
Abstract
Recent studies have reported the involvement of complement system proteins in the initiation and progression of islet autoimmunity (IA) in the study of Type 1 diabetes (T1D). However, the genetic factors of complement system proteins at the time of triggering of IA are unknown. Through complement system protein quantitative trait locus (pQTL) mapping discovery analysis of 170 participants from the Diabetes Autoimmunity Study in the Young (DAISY) and replication analysis of 385 IA cases from The Environment Determinants of Diabetes in the Young (TEDDY) study, we identify 68 significant pQTLs in total for C8A, C8B, CFB, C4A, and MBL2. Furthermore, all replicated pQTLs of CFB and C4A are previously reported to be associated with T1D risk. Our study provides evidence for the potential biological roles of complement system proteins in the etiology of IA and T1D for young children at high risk of developing T1D.
Similar content being viewed by others

Introduction
Type 1 diabetes (T1D) is a complex autoimmune disease where an autoimmune attack destroys the pancreatic islet beta cells, which eventually results in a complete dependency on exogenous insulin. There is no cure for T1D yet, and the disease progression is not well understood. Hence, the development of biomarkers that can help track disease progression and determine disease causes is important for T1D prevention and therapy. The islet antibodies are well-recognized and reliable diagnostic biomarkers that mark the onset of islet autoimmunity (IA), and persistent multi-positivity of islet autoantibodies can be used to predict individuals at high risk of developing T1D1,2. However, the genetic mechanisms of the initiation of IA, as well as the progression from IA to clinical diabetes, are still largely unknown.
The complement system, also known as the complement cascade, plays an important role in the innate immune system, which helps to fight bacterial or viral infections and promotes clearance of damaged cells3. Three interrelated pathways activate the complement system, specifically the classical, lectin, and alternative pathways. These activation pathways converge at the terminal pathway, where much of the effector function resides3. The complement system also modulates adaptive immunity through activation by antibodies, including autoantibodies4. Several studies have suggested that the dysregulation of the complement system contributes to the pathogenesis of autoimmune diseases, such as systemic lupus erythematosus5, rheumatoid arthritis6, and T1D7,8. More specifically, increasing studies have reported the involvement of the complement system in the initiation and progression of IA for the study of T1D9,10. Furthermore, recent studies observed lower levels of several complement system proteins in children with islet autoantibodies compared to healthy controls11,12,13,14, and this observation of reduced levels in complement proteins was persistent in both before and after the detection of autoantibodies14. Although the associations of genetic variants within several complement proteins with IA have been studied7, the genetic factors of complement system proteins at the time of triggering of IA are currently unclear15.
Circulating plasma proteins play a fundamental role in human biological processes and are frequently the targets of pharmaceutical interventions16. Protein quantitative trait locus (pQTL) mapping is an approach that can identify genetic variants underlying variation in protein expression levels. In prior studies, pQTL analysis has led to critical advances in knowledge of the genetic architecture of plasma proteins and their relationship to disease17,18,19,20,21.
In this study, we aim to improve our understanding of the genetic factors of complement system proteins at the time of triggering of IA and bring us closer to dissecting the genetic mechanisms underlying the development and progression of T1D. Here, we performed pQTL analysis of 170 participants in the Diabetes Autoimmunity Study in the Young (DAISY) to identify statistically significant pQTLs for complement system proteins. Complement system proteins for DAISY participants were measured by two different protein assay platforms, selected reaction monitoring (SRM)22, and Exsera Biolabs23 (hereinafter referred to as Exsera). Complement system proteins measured by the two platforms overlapped by approximately overlapped in half. Hence, using proteins from both platforms provides a more comprehensive list of complement system proteins for study. Replication analysis was then conducted on 385 IA cases in the Environment Determinants of Diabetes in the Young (TEDDY) study, and the proteins were measured by SRM for TEDDY participants. Finally, we used the T1D Knowledge Portal to examine replicated pQTLs for their association with T1D and other disease-relevant traits. We identified 68 statistically significant pQTLs in total for C8A, C8B, CFB, C4A, and MBL2. All replicated pQTLs of CFB and C4A were previously reported to be associated with T1D risk. Our study provides evidence for the potential biological roles of complement system proteins in the etiology of IA and T1D for young children at high risk of developing T1D.
Results
Participant characteristics
The demographic and clinical characteristics of the study samples are summarized in Table 1, which includes 170 samples with 131 IA cases and 39 controls for DAISY discovery analysis, and 385 IA cases for TEDDY replication analysis after combining available proteomics and genotype data (“Methods”). Overall, the participants in the DAISY study had a median age of 6.2 years, which was older than TEDDY participants with a median age of 1.8 years (Table 1). The percentage of first-degree relatives with T1D in DAISY was 54%, much higher than 22% from TEDDY (Table 1). For the other two clinical characteristics, female (%) and self-reported Non-Hispanic White (NHW) (%), the percentages were similar between the two studies. Females accounted for 46% and 44% in DAISY and TEDDY, respectively, and NHW was the most common population in both studies.
cis-pQTLs discovery analysis in DAISY
We first performed cis-pQTL mapping for DAISY samples in pooled (IA cases and controls) and IA case-stratified analyses, respectively (“Methods”). There were 19 and 16 complement system proteins measured by the SRM and Exsera platform, respectively, for DAISY samples, and 10 proteins overlapped between the two platforms (Table 2). We observed 14 proteins with false discovery rate (FDR)-significant (FDR < 0.1) cis-pQTLs (Table 2). MBL2 has the greatest number of FDR-significant cis-pQTLs with 264 and 211 cis-pQTLs identified from Exsera pooled and case-stratified analysis, respectively (Supplementary Data 1). However, no statistically significant cis-pQTLs of MBL2 from the SRM platform were identified. Additionally, MBL2 had the cis-pQTLs with the greatest strength of statistical significance (chr10:52771475-T, chr10:52773600-G, chr10:52775077-T with p-value = 3.12 × 10−19 and p-value = 5.22 × 10−16 from Exsera pooled and case-stratified analysis, respectively, Supplementary Data 1). Among other genes, CFH and C4B had FDR-significant cis-pQTLs from both SRM and Exsera platforms (Table 2).
For six proteins that had many FDR-significant cis-pQTLs (i.e., CFH, C8A, CFB, C4A, C4B, and MBL2), we performed fine-mapping to identify potential causal cis-pQTLs for each protein (“Methods”). For the proteins C8A, CFB, and C4B, we observed corresponding 95% credible sets (CS) containing 29, 26, and 9 fine-mapped variants, respectively, from the SRM pooled analysis. For CFH, we observed a single 95% CS with 69 fine-mapped variants in the pooled analysis of protein levels from the Exsera platform (Supplementary Figs. 1–4). MBL2 has fine-mapped variants from both Exsera analyses, where 14 variants were identified in two 95% CSs from pooled analysis, and 4 variants were identified in one 95% CS from case-stratified analysis (Supplementary Fig. 5). However, there were no fine-mapped variants for CFH and C4A from SRM pooled analysis (Supplementary Data 1). We used non-fine-mapped cis-pQTLs for these two proteins from the corresponding analysis. After fine-mapping analysis, we identified 240 FDR-significant (FDR < 0.1) cis-pQTLs in total from DAISY discovery analysis.
Replicated cis-pQTLs in TEDDY
To follow up on our discovery pQTL analysis in DAISY, we performed replication analysis in TEDDY for the 240 cis-pQTLs reported in Supplementary Data 1, which map to 14 proteins. A cis-pQTL was deemed replicated if it had an FDR-corrected p-value < 0.05. Of these, we observed 68 cis-pQTLs passing the replication threshold, including 27 for C8A, 23 for CFB, 12 for C4A, 4 for MBL2, and 2 for C8B, respectively. The summary of all replicated cis-pQTLs for C8B and MBL2, and top replicated cis-pQTLs for C8A, CFB, and C4A is presented in Table 3. The other replicated cis-pQTLs for C8A, CFB, and C4A are presented in Supplementary Data 2. The replicated cis-pQTLs of CFB, C4A, and MBL2 decreased protein expression levels, while the replicated cis-pQTLs of C8A and C8B increased protein levels. All replicated cis-pQTLs had the same direction of effect between DAISY and TEDDY. However, the replicated signals in TEDDY generally achieved a lower level of statistical significance compared to DAISY. One exception to this finding was the CFB pQTL (chr10:31562639-A: Beta (SE) = −0.45 (0.11) and p-value = 3.92 × 10−5 in TEDDY vs Beta (SE) = −0.34 (0.08) and p-value = 4.37 × 10−5 in DAISY; Supplementary Data 1 and 2). To investigate the impact of HLA genotypes on replicated pQTLs, we first examined the association between HLA genotypes and five proteins with replicated pQTLs (i.e., C8A, C8B, CFB, C4A, and MBL2). We found that the T1D high-risk HLA genotype DR4/4 was significantly associated with C4A in both DAISY (Beta (SE) = 0.27 (0.10) and p-value = 9.45 × 10−3) and TEDDY (Beta (SE) = 0.37 (0.15) and p-value = 0.01) (Supplementary Table 1). Based on this observation, we further carried out pQTL mapping of C4A in TEDDY with additional adjustment for HLA genotypes. We found that 11 out of 12 C4A replicated pQTLs were still nominally significant (Supplementary Data 2), which indicates minimal impact of HLA-DR-DQ genotypes on our reported pQTL associations for C4A.
Examination of association with T1D for replicated pQTL variants
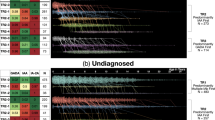
We further examined the association of replicated cis-pQTLs with T1D through publicly available resources. The T1D Knowledge Portal (https://t1d.hugeamp.org) enables searching of human genomic variants linked to T1D and other related phenotypes. We used the T1D Knowledge Portal and found that all 23 and 12 replicated cis-pQTLs for CFB and C4A, respectively, have been reported genome-wide significantly (p-value < 5 × 10−8) associated with T1D risk (Figs. 1 and 2 and Supplementary Data 2). The T1D association p-values were obtained from a previous T1D study24, and the p-values range from 1.11 × 10−37 (C4A: rs9262570) to 6.29 × 10−235 (CFB: rs114355928) (Supplementary Data 2). For the other replicated cis-pQTLs, we did not find any nominally significant association with T1D risk, however, we found that they were significantly associated with highlight scatter reticulocyte count (C8A and C8B, Supplementary Figs. 6 and 7), chronic kidney disease, and reticulocyte count (MBL2, Supplementary Figs. 8 and 9).

PheWAS results were obtained from T1D Knowledge Portal (https://t1d.hugeamp.org); the phenotypes in the Figure are sorted by the association p-value between the variant and the phenotype. The Figure shows the top 25 associations out of 232 phenotypes; Beta, effect size of association between rs150978802 and phenotype; error bar indicates 95% confidence interval.

PheWAS results were obtained from T1D Knowledge Portal (https://t1d.hugeamp.org); the phenotypes in the Figure are sorted by the association p-value between the variant and the phenotype. The Figure shows the top 25 associations out of 202 phenotypes; Beta, the effect size of the association between rs9263822 and phenotype. The error bar indicates a 95% confidence interval.
Discussion
Previous reports have examined the involvement of the complement system in the initiation and progression of IA for the study of T1D8,11,12,14, however, the genetic factors underlying the complement system proteins at the time of triggering of IA are poorly unknown. Our study investigated genetic variants that are associated with complement system proteins in DAISY, a prospective cohort of children from the general population who either had a first-degree relative with T1D or had a high-risk human leukocyte antigen (HLA) genotype. From pooled and IA case-stratified analyses with 170 participants in the DAISY, 240 significant (FDR < 0.1) cis-pQTLs were associated with 14 complement system proteins. We replicated 68 cis-pQTLs with statistical significance (FDR < 0.05) in the 385 IA cases from the TEDDY study. The 68 replicated cis-pQTLs represent C8A and C8B, CFB and C4A (within the HLA region), and MBL2. The CFB and C4A cis-pQTLs are known genetic associations with T1D. However, the strong linkage disequilibrium within the region has limited our understanding of the independent contribution of the complement system to T1D risk. Our study provides insight into the role of the complement system in disease progression.
CFB (Complement Factor B) is located on chromosome 6 and between the HLA class II and class I regions7. The HLA region is the single most important genetic determinant of T1D susceptibility. The variability in the HLA region has been estimated to explain approximately 60% of the genetic influence of T1D25. CFB is a component of the alternative pathway of complement activation. The detailed function of CFB has been reported in a previous study15. T1D-associated genetic variants in CFB have been reported in European ancestry26,27 and Northern India28. The 23 replicated cis-pQTLs of CFB identified in our study, which decrease protein level, are found to be significantly associated with increasing T1D risk in a large-scale genomic study of T1D24. The T1D association p-value ranges from 5.38 × 10−43 (rs115272033, EHMT2 intron) to 6.29 × 10−235 (rs114355928, TSBP1 intron) (Supplementary Data 2).
C4A (Complement Component 4A) is also located between the HLA class II and class I regions7 and is part of the classical and lectin pathways of activating the complement system. Two functionally distinct genes, C4A and C4B, code for the C4 protein together. Limited T1D association studies have been focused on the structural variation of this region15. The previously reported associations of genetic variants in C4A with T1D focused on European ancestry population29,30. Our study found that the 12 replicated cis-pQTLs of C4A were previously identified to be significantly associated with increasing T1D risk24. The T1D association p-value ranges from 1.11 × 10−37 (rs9262570, intergenic) to 1.33 × 10−88 (rs9263822, PSORS1C3 intron) (Supplementary Data 2).
Our study identified three genes outside of the HLA region (C8A, C8B, and MBL2) in addition to two genes in the HLA region. Both C8A (Complement C8 Alpha Chain) and C8B (Complement C8 Beta Chain) are located on chromosome 1 and encode Complement Component 8 (C8) protein. C8 participates in the formation of the membrane attack complex, which causes cell lysis and/or pro-inflammatory signaling15.
MBL2 (Mannose-Binding Lectin 2) is located on chromosome 10 and is the only single gene to encode human Mannose-Binding Lectin (MBL). MBL is a soluble lectin that activates the lectin complement pathway by recognizing microorganisms through the carbohydrate-recognition domain, thereby modulating inflammation31. Although our replicated cis-pQTLs of MBL2 were not associated with T1D, previous studies have identified their associations with type 2 diabetes and pneumonia. For example, the variant rs1800450 has been linked to type 2 diabetes in diverse populations, including the full-heritage Pima Indians and the Old Order Amish32, and the North Chinese Han population33. Additionally, Uysalol et al.34 reported that the rs1800450 genotypes associated with low MBL expression were significantly more common in patients with pneumonia and severe infections.
Strengths of this study include being one of the very few studies that examined pQTL mapping of complement system proteins in children with high risk of developing T1D and the use of independent study data to replicate significant pQTL signals. However, some limitations warrant mentioning. First, the statistical power of identifying significant cis-pQTLs in our study is still limited due to the sample size of the discovery analysis (Supplementary Table 2). Second, the protein assay platform in the replication analysis is different from that in the discovery analysis for the significant findings of MBL2. We only identified statistically significant cis-pQTLs in the Exsera platform for MBL2 in the DAISY discovery analysis, however, the replication analysis in TEDDY was only available to use measured protein levels from a different protein assay, the SRM platform. The lack of statistical significance in MBL2 in the SRM platform compared to the Exsera may result from a variety of reasons. The SRM method utilizes multiple reaction mass spectrometry to measure the abundance of specific peptides22, while Exsera is a standard immunology method that utilizes monoclonal antibody pairs to measure specific epitopes23, which might not overlap. Endogenous protein processing and genetic polymorphism might affect the assays differently as well. In the case of SRM, signal interference with chemical background or coeluting peptides can be an issue. However, the peptides measured by SRM in this study were pre-tested as previously described13,14, and any peptides with signal interference were excluded from the analysis. These differences in methodology will lead to differences in sensitivity and specificity, which might affect the detection of MBL2 pQTLs. Hence, future validation for the identified cis-pQTLs of MBL2 needs to be conducted on measured protein levels from the Exsera platform. Lastly, the participants in our study are predominantly composed of NHWs. Hence, our findings cannot be generalized to non-European ancestry populations yet. Future research focusing on non-European or diverse ancestries will help improve the understanding of genetic mechanisms underlying the complement system and provide a more comprehensive insight into the role of complement system proteins in the etiology of T1D.
Methods
Overview of approach
We performed cis-pQTL mapping for 170 samples (131 IA cases and 39 controls) in The DAISY as a discovery analysis to identify statistically significant cis-pQTL variants for complement system proteins. To follow up on our pQTL analysis in DAISY, we conducted replication analysis for 385 IA samples from the Environmental Determinants of Diabetes in the Young (TEDDY) study. Finally, we used the T1D Knowledge Portal (https://t1d.hugeamp.org/) to examine replicated cis-pQTLs for their association with T1D and other disease-relevant traits.
DAISY: study design, proteomic profiling, and genotyping
Study design
DAISY is a prospective cohort of 2547 children from the general population who either had a first-degree relative with T1D or had a high-risk HLA genotype. The participants were recruited in Denver, Colorado, between 1993 and 2004, and followed for up to 21 years35,36. Follow-up results are available through April 4, 2022. The primary goal of DAISY is to learn how genes and the environment interact to cause childhood T1D35.
Proteomic profiling
The peptides of complement system proteins were measured by two different assay platforms for DAISY participants, SRM-based22, and Exsera Biolabs-based (referred herein as Exsera)23. The Exsera targeted proteomics uses commercial immunoassays in a College of American Pathologists/Clinical Laboratory Improvement Amendments (CAP/CLIA)-accredited laboratory. The details of two protein assay platforms for DAISY participants are described in a previous study14.
Genotype quality control and imputation
Genome-wide genotyping was performed using the custom-designed Infinium TEDDY-T1D Exome array (Illumina) and was genotyped at the University of Virginia (UVA) Genome Sciences Laboratory following the manufacturer’s protocol (Illumina). The following quality control (QC) criteria were applied: (1) samples with a genotype call rate < 0.95 were removed; and (2) single nucleotide polymorphism (SNP) level QC included removal of monomorphic SNPs, and removal of SNPs that deviated from Hardy-Weiberg equilibrium (p-value < 1 × 10−20 at the HLA region or p-value < 1 × 10−6 otherwise). Genome-wide imputation used the Trans-Omics for Precision Medicine (TOPMed) multi-ancestry reference panel (version R2). The SNP was selected if the minor allele frequency (MAF) ≥ 0.05 and the imputation quality R2 ≥ 0.7. The details of the QC criteria of genomic variants used in this study are provided in a previous study37.
TEDDY: study design, proteomic profiling, and genotyping
Study design
TEDDY is an international prospective study that was designed to identify T1D-associated environmental factors in children who carry a high genetic risk for the disease38. The participating clinical centers include Colorado, Georgia/Florida, and Washington in the US, and Finland, Germany, and Sweden in Europe. We only reported summary statistics of self-reported NHW from US centers for TEDDY, as many missing or unknown race/ethnicity were reported from European centers.
Proteomic profiling and genotyping
Both proteomics and imputed genotype data were provided by the TEDDY Data Coordinating Center. The peptides of complement system proteins for TEDDY participants were measured by an SRM-based assay. The details of the protein assay for TEDDY are described in a previous study13. SNPs were genotyped using the ImmunoChip and/or the TEDDY-T1DexomeChip at the Center for Public Health Genomics at the UVA, US. GWAS imputation analysis was conducted using the TOPMed Version R2 (built from 97,256 deeply sequenced human genomes containing 308,107,085 genetic variants), the 1000 Genomes, and a subset of the TEDDY subjects (n = 1119) with the whole-genome sequencing data as reference panels. MetaMinimac2 was used to combine genotype data imputed against these three reference panels. For imputed genotype data, we retained rare variants with MAF > 0.05 in unrelated controls with European ancestry and with imputation quality R2 > 0.50.
cis-pQTL mapping in DAISY
A cis-pQTL mapping is to test the association between measured protein levels and cis-pQTL genomic variants via a statistical model. We defined a cis-pQTL genomic variant as a SNP within ±1 Mb of the transcription start site (TSS) of the corresponding protein-coding gene. We applied a linear mixed model adjusted for age, sex, self-reported race/ethnicity, protein plate effects, first-degree relative with T1D (yes or no), the first two principal components (PCs) of genetic ancestry, and the genetic relationship matrix (GRM) to perform cis-pQTL mapping in our study. The association analyses were conducted using R/GENESIS39. The mapping results were then filtered on (1) expected heterozygosity count (EHC) > 6 for WGS data, and expected heterozygosity count2) imputation quality > 0.3 and EHC > 6 for imputation genotypes by using R/EasyQC40.
We first performed cis-pQTL mapping on DAISY participants with IA, herein referred to as IA cases. Participants were considered to have IA if they were positive for one or more islet autoantibody tests on two or more consecutive visits or were autoantibody positive with a diagnosis of diabetes at the next visit by the American Diabetes Association criteria41. For protein levels of IA cases, (1) we averaged measured peptides if a participant had multiple peptides at one time point, and (2) we used the earliest time point if a participant had multiple visits for peptide measures. We then applied log2-transformation on the selected peptides and treated the transformed peptides as outcome variables for the linear mixed model. Due to limited IA cases in DAISY, we also conducted a case-control pooled analysis for DAISY with additional group adjustment (i.e., case or control) to increase the statistical power of cis-pQTL mapping. For protein levels of controls, we followed the same procedure as IA cases, except for the selection of time points for controls who had multiple peptides across different visits. To obtain a similar age as IA cases for most controls, we selected the closest time point to the median age of selected visits of IA cases for controls. Finally, we applied FDR correction (Benjamini-Hochberg) at 10% on cis-pQTL mapping results to identify statistically significant cis-pQTLs in DAISY by different protein assay platforms and by different stratified analyses, respectively. For proteins with many FDR-significant cis-pQTLs, we further performed fine-mapping using SuSiE42 to identify potential causal cis-pQTLs. The variant clustered in a 95% CS is considered a fine-mapped variant42.
Replication analysis in TEDDY
To follow-up on statistically significant cis-pQTLs identified in DAISY discovery analysis, we applied a linear mixed model adjusted for age, sex, protein plate effects, clinical centers, first-degree relative with T1D (yes or no), the first two PCs of ancestry, and GRM to test association between DAISY FDR-significant cis-pQTLs and log2-transformed peptides for TEDDY IA cases. We then applied FDR correction at 5% on TEDDY mapping results to identify replicated cis-pQTLs.
PheWAS of replicated cis-pQTLs
The T1D Knowledge Portal (https://t1d.hugeamp.org) enables searching of human genomic variants linked to T1D and other related phenotypes. We used the T1D Knowledge Portal to examine replicated cis-pQTLs for their association with T1D and other disease-relevant traits.
Ethical approval Informed consent
For DAISY cohort, written informed consent was obtained from participants and parents. The Colorado Multiple Institutional Review Board approved all protocols of DAISY cohort. For TEDDY study,written informed consentwas obtained for all study participants from a parent or a primary caretaker for both genetic screening and participation in the prospective follow-up for the TEDDY Study. All procedures were approved by the ethics committees/ institutional review boards including Colorado Multiple Institutional Review Board (04-0361);Medical College ofGeorgia Human Assurance Committee (2004–2010)/Georgia Health Sciences University Human Assurance Committee (2011–2012)/Georgia Regents University Institutional Review Board (2013–2017)/Augusta University Institutional Review Board (2017-present) (HAC 0405380); University of Florida Health Center Institutional Review Board (IRB201600277); Washington State Institutional Review Board (2004–2012)/Western Institutional Review Board (2013–present) (20130211); Ethics Committee of the Hospital District of Southwest Finland (Dnro168/2004); Bayerischen Landesärztekammer (Bavarian Medical Association) Ethics Committee (04089); and Regional Ethics Board in Lund, Section 2 (2004–2012)/Lund University Committee for Continuing Ethical Review (2013–present) (217/2004). In addition, TEDDY is monitored by an external evaluation committee formed by the National Institutes of Health, Bethesda, MD, U.S.A.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The TEDDY Immunochip (SNP) data that support the findings of this study have been deposited in NCBI’s database of Genotypes and Phenotypes (dbGaP) with the primary accession code phs001442.v4.p3. The TEDDY-T1DExome Array data that support the findings of this study have been deposited in NCBI’s database of Genotypes and Phenotypes (dbGaP) with the primary accession code phs001442.v4.p3. The TEDDY Whole Genome Sequencing (WGS) data that support the findings of this study have been deposited in NCBI’s database of Genotypes and Phenotypes (dbGaP) with the primary accession code phs001442.v4.p3. The TEDDY Proteomics Discovery Phase data that support the findings of this study are available from the Mass Spectrometry Interactive Virtual Environment (MassIVE) repository https://massive.ucsd.edu/ via its dataset identifier MSV000091560 (https://doi.org/10.25345/C5V11VW3F). The TEDDY Proteomics Validation Phase data that support the findings of this study are available from the Mass Spectrometry Interactive Virtual Environment (MassIVE) repository https://massive.ucsd.edu/ via its dataset identifier MSV000091562 (https://doi.org/10.25345/C5KH0F84V).
References
Eringsmark Regnéll, S. & Lernmark, A. The environment and the origins of islet autoimmunity and type 1 diabetes. Diabet. Med. J. Br. Diabet. Assoc. 30, 155–160 (2013).
Krischer, J. P. et al. Characteristics of children diagnosed with type 1 diabetes before vs after 6 years of age in the TEDDY cohort study. Diabetologia 64, 2247–2257 (2021).
Holers, V. M. Complement and its receptors: new insights into human disease. Annu. Rev. Immunol. 32, 433–459 (2014).
Radillo, O. et al. Complement-fixing islet cell antibodies in type-1 diabetes can trigger the assembly of the terminal complement complex on human islet cells and are potentially cytotoxic. Clin. Immunol. Immunopathol. 79, 217–223 (1996).
Chen, M., Daha, M. R. & Kallenberg, C. G. M. The complement system in systemic autoimmune disease. J. Autoimmun. 34, J276–J286 (2010).
Sturfelt, G. & Truedsson, L. Complement in the immunopathogenesis of rheumatic disease. Nat. Rev. Rheumatol. 8, 458–468 (2012).
Törn, C. et al. Complement gene variants in relation to autoantibodies to beta cell specific antigens and type 1 diabetes in the TEDDY Study. Sci. Rep. 6, 27887 (2016).
Ajjan, R. A. & Schroeder, V. Role of complement in diabetes. Mol. Immunol. 114, 270–277 (2019).
Zhang, Q. et al. Serum proteomics reveals systemic dysregulation of innate immunity in type 1 diabetes. J. Exp. Med. 210, 191–203 (2013).
Sarkar, S. et al. Systematic review of type 1 diabetes biomarkers reveals regulation in circulating proteins related to complement, lipid metabolism, and immune response. Clin. Proteom. 20, 38 (2023).
Moulder, R. et al. Serum proteomes distinguish children developing type 1 diabetes in a cohort with HLA-conferred susceptibility. Diabetes 64, 2265–2278 (2015).
von Toerne, C. et al. Peptide serum markers in islet autoantibody-positive children. Diabetologia 60, 287–295 (2017).
Nakayasu, E. S. et al. Plasma protein biomarkers predict the development of persistent autoantibodies and type 1 diabetes 6 months prior to the onset of autoimmunity. Cell Rep. Med. 4, 101093 (2023).
Webb-Robertson, B.-J. M. et al. Decrease in multiple complement proteins associated with development of islet autoimmunity and type 1 diabetes. iScience 27, 108769 (2024).
Onengut-Gumuscu, S. et al. Genetic variants in the complement system and their potential link in the aetiology of type 1 diabetes. Diabetes Metab. Res. Rev. 40, e3716 (2024).
Sun, B. B. et al. Genomic atlas of the human plasma proteome. Nature 558, 73–79 (2018).
Suhre, K. et al. Connecting genetic risk to disease end points through the human blood plasma proteome. Nat. Commun. 8, 14357 (2017).
Yao, C. et al. Genome-wide mapping of plasma protein QTLs identifies putatively causal genes and pathways for cardiovascular disease. Nat. Commun. 9, 3268 (2018).
Yang, Z. et al. Genetic landscape of the ACE2 coronavirus receptor. Circulation 145, 1398–1411 (2022).
Katz, D. H. et al. Whole genome sequence analysis of the plasma proteome in black adults provides novel insights into cardiovascular disease. Circulation 145, 357–370 (2022).
Jiang, M.-Z. et al. Whole genome sequencing based analysis of inflammation biomarkers in the Trans-Omics for Precision Medicine (TOPMed) consortium. Hum. Mol. Genet. 33, 1429–1441 (2024).
Nakayasu, E. S. et al. Tutorial: best practices and considerations for mass-spectrometry-based protein biomarker discovery and validation. Nat. Protoc. 16, 3737–3760 (2021).
Prohászka, Z., Nilsson, B., Frazer-Abel, A. & Kirschfink, M. Complement analysis 2016: Clinical indications, laboratory diagnostics and quality control. Immunobiology 221, 1247–1258 (2016).
Chiou, J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402 (2021).
Lambert, A. P. et al. Absolute risk of childhood-onset type 1 diabetes defined by human leukocyte antigen class II genotype: a population-based study in the United Kingdom. J. Clin. Endocrinol. Metab. 89, 4037–4043 (2004).
McCluskey, J. et al. HLA and complement allotypes in Type 1 (insulin-dependent) diabetes. Diabetologia 24, 162–165 (1983).
Alper, C. A., Awdeh, Z. L. & Yunis, E. J. Complotypes and extended haplotypes in laboratory medicine. Complement Inflamm. 6, 8–18 (1989).
Kumar, N., Kaur, G., Tandon, N. & Mehra, N. K. Allotyping human complement factor B in Asian Indian type 1 diabetic patients. Tissue Antigens 72, 517–524 (2008).
Bertrams, J., Hintzen, U., Schlicht, V. & Schoeps, S. C4: another marker for type I diabetes. Lancet Lond. Engl. 1, 41 (1982).
Bertrams, J. et al. Gene and haplotype frequencies of the fourth component of complement (C4) in type 1 diabetics and normal controls. Immunobiology 166, 335–344 (1984).
Choteau, L. et al. Role of mannose-binding lectin in intestinal homeostasis and fungal elimination. Mucosal Immunol. 9, 767–776 (2016).
Muller, Y. L. et al. Functional variants in MBL2 are associated with type 2 diabetes and pre-diabetes traits in Pima Indians and the old order Amish. Diabetes 59, 2080–2085 (2010).
Zhang, N. et al. Association of levels of mannose-binding lectin and the MBL2 gene with type 2 diabetes and diabetic nephropathy. PLoS One 8, e83059 (2013).
Uysalol, M. et al. Importance of mannose-binding lectin2 polymorphism (rs1800450) in infections in children. Biomarkers 27, 44–49 (2022).
Rewers, M. et al. Newborn screening for HLA markers associated with IDDM: diabetes autoimmunity study in the young (DAISY). Diabetologia 39, 807–812 (1996).
Frohnert, B. I. et al. Late-onset islet autoimmunity in childhood: the diabetes autoimmunity study in the young (DAISY). Diabetologia 60, 998–1006 (2017).
Buckner, T. et al. Genome-wide analysis of oxylipins and oxylipin profiles in a pediatric population. Front. Nutr. 10, 1040993 (2023).
TEDDY Study Group The environmental determinants of diabetes in the young (TEDDY) study: study design. Pediatr. Diabetes 8, 286–298 (2007).
Gogarten, S. M. et al. Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
American Diabetes Association. 2. Classification and Diagnosis of Diabetes. Diabetes Care 39, S13–S22 (2015).
Wang, G., Sarkar, A., Carbonetto, P. & Stephens, M. A simple new approach to variable selection in regression, with application to genetic fine mapping. J. R. Stat. Soc. Ser. B Stat. Methodol. 82, 1273–1300 (2020).
Acknowledgements
This work was funded by Leona M. and Harry B. Helmsley Charitable Trust grants 2018PG-T1D017 and G-2103-05121. The TEDDY Study is funded by U01 DK63829, U01 DK63861, U01 DK63821, U01 DK63865, U01 DK63863, U01 DK63836, U01 DK63790, UC4 DK63829, UC4 DK63861, UC4 DK63821, UC4 DK63865, UC4 DK63863, UC4 DK63836, UC4 DK95300, UC4 DK100238, UC4 DK106955, UC4 DK112243, UC4 DK117483, U01 DK124166, U01 DK128847, and contract no. HHSN267200700014C from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institute of Allergy and Infectious Diseases (NIAID), Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), National Institute of Environmental Health Sciences (NIEHS), Centers for Disease Control and Prevention (CDC), and Breakthrough T1D (formerly JDRF). This work is supported in part by the NIH/NCATS Clinical and Translational Science Awards to the University of Florida (UL1 TR000064) and the University of Colorado (UL1 TR002535). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors acknowledge Research Computing at The University of Virginia for providing computational resources and technical support that have contributed to the results reported within this manuscript. URL: https://rc.virginia.edu.
Author information
Authors and Affiliations
Contributions
X.H., A.M., and M.J.R. designed the study. B.-J.M.W.-R., A.F.-A., E.S.N., T.O.M., and M.J.R. provided DAISY proteomics data. S.O.-G., W.-M.C., and S.S.R. provided DAISY genotype data. H.M.P. provided TEDDY proteomics and genotype data. XW performed statistical analyses and wrote the manuscript. X.W., A.M., B.-J.M.W.-R., E.S.N., S.O.-G., S.S.R., and M.J.R. interpreted results. All authors read, reviewed, and approved the final manuscript for publication.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests. Dr. Ani Manichaikul is an Editorial Board Member for Communications Biology, but was not involved in the editorial review of, nor the decision to publish this article.
Peer review
Peer review information
Communications Biology thanks Shuoming Luo and Rachida Raache for their contribution to the peer review of this work. Primary Handling Editors: Min Zhang and Mengtan Xing. [A peer review file is available].
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Hu, X., Webb-Robertson, BJ.M., Parikh, H.M. et al. Genetic mapping of complement system proteins for islet autoimmunity in children with high risk of T1D. Commun Biol 8, 1366 (2025). https://doi.org/10.1038/s42003-025-08739-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42003-025-08739-9
