
Abstract
Cathepsin B (CatB), a protease in endosomal and lysosomal compartments, plays a key role in neuronal protein processing and degradation, but its function in brain development remains unclear. In this study, we found that CatB is highly expressed in the cortex of E12.5–E16.5 mice. Morphological analysis revealed significant defects in cortical development in CatB knockout (KO) mice, particularly in layer 6. In vitro experiments showed that CatB deficiency notably impaired neuronal migration and development. Behaviorally, CatB KO mice displayed prominent depressive-like behaviors, and electrophysiological recordings demonstrated significantly reduced neuronal activity in layer 6 of the medial prefrontal cortex. Mechanistically, proteomics analysis revealed that CatB KO affected neuronal migration and axonal growth, and decreased the expression of key transcription factors involved in neuronal development, particularly PEG3. Deficiency of PEG3 also significantly impaired neuronal migration and development. Our findings uncover a role for CatB in cortical development and suggest a mechanism linking CatB deficiency with depression and developmental defects through the destabilization of PEG3.
Similar content being viewed by others


Introduction
The development of the cerebral cortex is a dynamic and precisely orchestrated process involving neurogenesis, neuronal migration, and circuit formation. During embryonic development, newborn neurons form within the neuroepithelium, a proliferative layer of the neural tube, and subsequently migrate throughout the central nervous system (CNS), finally integrating into an appropriate layer and forming part of a neuronal circuit1. In mice, newborn neurons originate from the ventricular zone (VZ) from embryonic day 10 (E10) through E17 and then migrate in an inside-out pattern from the VZ toward the pial surface, first forming deep cortical layers VI and V, and then progressively seeding the upper layers after postnatal day 8 (P8)2. Given that the cerebral cortex is responsible for higher cognitive functions and emotional processing, abnormal neuronal migration is an important cause of many neurodevelopmental conditions, including autism spectrum disorders, schizophrenia, epilepsy, and intellectual disability3,4,5.
Cathepsin B (CatB, EC 3.4.22.1) was the first member of the C1 family of papain-like, lysosomal cysteine peptidases to be identified, and is known to be expressed in most cell and tissue types. CatB is synthesized as an inactive pre-proenzyme and is subsequently modified in its N-glycosidically linked oligosaccharide chains with mannose-6 phosphate residues in the Golgi apparatus before being transferred to endo/lysosomes. Within the acidic environment of the endo/lysosome, inactive CatB is processed via autocatalysis or by other proteases into a mature, active, two-chain form6,7. Over the last two decades, dysregulated CatB synthesis and activity have been implicated in the pathology of several diseases, such as cancer8,9,10,11, pancreatitis12, liver fibrosis13, rheumatoid arthritis14,15, inflammatory pain16, traumatic brain injury17, hypoxic-ischemic brain injury18, Alzheimer’s disease19,20,21,22 and SARS-CoV-2 infection23,24. While the pathological functions of CatB are gradually being elucidated, its physiological functions, other than that associated with lysosomal cargo remain to be clarified.
CatB knockout mice were reported to be phenotypically indistinguishable from their wild-type (WT) siblings and did not display any organ development or immune system abnormalities6. Owing to overlapping substrate specificities, the cysteine cathepsin network can compensate for the loss of function of individual enzymes. However, CatB−/−/CatL−/− double-knockout mice die from brain atrophy shortly after birth, which is not observed in corresponding single-knockout animals25. This suggests that CatB has irreplaceable physiological functions during development. Recently, using CRISPR technology, we deleted the CatB-encoding gene (Ctsb) in differentiated N2a cells and found that these cells failed to produce neurite outgrowths, unlike their WT counterparts26. These findings provided valuable insight into the physiological functions of CatB in neural development. The role of CatB in brain development has yet to be characterized.
In this study, we found that CatB-deficient mice have normal cognitive function, but exhibit depressive behaviors. However, the knockdown of CatB in adult WT mice did not result in similar depression-like behaviors. Additionally, mice lacking CatB showed decreased neuronal activity in depression-related brain regions, including the paraventricular thalamic nucleus (PVT). Fluorescence retrograde tracing indicated that layer 6 of the prefrontal and insular cortices mediated the effects of CatB deficiency on the anterior PVT (aPVT). Coincidentally, CatB was found to be highly expressed in the embryonic cerebral cortex, especially from E12.5 to E16.5, which is the critical stage in the development of cortical layer 6. Mechanistically, we found that CatB is involved in neuronal migration and differentiation through the stabilization of paternally expressed gene 3 (PEG3), a key transcription factor in neuron development. Together, our results revealed that CatB plays a vital regulatory role in cortical development through the stabilization of a related transcriptional network.
Materials and methods
Animals
Wild-type and CatB knock-out (CatB+/−) mice of C57BL/6 background were kept and bred in the Animal room of Beijing Institute of Technology. The selection of CatB−/− mice of homozygous mice from their littermates was performed by examining the template genomic DNA isolated from tail biopsies, using a CatB-exon 4-specific PCR with MCB11 primers (5’-GGTTGCGTTCGGTGAGG-3’) and MCBGT (5’-AACAAGAGC CGC AGG AGC-3’)18,27. The primers for genotyping were listed in supplementary table 1. All behavioral tests were performed using 2-month-old mice. Both male and female mice were included in equal numbers. All animal studies were carried out following the guidelines contained in the Regulation of Laboratory Animals (Beijing Institute of Technology) and under the protocols approved by the Institutional Animal Care and Use Committee review panels at Beijing Institute of Technology.
Cell culture
The mouse neuroblastoma cell line N2a (CCL-131, ATCC) was maintained in DMEM (Gibco, Cat# 11965092) containing 10% fetal bovine serum (Gibco, Cat#16010142) supplemented with 1% penicillin-streptomycin (Gibco, Cat#15140122) at 37 °C in a humidified atmosphere with 5% CO2. The maximum number of passages for cell lines was 20.
Single-cell suspensions were prepared from the mouse neonatal cortex following the previously described methods28. In brief, there were 3 independent dissections in total, resulting in 9 wells per treatment. Neonatal C57BL/6J mice of P0 were used in the present study. Neocortices from 30 pups were pooled together. The pups were euthanized by decapitation after cold-induced anesthesia, and their brains were rapidly removed. Neocortical tissues were dissected in ice-cold Hank’s buffer and subjected to enzymatic digestion using the papain (Solarbio, Cat#G8430). The single-cell suspensions were obtained after application to a 30-mm cell strainer.
Cells were counted and seeded on glass coverslips pre-coated with 500 μg/mL poly-L-lysine (Sigma-Aldrich, Cat#P1399) and were cultured with MEM (Gibco, Cat#11090-081) containing 10% horse serum (ThermoFisher, Cat#16050122) supplemented with 1 mg/mL glucose (Sigma, Cat#07-0680-5), 1% penicillin-streptomycin (Gibco, Cat#15140122) and 100 mM sodium pyruvate (Gibco, Cat#11360-070) overnight. On the next day after seeding, the medium was replaced with Neurobasal (Gibco, Cat#21103-049) supplemented with 2% B27 (Gibco, Cat#17504-044), 1 mg/mL glucose (Sigma, Cat#07-0680-5), 1% penicillin-streptomycin (Gibco, Cat#15140122) and 200mM L-glutamine (Gibco, Cat#25030-081). The cells will be continued to culture for indicated time points.
The single-cell suspensions were also cultured in a 100 mm cell culture dish for neurosphere proliferation. Neurospheres were passaged every three days. The aggregated neurospheres were collected into a centrifuge tube and centrifuged at 2500 rpm for 5 min, and the supernatant was sucked out. The supernatant was resuspended with accutase (Gibco) and digested in a 37 °C incubator for about 10 min, gently and repeatedly blown to single cells. The digestion was terminated with FBS, and the single cells were evenly spread in the dish, and the medium was changed by half volume. The proliferation medium was composed of DMEM/F12 (Gibco, Cat# 11320033), 1% Penicillin-Streptomycin Liquid, 2% B27 supplement, 20 ng/mL EGF (Peprotech, Cat#315-09), 20 ng/mL bFGF (Peprotech, Cat# 450-33). After the third passage digestion, the cells were evenly spread on 14 mm poly-L-lysine (Sigma Aldrich, Cat# 32160801) treated cell slides in a 24-well plate at a volume of 2 × 105 cells /mL for differentiation. The differentiation medium was composed of Neuobasal (Gibco, Cat#21103-049), 1% L- glutamine, 2% B27 supplement, 1%NEAA (Gibco, Cat#11140050), 0.1%AA (Gibco, Cat#11130051), 1%Penicillin-Streptomycin Liquid, 10 ng/mL BDNF (Peprotech, Cat#450-02). 1/3 of the medium was changed every three days, and cell slides were collected according to day (D) 1, D3, D5, D8, D11, and D14, respectively. 10 μM CA-074Me (Med Chem Express, HY-100350) was dissolved in DMSO and applied to the cell culture.
hiPSCs differentiation into forebrain neurons
hiPSCs line (907005-CBP35, passages 16, Precision BioMedicals, Tianjing, China) was maintained under feeder-free conditions. Culture plates were coated with Matrigel matrix 2 h at 37 °C (Corning, NY, 1/100, diluted in DMEM F12 medium,). hiPSCs were cultured in mTESR1 plus (STEMCELL technologies, Cat#05825) complemented with 1% penicillin/streptomycin. Cultures were fed daily and passaged every 5 to 7 days. Neural induction was carried out in STEMdiff™ SMADi Neural Induction Kit (STEMCELL technologies, Cat#08581) according to the manufacturer’s instructions. Induction efficiency was identified by immunofluorescence staining after the third passage. Then, differentiation of NSCs was achieved using the STEMdiff™ Forebrain Neuron Differentiation Kit (STEMCELL technologies, Cat #08600). Five days later, seed neuronal precursors onto a warm (37 °C) coated 24-well plate at a density of 1.5 × 104 - 3 ×104 cells/cm2 in STEMdiff™ Forebrain Neuron Maturation Medium (STEMCELL technologies, Cat#08605). 12 h later, transfect PEG3 siRNA or negative control siRNA with Lipofectamine RNAiMAX (Invitrogen, Cat#13778075) or add 10 μM CA-074Me. The full medium was changed every 3 days.
Cell viability assay
Neural stem cells were seeded in 96-well plates for differentiation (2000 cells/well), Using the above-mentioned neural stem cell differentiation medium. 1/3 of the medium was changed every three days, and the treatment group was supplemented with CA-074Me. Cell viability assays were performed using a cell counting kit (CCK-8, Beyotime). The optical density was read at a wavelength of 450 nm with a microplate reader. Cell viability was calculated using the formula: optical density of the treated group/control group.
Generation of CatB knock-out cells
A neuronal CTSB knockout (CTSB KO) cell line was established by using N2a cells and a commercially available clustered regularly interspaced short palindromic repeats (CRISPR) and homology-directed repair (HDR) (ORIGEN, Cat#KN303984) on N2a cells via CRISPR and HDR. Positive cells were selected with 2 μg/ml puromycin (InvivoGen, Cat#ant-pr) for 15 days. A single cell clone was picked up and continued to culture. CTSB expression was assessed by Q-PCR and immunoblotting. During the generation, we also included scramble control gRNA (SKU GE100003, ORIGENE, Cat#KN303984) to serve as a negative control. Positive clones were collected and cultured. However, some positive cell gene KO may occur in one allele (heterozygous). To exclude heterozygous cells, 10 positive single clones were subjected to RT-PCR and Western blotting for further selection. The homozygous cells were confirmed and used in the subsequent experiments.
Immunofluorescent staining
The brains sections from embryos, post-born neonatal, and adults of WT and CatB−/− mice were incubated with primary antibodies against CatB (1:1000, AF965, R&D system), Ctip2 (1:1000, ab18465, Abcam), Tbr1 (1:500, ab183032, Abcam), Pax6 (1:500, ab5790, Abcam), MAP2 (1:500, ab11267, Abcam), Nestin (1:1000, MA1-110, Invitrogen) and cfos (1:1000, sc-166940, Santa Cruz) at 4 °C overnight. After washing with PBS, the sections were incubated with second antibodies (1:1000, ab150077&ab150115&ab150080&ab150116&ab150165&ab15704, Abcam) at 4 °C for 2 h. The sections were mounted in Antifade Mounting Medium (Beyotime, Cat#P0126). Fluorescence images were taken using a Nikon confocal microscope (Nikon A1R).
The cultured N2a cells, primary neurons or differentiated neurons were fixed with 4% paraformaldehyde (Biosharp, Cat#BL539A), permeabilized with 0.3% Triton X-100 (Biosharp, Cat#BS084) in 1% PBS, and blocked with 2% bovine serum albumin (Solarbio, Cat#A8850) and 2% donkey serum (Solarbio, Cat#SL050) in PBS. They were then incubated with the primary antibodies overnight at 4 °C. After washing with PBS, the sections were incubated with second antibodies, then incubated with Hoechst 33258 (1:500, Sigma-Aldrich, Cat#94403) and mounted in Antifade Mounting Medium. Fluorescence images were taken using a Nikon confocal microscope (Nikon A1R).
Immunoblotting analysis
Cortices from mice at embryonic day (E12.5, E14.5, and E16.5), postnatal day (P0 and P10) and adult (P60) were dissected and homogenized in RIPA buffer containing protease inhibitors. Cultured cells were also homogenized as the above method. The samples were quickly frozen and stored until use at −80 °C. The immunoblotting analyses were conducted29. In brief, each sample was lysed and electrophoresed using 15, 12, or 7.5% SDS-polyacrylamide gels. The proteins on the SDS gels were then electrophoretically transferred to nitrocellulose membranes. Following the blocking, the membranes were incubated at 4 °C overnight under gentle agitation with primary antibodies against CatB (1:1000, AF965, R&D system), PEG3 (1:1000, 84030-5-RR, Proteintech), Foxo1 (1:1000, ab179450, Abcam), Foxc1 (1:500, ab227977, Abcam), Ubiquitin (1:600, 10201-2-AP, Proteintech), β-actin (1:10000, ab6276, Abcam) and LC3B (1:2000, 2775S, Cell signaling technology). After washing, the membranes were incubated with horseradish peroxidase (HRP)-labeled second antibodies (1:10000, ab6721&ab6728&ab6885, Abcam) for 2 h at room temperature. Subsequently, the membrane-bound, HRP-labeled antibodies were detected using an enhanced chemiluminescence detection system (ECL kit; Millipore, Cat# WBKLS0500) with an image analyzer (LAS-4000; Fuji Photo Film).
Whole-cell patch-clamp recordings
Coronal brain slices (300 μm) containing aPVT, and layer VI of the mPFC and insular cortex were prepared from 2-month-old CatB KO and WT mice. Neurons were recorded at −70 mV using a MultiClamp 700B amplifier (Molecular Devices). Pipettes (3–6 MΩ) were filled with a K-gluconate-based internal solution. Spontaneous EPSCs and action potentials were recorded in voltage- and current-clamp modes, respectively. Signals were filtered at 2 kHz, digitized at 10 kHz, and analyzed with Mini Analysis (Synaptosoft).
Scratch wound assay
The scratch wound assay is a common in vitro method used to measure cell migration/proliferation30. The capacity of the cultured neurons to induce repopulation of the wound was examined by scratching confluent cell monolayers. The wound was induced by dragging a sterile pipette tip (200 μL) across the surface of N2a and N3a monolayers. The detached cells and debris were removed immediately by washing with PBS. The cells were maintained for an additional 72 h in the presence or absence of inhibitors. Bright-field images at the region of interest were taken at 0 and 72 h after incubation under an inverted microscope (Nikon A1R). The cells were then fixed and stained for F-actin to further examine the effects of CatB deficiency or inhibitors on the migration/proliferation of astrocytes.
Inhibitors
10 μM pepstatin (PEPTIDE, Cat#4397) was used to inhibit CTSD, 10 μM Z-FF-FMK (Cat#sc-364671) was used to inhibit CTSL and 10 μM CA-074Me (PEPTIDE, Cat#4323-v) was used to inhibit CTSB. It was noted that CA-074Me was reported to be selective for CTSB at concentrations below 1 μM and concentrations (more than 10 μM) will be needed to inhibit CTSB; However, high concentration of CA-074Me inhibited CTSs broadly31.
Chronic restraint stress (CRS)
Mice were individually restricted in 50-ml conical tubes with holes for air for 2 to 3 h per day in the 2p.m–5p.m for 10 consecutive days. Behavior tests were performed before CRS, CRS day 5, and day 10. On the test day, mice were transferred to the testing room to acclimate to the experimental environment for at least 1 h. The apparatus was thoroughly cleaned with 70% alcohol to remove the previously tested animal’s odor and trace after each test session.
Morris water maze test
The Morris water maze test was performed to measure the long-term memory of mice. The water maze model was used in a circular tank (diameter 1.2 m) filled with water which temperature was kept at 22–23 °C. The experiments were performed on 6 consecutive days, including the cued platform task on day 1, the training task on days 2–5, and the test on day 6. In the cued task, the platform was located above the surface of the water and marked by an attached flag. Throughout the training and test days, the platform was located 1 cm under the surface of the water. On the test day, the platform was removed from the water. Throughout the experiment, mice were trained for 4 trials per day, with an intertrial interval of 20 min. Each mouse was allowed 90 s to find the platform. If the mice found the platform, they were allowed to stay on the platform for 15 s before being transferred to the home cage. The number of platform crossings, latency to reach the original platform location, swim speed, and total distance traveled were recorded using a video tracking system (Smart 3.0, Panlab, Spain).
Y maze test
The short-term memory was evaluated using a Y maze as previously reported32. Briefly, the Y maze made of black Plexiglas (5 cm width, 35 cm length, 10 cm height) consisted of identical three arms with 120°. Each mouse was placed at the end of one fixed arm and allowed to move freely through the maze during a 5-min session and its behavior was recorded with a camcorder. Three consecutive choices were defined as an alternation. The percentage of alternations was calculated as (Number of alterations/Number of entries) × 100. In addition, the total number of arms entered during the session was also determined.
Open field test
Mice were placed in the center of an open chamber (50 cm × 50 cm × 50 cm) and allowed to explore their surroundings freely for 10 min. The arena was divided into 16 squares via an automated grid system using Smart 3.0 software (Panlab, Spanish), which was also used to record data. Time in the center squares of the arena, distance traveled, and speed were recorded.
Elevated plus maze
The EPM apparatus comprised two open arms (30 × 5 cm), two closed arms (30 × 5 × 15 cm), and a central platform (5 × 5 cm) elevated 100 cm above ground. Mice were placed in the center facing one of the two open arms and allowed to explore for 10 min, and their behavior was monitored with a camera (Smart 3.0, Panlab, Spanish) placed on the top of the maze. The amount of time spent in the open arms was used as an indicator of anxiety-like behavior.
Tail-suspension test
Mice were suspended 30 cm above the floor by adhesive tape placed approximately 1 cm from the tip of the tail. A 4 cm long hollow cylinder (with an inner diameter of about 1.3 cm) was placed at the base of the mouse tail to prevent mice climb or hang on to their tail. The test lasted 6 min and the animal’s immobility time was calculated during the last 5 min.
Forced swimming test
Mice were individually placed in a Plexiglas cylinder (height of 30 cm, diameter of 19 cm) containing 19 cm water (23 ± 1 °C). The test lasted for 6 min. The duration of immobility was recorded during the final 4 min with a video tracking system (Smart 3.0, Panlab, Spanish). Immobility was defined as the absence of active escape behaviors such as swimming or climbing, with only minimal movements to keep the head above water.
Sucrose preference test
Mice were individually housed and given continuous exposure to a water bottle containing regular water and a second with 1% sucrose (Sigma Aldrich, Cat#100892). The positions of the sucrose water bottle and the regular water bottle were swapped every 12 h. After 48 h of habituation, mice were deprived of both food and water for 24 h. Immediately after deprivation, mice were given 24 h access to one bottle of 1% (wt/vol) sucrose solution and one bottle of regular water with switching the positions of the sucrose and the regular water bottle every 12 h. Each bottle was weighed before and after the test and sucrose preference was expressed as (Δweightsucrose)/(Δweightsucrose + Δweightwater) × 100.
Real-time quantitative PCR analysis
The mRNA isolated from cells was subjected to a real-time quantitative PCR. The total RNA was extracted with RNAiso Plus (Takara, Japan, Cat#9108) according to the manufacturer’s instructions. 1 μg RNA was reverse transcribed to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen, Japan, Cat#205311). After an initial denaturation step at 95 °C for 5 min, temperature cycling was initiated. Each cycle consisted of denaturation at 95 °C for 5 s, annealing at 60 °C for 10 s, and elongation for 30 s. In total, 40 cycles were performed. The cDNA was amplified in duplicate using a Rotor-Gene SYBR Green RT-PCR Kit (Qiagen, Japan, Cat#204174) with a Corbett Rotor-Gene RG-3000A Real-Time PCR System. The data were evaluated using the RG-3000A software program (version Rotor-Gene 6.1.93, Corbett). The sequences of primer pairs are described below. CatB: 5’-GCAGCCAACTCTTGGAACCTT-3’ and 5’-GGATTCCAGCCACAATTTCTG-3’, mouse PEG3: 5’- GAGGTCCAAGAGAAC
TGCCTAC-3’ and 5’-GAGAAGACTCGTCCTCACAGATC-3’, mouse Actin: 5’-A
GAGGGAAATCGTGCGTGAC-3’ and 5’- CAATAGTGATGACCTGGCCGT-3’. human PEG3: 5’- CGGAACAGAAGAGAGTCCTCAC-3’ and 5’- TGCTTCTTGGG
TTCCTGGTGTG-3’, human Actin: 5’- CACCATTGGCAATGAGCGGTTC-3’and
5’-AGGTCTTTGCGGATGTCCACGT-3’. For normalization, an endogenous control (actin) was assessed to control for the cDNA input, and the relative units were calculated by a comparative Ct method. All the real-time qPCR experiments were repeated three times, and the results are presented as means of the ratios ± SEM.
Label-free quantitative LC-MS/MS analysis
Cortical tissues from WT (n = 3) and CatB KO (n = 4) mice at embryonic day 12.5 (E12.5) were collected and lysed in PASP buffer containing 100 mM ammonium bicarbonate and 8 M urea (pH 8.0). Protein concentrations were determined using the Bradford assay. Proteins were enzymatically digested with trypsin, allowing up to two missed cleavage sites. Peptide separation was performed using a nano Elute UHPLC system (Bruker Daltonics) equipped with an in-house packed analytical column (15 cm × 100 μm, 1.9 μm C18 resin), coupled to a timsTOF Pro2 mass spectrometer (Bruker Daltonics) via a Captive Spray ion source (electrospray voltage: 1.5 kV). MS data were acquired in PASEF mode, with a scan range of m/z 100–1700, an ion mobility (1/K₀) range of 0.85–1.3 V s/cm², a ramp time of 100 ms, 10 MS/MS scans per cycle, a total cycle time of 1.17 s, an intensity threshold of 2500, and a target intensity of 20,000.
Raw data were processed using Proteome Discoverer 2.4 (Thermo Fisher Scientific) and MaxQuant software. Peptide and protein identification was performed using the UniProt Mus musculus FASTA database (downloaded on January 27, 2022; 86,515 sequences). Search parameters included a precursor mass tolerance of 20 ppm and a fragment mass tolerance of 0.05 Da. Carbamidomethylation of cysteine residues was specified as a fixed modification, while oxidation of methionine and protein N-terminal acetylation were set as variable modifications. All mass spectrometry experiments were performed by Novogene Co. Ltd. (Tianjin, China). The false discovery rate (FDR) was controlled at 1% at both peptide-spectrum match (PSM) and protein levels. Label-free quantification was based on precursor intensities. Differential expression analysis was conducted using the limma package in R, with a significance threshold of p < 0.05 and a fold change ≥ 1.5. Gene Ontology enrichment was performed using the R packages clusterProfiler (v4.8.1), simplifyEnrichment (v1.12.0), and org.Mm.eg.db (v3.17.0).
Transmission electron microscope
N2a cells were incubated with vehicle and CA074me for 12 hours and fixed with 2.5% glutaraldehyde in 0.01 mol/l sodium phosphate buffer (pH 7.4) for 1 h, followed by 1% osmium tetroxide for 2 h. After dehydration, thin sections were cut using a Leica EM UC7 Ultramicrotome (Leica) and stained with 2% uranyl acetate and lead citrate. The ultrastructural analysis was performed using a HITACHI HT 7800 80 kV electron microscope (Hitachi High-Tech).
Statistics and Reproducibility
All data analysis and statistics were calculated with GraphPad Prism 9.0 (GraphPad Software) and Origin 8.5 software. Two groups were compared using an unpaired two-sided Student’s t-test. In more than two experimental groups, data were analyzed by a one-way ANOVA test. When the one-way ANOVA tests were statistically significant, Bonferroni’s and Dunn’s multiple-comparison post hoc analyses, respectively, were used to compare the differences between individual groups. P values less than 0.05 were considered statistically significant and are indicated by *P < 0.05, **P < 0.01, and ***P < 0.001 in the figures. Data are shown as means ± standard errors (SEM).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
CatB was highly expressed in the developing cortex of mice
To determine whether CatB is associated with brain development, we quantified CatB levels in the embryonic (E12.5, E14.5, and E16.5), neonatal (P0), and postnatal (P10) cerebral cortex of WT mice by western blot (Fig. 1A). The results showed that the expression of both pro- and mature CatB decreased during embryonic development, with the highest abundance being detected at E12.5 (Fig. 1B, C).

A CatB protein levels in mouse cortex at indicated embryonic (E), postnatal (P), or adult stages assessed by Western blot. Representative blots from three independent experiments (n = 3 mice per time point). B, C Quantification of pro-form (B) and mature-form (C) CatB expression normalized to Actin (n = 3 mice per group). D Schematic illustration of cell migration from ventricular zone (VZ) to preplate (PP) in E12.5 embryos. E Fluorescent images of CatB (magenta) and Hoechst (blue) in WT mice at E12.5, E14.5 and P0. Scale bar, 100 μm. F Quantification of CatB staining intensity in (E), normalized to VZ/SVZ at each time point. Images were taken from 3 mice per stage, 3 brain sections per mouse. G Immunofluorescent staining of CatB (magenta) with Ctip2 (green) or Trb1(green) in the cortex of WT mice at E12.5. Scale bar, 50 μm. *P < 0.05, **P < 0.01. Error bars represent mean ± SEM.
In the mouse forebrain, the cerebral cortex is visible from E9.5, when it consists of a unique layer of proliferating neuroblasts, termed the germinative neuroepithelium or ventricular zone (VZ). Cortical plate neurons are generated in an inside-out pattern, that is, layer 6 neurons are born first, followed by layer 5 neurons, with neurons of layers 4, 3, and 2 being born last. In the VZ, neurogenesis starts at E11.0 ventrolaterally and at E12.0 dorsomedially, peaks around E12.5–E13.5, and continues at lower levels until P17; neuronal differentiation and migration continue after birth33. At around E12.5, a first layer (layer 6) differentiates in an area above the VZ, called the preplate (PP) (Fig. 1D). To investigate whether high CatB expression at E12.5 correlates with brain development, we performed immunofluorescence staining for CatB at E12.5, E14.5, and P0 in WT mice (Fig. 1E). The results showed that CatB is mainly expressed in the PP at E12.5. However, there was no difference in CatB expression between layer 2–6 and layer 1 at P0 (Fig. 1F). The results of co-staining for specific markers of layers 5 and 6 (Ctip2 and Tbr1, respectively) showed that CatB was mainly distributed around these layers of the cortex (Fig. 1G). These results suggested that CatB may also be involved in neuronal differentiation. In addition, we found that CatB deletion decreased the area of cortical layers 5 and 6 (Fig. S1A, B) as well as the number of cells in layer 6, but not layer 5 (Fig. S1C, D), in P7 mice. Together, these observations indicated that CatB is involved in cortical neuronal development, especially in layer 6.
CatB deficiency blocked neuronal migration and differentiation
To investigate whether CatB regulates the migration of neurons, we conducted a scratch wound healing assay in N2a (a mouse-derived neuronal cell line) cells. The results showed that the migration of N2a cells was significantly inhibited by CA-074Me, a cell-permeable CatB inhibitor, but not pepstatin, an aspartyl peptidase inhibitor, or Z-FF-FMK, a specific CatL inhibitor (Fig. 2A, B). CatB has been reported to have enzymatic activity both inside and outside of neurons34,35. To determine whether intracellular or extracellular CatB is involved in N2a cell migration, we applied CA-074, a non-cell permeable CatB inhibitor, to N2a cell cultures after wounding, and found that extracellular CatB did not inhibit N2a cell migration to the same extent as intracellular CatB (Fig. 2C, D). To verify these results, we knocked out the Ctsb gene in N2a cells (designated as N3a cells) using CRISPR-mediated homology-directed repair (HDR). The complete knockout of CatB in N3a cells was confirmed by both qPCR (Fig. S2A). Consistent with the results obtained with CA-074Me-treated N2a cells, both migration and scratch wound closure were impaired in N3a cells (Fig. 2C, D; Fig. S2B). These results indicated that CatB is involved in neuronal migration.

A Fluorescent images of Filamentous actin (F-actin, magenta) in N2a cells at 72 h after damage by scratch. The N2a cells were treated with 10 μM Pepstatin (aspartyl proteases inhibitor), 10 μM Z-FF-FMK (CatL inhibitor), 10 μM CA-074 (cell none-permeable CatB inhibitor) or 10 μM CA-074Me (cell-permeable CatB inhibitor) 1 h scratch. Scale bars were 5 μm. The right images are the close-ups of the boxes in the corresponding left images. B Quantification of the migration of N2a cells shown in (A). The gap width was quantitatively evaluated using Image J software. C Bright-field images of N2a and N3a cells at 72 h after damage by scratch. Scale bars were 50 μm. D Quantification of the migration of N2a and N3a cells shown in (C). E Fluorescent images of F-actin (green), MAP2 (magenta) with Hoechst (blue) in primary cortical neurons with CatB siRNA or CA-074Me treatment. Scale bars were 10 μm. F Immunofluorescent staining of Nestin (green) in NSCs at day 1, 3, 5, 8 and 11 after differentiation. Scale bars were 20 μm. G Immunofluorescent staining of MAP2 (green) with CatB (red) in neuron stem cells (NSCs) at day 1, 3, 5, 8, and 11 after differentiation. Scale bars were 20 μm. H Quantitative analysis of the fluorescent density of MAP2, Nestin and CatB shown in (E, F). I Immunofluorescent staining of MAP2 in WT NSCs, WT NSCs treated with CA-074Me, and NSCs of CatB−/− mice at day 5 and 8 after differentiation. J, K Quantitative analysis of the number and the mean branch number of MAP2-positive cells. All data were obtained from three independent experiments. **P < 0.01, ***P < 0.001. Error bars represent mean ± SEM.
We previously found that CatB expression was markedly increased in N2a cells after 24–48 h of treatment with retinoic acid (RA), which can induce the neuronal differentiation of N2a cells26. To verify whether CatB can influence neurite outgrowth, we treated N2a and N3a cells with RA and stained their neurites with the Neuro-Chrom Pan Neuronal Marker antibody. After 72 h of RA treatment, neurite length was noticeably increased in N2a cells, whereas the opposite effect was observed in N3a cells (Fig. S2C, D). Furthermore, the knockdown of CatB with siRNA or its inhibition with CA-074Me also blocked neurite outgrowth in primary neurons during the differentiation stage (Fig. 2E). Collectively, these findings demonstrated that CatB plays an important role in neurite outgrowth, which is reflected in the inhibition of growth cone creation in the initiation stage of neurite outgrowth.
Here, we found that CatB-mediated neurite growth is closely related to neuronal differentiation. We further found that CatB is highly expressed in the outer cortex from E12.5 to E14.5, which is the critical period for neural progenitor cell migration and differentiation. These observations implied that CatB may be closely related to the process of neural progenitor cell differentiation. To test this possibility, we harvested neural stem cells from the cortex of neonatal mice and induced their neuronal differentiation. Immunofluorescence staining showed that the expression of nestin (a specific marker of neural stem cells) was highest in the first 3 days of differentiation and subsequently decreased (Fig. 2G), which was consistent with the trend observed for CatB expression (Fig. 2G, H). However, the expression of MAP2, a marker of mature neurons, gradually increased and eventually stabilized during the differentiation of the neural stem cells, indicating that they had differentiated into mature neurons (Fig. 2G, H). The inhibition of CatB activity with CA-074Me on day 1 of differentiation decreased the number of MAP2-positive cells as well as the number of branches in mature neurons (Fig. 2G, H). To exclude possible deleterious effects of CA-074Me on the differentiation of neural stem cells, we examined their viability following 48 h of treatment with CA-074Me. Meanwhile, cortical primary neural stem cells extracted from neonatal CatB−/− mice also exhibited impaired differentiation (Fig. 2I–K). These data indicated that CatB is highly expressed in the early stages of neural stem cell differentiation and participates in the regulation of this process.
CatB knockout mice exhibited depressive-like behaviors
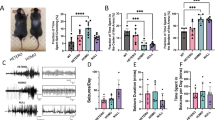
To investigate the physiological function of CatB in vitro, we conducted a series of behavioral experiments to explore the behavioral characteristics of CatB knockout (CatB−/−) mice (Fig. 3A). First, we performed the tail suspension test (TST), the forced swim test (FST), and the sucrose preference test (SPT) to assess depression-related behaviors in CatB−/−, CatB knockdown mice (CatB+/−), and their WT littermates (controls). We found that the immobility time of CatB−/− mice was significantly increased compared with that of WT animals in both the TST (Fig. 3B) and the FST (Fig. 3C). Moreover, CatB−/− mice displayed reduced sucrose preference relative to their WT littermates (Fig.3D). Subsequently, mice in the three groups were exposed to 2 weeks of chronic restraint stress (CRS) to investigate whether CatB deficiency represented more vulnerable to major risk factor for depression36. No difference in immobility time in the TST was observed among the three groups after 5 days of exposure to CRS (Fig. S3A); however, on day 10 of CRS, a significant increase in immobility time was observed among WT but not CatB−/− mice (Fig. 3E). These data suggested that CatB deficiency can induce depressive-like behaviors in mice. The elevated plus maze and open field tests showed no differences in anxiety-related behaviors among the groups of mice (Fig. S3B–D). Additionally, the Morris water maze and Y maze tests revealed no impact of CatB knockout on learning and memory (Fig. S3E–K). Together, these findings suggest that CatB knockout is associated with depressive-like behaviors in mice, with differential effects observed across various behavioral tests, but did not promote anxiety or affect learning and memory.

A Experimental timeline. B–D Immobility time in tail suspension test (TST), forced swim test (FST), and percent of sucrose consumption in 2-month-old WT, CatB+/−, and CatB−/− mice. n = 12, 9, 9 for (B); n = 8, 7, 8 for (C); n = 10, 8, 8 for (D). E Behavioral assessment of mice subjected to 0 days and 10 days of chronic restraint stress (CRS) on mean time spent immobile in TST. Before CRS: n = 12 (WT), 9 (CatB+/− and CatB−/−); After CRS: n = 10 (WT), 7 (CatB+/−), 6 (CatB−/−). F Representative images of cfos expression in the PVT of WT and CatB−/− mice. Scale bars were 100 μm. G Quantification of c-Fos–positive cells in the PVT (n = 6 WT, 5 KO). H Schematic diagram of retrograde neuronal circuit tracing. I, J CTB-555–labeled cells in PVT (I) and in layer 6 of the insular cortex and PFC (J). Scale bars, 1 mm. n = 5 mice. Scale bars were 1 mm. K Schematic illustration of recordings from layer 6 pyramidal neurons in the mPFC in acute brain slices. L Frequencies of induced APs at different current steps. M Raw traces and averaged traces of sEPSC. N, O Summary result of averaged sEPSC frequency and amplitude. n = 5 mice per group; 21 cells (WT), 20 cells (CatB−/−). *P < 0.05, ***P < 0.001. Error bars represent mean ± SEM.
Because CatB knockout mice exhibited notable depressive-like behaviors in the absence of any stimulus, we speculated that the deletion of CatB may contribute to developmental deficits that subsequently result in depressive-like behaviors. To verify whether CatB deletion in adulthood causes depression, we knocked out CatB in the whole brain of CatB-floxed adult mice and then tested for depressive-like behavior in the animals. To knock out CatB, we injected Cre-dependent (AAVcap-B10-CMV-Cre-EGFP) or Cre-independent (AAVcap-B10-CMV-EGFP) AAVs capable of crossing the blood-brain barrier37 into the animals via the tail vein (Fig. S4A). The efficiency of CatB knockout was validated by immunofluorescence staining. The results showed that the AAVs were abundantly expressed in the brain, achieving a CatB knockout efficiency of approximately 90% (Fig. S4B–D). One month after AAV delivery, we subjected the mice to the FST, SPT, and TST. Unlike mice lacking CatB from conception, adult CatB-deficient mice showed no depression-like behavior (Fig. S4E–G). These data suggested that the loss of CatB in adulthood does not induce depression-related behaviors in mice.
CatB deletion impaired the development and activity of neurons in layer 6 of the medial prefrontal cortex and insular cortex
To explore how CatB deficiency contributes to depressive-like behaviors, we performed c-Fos-staining and quantified the number of c-Fos-positive cells in depression-related brain regions including PVT, hippocampus, nucleus accumbens (NAc), basolateral amygdala (BLA) and (Fig. 3F, Fig. S5A–E). Among the regions assessed, the most significant decrease in the number of c-Fos-positive cells in CatB−/− mice relative to WT mice was observed in the PVT (Fig. 3G), but not in the hippocampus, NAc and BLA (Fig. S5F–H). Additionally, there are no differences in c-Fos-positive cells in PVT of WT and CatB−/− mice in the home cage (Fig. S5I, J). The PVT is an elongated nucleus that spans the anterior–posterior extent of the dorsal midline thalamus and forms extensive connections throughout the brain38. It was initially found to be associated with neural circuits relating to arousal and motivation-related behaviors and has more recently also been implicated in depression39,40,41. Consistent with our findings, recent research has shown that inhibition of the anterior PVT elicits antidepressant-like effects, further implicating PVT hypoactivity in depression-related behaviors42. Next, we prepared acute brain slices from CatB−/− and WT mice for whole-cell electrophysiology and then monitored spontaneous excitatory postsynaptic currents (sEPSCs). No differences in sEPSC amplitude, frequency, rise time, and decay time were observed between CatB−/− mice and their WT littermates (Fig. S5K–N). These results suggested that CatB deletion may not directly affect the activity of c-Fos-expressing cells in the PVT.
Given that the PVT has extensive neural circuit connections with other brain regions, we hypothesized that CatB knockout might affect brain regions upstream of the PVT, resulting in a decrease in the c-Fos response. We injected cholera toxin subunit B (CTB)-Alexa Fluor 555 conjugates into the aPVT and examined retrograde labeling in WT mice (Fig. 3H). We first confirmed that CTB expression was confined to the aPVT after 30 days of injection (Fig. 3I). Subsequently, we counted the numbers of CTB-positive cells through a series of coronal sections. Interestingly, we found a large number of CTB-positive cells in layer 6 of the insular cortex and layer 6 of the medial prefrontal cortex (mPFC) (Fig. 3J). Both the mPFC and the insular cortex have been associated with depression. Next, we measured the c-Fos expression levels in layer 6 of both cortices and found that there were no differences in the number of c-Fos-positive cells in these two regions between CatB−/− and WT mice (Fig. S6A–C). Then, we stained for the layer 6-specific marker, Tbr143, and found that the area of layer 6 of the mPFC, but not that of layer 6 of the insular cortex, was significantly reduced in CatB knockout mice (Fig. S6D–G). However, there were no marked changes in the number of cells per mm2 in layer 6 of either the mPFC or the insular cortex (Fig. S6D, F, H). These observations indicated that the loss of CatB may affect the development of layer 6 of the mPFC. Next, we performed whole-cell patch-clamp recordings on layer 6 pyramidal neurons in the mPFC and insular cortex to evaluate whether CatB deletion affected the electrophysiological properties of neurons in these two regions (Fig. 3K and Fig. S7A). To examine intrinsic excitability, the firing responses of layer 6 pyramidal neurons in the mPFC and insular cortex to depolarizing current injections were analyzed under a current-clamp mode. No differences in generated action potentials (APs) were detected between neurons of CatB−/− mice and those of their WT littermates in response to injection of the same amount of current (Fig. 3L and Fig. S7B). To determine the impact of the loss of CatB on excitatory synaptic transmission in layer 6 pyramidal neurons of the mPFC and insula, we recorded spontaneous excitatory postsynaptic responses. The amplitude and average firing frequency of sEPSCs of layer 6 pyramidal neurons in the mPFC and insular cortex was significantly lower in CatB−/− mice than in their WT counterparts (Fig. 3M–O and Fig. S7C–E). No difference in the rise time or the decay time of sEPSCs in either region was noted between the two groups of mice (Fig. S7F–I). These results suggested that the loss of CatB disrupts the development and excitatory synaptic transmission of neurons in cortical layer 6.
CatB knockout decreased the expression of development-related transcription factors
Having demonstrated the effects of CatB deficiency on cortical development and neuronal migration and differentiation, we next sought to define the underlying molecular mechanisms. To this end, we performed a label-free, quantitative liquid chromatographic–tandem mass spectrometric (LC-MS/MS) analysis to determine the protein expression profiles in E12.5 WT and CatB−/− cortices. A total of 6,286 proteins were identified, 119 of which were found to be differentially expressed using WT/CatB−/− ratio >1.5, P < 0.05 as the selection criteria (Fig. 4A). Gene Ontology (GO) term similarity analysis for these 119 proteins identified 23 that were associated with nervous system development, neuronal differentiation, and neuronal projection (Fig. 4A, B). GO term analysis of these 23 proteins further showed enrichment of transcription-related terms, in addition to cell development, migration, and projection (Fig. 4C). Cellular identity is established by the interplay between transcriptional factors, cis-regulatory elements, and the chromatin landscape. These molecular interactions form the basis of gene regulatory networks, ensuring the precise temporal and spatial regulation of gene expression44. By comparing the 23 differentially expressed proteins related to cortical or neuronal development with the transcription factor database (Animal TFDB3.0), we screened out four transcription factors, including PEG3, forkhead box C1 (FOXC1), forkhead box protein O1 (FOXO1), and homeobox protein aristaless-like 4 (ALX4) (Fig. 4D). PEG3 is involved in neuronal development and modulates maternal and social behavior45,46; FOXC1 regulates neural crest-derived brain structures and cell migration47,48; FOXO1 is required for neurogenesis and neuronal survival49; and ALX4 contributes to forebrain development and is implicated in neurodevelopmental disorders such as autism50. Subsequently, we extracted cortical proteins from WT and CatB−/− mice for immunoblotting. Quantitative analysis revealed significant downregulation of three detectable transcription factors in CatB−/− mice compared to WT littermates (Fig. 4E–H). ALX4 protein levels fell below the assay’s detection threshold in both genotypes and were therefore excluded from statistical comparison. Peg3, an imprinted gene, displays parent-of-origin-dependent monoallelic expression and regulates complex mammalian traits, including growth and behavior. Peg3 is expressed in embryos and the adult brain from the paternal allele only and plays an important role in brain development and the migration of oxytocin neurons. Mutation of the Peg3 gene results in growth retardation as well as a striking impairment of maternal behavior that frequently results in the death of offspring51,52,53,54.

A Similarity heatmap from 163 GO terms enriched in differentially expressed proteins by CatB KO that have been clustered and annotated with word clouds. B Representative developmental and differential related-GO terms enriched in differentially expressed proteins by CatB KO. C The volcano plot of the proteins identified from WT and CatB KO cortices by label-free quantitative LC-MS/MS. The proteins are plotted according to their log2 fold changes (x axis) and log10 P values (y axis). Proteins down-regulated and up-regulated in CatB KO are shown as blue and red dots, respectively. Among them, proteins with known functions in transcription and neuronal differentiation or nervous system development are marked. D Venn diagram of the number of differentially expressed proteins by CatB KO overlapping with transcriptional factors of mouse. The overlapping proteins related to cortical development and neuronal differentiation are labeled red. E Western blotting analysis of the expression of PEG3, Foxo1, and Foxc1 in cortex from E12.5 WT and CatB KO mice. F–H Quantification of the expression of PEG3 (F), Foxo1 (G), and Foxc1 (H) in cortex from E12.5 WT and CatB KO mice. PEG3 and Foxc1: n = 4 mice per group; Foxo1: n = 3 mice per group; all with 3 independent biological replicates. *P < 0.05, ***P < 0.001. Error bars represent mean ± SEM.
To examine whether the migration of N2a cells was regulated by PEG3. We knocked down the expression of PEG3 by transfected N2a cells with siRNA targeting PEG3, and validated by qPCR (Fig. S8A). The migration of N2a cells was significantly inhibited by the knockdown of PEG3 (Fig. S8B, C), suggesting the involvement of PEG3 in neuronal migration. Furthermore, we induced neural stem/progenitor cells (NSC/NPC) and neurons from human pluripotent stem cells (hiPSCs). The induced NSCs were validated by staining nestin and pax6, which are the markers of NSCs (Fig. S8D, E). Next, we examined whether inhibition of PEG3 or CatB could inhibit the differentiation of NSCs into neurons. PEG3 siRNA 1 was validated to knock-down the expression of PEG3 (Fig. S8F). Moreover, Nestin−/Map2+ staining indicated the maturation of neurons (Fig. S8G). However, neither inhibition of PEG3 expression nor inhibition of CatB enzymatic activity could induce long neurites in Nestin−/Map2+ cells (Fig. S8G). Given that CatB knockout markedly reduces PEG3 expression, we propose a potential regulatory link between CatB and cortical development via PEG3, although further in vivo functional studies are needed to substantiate this hypothesis.
CatB deficiency promoted the degradation of PEG3
To determine whether CatB deletion affects the production or degradation of PEG3, we first measured the mRNA levels of Peg3 in cortical samples of WT and CatB−/− mice at E12.5 using qPCR. We found that Peg3 mRNA expression levels were similar between WT and CatB knockout mice (Fig. 5A), indicating that CatB regulates PEG3 expression at the protein level. Next, we treated N2a cells with CA-074Me for 12 h, and found that PEG3 expression was significantly reduced compared with that in control cells (Fig. 5B, C), suggesting that CatB enzymatic activity may contribute to PEG3 protein stabilization. Accordingly, we subsequently performed a series of cycloheximide (CHX)-chase assays to determine whether the loss of CatB enzymatic activity promotes PEG3 degradation. The results showed that PEG3 expression was slightly decreased after 6 h and diminished after 12 h of CHX treatment. However, PEG3 expression was almost undetectable in N2a cells after 6 h of treatment with CHX in the presence of CA-074Me, indicating that the inhibition of CatB accelerated the degradation of PEG3 (Fig. 5D, E). We then explored why the loss of CatB enzymatic activity accelerated PEG3 protein degradation. The ubiquitin-proteasome (UPS) and autophagy-lysosome pathways are two major routes for protein degradation. To evaluate the effects of CatB inhibition on autophagy-lysosome activity, we analyzed the LC3II/LC3I ratio. CatB inhibition led to a significant increase in the LC3II/LC3I ratio (Fig. 5F, G). This increase can be interpreted in two ways: (1) an upregulation of autophagosome formation, reflecting enhanced autophagic flux, or (2) a blockade in autophagosome degradation due to impaired autophagosome-lysosome fusion, resulting in defective autophagic turnover. To clarify this, we utilized the mCherry-GFP-LC3 fluorescence system and found that CatB inhibition significantly impaired the fusion of autophagosomes with lysosomes (Fig. 5H, I). These results were further confirmed by transmission electron microscopy, which revealed an accumulation of autophagosomes and impaired lysosome fusion in CA-074Me-treated N2a cells (Fig. 5J–L). Inhibiting CatB reduces autophagy–lysosomal pathway activity which may, in turn, increase UPS activity. To determine whether this enhanced UPS activity contributes to PEG3 degradation, we treated N2a cells with MG132, a UPS inhibitor, and observed a significant increase in PEG3 protein levels. Furthermore, MG132 treatment abolished the pro-degradation effects of CA-074Me on PEG3 (Fig. 5O, P). Collectively, these findings demonstrate that CatB plays a crucial role in stabilizing PEG3 protein expression, primarily by regulating the balance between autophagy-lysosome and ubiquitin-proteasome pathways.

A Relative mRNA expression of PEG in the cortex of E12.5. WT (n = 4) and CatB−/− (n = 3) mice. B Immunoblot analysis of PEG3 in the N2a cells without or with 10 μM CA-074Me for 12 h. C Quantification of PEG3 in the immunoblots shown in (B). E Immunoblot analysis of PEG3 in the N2a cells exposed or not to 100 μg/ml CHX, 10 μM CA-074Me or a combination of both for 0, 1, 6, and 24 h. E Quantification of PEG3 in the immunoblots shown in (D). F Immunoblot analysis of LC3B in the N2a cells exposed or not to 10 μM CA-074Me. G Quantification the ratio of LC3B II/LC3B I in the immunoblots shown in (F). H Confocal image of the mCherry-GFP -LC3B signal in N2a cells exposed or not to 10 μM CA-074Me. The scale bar was 15 μm. I Quantification of the GFP-mCherry+ (red dots) and GFP+mCherry+ (yellow dots) positive LC3B puncta in (H) (n = 4 cells per group, the data were obtained from three independent experiments.). J Transmission electron microscope images of the autophagosome (red arrow) and autolysosome (yellow arrow) in N2a cells exposed or not to 10 μM CA-074Me. Scale bars were 1 μm and 500 nm. K, L Quantification of autophagosome (K) and autolysosome (L) shown in (H) (n = 9 cells per group). M Immunoblot analysis of ubiquitin in the N2a cells exposed or not to 10 μM CA-074Me. G Quantification of ubiquitin in the immunoblots shown in (M) (n = 3 per group, the data were obtained from three independent experiments. O Immunoblot analysis of PEG3 in the N2a cells exposed or not to 100 nM MG132, 10 μM CA-074Me or a combination of both for 12 h. P Quantification of PEG3 in the immunoblots shown in (O). (n = 3 per group, the data were obtained from three independent experiments.) *P < 0.05, ***P < 0.01, ***P < 0.001. Error bars represent mean ± SEM.
Discussion
In this study, we have provided evidence for the physiological roles of CatB in cortical development. Our findings suggest that CatB regulates the differentiation of neurons and their subsequent migration to cortical layer 6 through the stabilization of PEG3. We showed that CatB-deficient mice have normal cognitive function but exhibit depressive behaviors. However, the knockdown of CatB in adult WT mice did not result in similar depression-related behaviors. CatB-deficient mice exhibited impaired neuronal development and activity in layer 6 of the mPFC, which subsequently decreased neuronal activity in the aPVT, a depression-related brain region. Coincidentally, CatB was found to be highly expressed in the embryonic cerebral cortex, especially from E12.5 to E16.5, a critical stage in the development of cortical layer 6. At the intracellular level, we found that neither the deletion nor the inhibition of CatB affected Peg3 mRNA expression, but instead promoted PEG3 protein degradation. Mutations in PEG3 have been reported to induce defective neuronal connectivity55. Our findings enhance the understanding of the molecular and cellular mechanisms involved in the regulation of cortical development and highlight the critical role of CatB in neuronal differentiation and migration.
CatB expression is elevated in many pathological conditions and the pathological functions of CatB have been extensively reported7. However, studies relating to its physiological functions are relatively rare, potentially owing to the compensatory effect of CatL in CatB−/− mice25. Padamsey et al.35 found that lysosomal Ca2+ signaling in the brain regulates CatB exocytosis from lysosomes, which modulates long-term structural plasticity in dendritic spines by enhancing the activity of matrix metalloproteinase 9, an enzyme involved in extracellular matrix remodeling and synaptic plasticity. Extracellular CatB has also been reported to enhance axonal outgrowth in neurons by degrading chondroitin sulfate proteoglycans, major axon growth-inhibitory extracellular matrix components56. In addition to exerting direct effects on the brain, exercise has been shown to regulate brain function through the secretion of skeletal muscle-derived factors, called myokines. CatB has been identified as a myokine that enhances adult hippocampal neurogenesis and spatial memory function in response to exercise57,58. Here, we found that CatB deletion significantly inhibits neurite outgrowth in both primary cultured neurons and N2a cells. The punctate staining signals of CatB and LAMP2 were well co-localized at the neuritic edge of neurons, which suggests that CatB is involved in the initiation of neurite outgrowth26. Furthermore, we found that while learning and memory abilities did not differ between CatB−/− and WT mice, CatB knockout induced depressive behaviors in mice; however, the knockdown of CatB in adult WT mice did not induce similar effects. One explanation may be that, in mice, the loss of CatB causes structural abnormalities in the brain in early development but not in adulthood. Similar results have been reported for urokinase-type plasminogen activator receptor (uPAR), which participates in the functioning and development of the speech cortex. uPAR−/− mice exhibit enhanced susceptibility to epileptic seizures and cortical abnormalities consistent with altered neuronal migration and maturation59. Furthermore, we noted that CatB was relatively highly expressed in the embryonic cerebral cortex, especially from E12.5 to E16.5, a stage that is critical for cortical development60. Importantly, mice heterozygous for CatD deficiency showed susceptibility to stress-induced depression61; however, whether CatD and CatB induce depressive-like behaviors through similar mechanisms remains to be determined.
Abnormal development of the cortex can lead to impaired neural circuit connections. We found that there was a significant increase in the number of c-Fos-positive neurons in the PVT of CatB−/− mice in response to depression-like behavior, whereas the spontaneous electrophysiological properties of aPVT neurons were not affected. CTB retrograde tracing revealed that the aPVT received a large number of projections from layers 6 of the mPFC and insular cortex. In addition, we observed that the mPFC layer 6–aPVT circuit functions as a top-down pathway that underlies the pathology of CatB deficiency-induced depressive-like behavior. Substantial evidence supports that the mPFC is a major contributor to the pathophysiology of depression62. Changes in both mPFC architecture and the function of mPFC neurons have been reported in depression, including neuronal volume reduction63, synapse loss64, altered synaptic transmission65, and downstream structures66. Of note, mPFC thinning is closely related to the onset of MDD67, which is consistent with our findings. The mPFC and the PVT exhibit two-way projection68,69,70. The mPFC–PVT circuit is a critical higher-order cortico–thalamo–cortical integration site that plays a vital role in various behaviors, including reward seeking, cue saliency, and emotional memory69,71. Owing to a loose definition of the boundaries that separate the anterior and posterior PVT (aPVT and pPVT, respectively), as well as a failure to recognize the existence of a transitional middle region, many studies on PVT subregions have yielded conflicting results. Yamamuro et al. reported that the mPFC–pPVT circuit is closely related to social memory70. Li et al. showed that this circuit determines chronic social defeat stress-induced anhedonia, which is a key characteristic of depression72. Additionally, the sustained antidepressant-like effects of (2S,6S)-hydroxynorketamine in stressed animal models involve neurobiological changes in the aPVT42. Most recently, Xu and colleagues reported that Glutamatergic neurons in the anterior paraventricular thalamus (aPVT) responded to visceral pain, while those in the posterior PVT (pPVT) were more responsive to anxiety73. In this study, we found that CatB−/− mice did not display a preference for sucrose-containing water over normal water, which is a notable indication of anhedonia. Gao et al. defined two types of neurons in the PVT that differ markedly in gene expression, anatomy, and function74. These neurons are not restricted to specific antero–posterior locations, but rather are biased to opposite ends of a gradient, and have unique response patterns to different behaviors. In future work, we will explore whether CatB deficiency leads to abnormal mPFC–aPVT connections, whether this is the key feature leading to depressive-like behavior in CatB−/− mice and whether the enhancement of the mPFC–aPVT circuit can rescue this behavior. In addition, it is worth exploring if there is feedback regulation in the bidirectional projection between the mPFC and the aPVT under depressive-like behavior.
The selective degradation of proteins requires fine-tuned coordination between the proteasome and the lysosome, two independent but interconnected degradation systems75. In general, lysosomes are responsible for the degradation of long-lived proteins, insoluble protein aggregates, and even entire organelles via endocytosis, phagocytosis, or autophagy76. Meanwhile, proteasomes eliminate short-lived proteins and soluble misfolded proteins via the UPS77. In neuronal lysosomes, CatB has been reported to be involved in the degradation of amyloid beta (Aβ); indeed, CatB deficiency aggravates Aβ pathology in Alzheimer’s disease19,78, suggesting that CatB plays a key role in the maintenance of lysosomal function. Genetic studies in mice have demonstrated that the inactivation of autophagy by the knockout of essential autophagic genes results in the accumulation and aggregation of ubiquitylated proteins79,80, implying that the impairment of autophagy may be compensated for by the upregulation of the UPS. We found that PEG3 protein degradation was enhanced in the cortex of E12.5 CatB knockout mice as well as in cultured neurons pretreated with CatB inhibitors, and that CatB deficiency enhanced UPS-mediated degradation of PEG3. There are two possible interpretations for this result. Therefore, CatB deficiency-induced lysosome impairment may impact substrate flux through the UPS.
During development, newborn neurons migrate from their germinal zone and disperse throughout the CNS, finally integrating into an appropriate layer and forming part of a neuronal circuit60. Migrating neurons are defined by the presence of a leading process, a structure similar to the growth cones of growing axons. We previously found that the genetic deletion or pharmacological inhibition of CatB blocked lamellipodia, filopodia, or growth cone formation in cultured neurons26. In this study, we further observed that the physiological function of CatB includes the regulation of neuronal migration and cortical layer formation in mice. Mutations in another lysosomal enzyme, acid phosphatase 2, result in cerebellar defects, characterized by the excessive migration of Purkinje cells to the molecular layer81. In addition, the genetic deletion of vacuole protein sorting 18, a core protein in intracellular vesicle transport, leads to impaired neuronal migration due to the disruption of multiple vesicle transport pathways involved in lysosome production. These findings highlight the importance of lysosomes in neuronal migration. However, it remains unclear why CatB specifically affects the development of layer 6 of the mPFC. It has been characterized that specific neuronal transcription factors facilitated brain development44. We postulate that some key transcription factors that are highly expressed during cortical layer 6 development may also regulate CatB transcription. However, how CatB expression is regulated during cortical layer 6 development remains to be elucidated.
In conclusion, our study demonstrated that CatB plays a critical role in cortical layer 6 development and that CatB deficiency induces mPFC–PVT circuit-dependent depressive behavior in adult mice. Moreover, we observed that CatB is highly expressed between E12.5 and E16.5, which is the key stage in cortical layer 6 development. The inhibition of CatB enhanced PEG3 proteasomal degradation, resulting in reduced neuronal migration and differentiation. The mechanistic insights provided in this study offer a valuable background for understanding the function of CatB in brain development and may explain the association between CatB mutation and neurological disorders.
Data availability
All data are available in the main text or the supplementary materials.
References
Cooper, J. A. Cell biology in neuroscience: mechanisms of cell migration in the nervous system. J. Cell Biol. 202, 725–734 (2013).
Zhang, T. et al. Loss of Lgl1 disrupts the radial glial fiber-guided cortical neuronal migration and causes subcortical band heterotopia in mice. Neuroscience 400, 132–145 (2019).
Yang, D. Y., Beam, D., Pelphrey, K. A., Abdullahi, S. & Jou, R. J. Cortical morphological markers in children with autism: a structural magnetic resonance imaging study of thickness, area, volume, and gyrification. Mol. Autism 7, 11 (2016).
Birnbaum, R. & Weinberger, D. R. Genetic insights into the neurodevelopmental origins of schizophrenia. Nat. Rev. Neurosci. 18, 727–740 (2017).
Penagarikano, O. et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell 147, 235–246 (2011).
Halangk, W. et al. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Invest. 106, 773–781 (2000).
Xie, Z. et al. Cathepsin B in programmed cell death machinery: mechanisms of execution and regulatory pathways. Cell Death Dis. 14, 255 (2023).
Gondi, C. S. & Rao, J. S. Cathepsin B as a cancer target. Expert Opin. Ther. Targets 17, 281–291 (2013).
Palermo, C. & Joyce, J. A. Cysteine cathepsin proteases as pharmacological targets in cancer. Trends Pharm. Sci. 29, 22–28 (2008).
Yan, S. & Sloane, B. F. Molecular regulation of human cathepsin B: implication in pathologies. Biol. Chem. 384, 845–854 (2003).
Kos, J., Mitrovic, A. & Mirkovic, B. The current stage of cathepsin B inhibitors as potential anticancer agents. Future Med. Chem. 6, 1355–1371 (2014).
Van Acker, G. J. et al. Cathepsin B inhibition prevents trypsinogen activation and reduces pancreatitis severity. Am. J. Physiol. Gastrointest. Liver Physiol. 283, G794–G800 (2002).
Canbay, A. et al. Cathepsin B inactivation attenuates hepatic injury and fibrosis during cholestasis. J. Clin. Invest. 112, 152–159 (2003).
Hashimoto, Y. et al. Significance of cathepsin B accumulation in synovial fluid of rheumatoid arthritis. Biochem. Biophys. Res Commun. 283, 334–339 (2001).
Yoshifuji, H. et al. Amelioration of experimental arthritis by a calpain-inhibitory compound: regulation of cytokine production by E-64-d in vivo and in vitro. Int Immunol. 17, 1327–1336 (2005).
Sun, L. et al. Microglial cathepsin B contributes to the initiation of peripheral inflammation-induced chronic pain. J. Neurosci. 32, 11330–11342 (2012).
Hook, G., Jacobsen, J. S., Grabstein, K., Kindy, M. & Hook, V. Cathepsin B is a new drug target for traumatic brain injury therapeutics: evidence for E64d as a promising lead drug candidate. Front Neurol. 6, 178 (2015).
Ni, J. et al. The Critical Role of Proteolytic Relay through Cathepsins B and E in the Phenotypic Change of Microglia/Macrophage. J. Neurosci. 35, 12488–12501 (2015).
Sun, B. et al. Cystatin C-cathepsin B axis regulates amyloid beta levels and associated neuronal deficits in an animal model of Alzheimer’s disease. Neuron 60, 247–257 (2008).
Hook, V. Y., Kindy, M., Reinheckel, T., Peters, C. & Hook, G. Genetic cathepsin B deficiency reduces beta-amyloid in transgenic mice expressing human wild-type amyloid precursor protein. Biochem Biophys. Res Commun. 386, 284–288 (2009).
Kindy, M. S. et al. Deletion of the cathepsin B gene improves memory deficits in a transgenic ALZHeimer’s disease mouse model expressing AbetaPP containing the wild-type beta-secretase site sequence. J. Alzheimers Dis. 29, 827–840 (2012).
Embury, C. M. et al. Cathepsin B improves ss-amyloidosis and learning and memory in models of Alzheimer’s disease. J. Neuroimmune Pharm. 12, 340–352 (2017).
Pislar, A. et al. The role of cysteine peptidases in coronavirus cell entry and replication: the therapeutic potential of cathepsin inhibitors. PLoS Pathog. 16, e1009013 (2020).
Padmanabhan, P., Desikan, R. & Dixit, N. M. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS Comput. Biol. 16, e1008461 (2020).
Felbor, U. et al. Neuronal loss and brain atrophy in mice lacking cathepsins B and L. Proc. Natl Acad. Sci. USA 99, 7883–7888 (2002).
Jiang, M. et al. Cathepsin B inhibition blocks neurite outgrowth in cultured neurons by regulating lysosomal trafficking and remodeling. J. Neurochem. 155, 300–312 (2020).
Wu, Z. et al. Cathepsin B plays a critical role in inducing Alzheimer’s disease-like phenotypes following chronic systemic exposure to lipopolysaccharide from Porphyromonas gingivalis in mice. Brain Behav. Immun. 65, 350–361 (2017).
Meng, J. et al. Nucleus distribution of cathepsin B in senescent microglia promotes brain aging through degradation of sirtuins. Neurobiol. Aging 96, 255–266 (2020).
Ni, J. et al. Increased expression and altered subcellular distribution of cathepsin B in microglia induce cognitive impairment through oxidative stress and inflammatory response in mice. Aging Cell 18, e12856 (2019).
Ni, J. et al. Cathepsin H deficiency decreases hypoxia-ischemia-induced hippocampal atrophy in neonatal mice through attenuated TLR3/IFN-beta signaling. J. Neuroinflammation 18, 176 (2021).
Orlowski, G. M. et al. Multiple cathepsins promote pro-IL-1beta synthesis and NLRP3-mediated IL-1beta activation. J. Immunol. 195, 1685–1697 (2015).
Xie, Z. et al. Microglial cathepsin E plays a role in neuroinflammation and amyloid beta production in Alzheimer’s disease. Aging Cell 21, e13565 (2022).
Cecchi, C. & Boncinelli, E. homeogenes and mouse brain development. Trends Neurosci. 23, 347–352 (2000).
Mueller-Steiner, S. et al. Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer’s disease. Neuron 51, 703–714 (2006).
Padamsey, Z. et al. Activity-dependent exocytosis of lysosomes regulates the structural plasticity of dendritic spines. Neuron 93, 132–146 (2017).
Bickle, J. G. et al. 5-HT(1A) receptors on dentate gyrus granule cells confer stress resilience. Biol. Psychiatry 95, 800–809 (2023).
Goertsen, D. et al. AAV capsid variants with brain-wide transgene expression and decreased liver targeting after intravenous delivery in mouse and marmoset. Nat. Neurosci. 25, 106–115 (2022).
Flagel, S. B. The paraventricular nucleus of the thalamus and its potential role in psychopathology. Neuropsychopharmacology 47, 385–386 (2022).
Ito, H. T., Zhang, S. J., Witter, M. P., Moser, E. I. & Moser, M. B. A prefrontal-thalamo-hippocampal circuit for goal-directed spatial navigation. Nature 522, 50–55 (2015).
Kasahara, T. et al. Depression-like episodes in mice harboring mtDNA deletions in paraventricular thalamus. Mol. Psychiatry 21, 39–48 (2016).
Ren, S. et al. The paraventricular thalamus is a critical thalamic area for wakefulness. Science 362, 429–434 (2018).
Kawatake-Kuno, A. et al. Sustained antidepressant effects of ketamine metabolite involve GABAergic inhibition-mediated molecular dynamics in aPVT glutamatergic neurons. Neuron 112, 1265–1285.e10 (2024).
Chiang, S. Y. et al. Usp11 controls cortical neurogenesis and neuronal migration through Sox11 stabilization. Sci. Adv. 7, eabc6093 (2021).
Noack, F. et al. Multimodal profiling of the transcriptional regulatory landscape of the developing mouse cortex identifies Neurog2 as a key epigenome remodeler. Nat. Neurosci. 25, 154-+ (2022).
Tanaka, K. et al. Paternally expressed gene 3 promotes sexual dimorphism in metabolism and behavior. PLoS Genet. 18, e1010003 (2022).
Broad, K. D., Curley, J. P. & Keverne, E. B. Increased apoptosis during neonatal brain development underlies the adult behavioral deficits seen in mice lacking a functional. Dev. Neurobiol. 69, 314–325 (2009).
Xu, P. F. et al. Foxc1 establishes enhancer accessibility for craniofacial cartilage differentiation. Elife 10, e63595 (2021).
Biyani, S., Patil, A. S. & Swami, V. The influence of FOXC1 gene on development, organogenesis, and functions. Clin. Transl. Metab. 22 (2024).
Kim, D. Y., Hwang, I., Muller, F. L. & Paik, J. H. Functional regulation of FoxO1 in neural stem cell differentiation. Cell Death Differ. 22, 2034–2045 (2015).
Zhang, D. M., Liu, C., Li, H. & Jiao, J. W. deficiency of STING signaling in embryonic cerebral cortex leads to neurogenic abnormalities and autistic-like behaviors. Adv Sci. 7 (2020).
Li, L. et al. Regulation of maternal behavior and offspring growth by paternally expressed Peg3. Science 284, 330–333 (1999).
Shi, Y. et al. The effect of macrophage polarization on the expression of the oxytocin signalling system in enteric neurons. J. Neuroinflammation 18, 261 (2021).
Seifinejad, A. et al. Molecular codes and in vitro generation of hypocretin and melanin concentrating hormone neurons. Proc. Natl Acad. Sci. USA 116, 17061–17070 (2019).
Adnani, L. et al. Zac1 regulates the differentiation and migration of neocortical neurons via Pac1. J. Neurosci. 35, 13430–13447 (2015).
Li, L. L. et al. Regulation of maternal behavior and offspring growth by paternally expressed. Science 284, 330–333 (1999).
Tran, A. P., Sundar, S., Yu, M., Lang, B. T. & Silver, J. Modulation of receptor protein tyrosine phosphatase sigma increases chondroitin sulfate proteoglycan degradation through cathepsin B secretion to enhance axon outgrowth. J. Neurosci. 38, 5399–5414 (2018).
Moon, H. Y. et al. Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab. 24, 332–340 (2016).
Ni, J., Lan, F., Xu, Y., Nakanishi, H. & Li, X. Extralysosomal cathepsin B in central nervous system: Mechanisms and therapeutic implications. Brain Pathol. 32, e13071 (2022).
Royer-Zemmour, B. et al. Epileptic and developmental disorders of the speech cortex: ligand/receptor interaction of wild-type and mutant SRPX2 with the plasminogen activator receptor uPAR. Hum. Mol. Genet. 17, 3617–3630 (2008).
Hevner, R. F. et al. Tbr1 regulates differentiation of the preplate and layer 6. Neuron 29, 353–366 (2001).
Zhou, R. et al. Mice heterozygous for cathepsin D deficiency exhibit mania-related behavior and stress-induced depression. Prog. Neuro-Psychoph. 63, 110–118 (2015).
Krishnan, V. & Nestler, E. J. The molecular neurobiology of depression. Nature 455, 894–902 (2008).
Rajkowska, G. et al. Morphometric evidence for neuronal and glial prefrontal cell pathology in major depression. Biol. Psychiat 45, 1085–1098 (1999).
Kang, H. J. et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat. Med. 18, 1413-+ (2012).
Duman, R. S. & Aghajanian, G. K. Synaptic dysfunction in depression: potential therapeutic targets. Science 338, 68–72 (2012).
Duman, R. S., Sanacora, G. & Krystal, J. H. Altered connectivity in depression: GABA and glutamate neurotransmitter deficits and reversal by novel treatments. Neuron 102, 75–90 (2019).
Treadway, M. T. et al. Illness progression, recent stress, and morphometry of hippocampal subfields and medial prefrontal cortex in major depression. Biol. Psychiat. 77, 285–294 (2015).
Viena, T. D., Rasch, G. E. & Allen, T. A. Dual medial prefrontal cortex and hippocampus projecting neurons in the paraventricular nucleus of the thalamus. Brain Struct. Funct. 227, 1857–1869 (2022).
Kato, T. M. et al. Presynaptic dysregulation of the paraventricular thalamic nucleus causes depression-like behavior. Sci. Rep.—UK 9, 16506 (2019).
Yamamuro, K. et al. A prefrontal-paraventricular thalamus circuit requires juvenile social experience to regulate adult sociability in mice. Nat. Neurosci. 23, 1240 (2020).
Lucantonio, F. et al. Aversive stimuli bias corticothalamic responses to motivationally significant cues. Elife 10, e57634 (2021).
Li, H. et al. Discrete prefrontal neuronal circuits determine repeated stress-induced behavioral phenotypes in male mice. Neuron 112, 786–804.e8 (2024).
Li, D. et al. The paraventricular thalamus mediates visceral pain and anxiety-like behaviors via two distinct pathways. Neuron 113, 1–15 (2025).
Gao, C. et al. Two genetically, anatomically and functionally distinct cell types segregate across anteroposterior axis of paraventricular thalamus. Nat. Neurosci. 23, 217 (2020).
Zhao, L., Zhao, J., Zhong, K. H., Tong, A. P. & Jia, D. Targeted protein degradation: mechanisms, strategies and application. Signal Transduct Target Ther. 7 (2022).
Martini-Stoica, H., Xu, Y., Ballabio, A. & Zheng, H. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 39, 221–234 (2016).
Pohl, C. & Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 366, 818-+ (2019).
Wang, C., Sun, B. G., Zhou, Y. G., Grubb, A. & Gan, L. Cathepsin B degrades amyloid-β in mice expressing wild-type human amyloid precursor protein. J. Biol. Chem. 287, 39834-41 (2012).
Hara, T. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006).
Komatsu, M. et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006).
Bailey, K., Balaei, M. R., Mehdizadeh, M. & Marzban, H. Spatial and temporal expression of lysosomal acid phosphatase 2 (ACP2) reveals dynamic patterning of the mouse cerebellar cortex. Cerebellum 12, 870–881 (2013).
Acknowledgements
The authors thank the Biological and Medical Engineering Core Facilities of Beijing Institute of Technology for supporting experimental equipment. This work was supported by the following grants: Beijing Life Science Academy (BLSA) 2024300CD0080 and 2023000CB0010, Alzheimer’s Disease Pathogenesis and Drug Development Innovation Team, Innovation Team Project of Guangdong General Colleges and Universities (Natural Science) 2024KCXTD016, National Natural Science Foundation of China grant 32070954, Chunhui project of Ministry of Education of the People’s Republic of China HZKY20220041, and Key Science and Technology Research Program of Hebei Provincial Health Commission Grant ZF2024142. Schematic diagrams were created with BioRender.com. We gratefully acknowledge the use of this tool for figure preparation.
Author information
Authors and Affiliations
Contributions
Z.X. and Y.Q. performed experiments, analyzed data, and wrote the paper. F.L., W.K., and S.Z. performed some experiments. J.S., Y.Y., and Z.Q. analyzed data and provided samples or reagents. Z.B., H.Q., and J.M. designed the experiments. J.N. supervised this research, analyzed data, and wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editors: Christian Wozny and Benjamin Bessieres. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xie, Z., Yang, Q., Lan, F. et al. Cathepsin B deficiency disrupts cortical development via PEG3, leading to depression-like behavior. Commun Biol 8, 1097 (2025). https://doi.org/10.1038/s42003-025-08508-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-025-08508-8
