavirus antibodies. Cell Rep. 35, 108962 (2021).
Horwitz, J. A. et al. Non-neutralizing antibodies alter the course of HIV-1 infection in vivo. Cell 170, 637–648 (2017).
Abreu-Mota, T. et al. Nonneutralizing antibodies elicited by recombinant Lassa-Rabies vaccine are critical for protection against Lassa fever. Nat. Commun. 9, 4223 (2018).
Chung, K. M. et al. Antibodies against West Nile Virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J. Virol. 80, 1340–1351 (2006).
Durie, I. A. et al. Structural characterization of protective non-neutralizing antibodies targeting Crimean-Congo hemorrhagic fever virus. Nat. Commun. 13, 7298 (2022).
Clark, J. J. et al. Protective effect and molecular mechanisms of human non-neutralizing cross-reactive spike antibodies elicited by SARS-CoV-2 mRNA vaccination. Cell Rep. 43, 114922 (2024).
Meyer, K., Ait-Goughoulte, M., Keck, Z. Y., Foung, S. & Ray, R. Antibody-dependent enhancement of hepatitis C virus infection. J. Virol. 82, 2140–2149 (2008).
Tarr, A. W. et al. Hepatitis C patient-derived glycoproteins exhibit marked differences in susceptibility to serum neutralizing antibodies: genetic subtype defines antigenic but not neutralization serotype. J. Virol. 85, 4246–4257 (2011).
Tarr, A. W. et al. Genetic diversity underlying the envelope glycoproteins of hepatitis C virus: structural and functional consequences and the implications for vaccine design. Viruses 7, 3995–4046 (2015).
Flyak, A. I. et al. HCV broadly neutralizing antibodies use a CDRH3 disulfide motif to recognize an E2 glycoprotein site that can be targeted for vaccine design. Cell Host Microbe 24, 703–716 (2018).
Tzarum, N. et al. Genetic and structural insights into broad neutralization of hepatitis C virus by human VH1-69 antibodies. Sci. Adv. 5, eaav1882 (2019).
Tzarum, N. et al. An alternate conformation of HCV E2 neutralizing face as an additional vaccine target. Sci. Adv. 6, eabb5642 (2020).
Chen, F. et al. Functional convergence of a germline-encoded neutralizing antibody response in rhesus macaques immunized with HCV envelope glycoproteins. Immunity 54, 781–796 (2021).
Weber, T. et al. Analysis of antibodies from HCV elite neutralizers identifies genetic determinants of broad neutralization. Immunity 55, 341–354 (2022).
Ogega, C. O. et al. Convergent evolution and targeting of diverse E2 epitopes by human broadly neutralizing antibodies are associated with HCV clearance. Immunity 57, 890–903 (2024).
Flyak, A. I. et al. An ultralong CDRH2 in HCV neutralizing antibody demonstrates structural plasticity of antibodies against E2 glycoprotein. eLife 9, e53169 (2020).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
Keck, Z. Y. et al. Human monoclonal antibodies to a novel cluster of conformational epitopes on HCV E2 with resistance to neutralization escape in a genotype 2a isolate. PLoS Pathog. 8, e1002653 (2012).
Keck, Z. Y. et al. Affinity maturation of a broadly neutralizing human monoclonal antibody that prevents acute hepatitis C virus infection in mice. Hepatology 64, 1922–1933 (2016).
Hadlock, K. G. et al. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J. Virol. 74, 10407–10416 (2000).
Keck, Z. Y. et al. Definition of a conserved immunodominant domain on hepatitis C virus E2 glycoprotein by neutralizing human monoclonal antibodies. J. Virol. 82, 6061–6066 (2008).
Pierce, B. G. et al. Global mapping of antibody recognition of the hepatitis C virus E2 glycoprotein: Implications for vaccine design. Proc. Natl. Acad. Sci USA 113, E6946–E6954 (2016).
Keck, Z. Y. et al. Immunogenic and functional organization of hepatitis C virus (HCV) glycoprotein E2 on infectious HCV virions. J. Virol. 81, 1043–1047 (2007).
Sevvana, M., Keck, Z., Foung, S. K. & Kuhn, R. J. Structural perspectives on HCV humoral immune evasion mechanisms. Curr. Opin. Virol. 49, 92–101 (2021).
Keck, Z. Y. et al. Analysis of a highly flexible conformational immunogenic domain in hepatitis C virus E2. J. Virol. 79, 13199–13208 (2005).
Keck, Z. Y. et al. A point mutation leading to hepatitis C virus escape from neutralization by a monoclonal antibody to a conserved conformational epitope. J. Virol. 82, 6067–6072 (2008).
Owsianka, A. M. et al. Broadly neutralizing human monoclonal antibodies to the hepatitis C virus E2 glycoprotein. J. Gen. Virol. 89, 653–659 (2008).
Wu, S. et al. Fabs enable single particle cryoEM studies of small proteins. Structure 20, 582–592 (2012).
Mishra, A. K. et al. CryoEM structure of a therapeutic antibody (favezelimab) bound to human LAG3 determined using a bivalent Fab as fiducial marker. Structure 31, 1149–1157 (2023).
Shahid, S. et al. Crystal structure of a bivalent antibody Fab fragment. J. Mol. Biol. 433, 166714 (2021).
Lee, R. A., Razaz, M. & Hayward, S. The DynDom database of protein domain motions. Bioinformatics 19, 1290–1291 (2003).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Lesk, A. M. & Chothia, C. Elbow motion in the immunoglobulins involves a molecular ball-and-socket joint. Nature 335, 188–190 (1988).
Chen, F., Tzarum, N., Wilson, I. A. & Law, M. VH1-69 antiviral broadly neutralizing antibodies: genetics, structures, and relevance to rational vaccine design. Curr. Opin. Virol. 34, 149–159 (2019).
Chen, F. et al. Antibody responses to immunization with HCV envelope glycoproteins as a baseline for B-cell-based vaccine development. Gastroenterology 158, 1058–1071 (2020).
Augestad, E. H. et al. The hepatitis C virus envelope protein complex is a dimer of heterodimers. Nature 633, 704–709 (2024).
Lawrence, M. C. & Colman, P. M. Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 (1993).
Kumar, A. et al. Structural insights into hepatitis C virus receptor binding and entry. Nature 598, 521–525 (2021).
Torrents de la Pena, A. et al. Structure of the hepatitis C virus E1E2 glycoprotein complex. Science 378, 263–269 (2022).
Metcalf, M. C. et al. Structure of engineered hepatitis C virus E1E2 ectodomain in complex with neutralizing antibodies. Nat. Commun. 14, 3980 (2023).
Prentoe, J. et al. Hypervariable region 1 and N-linked glycans of hepatitis C regulate virion neutralization by modulating envelope conformations. Proc. Natl. Acad. Sci. USA 116, 10039–10047 (2019).
Prentoe, J., Velazquez-Moctezuma, R., Foung, S. K., Law, M. & Bukh, J. Hypervariable region 1 shielding of hepatitis C virus is a main contributor to genotypic differences in neutralization sensitivity. Hepatology 64, 1881–1892 (2016). (2016).
Bailey, J. R. et al. Naturally selected hepatitis C virus polymorphisms confer broad neutralizing antibody resistance. J. Clin. Investig. 125, 437–447 (2015).
Dacon, C. et al. Broadly neutralizing antibodies target the coronavirus fusion peptide. Science 377, 728–735 (2022).
Yin, R. & Pierce, B. G. Evaluation of AlphaFold antibody-antigen modeling with implications for improving predictive accuracy. Protein Sci. 33, e4865 (2024).
Lensink, M. F. et al. Impact of AlphaFold on structure prediction of protein complexes: the CASP15-CAPRI experiment. Proteins 91, 1658–1683 (2023).
Kong, L. et al. Structural basis of hepatitis C virus neutralization by broadly neutralizing antibody HCV1. Proc. Natl. Acad. Sci. USA 109, 9499–9504 (2012).
Potter, J. A. et al. Toward a hepatitis C virus vaccine: the structural basis of hepatitis C virus neutralization by AP33, a broadly neutralizing antibody. J. Virol. 86, 12923–12932 (2012).
Aleman, F. et al. Immunogenetic and structural analysis of a class of HCV broadly neutralizing antibodies and their precursors. Proc. Natl. Acad. Sci. USA 115, 7569–7574 (2018).
Li, Y. et al. Structural basis for penetration of the glycan shield of hepatitis C virus E2 glycoprotein by a broadly neutralizing human antibody. J. Biol. Chem. 290, 10117–10125 (2015).
Meola, A. et al. Structural flexibility of a conserved antigenic region in hepatitis C virus glycoprotein E2 recognized by broadly neutralizing antibodies. J. Virol. 89, 2170–2181 (2015).
Lee, Y. et al. Cryo-EM structure of the human L-type amino acid transporter 1 in complex with glycoprotein CD98hc. Nat. Struct. Mol. Biol. 26, 510–517 (2019).
Bloch, J. S. et al. Structure and mechanism of the ER-based glucosyltransferase ALG6. Nature 579, 443–447 (2020).
Kim, J. et al. Structure and drug resistance of the Plasmodium falciparum transporter PfCRT. Nature 576, 315–320 (2019).
Bailey, L. J. et al. Locking the elbow: improved antibody Fab fragments as chaperones for structure determination. J. Mol. Biol. 430, 337–347 (2018).
Wu, X. & Rapoport, T. A. Cryo-EM structure determination of small proteins by nanobody-binding scaffolds (Legobodies). Proc. Natl. Acad. Sci. USA 118, e2115001118 (2021).
Zimmermann, I. et al. Synthetic single domain antibodies for the conformational trapping of membrane proteins. eLife 7, e34317 (2018).
McMahon, C. et al. Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 (2018).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature 630, 493–500 (2024).
Urbanowicz, R. A. et al. Antigenicity and immunogenicity of differentially glycosylated hepatitis C virus E2 envelope proteins expressed in mammalian and insect cells. J. Virol. 93, e01403–e01418 (2019).
Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. HKL-3000: the integration of data reduction and structure solution-from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 (2006).
Storoni, L. C., McCoy, A. J. & Read, R. J. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 60, 432–438 (2004).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Murshudov, G. N. et al. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26, 283–291 (1993).
Williams, C. J. et al. MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 (2018).
Punjani, A., Rubinstein, J. L., Fleet, D. J. & Brubaker, M. A. cryoSPARC: algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017).
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 (1994).
Kuiken, C., Yusim, K., Boykin, L. & Richardson, R. The Los Alamos hepatitis C sequence database. Bioinformatics 21, 379–384 (2005).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Phillips, J. C. et al. Scalable molecular dynamics on CPU and GPU architectures with NAMD. J. Chem. Phys. 153, 044130 (2020).
Huang, J. et al. CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 (2017).
Mirdita, M. et al. ColabFold: making protein folding accessible to all. Nat. Methods 19, 679–682 (2022).
Adolf-Bryfogle, J. et al. Growing glycans in Rosetta: accurate de novo glycan modeling, density fitting, and rational sequon design. PLoS Comput. Biol. 20, e1011895 (2024).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: a web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Leman, J. K. et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods 17, 665–680 (2020).
Evans, R. et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021.2010.2004.463034 (2021).
Acknowledgements
This work was supported by National Institutes of Health Grant AI168048 to R.A.M., B.G.P., E.A.T., and T.R.F., GM144083 to B.G.P., and U19 AI159840 to S.H.H.F. X-ray diffraction data were collected at the GM/CA beamline at the Advanced Photon Source of Argonne National Laboratory, which is funded by the National Cancer Institute (ACB-12002) and the National Institute of General Medical Sciences (AGM-12006, P30GM138396). Cryo-EM data were collected at the National Cryo-EM Facility (NCEF) at the Frederick National Laboratory for Cancer Research under contract HSSN261200800001E. We thank Adam Wier and Tara Fox (NCEF) for cryo-EM data collection. We also thank Edwin Pozharski (University of Maryland) for advice on cryo-EM image analysis. S.S.H. acknowledges support from the University of Maryland School of Medicine (UMSOM), UMSOM Center for Biomolecular Therapeutics, and the University of Maryland Marlene and Stewart Greenebaum Cancer Center. High-performance computing resources from the University of Maryland Zaratan cluster were used in this study.
Author information
Authors and Affiliations
Contributions
S.S., S.S.K., R.Y., L.J., Y.L., N.F., L.K., E.A.T., and Z.K. performed the experiments and data analyses. S.S.H., S.K.H.F, T.R.F, B.G.P., and R.A.M. supervised the project. All authors prepared the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Alba Torrents de la Peña, Alexey Rak, and the other, anonymous, reviewer for their contribution to the peer review of this work. Primary Handling Editors: Laura Rodríguez Pérez. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shahid, S., Karade, S.S., Hasan, S.S. et al. Cryo-EM structures of HCV E2 glycoprotein bound to neutralizing and non-neutralizing antibodies determined using bivalent Fabs as fiducial markers. Commun Biol 8, 825 (2025). https://doi.org/10.1038/s42003-025-08239-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08239-w
