
Abstract
The gut microbiota offers an extensive resource of enzymes, but many remain uncharacterized. To distinguish the activities of similar annotated proteins and mine the potentially applicable ones in the microbiome, we applied an effective Activity-Based Metaproteomics (ABMP) strategy using a specific activity-based probe (ABP) to screen the entire gut microbiome for directly discovering active enzymes and their potential applications, not for exploring host-microbiome interactions. By using an activity-based cyclophellitol aziridine probe specific to α-galactosidases (AGAL), we successfully identified and characterized several gut microbiota enzymes possessing AGAL activities. Cryo-electron microscopy analysis of a newly characterized enzyme (AGLA5) revealed the covalent binding conformations between the AGAL5 active site and the cyclophellitol aziridine ABP, which could provide insights into the enzyme’s catalytic mechanism. The four newly characterized AGALs have diverse potential activities, including raffinose family oligosaccharides (RFOs) hydrolysis and enzymatic blood group transformation. Collectively, we present a ABMP platform that facilitates gut microbiota AGALs discovery, biochemical activity annotations and potential industrial or biopharmaceutical applications.

Similar content being viewed by others
Introduction
The gut microbiota is a complex microsystem and comprises tens of trillions of microorganisms inhabiting the gastrointestinal tract of humans and animals, providing an abundant natural enzyme resource1,2. However, linking these microbial enzymes to their biological roles and the chemical transformation of xenobiotics presents a challenge3. Despite significant advances in DNA sequencing technologies and bioinformatics approaches, a large proportion of genes in the gut microbiome remains uncharacterized4, making it difficult to elucidate the enzyme roles in the gut microbiota. To overcome this challenge and discover novel enzymes in the gut microbiome, several strategies have been employed, including metagenomics5, metatranscriptomics6, and protein structural classification7, have been employed. Recently, structural alignment based on Alphafold prediction can also be used for functional classification and annotation of uncharacterized proteins8,9. However, these methods can only identify a correlation between gene abundance and predict functional annotation, failing to account for differences in enzyme expression or differing activities between similarly annotated enzymes, which restricts their applications10. In addition, the abundance of gene transcripts or proteins cannot reflect the activities of functional proteins in their native environment11,12.
Activity-based protein profiling (ABPP) has emerged as a potent chemical proteomic strategy to directly characterize enzyme function with activity-based probes (ABPs) in native biological systems13,14. The potential of ABPP for protein discovery was early recognized by Cravatt group, who identified over 80% of human/mouse serine hydrolases using fluorophosphonate ABPs15,16. The feature of ABPP makes it suitable for use in the complex gut microbiota as this technology directly screens specific enzymes at the molecular level without the need for strain isolation and in vitro culture17,18,19. Thus far, ABPP approaches have detected gut microbial enzymes such as serine hydrolases20, bile salt hydrolases21,22, β-glucuronidases23,24,25, and serine-type endopeptidases26 in microbiome. Metaproteomics has rapidly become a key tool to study the spatiotemporal characterization of microbial communities27,28. By combining metaproteomics with ABPP, a strategy named activity-based metaproteomics (ABMP) can identify and quantify interesting proteins and species present in gut microbial samples at the functional level. But the ABMP strategy was mainly applied in elucidating interactions between dysregulation of host and microbial proteins’ identity in previous reports25,26,29, its use for highly active enzyme mining and further characterization or potential applications of these microbial enzymes in gut microbiome study has been poorly reported. Additionally, the selectivity of ABPs for target proteins is an important factor in discovering highly active enzymes or enzyme families, aiding in the exploitation and identification of enzymes activities30. Therefore, specific ABPs can be used to mine and identify the treasure trove of the gut microbiota enzymes, which may have better applications in host health and industrial technology studies.
α-Galactosidase (AGAL) is an exo-glycosidase responsible for the hydrolytic cleavage of terminal non-reducing α-galactose residues from oligosaccharide, polysaccharides, and glycoconjugate substrates, which are ubiquitous in all kingdoms of life31. AGALs have garnered considerable interest due to their numerous potential biotechnological and medical applications. They have been extensively studied in the food industry for the removal of anti-nutritional factors or for the improvement of human and animal diets32. AGALs are also crucial for the treatment of Fabry disease33 and blood group conversion34,35. Nonetheless, the industrial production of AGALs is currently insufficient to meet the increasing demand of AGALs usage36, emphasizing the need to discover more active enzymes. Gut microbial AGALs have been identified for their metabolism of raffinose, stachyose, and other raffinose family oligosaccharides (RFOs), which have long been considered anti-nutritional factors as they cause flatulence in humans and animals. Recently studies have also highlighted their prebiotic potential in promoting the growth of beneficial bacteria in the large intestine37. This suggests that gut microbiota is an ideal resource for discovering novel AGALs. Therefore, based on their physiological significance and vast industrial and biomedical potential, we have targeted the AGAL enzyme family for mining from gut microbiota.
In this study, we utilized the ABMP strategy, employing a specific AGAL ABP, to screen the entire mouse gut microbiota. Using this technique, we discovered and identified active members of the AGAL family from complex protein extracts isolated from the gut microbiota. We successfully overexpressed four new recombinant AGALs that were shown to possess different glycoside hydrolysis and transglycosylation activities in biochemical assays. Notably, one AGAL exhibited distinct enzyme activity characteristics, and was strongly inhibited by CaCl2. Therefore, its structure, with and without the cyclophellitol aziridine probe molecule, was solved using cryo-electron microscopy to interpret the mechanisms of the method. The discovery and identification of gut microbial AGALs, along with structural information and functional activities provide new insights into the study of the glycoside hydrolase family. Our study not only provides an advanced technique for discovering gut microbial AGALs but also serves as an example of a typical gut microbial enzyme biochemical characterization.
Results
Efficient enrichment of bacterial α-galactosidases by ABMP screening in the gut microbiome
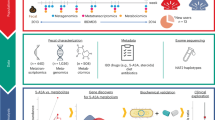
To achieve high-throughput enrichment of active α-galactosidases from the mouse gut microbiome, we developed an ABMP screening approach, as illustrated in Fig. 1a. Our ABMP strategy involved the incubation of AGAL-specific ABPs with fecal samples. Subsequently, the ABP-labeled active proteins were enriched using streptavidin-magnetic beads, visualized by silver staining, and analyzed by on-beads digestion or in-gel digestion LC-MS/MS. Our previously reported AGAL-ABPs (ABP1 and ABP2, Fig. 1b) contained a α-galactoside cyclophellitol-aziridine scaffold with high selectivity for active AGALs38,39. Upon labeling an active AGAL, the cyclophellitol-aziridine specifically labels the enzyme catalytic nucleophile in a conformation resembling the covalent intermediate of a hydrolysis reaction (Fig. 1c). This ABMP platform provides a high-throughput approach for directly identifying active AGALs from gut microbiome samples. We aim to directly discover active α-galactosidases (AGALs) in the mouse gut microbiota using on-beads digestion and in-gel digestion for mass spectrometry-based analysis. The ABMP method was performed on feces from two groups of mice (four mice in per group) as biological replicates (Rep1 and Rep2). Gut microbial proteins isolated from mouse feces were incubated with 1.0 μM ABP1 at 37 °C and pH 6.0 for 1 h. Next, LC-MS/MS was performed using an Orbitrap Fusion Lumos mass spectrometer at a resolution of 60,000 in profile mode, equipped with an Ultimate™ 3000 RSLCnano system in a 60-min gradient (buffer A: 0.1% FA in water, buffer B: 0.1% FA in 80% MeCN) at a flow rate of 300 nL/min.

a Workflow of the ABMP approach for high-throughput screening of gut microbiome samples. b The AGAL activity-based probe (ABP1 and ABP2) structure with a cyclophellitol-aziridine scaffold for selective labeling of active AGALs. c Mechanism-based inhibition of active AGALs by cyclophellitol-derived ABPs.
The LC-MS/MS analysis produced eight raw data files, corresponding to the samples collected from the Rep1 and Rep2 mouse cohorts, respectively. These files were then analyzed using MaxQuant software, resulting in the identification of 1519 peptides and 808 protein groups (groups of proteins sharing the same set of identified peptides) after the removing contaminants. Since there is no specific library for metagenomics, the overall MS/MS identification rate was 4.4%. This rate represents the proportion of MS spectra matched to peptide sequences (4822 MS) out of the number of tandem MS spectra submitted for analysis (109507 MS/MS) in MaxQuant. Out of the 808 identified protein groups, 31 groups were classified as α-galactosidase groups. These 31 groups included a total of 149 α-galactosidases (Supplementary Data 1). For subsequent analysis, the first ID (represents the α-galactosidase enzyme) of the ‘majority protein ID’ in each protein group was selected as the representative protein, ordered by their ‘peptide counts (all)’.
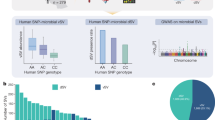
In order to investigate potential background protein contamination resulting from non-specific binding to the beads, we utilized a Venn diagram to visually represent the proteins identified in both the DMSO control group and the probe group. For on-beads digestion using ABP1, we identified and quantified a total of 775 protein groups in Rep1, with 30 protein groups classified as AGAL. In Rep2, we identified 314 protein groups, with 24 belonging to the AGAL group (Fig. 2a). Notably, a substantial portion of the AGALs identified in Rep1 were also detected in Rep2 (Fig. 2a). Among the protein groups from probe on-bead digested samples, 42 protein groups were found to be shared between replicates (Supplementary Fig. 1a). Out of these, 24 protein groups in Rep1 and 24 protein groups in Rep2 were classified as AGAL (Supplementary Fig. 1a). Through a comparison of protein abundances and distributions in both replicates, it was evident that the 1 μM probe significantly increased the identification count and the intensity of AGALs compared to the DMSO group (Fig. 2b, Supplementary Fig. 1). In-gel digestion yielded a total of 134 protein groups in Rep1, of which 23 groups were classified as AGAL protein groups. In Rep2, 25 protein groups were identified, with eight groups belonging to the AGAL group (Supplementary Fig. 1b, c). Considering the potential subjective errors associated with gel excision during in-gel digestion, which can lead to variations in AGAL identification counts, we primarily focused on the data obtained from on-bead digestion.

a Venn diagram showing the number of protein groups identified by on-bead digestion in two biological replicates and the number of intersecting proteins (black number) and AGALs (red number) in the replicate probe groups. b Protein abundance distribution calculated from MS intensities of quantified peptides of each protein. c From top to bottom, it shows dendrogram representation of protein sequence similarity; tile plot indicating the taxonomic classifications (phylum, genus); histogram representing the number of identified proteins in each protein group; bubble plot of identified AGAL protein groups organized by sequence similarity, with bubble size indicating identified peptide counts and colored coding indicating abundance. AGAL abundance is represented by intensities, which are peptide signal intensities determined in MaxQuant.
To assess the reproducibility of the ABMP method, correlation analysis was performed on the standardized data (Supplementary Fig. 2a). Within the same probe-enriched groups, the correlation coefficient between different replicate samples exceeded 0.4 (p < 0.01), while the correlation within individual samples surpassed 0.8 (p < 0.01) (Supplementary Fig. 2b). Moreover, the correlation between the target proteins identified by the probe in the two replicate samples was determined to be 0.63 (p < 0.01) (Supplementary Fig. 2b). These findings indicate a reasonable level of reproducibility for the ABMP method in capturing target proteins.
In the probe-enriched group, we discovered 31 protein groups classified as AGAL groups, which exhibited protein lengths ranging from 614 to 763 amino acids (Fig. 2c) and sequence similarity range from 7.06% to 89.71% (Supplementary Data 1). Taxonomic classification revealed that these AGAL proteins belong to 15 genera and three prevalent phyla in the gut microbiota: Actinomycetota, Bacillota, and Bacteroidota (Fig. 2c). Due to the high similarity of AGALs from Actinomycetota, the identified peptides are shared among different proteins and the protein group contains multiple proteins. For instance, AH67_08535 was found to have 53 AGALs in the same protein group (Fig. 2c). To evaluate whether gut microbial abundance affects the Animal Microbiome Database (AMDB), we performed a Pearson correlation analysis between the intensity of the proteins and the relative abundance of corresponding gut bacterial taxa in AMDB (Supplementary Fig. 3). The results indicated that the intensity of the target proteins did not exhibit a significant correlation with the abundance of the corresponding bacteria. Based on this finding, we conclude that the enrichment of target proteins by ABMP is not significantly affected by the relative abundance of gut microbiota.
To further validate the enrichment of target proteins achieved by the ABMP method, we performed a Games-Howell test on the data comparing the AGAL group treated with DMSO (control group) and the probe group in the on-bead digestion (Supplementary Fig. 2d). This analysis confirmed that the addition of probes significantly enriched the target proteins compared to the control group (p < 0.05). Among the AGAL proteins, 15 of them were found to have more than two unique peptides (Fig. 2c). Based on the fold change (FC) resulting from the addition of the probe in both replicate samples, we observed that the top ten enriched proteins were all AGAL proteins (Supplementary Fig. 2e, Supplementary Data 1). This observation supports our assertion that our method effectively enriches AGALs from the gut microbiota. For subsequent protein functional characterization research, we selected six AGAL proteins that had a minimum of two unique peptides out of the initially identified 15 AGALs based on their higher intensity ratio between the probe group and DMSO group in duplicate results. These six AGALs (AGAL1-6) were selected from six genera in the phylogenetic tree, and the additional information is shown in Supplementary Table 1. In addition, all six AGALs share sequence similarity, range from 43.92% between AGAL1 and AGAL2 to as high as 73.52% between AGAL3 and AGAL5 (Supplementary Fig. 4). Phylogenetic analysis and sequence similarity analysis revealed notable distinctions between our protein and the previously published AGALs (Supplementary Fig. 4). Comparing to the two known AGALs from Geobacillus stearothermophilus, named AgaA and AgaB40, all AGAL3, AGAL5 and AGAL6 have less than 70% sequence similarity respectively. Interestingly, all six AGALs share even less than 24% sequence similarity with AGALs from human, coffee bean and Escherichia coli (Supplementary Fig. 4b). These findings indicate variations and divergence between our identified proteins and the known AGALs. Evolutionary analysis of these AGALs draw similar conclusions to sequence similarity alignment (Supplementary Fig. 4b), which was inferred by using the Maximum Likelihood method and JTT matrix-based model41, and was conducted in MEGA-X42.
Enzymological characterization of recombinant AGALs
We selected four α-galactosidases, AGAL1, AGAL3, AGAL5, and AGAL6, for large-scale purification and subsequent characterization due to their robust expression in E. coli and high yields of purified protein (Supplementary Fig. 5, Supplementary Data 2). Since the expression of AGAL2 and AGAL4 did not yield adequate amounts of purified proteins, they were thus excluded from further characterization. We then determined the optimal pH for the cleavage of the galactose activity-specific substrate pNP-αGal for the four enzymes in various buffers such as Mcllvaine buffer pH 4.0–8.0, Tris buffer pH 7.0–9.0, phosphate buffer pH 6.0–8.0, and HEPES buffer pH 7.0–8.0. All enzymes preferring with pH 6.0–7.0, though there was some variety within this range between the enzymes regarding the optimal pH (Fig. 3a). Notably, none of the four enzymes exhibited activity towards the β-pNP-galactopyranoside or the α-pNP-glucopyranoside substrates, indicating specificity for the α linkage and galactose moiety of the preferred pNP-αGal substrate. Kinetic constants (Km) values and specific activities of these AGALs are shown alongside Michaelis Menten kinetics graphs in Fig. 3b. AGAL3 and AGAL5 displayed higher enzyme activity, with specific activities of 569.3 U/mg and 465.1 U/mg protein, respectively. The activity of these two AGALs is about equal to the known rGal27B produced by Thermophilic Neosartorya fischeri P1 with a specific activity of 477 U/mg43, and is much higher than that previously reported for GalC from Aspergillus oryzae YZ1 at 76.9 U/mg specific activity44, but lower than that reported for the TtGal27A from Thielavia terrestris strain with a specific activity of 752 U/mg towards pNP-α-Gal45. Thus, our results provide valuable information regarding the biochemical properties of these newly characterized AGALs, which may have potential applications in various industries.

a Optimal pH for hydrolysis of pNP-αGal over a range of pH values from 4.0 to 9.0. b AGALs hydrolysis process kinetics, showing Km (mM), and specific activity (U/mg Protein). c Effect of CaCl2 on AGALs activity.
To ensure the validity of our findings, we investigated the impact of potential contaminants and additives on the activity of these AGALs, as many α-galactosidases are known to be inhibited by free galactose or glucose molecules, as well as various metal ions46,47. We observed that maltose had an inhibitory effect on AGAL3 and AGAL5 (Supplementary Fig. 6a). Notably, we found that Ca2+ could significantly inhibit AGALs activity, particularly for AGAL5, with the enzyme’s activity almost completely blocked in the presence of 2 mM CaCl2 (Fig. 3c). In contrast, none of the enzymes, except for AGAL5, were inhibited by reducing agents such as β-mercaptoethanol (BME) or dithiothreitol (DTT) (Supplementary Fig. 6b), suggesting that sulfhydryl groups are not essential for the enzyme activity of AGAL1, AGAL3, and AGAL6. The inhibitory effect of reducing agents on AGAL5 may indicate that thiol groups play a role in the enzyme’s activity, possibly by forming disulfide bridges to stabilize its structure. Interestingly, we observed that Tween-20 and Triton X-100 activated AGAL1, AGAL3, and AGAL6, possibly by improving their solubility. This finding is consistent with previous reports demonstrating that detergents can promote optimal activity of AGALs to a certain extent48,49. However, we also found that AGAL5 exhibited considerable sensitivity to Triton X-100 detergents (Supplementary Fig. 6b), but no effect by Tween-20. In addition, we observed that a small percentage of DMSO reagents had no impact on any of the AGALs. Our findings provide important insights into the potential factors that may impact the activity of these AGALs, which may have implications for their use in various applications.
AGAL structural and catalytic mechanism studies by cryo-EM
We selected AGAL5 for structural elucidation by cryo-electron microscopy (cryo-EM) due to its high enzymatic activity and ease of production in E. coli. Our enzyme activity assays revealed that CaCl2 and ABP2 inhibit the hydrolysis activity of AGAL5. To gain insight into the mechanism of enzymatic activity and the impact of these inhibitors, we solved the structures of AGAL5 at three different conditions: native, incubated with CaCl2, and incubated with ABP2, at near-atomic resolution (3.28 Å, 3.37 Å, and 3.17 Å, respectively). The model refinement and validation statistics were shown in Supplementary Table 2; they were deposited as PDB 8K1A, 8K7U, and 8K7V. All three structures are tetramers, consistent with the published crystal structures of the other AGALs (PDB ID: 2XN050, 2YFN51, 4FNQ40). Differences in root-mean-squared deviation (RMSD) values between the various structures are listed in Supplementary Table 3. Each protomer of all structures comprises three domains: N-terminal domain, C-terminal domain and catalytic (β/α)8-barrel domain, which has 8 helixes and strands (Fig. 4d–f). We did not impose D2 symmetry during reconstruction, as we found minor differences among the four protomers of the tetramer, reflecting the solution state of the protein. This aspect of cryo-EM is particularly advantageous, as it allows for the visualization of different states of the molecule without the need for a crystal sample. Notably, the cryo-EM native AGAL5 structure appears dilated, with a more flexible catalytic (β/α)8-barrel (fewer secondary structures, Supplementary Fig. 7d) when compared to the crystal structure (PDB: 4FNQ) of the AgaB40. No Ca2+ density was observed when we aligned the two maps of native AGAL5 and CaCl2 treated AGAL5 (Fig. 4a, light gray and blue). However, several regions in the (β/α)8 barrel domain of AGAL5 became more flexible after CaCl2 treatment, which was too weak to model (Fig. 4b, c). CaCl2 treated AGAL5 exhibited significantly more flexible compared to the other structures, including AGAL5 bound with ABP2 (Supplementary Fig. 7e). These findings provide new insights into the structural basis of AGAL5 enzymatic activity and its inhibition by CaCl2 and ABP2.

a Superposition of the electron density maps of native AGAL5 (light gray) and CaCl2 treated AGAL5 (blue). b Models from (a), highlighting structural differences, particularly the loops in CaCl2 treated AGAL5, which cannot be resolved. c Red circles mark the missing regions in CaCl2 treated AGAL5 in the zoom-in view of a protomer from (b). d The N-terminal, (β/α)8-barrel, and C-terminal domains of one protomer in the native AGAL5 tetramer model are highlighted, with the active site Asp480 shown as a sphere. e Another orientation of the native AGAL5 tetramer model, rotated 90° along with x axis. f Secondary structure and critical catalytic residue are labeled and numbered in a zoom-in view of the catalytic region ((β/α)8-barrel domain).
ABP2, initially employed to capture and enrich AGAL, has been shown to covalently bind to the active sites of target enzymes38. However, the precise mechanism by which ABP2 inhibits the activity of the newly identified AGALs in this study remains elusive. By comparing the electron density maps of AGAL5 and AGAL5 bound with ABP2, we discovered that the latter exhibited a more defined and stable structure. This was evident from the more distinct side chain density of amino acid residues in the presence of ABP2. Notably, the local resolution map of AGAL5 bound with ABP2 was largely uniform at a resolution of 3.2 Å. In contrast, the local resolution maps of native AGAL5 and CaCl2 treated AGAL5 varied between 3.2 to 3.8 Å (Supplementary Fig. 7b). To further understand the inhibition mechanism, we examined the four protomers in AGAL5 bound with ABP2. Figure 5 shows that ABP2 covalently binds to all four protomers, with the ligands superimposed (see Supplementary Fig. 8). Figure 5b details the covalent bond between the hydrolyzed ABP2 and Asp480 in chain A, where ABP2 adopts a ‘boat’ conformation upon binding to AGAL5. This finding supports the mechanism of cyclophellitol aziridine compounds hydrolyzing via retaining AGAL, as shown in Fig. 1c. LigPlot+52 analysis of all four AGAL5 protomers highlighted the specific hydrogen bond interactions (Fig. 5c), revealing variations in the number and types of hydrogen bonds formed between ABP2 and the protomers. Figure 6a displays the electron density of ABP2 bound in chains A, B, C, and D. Interestingly, the head region of ABP2 is stable across all chains, while the tail regions exhibit flexibility. As shown in Fig. 6a, the tail density of ABP2 in chains A and C is stable, whereas in chains B and D, the tail density of ABP2 show three different alternative conformations, indicating flexibility. These observations suggest that various states of ABP2 bound to AGAL5 were captured by cryo-EM (Figs. 5c and 6a). Furthermore, the surface electrostatic potential of AGAL5 bound with ABP2 revealed negatively charged regions (Fig. 6b), which may facilitate ABP2 binding. Comparative analysis with structures from GH36 family (PDB 6GWG53 and 5M1I54) and our structure of AGAL5 bound with ABP2, where ligands covalently bind (Fig. 6c, d), showed conserved binding to Asp residues (Asp327 for 6GWG and 5M1I; Asp480 for our AGAL5).

a Full model of AGAL5 bound with ABP2 (yellow). b A close-up view of ABP2 covalently bound to AGAL5 in one protomer, highlighting the covalent bond and the ‘boat’ conformation of ABP2. c Detailed interaction analysis for each protomer.

a The electron density of ABP2 was located in chains A, B, C, and D, the tail density in chain B and chain D exhibit three alternative regions (red boxes). b The electrostatic surface potential of AGAL5 bound to ABP2 reveals negatively charged regions (red) on the enzyme surface around the ABP2 (green). c The crystal structures (PDB 6GWG53 and 5M1I54) from GH36 family were compared to AGAL5 bound with ABP2. Their structures are conserved (6GWG: gray; 5M1I: purple; 8K7V (AGAL5): cyan). d The ligand FEK in 6GWG and 7D8 in 5M1I bind to Asp327 residue, whereas ABP2 binds to Asp480 residue in AGAL5, as shown in the superimposed structures.
The hydrolysis and transglycosylation activities of novel AGALs towards natural sugars
To explore the potential applications of the AGALs identified through the ABMP strategy, we evaluated their cleavage abilities against several natural substrates. Specifically, we examined the hydrolysis activity of the recombinant AGALs towards raffinose family oligosaccharides (RFOs), which are commonly removed from soymilk and during sugar crystallization in the food industry. Within 10 min, all four AGALs exhibited hydrolysis activity towards melibiose, raffinose, and stachyose, resulting in glucose and galactose breakdown products for melibiose, sucrose and galactose for raffinose, and raffinose and galactose for stachyose (Supplementary Fig. 9). Notably, AGAL1 and AGAL3 completed melibiose hydrolysis in 30 min, while AGAL5 and AGAL6 required 60 min and 10 min, respectively, to achieve the same level of hydrolysis. In addition, AGAL1 and AGAL3 exhibited superior hydrolysis activity towards raffinose, breaking it down into sucrose and galactose within 30 min, compared to AGAL5 and AGAL6. These findings suggest that the newly discovered AGALs have potential applications in the food industry for the efficient breakdown of RFOs. Hydrolysis of stachyose by AGALs is a two-step process that initially produces raffinose and galactose, with raffinose then further hydrolyzed into monosaccharides, as depicted in Fig. 7a. However, we observed that stachyose hydrolysis by AGAL1, AGAL3, and AGAL6 was relatively slow, with detectable amounts of starting material still present after 3 h as assessed by TLC (Supplementary Fig. 9). Our findings suggest that the most efficient hydrolysis can be achieved by matching the appropriate substrate to the corresponding AGAL. Interestingly, these AGALs exhibited varying activities towards the same substrate, despite being classified within the same glycoside hydrolase family and identified through the same ABMP strategy.

a Reaction process and TLC analysis of stachyose hydrolysis by AGAL1. b Reaction process and TLC analysis of blood group antigen trisaccharide hydrolysis by AGAL3. c LC-MS SCAN and SRM analysis of blood group B antigen trisaccharide: Blood group B trisaccharide, [M + H]+ = 489.3, [M+Na]+=511.2, [M + K]+ = 533.1; Galactose, [M + 2PMP-H2O + H]+ = 511.3, [M + 2PMP-H2O + Na]+=533.2, [M + 2PMP-H2O + K]+ = 549.1; Disaccharide, [M + 2PMP-H2O + H]+ = 657.3, [M + 2PMP-H2O + Na]+=679.3, [M + 2PMP-H2O + K]+ = 695.2. d Reaction process and TLC analysis of transglycosylation towards monosaccharides and polysaccharides by AGAL6.
We also evaluated the potential of AGALs to cleave the trisaccharide group of blood type A and B antigens, which could have implications for their application as blood group transformers. After 1 h, hydrolysis of the blood group B antigen trisaccharide was observed for AGAL3, AGAL5, and AGAL6, but not for AGAL1. After 3 h, complete cleavage of the galactose from the B antigen trisaccharide was observed for AGAL3, and majority of the B antigen trisaccharide showed galactose cleavage for AGAL5 and AGAL6 (Fig. 7b, Supplementary Fig. 10). Notably, no hydrolysis was observed on the blood group A antigen trisaccharide by any of the AGALs, indicating that the enzymes cannot cleave the GalNAc moiety. The lack of activity maybe due to the acetyl group on GalNAc interfering with enzyme recognition of the substrate. Collectively, our findings suggest that AGAL3, AGAL5, and AGAL6 exhibit high specificity for the galactose moiety and have potential medical applications for converting the blood group B antigen trisaccharide to the universal blood group O antigen (H antigen) disaccharide.
AGAL3 exhibits promise for blood group B antigen trisaccharide cleavage, as supported by our data. However, additional experiments with longer reaction time may demonstrate complete cleavage by AGAL5 and AGAL6 and warrant further investigation. To confirm the hydrolysis activity of AGAL3 towards the blood group B antigen trisaccharide, we employed an LC-MS-based assay. Saccharide mass spectrum peaks were determined using a selected reaction monitoring (SRM) method. Notably, after 3 h reaction, the blood group B antigen trisaccharide peak had completely disappeared, while the H disaccharide and galactose peaks were clearly present after only 1 h of reaction (Fig. 7c).
Several types of AGALs have been reported to catalyze transglycosylation reactions, making them promising catalysts for the synthesis of novel oligosaccharides with potential medical applications36. To explore the transglycosylation activity of the newly identified AGALs, we examined their activity towards various substrates, including glucose, galactose, xylose, fructose, lactose, sucrose, raffinose, stachyose, and melibiose, using pNP-galactose as a donor substrate. TLC analysis revealed some reactions exhibiting a different spot below the control without pNP-galactose substrate (Supplementary Fig. 11). Specifically, AGAL1 glycosylated raffinose, AGAL3 glycosylated fructose, and AGAL6 glycosylated galactose, fructose, sucrose, and raffinose. Although some of the corresponding glycosyl product dots were relatively minute, they were indicated by the red arrow and pane on the TLC plates (Supplementary Fig. 11). Notably, no transglycosylation activity was observed for AGAL5. Of the AGALs examined, AGAL6 exhibited the broadest spectrum of transglycosylation activity towards the selected substrates (Fig. 7d, Supplementary Fig. 11). We also observed that all 4 AGALs hydrolyzed raffinose, stachyose, and melibiose, consistent with our previous hydrolysis assay (Fig. 7a, Supplementary Fig. 9). Interestingly, TLC analysis revealed a combination of both hydrolysis and transglycosylation towards raffinose by AGAL6, explaining its relatively low hydrolysis activity in our assay. Overall, our findings demonstrate the diverse hydrolysis and transglycosylation activities of the same family of enzymes in the intestinal flora.
Discussion
The gut microbiome is a rich source of enzymes, but the complex biological context and difficulty in cultivating anaerobic gut bacteria present challenges in characterizing useful enzymatic activities for hosts. Metagenomic, metatranscriptomic, and structural alignment strategies have been the primary approaches for novel proteins discovery and annotation from gut microbiomes5,6,7,8,9. However, the abundance of data generated by omics approaches can make it difficult to link biological information with specific enzymatic activities, mainly due to the daunting task of protein annotation. Activity-based metaproteomics profiling (ABMP) strategy could overcome these obstacles by employing well-designed ABPs to capture specific enzymes in mouse gut microbiota and identify their biochemical activities. We used mouse gut microbiota instead of human as research object, because the mouse microbiota is a common model of organism used to simulate many complex diseases in biomedical research. In addition, mouse gut microbiome is easy to obtain and characterize for new enzyme discovery study55.
Studying gut microbiota proteins based on ABMP strategy does have considerable advantages due to the complex intestinal physiological environment. Many specific enzymes, such as serine hydrolases, bile salt hydrolases, β-glucuronidases, and serine-type endopeptidases were explored for the relationship between host health and target enzymes from gut microbiome20,21,22,23,24,25,26. For instance, Redinobo and colleagues recently identified gut microbial β-glucuronidases using ABMP and indicated that these enzymes not only efficiently reactivate xenobiotics substrates such as anti-tumor drug irinotecan, which induces gastrointestinal toxicity 24, but also chemically metabolize endobiotics such as neurotransmitters or hormones, thereby altering host homeostasis56. In addition, the Wright group applies this method to discover functional activity of multiple gut microbial proteins, including glycoside hydrolases, polysaccharide lyases, and sugar transport proteins which respond with the soluble dietary fiber deprivation57. But the ideas and research objectives of our study are very different from those work towards the relationship between host disease and gut microbiota enzymes. We would address the purpose of our study is to discover one type active enzyme from gut microbiome and use these enzymes to perform some potential applications. Thus, in our opinion there are still many unsolved mysteries in the research of gut microbiota proteins, either to explore the relationship between these proteins and host health or to discover new enzymes for developing products, both of which are very promising and potentially fruitful areas of study. In previous work, Overkleeft’s group developed α-galactoside-scaffold cyclophellitol-type ABPs and used them to visualize and identify human and Nicotiana benthamiana AGAL activities38,39. These ABPs exhibit high potency and selectivity for AGAL activities, making them ideal for exploring novel enzymes in the gut microbiota. Our use of ABP1 in this context provides a case study for activity-based proteome mining, which can be extended to other specific ABPs for discovering functional tools for synthetic biology and enzyme-assisted synthetic chemistry. The success of selective enrichment for target proteins by ABMP relies heavily on the selectivity of the chemical probe, making the choice of ABP a critical step in this approach. While most available probes lack sufficient specificity and selectivity, the mechanism-based specific ABPs will bridge this gap, and the ABMP methodology, together with specific ABPs, will become a powerful toolkit for functional proteome mining in systems biochemistry.
In metaproteomic research, managing large database files is a prevalent issue. Researchers often apply strict filtering to minimize false positives that large databases may generate58. We have implemented a two-tiered or iterative method59, leveraging data characteristics to condense the database and refine the search parameters, thereby increasing the sensitivity of peptide sequence identification. However, this approach can potentially result in unchecked false-discovery rates (FDR) and inaccurate peptide-spectrum matches (PSM), necessitating meticulous evaluation by researchers60,61. In our experiments, the rigorously chosen and verified proteins exhibited considerable activity. Moreover, performing comprehensive metagenomic or 16S rRNA sequencing before initiating research can significantly reduce such errors by informing the construction of a more targeted search database62.
Phylogenetic analysis based on the protein sequences of the 31 major AGALs identified by ABMP, revealed that the degree of probe-mediated enrichment was not significantly affected by sequence similarity or taxonomic distribution. For subsequent analysis, we selected the first protein ID from the top few abundance groups as the representative protein. Based on their amino acid sequence homology, enzymes with α-galactosidase activity are categorized into glycosyl hydrolase (GH) families 4, 27, 31, 36, 57, 97, and 110 (Carbohydrate Active Enzymes Database (CAZy), http://www.cazy.org)63. So far, the GH27 and GH36 families are extensively studied elaborately and biochemically characterized for their potential industrial applications. Generally, the eukaryotic AGALs are mostly acidic enzymes and belong to GH27 family, while prokaryotic AGALs are neutral enzymes and belong to GH36 family. Based on our data, we primarily identified AGALs from the GH36 family, along with a fewer AGALs representation from GH97. Notably, different AGALs exhibited large variations in enzyme activity towards substrate hydrolysis, metal ions, surfactants, buffer propensities, and optimal pH, underscoring their functional diversity. From this diverse group, we identified AGAL5 as a particularly promising candidate, exhibiting a specific activity higher than many previously reported microbial AGALs36,64, which motivated us to delve deeper into its mechanism.
Cryo-EM offers distinct advantages over X-ray crystallography, notably the ability to study molecules without the need for crystallization. Instead, the protein solution is rapidly forozen, preserving different states and avoiding packing force. Consequently, our cryo-EM structure of native AGAL5 provides a more dynamic representation of the protein, with fewer constraints and more conformational states compared to the crystal structure. The primary objective was to investigate the mechanism of enzymatic activity, starting with the effect of CaCl2 on AGAL5. These ions were shown to decrease the biochemical activity of AGALs in selective pNP-α-Gal substrate hydrolysis assays. The electron density map of CaCl2 treated AGAL5 was less stable compared to the native cryo-EM AGAL5 structure, particularly in the region surrounding the β-barrel. Increased flexibility made some helices and loops undetectable. Unfortunately, we were unable to locate the density for Ca2+ in the map, possibly due to the lack of fixation of the ion in a specific location. One possible explanation is that the positive Ca2+ ion coordinates with a negative ion in the catalytic domain, causing some helices and loops of the (β/α)8-barrel to become more flexible, thereby obstructing substrate binding.
Analysis of AGAL5 bound with ABP2 revealed the cryo-EM method’s ability to capture the states of the protomers. The ring opening cyclophellitol aziridine scaffold of ABP2 fitted perfectly into the density map, and the covalent binding states of ABP2 with AGAL5 protomers were observed. Interestingly, the flexible long chain tail of ABP2 exhibited different density maps among four protomers of AGAL5. Based on these results, we hypothesized that ABP2 entered AGAL’s shallow negative charge pocket through charge attraction, gradually approaching the active center via hydrogen bond interaction, and forming covalent bonds with AGALs through nucleophilic reactions, ultimately transitioning from a ‘chair’ to a ‘boat’ conformation65. AGAL5 belongs to the GH36 family, which, like GH4, GH27, GH57, and GH97 AGALs family, follows a catalytic mechanism that processes their substrates with net retention in stereochemistry at the anomeric center of the released galactoside through the classical Koshland model of double displacement reaction66.
AGALs are of significant interest for applications in the food and animal feed industries, as well as biomedical applications. In the food industry, AGALs have the potential to enhance the digestibility of the raffinose oligosaccharides family, which can lead to gas production in monogastric animal diets and the human digestive tract due to the lack of AGALs31,32. Moreover, AGALs from rice and coffee have been shown to cleave terminal α-1,3-linked galactose residues from blood group type B antigens on red blood cells, converting them to blood group type O antigen67. Given these findings, we sought to evaluate the potential activity of newly identified AGALs from the gut microbiome for potential applications in the food industry or biomedical field. Specifically, we assessed the hydrolysis and transglycosylation activities of our panel of four AGALs using various natural substrates. Our newly discovered AGALs demonstrated relatively high hydrolytic activity but limited transglycosylation activity. The selection of appropriate enzymes is dependent on the specific sugar substrate hydrolysis or glycosylation required. Of note, AGAL3 exhibited potent activity in breaking down raffinose and blood group B antigen trisaccharide, making it a promising candidate for hydrolysis application studies. Conversely, AGAL6’s broad-spectrum transglycosylation activity made it better suited for glycosyl application.
Conclusions
In summary, our study presents an effective and promising ABMP platform for the identification of active AGALs from the gut microbiota. By characterizing several AGALs based on their specificity towards the cyclophellitol aziridine ABP, we demonstrate this technique’s ability to target specific activities. Furthermore, we provide valuable protein activities through biochemical characterizations of these AGALs and cryo-EM structural studies. The discovery of active AGALs illustrates the exciting potential of the ABMP platform for the identification of active enzymes and their possible application values in biomedical research, diagnostics, and the food processing industry. Our study showcases the high-throughput, rapid, and accurate identification of active proteins from previously uncharacterized enzyme sources using this cost-effective approach for gut microbiota enzyme mining. These findings highlight the immense potential of our ABMP platform for future studies in this field.
Methods
Reagents and general procedures
All chemical reagents and solvents used were reagent grade or higher purity and obtained from commercial sources without further purification. Probe ABP1 and ABP2 were obtained from the Bio-organic Synthesis Group at Leiden University. Analytical thin-layer chromatography (TLC) was performed on silica gel 60 F254 plates (Merk, Supelco, 105554.0001) and analyzed by fluorescence detection with UV light (λ = 254 nm). Staining reagent containing 10% H2SO4 in ethanol was used, followed by heat treatment for visualization.
Animal work and stool sample preparation
All animal procedures were performed in accordance with protocols approved by The Institutional Animal Care and Use Committee at Shenzhen Institute of Advanced Technology (Protocol 2015-14). Two sets of 4 female C57BL/6J littermate mice aged 6–8 weeks were obtained from Guangdong Medical Laboratory Animal Center (GDMLAC) and maintained in a 12 h light/12 h dark cycle with ad libitum access to food (GDMLAC) and water. Stool samples were collected twice daily from each mouse, immediately frozen at −20 °C, and stored until microbial extraction. The samples were thawed to room temperature, diluted in cold PBS (pH 7.4), and vortexed to yield slurries, which were then centrifuged at 100 × g for 1 min. The upper layer was removed, and the pellet was rinsed twice with PBS before being resuspended in HEPES extraction buffer (100 mg pellet per 0.5 mL buffer) and subjected to ultrasonic fragmentation at 0 °C (12% power, 3s-on, 5s-off, Scientz-IID) for 30 min. The mixtures were centrifuged at 12,000 rpm for 5 min at 4 °C, and the supernatant was collected for BCA assay to determine concentration.
Pull down
Pull down experiments were performed following a previously reported protocol68. Stool microbiome protein samples (1.0 mg for each sample in 200 μL) was incubated with DMSO/ABP1 for 1 h at 37 °C and extracted by chloroform/methanol precipitation. The protein pellet was air-dried and samples were treated with urea buffer, dithiothreitol (DTT) and Tris-2-carboxyethylphosphine hydrochloride (TCEP) for reduction and alkylation. The samples then underwent another precipitation assay as described above. Next, after pull down buffer washing, the treated proteins were performed with magnetic bead separation and digestion: The magnetic bead samples were divided into two parts: 1/3 for on-bead digestion and 2/3 for in-gel digestion. Finally, the obtained peptides were extracted and dissolved in 0.1% formic acid for LC-MS/MS analysis.
LC-MS/MS analysis
All peptides were reconstituted in 0.1% formic acid (vol/vol) and separated on reversed-phase columns. The trapping column had a particle size of 3 μm, C18, and a length of 20 mm (Thermo Fisher Scientific, P/N 164535), while the analytical column had a particle size of 2 μm, C18, and a length of 150 mm (Thermo Fisher Scientific, P/N 164534). The separation was performed using an Ultimate™ 3000 RSLCnano system (Thermo Fisher Scientific, San Jose, CA, USA) with a 60-min gradient (buffer A: 0.1% FA in water, buffer B: 0.1% FA in 80% MeCN) at a flow rate of 300 nL/min. The peptides were then analyzed by Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific) in a data-dependent mode. Orbitrap Fusion Lumos mass spectrometer (ThermoFisher Scientific) mass spectrometer was operated in positive ion mode with an ion transfer tube temperature of 275 °C and a positive ion spray voltage of 2.1 kV. One full scan in the Orbitrap (at 60,000 resolution in profile mode with an AGC target of 4 × 105 and maximum injection time of 50 ms) was followed by as many MS/MS scans as could be acquired on the N most abundant ions in 1 s in the ion trap (rapid scan type, HCD collision energy of 30%, AGC target of 5 × 104, maximum injection time of 50 ms). Singly and unassigned charge states were rejected. Dynamic exclusion was enabled, an exclusion duration of 60 s, and an exclusion mass width of ±10 ppm.
Metaproteomics data processing
MetaLab software (version 1.1.1) was used to generate a sample-specific protein database from original large databases69. The mouse-specific microbiome database comprised 44,478 proteins, which were constructed using a MS/MS clustering strategy based on a non-redundant microbial protein database containing 1,761,33 protein sequences combined with the mouse proteome from Uniprot. Non-redundant microbial protein database was generated by clustering protein sequences of genomes assembled at the chromosomal level (retrieved in August 2018) from NCBI70 using CD-hit71. The MS/MS data were searched against the mouse-specific database using MaxQuant 2.0 with a precursor mass tolerance of 20 ppm and a fragment mass tolerance of 0.5 Da72. The “match between runs” option was enabled to match identifications across different replicates. Enzyme specificity was used, and only tryptic peptides with up to 2 mis-cleavages were allowed in the final data sets. Cysteine carboxamidomethylation was specified as a static modification, and oxidation of methionine residue and acetylation (protein-N) were allowed as variable modifications. Reverse decoy databases were included for all searches to estimate false-discovery rates. Peptide and protein identifications were quantified and filtered for less than 1% false-discovery rate (FDR). In order to facilitate further analysis, the protein group originating from the host mouse was excluded.
Quantitative taxonomic analyses were performed by assigning identified proteins to their corresponding taxonomic lineage in Genebank and summing up their intensities. To establish the phylogenetic relationship between different α-galactosidases, multiple alignment of protein sequences was performed using MAFFT v.773. For inferring the maximum-likelihood phylogenetic tree, the best-fitting substitution model (LG + G4) was selected using the Bayesian information criterion in IQ-TREE web server74.
Venn diagrams were generated using the online tool available at (http://bioinformatics.psb.ugent.be/) webtools/Venn/. The resulting diagrams were exported as PDF files and further customized by adjusting the color scheme and transparency. Data processing procedures primarily relied on the R programming language, with the ggplot2 package being the main tool for data visualization. Protein sequence similarity was calculated using the Biostrings package. Z-scores for the data were obtained utilizing the scale function. The scatter plot included the calculation of the Pearson correlation coefficient and the fitting curve, which were performed and added using the ggpubr package. The Pearson correlation coefficient between samples was analyzed using the PerformanceAnalytics package. The Games-Howell test, applied to compare groups, was conducted and visualized using the ggstatsplot package. Animal intestinal microbiome data were retrieved from the Animal Microbiome Database (AMDB) at http://leb.snu.ac.kr/amdb. Subsequently, the data were normalized. The fold change (FC) was determined by comparing the intensity of the protein group in the experimental group (probe group) to that of the control group (DMSO group), and any missing values were imputed with a value of 0.
Cloning
The genes encoding AGAL1 to AGAL5 (Gene names A4V02_01335, A4V02_08955, AT726_08105, AT726_08965, A4V09_19485) (Supplementary Data 2) were optimized and synthesized by GenScript, and were cloned using CloneEZ into a pET-30a (+) expression vector by NdeI/HindIII, respectively, so that a fusion gene with C-terminal His6-tag was obtained. AGAL6 (Gene name N134_04935) was additionally cloned into a pET-30a (+) vector by BamHI/XhoI with a C-terminal His6-tag, respectively. All cloning strains are TOP10. The antibiotic in the plasmid is kanamycin.
Expression trials
To determine ideal expression conditions, the recombinant plasmid was transformed into E. coli BL21 DE3, C41, or Rosetta using the heat-shock method and plated on LB-Agar plates containing 50 μg/ml kanamycin. After overnight growth at 37 °C, a colony was selected and placed in 5 ml of LB medium containing 50 μg/ml kanamycin and grown overnight at 37 °C with shaking at 200 rpm as a starter culture. The following day, the starter culture was added to 100 mL of LB medium containing 50 μg/ml kanamycin in a 250 ml flask and grown at 37 °C with shaking at 200 rpm until the OD600 nm reached 0.6. To test different expression conditions, flasks were induced with either 0.1 mM, 0.5 mM, or 1.0 mM IPTG and placed at 20 °C, 24 °C, 30 °C, or 37 °C. Aliquots of 10 ml were removed at 2, 4, 6, or 16 h, and the bacteria were pelleted by centrifugation at 4000 × g for 10 min. The pellets were then frozen at −20 °C for further analysis.
Large-scale expression and purification
A single colony of C41 transformed with the recombinant plasmid was grown in 5 mL of LB medium containing 50 μg/ml kanamycin at 37 °C with shaking at 200 rpm overnight as a starter culture. The next day, the starter culture was added to 500 ml of LB medium containing 50 μg/ml kanamycin in a 1 L flask and grown at 37 °C with shaking at 200 rpm until the OD600 nm reached 0.6. Once the OD600 nm reached 0.6, the bacteria were induced with 1 mM IPTG and grown at 24 °C with shaking at 200 rpm for 16 h. The cells were harvested by centrifugation at 4000 × g for 10 min, and the pellets were resuspended in a lysis buffer (20 mM HEPES pH 7.5, 5% glycerol, 250 mM NaCl) containing 1 mg/ml of lysozyme. The cells were sonicated, pulsing for 3 s with 5 s off for 10 min at 70% maximum power, and then passed through a French press twice. The cells were centrifuged at 30,000 × g at 4 °C for 30 min to remove insoluble components and membrane fractions. The proteins were then concentrated and filtered using Amicon filters with a 30,000 Dalton molecular weight cut off. The samples were filtered through a 0.2 μm filter and loaded onto a HisTrap HP 5 ml Ni-NTA column with wash buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 25 mM imidazole). The column was washed with 10 column volumes of wash buffer and then eluted with elution buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 300 mM imidazole) using a gradient. Peak fractions were collected and dialyzed overnight with 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. The dialyzed protein was concentrated again using Amicon filters with a 30,000 Dalton molecular weight cut off. The sample was then loaded onto a Superdex size exclusion column (GE) pre-equilibrated with 20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT. The peaks were pooled, and the purification was confirmed using SDS-PAGE. The protein samples were concentrated using an Amicon filter with a 30,000 Dalton molecular weight cut off until reaching 1 mg/ml. Finally, the proteins were flash-frozen in 50 μl aliquots in liquid nitrogen for further use.
Enzyme activity assays
Galactosidase activity was assayed using a colorimetric assay with pNP-α-galactopyranoside (Aladdin, Cat. N100668) as the substrate to measure released PNP. Each assay contained 2.5 mM α-galactopyranoside and 70 mM potassium phosphate buffer pH 6.5, and to ensure values within the linear range, either 100 μg, 10 μg, or 1.0 μg of enzyme or no enzyme as a negative control, in a 96-well plate. The plate was incubated at 37 °C for 5 min with gentle agitation. The reaction was stopped by adding 140 mM sodium borate buffer pH 9.8, and the absorbance at 407 nm was measured. The molar extinction coefficient of PNP, 18.6 mM−1 cm−1, was used to calculate the amount of released pNP.
pH dependence and different additives effect on enzyme activity
To determine the optimal pH for AGALs activity, a colorimetric assay was performed with α-galactopyranoside as the substrate using different buffers. HEPES buffer was used for pH 7–8, phosphate buffer was used for pH 6–8, McIlvaine buffer (Na2HPO4 and citric acid) was used for pH 4–8, and Tris-HCl buffer was used for pH 7–9. To assess the effect of different sugars, metal components, or other modifiers on AGALs activity, a colorimetric assay was performed with PNP-α-galactopyranoside as the substrate. The additives included 5 mM glucose, galactose, maltose, melibiose, stachyose, lactose, raffinose, EDTA, MgCl2, KCl, CaCl2, or 1 mM DTT, or 1% Triton X-100, Tween-20, or SDS.
Enzyme kinetics
To determine the kinetic constants (Km, Vmax) of the four enzymes, an enzyme assay was performed with increasing concentrations of substrate pNP-α-galactopyranoside ranging from 0 mM to 3.0 mM at pH 6.5 in 70 mM potassium phosphate buffer as described in Enzyme Activity Assays above. The enzyme activity was then plotted against the substrate concentration on GraphPad Prism 8.0. Non-linear regression analysis for Michaelis-Menten enzyme kinetics was performed using the software to calculate Vmax and Km values for each enzyme.
Negative-stain electron microscopy
Negative-staining electron microscopy was used to investigate AGAL5, CaCl2 treated AGAL5, and AGAL5 bound with an inhibitor. A 5 µL protein solution was applied to a glow-discharged grid with a continuous carbon film, followed by staining with 0.75% w/v uranyl acetate for 10 s and 60 s, respectively. After each staining step, 4 µL of uranyl acetate was added and removed. The processed sample was then examined using a 120 kV electron microscope from FEI Company. The images obtained from the electron microscope were used to analyze the structure and interactions of AGAL5 with CaCl2 and an inhibitor.
Cryo-EM grid preparation
Three frozen samples were prepared for cryo-EM single-particle analysis. The first sample contained AGAL5 at a concentration of 1.4 mg/ml. The second sample contained AGAL5 incubated with 5 mM CaCl2 for 1 h on ice at a concentration of 1.3 mg/ml. The third sample contained AGAL5 incubated with ABP2 at a ratio of 1:2 for 30 min at a concentration of 1.3 mg/ml. All samples were prepared using a Vitrobot operated at 4 °C and 100% humidity. They were applied to a glow-discharged 300 mesh Quantifoil Cu R1.2/1.3 grid, blotted for 2–3 s with a nominal blot force 1 of the Vitrobot, and immediately plunged into pre-cooled liquid ethane for vitrification.
Cryo-EM data processing
We collected 3277 micrographs of AGAL5 wild type, 3199 micrographs of CaCl2 treated AGAL5, and 8959 micrographs of AGAL5 bound with an inhibitor, respectively. The micrographs were motion-corrected using MotionCor275, and CTF parameters were estimated by CTFFIND4.176. For wild-type AGAL5 and CaCl2 treated AGAL5, micrographs were denoised using the TOPAZ program77. High-quality training data of protein particle were filtered from a small manually picked dataset through 2D classification and used as input for TOPAZ program. The trained model then picked a large number of protein particles from all the micrographs. For AGAL5 bound with ABP2, particles were picked manually from selected micrographs, and a subset of these particles was selected through 2D classification. This subset was used as a reference to pick all the particles from all the micrographs using the auto-pick method of Laplacian-of-Gaussian blob in Relion program78. After several rounds of 2D classification, high SNR (signal to noise) 2D classes were retained for further process. In total, 261,006 particles of wild-type AGAL5, 7,720,604 particles of CaCl2 treated AGAL5, and 240,999 particles of AGAL5 bound with ABP2 were selected. 3D initial models generated from good SNR 2D classes served as references in the 3D refinement and classification procedure. The best class of 3D classification was used as an updated reference for 3D auto-refinement. After several rounds of 3D classification and 3D refinement, we obtained consensus particles that were refined in the final reconstruction. As a result, we obtained a 3.28 Å wild-type AGAL5 map, a 3.37 Å map for CaCl2 treated AGAL5 map, and a 3.17 Å map for AGAL5 bound with ABP2 map through CTF refinement and post-processing. Local resolution maps showed that the inner resolution was higher than the surrounding resolution, respectively, and the density map of CaCl2 treated AGAL5 was incomplete. The reported resolution based on the gold-standard Fourier shell correlation at 0.143 criteria (see Supplementary Fig. 7a–c, Supplementary Table 2). The atomic models of AGAL5 were built using COOT79 and refined using the Phenix program. The crystal reference model (PDB: 4FNQ) was used as a starting point for the model building of AGAL5 atomic model. For CaCl2 treated AGAL5 and AGAL5 bound with ABP2, the same procedure was followed, except that the initial model was AGAL5 instead of the crystal model. Refinement was carried out using phenix.real_space_refine80 (Supplementary Table 2).
TLC to show hydrolysis and transglycosylation
To demonstrate RFOs hydrolysis, a 50 μL enzyme reaction mixture containing 1.14 μM of enzyme, 50 mM acetate buffer (pH 6.5), and 85 mM of the substrate (melibiose, raffinose, or stachyose) was incubated at 37 °C for 3 h in duplicate with a control containing no enzyme. At every 10-min interval within the 1st hour, 2nd hour, and 3rd hour time points, 10 μL of the reaction mixture was removed and boiled for 10 min to stop the reaction. A silica gel 60 F 254 plate (Sigma) was used for thin-layer chromatography (TLC) analysis, with 2.5–5 μL of the reaction mixture or standards spotted 1 cm from the bottom. The mobile phase used was butanol:ethanol:water (5:3:2), and a mixture of glucose + galactose, melibiose, sucrose, galactose, and raffinose standards were used for comparison. Once the mobile phase reached near the top of the plate, it was dried and developed with orcinol-sulfuric acid reagent (0.1% orcinol m/v and 70% sulfuric acid v/v), and the sugars were visualized by charring using a hotplate (Fig. 7a, Supplementary Fig. 9). The duplicate hydrolysis experiment was repeated three times.
To demonstrate blood group antigen trisaccharide hydrolysis, a 15 μL enzyme reaction mixture containing 1.5 μM of enzyme, 1.5 μL substrate (10 mg/ml) blood group antigen A or B, and 12 μL McIvaine buffer (pH 7.0) was incubated at 37 °C for 5 h in duplicate with a control containing no enzyme. After 1 h and 3 h time points, 5 μL reaction mixture was removed and boiled for 10 min to stop the reaction. A silica gel 60 F 254 plate (Sigma) was used for thin-layer chromatography (TLC) analysis, with 2.5–5 μL of the reaction mixture spotted 1 cm from the bottom. The mobile phase used was butanol:ethanol:water (5:3:2). Once the mobile phase reached near the top of the plate, it was dried and developed with orcinol-sulfuric acid reagent (0.1% orcinol m/v and 70% sulfuric acid v/v), and the sugars were visualized by charring using a hotplate (Fig. 7b, Supplementary Fig. 10). The duplicate hydrolysis experiment was repeated three times.
To test transglycosylation, a 50 μL enzyme reaction mixture containing 1.14 μM of enzyme, 50 mM acetate (pH 6.5), 15 mM of donor substrate (pNP-α-galactopyranoside), and 85 mM of acceptor substrate (stachyose, raffinose, or melibiose) was incubated at 37 °C for 24 h in duplicate. Reactions without acceptor substrate or donor substrate were used as controls. After each hour, 10 μL of the assay mixture was removed and boiled for 5 min to stop the reaction. A TLC analysis was performed using a silica gel 60 F 254 plate (Merk, Cat. 1.05554.0001), with 2.5–5 μL of the reaction mixture or standards spotted 1 cm from the bottom. The mobile phase used was t-butanol:ethanol:water (5:3:2), and the sugars were visualized by charring using a heat gun after developing the plate with orcinol-sulfuric acid reagent (Fig. 7d, Supplementary Fig. 11). The whole experiment was repeated three times.
LC-MS for blood group B antigen trisaccharide analysis
An HPLC-MS system equipped with Electron Spray Ionization (1260-ultivo, Agilent Technologies, Santa Clara, CA, USA) was used for analysis. A Waters Xbridge Amide analytical column (2.1 × 100 mm, 1.7 μm) was used for the separation. Samples were eluted with solvent A [30/70 ACN/H2O with 0.1% NH4OH] and solvent B [80/20 ACN/H2O with 0.1% NH4OH] using the following gradient program at a flow rate of 0.4 mL/min; 0–10 min, 100%–80% solvent B; 10–15 min, 60% solvent B; The injected volume was 1 μL and the column temperature was set at 30 °C. The gas flow and temperature were set to 7 mL/min and 300 °C, respectively. The pressure of the nebulizer was 40 psi, the capillary voltage was set to 4000 V for the positive ionization mode. Selected reaction monitoring (SRM) was chosen as the scan mode, detecting precursor to product ion transitions. Thus, the m/z transitions were 511 → 365 (CE: 33) for the B antigen trisaccharide.100 μL sample, 100 μL PMP methanolic solution (0.5 mol/L) and 100 μL ammonia solution were mixed in a centrifuge tube. The mixture was heated to 70 °C for 30 min. Subsequently, 10% acetic acid aqueous solution and CHCl3 (1.0 mL each) were added. The upper aqueous phase was filtered through a 0.22 um membrane for further analysis. A HPLC-MS system equipped with an Electron Spray Ionization (1260-ultivo, Agilent Technologies, Santa Clara, CA, USA) was used for analysis. Separation was performed on an Agilent Poroshell 120 EC-C18 column (3 × 100 mm, 2.7 μm). Samples were eluted with solvent A [Water with 5 mM ammonium acetate] and solvent B [Acetonitrile with 5 mM ammonium acetate] using the following gradient program at a flow rate of 0.3 mL/min; start with 5% solvent B, change from 5 to 50% solvent B in 15 min and constant 100% solvent B for 5 min. A post-run of 5 min was programmed to equilibrate the column between analyses. The injected volume was 5 μL and column temperature was set at 30 °C. The gas flow and temperature were set at 10 mL/min and 350 °C, respectively. The pressure of the nebulizer was 40 psi, the capillary voltage was set at 4000 V for the positive ionization mode. Selected reaction monitoring (SRM) was chosen as scan mode, detecting precursor to product ion transitions. Thus, the m/z transitions were 657.3 → 373.4 (Fragmentor: 140 V; CE: 40) and 511 → 217.2 (Fragmentor: 150 V; CE: 29) for the hydrolysis product (Fig. 7c).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Cryo-EM maps of AGAL5, CaCl2 treated AGAL5, and AGAL5 bound with ABP2 have been deposited in the EMDB with the accession codes EMD-36790, EMD-36943, and EMD-36944. Coordinate files for these structures have been deposited in the PDB with the accession codes 8K1A, 8K7U, and 8K7V. The gene sequences were provided in a single Excel file for each figure/table in a separate sheet in Supplementary Data 2. The proteomics (LC/MS-MS) raw data has been deposited to the PRIDE database (https://www.ebi.ac.uk/pride/) with the dataset identifier PXD046454.
References
Schmidt, T. S. B., Raes, J. & Bork, P. The human gut microbiome: from association to modulation. Cell 172, 1198–1215 (2018).
Zhu, J. et al. An expanded gene catalog of mouse gut metagenomes. mSphere 6, e01119-20 (2021).
Koppel, N., Maini Rekdal, V. & Balskus, E. P. Chemical transformation of xenobiotics by the human gut microbiota. Science 356, eaag2770 (2017).
Lobb, B., Tremblay, B. J., Moreno-Hagelsieb, G. & Doxey, A. C. An assessment of genome annotation coverage across the bacterial tree of life. Microb. Genom. 6, e000341 (2020).
Yoon, S. S., Kim, E. K. & Lee, W. J. Functional genomic and metagenomic approaches to understanding gut microbiota-animal mutualism. Curr. Opin. Microbiol. 24, 38–46 (2015).
Zhang, Y. et al. Metatranscriptomics for the human microbiome and microbial community Functional Profiling. Annu. Rev. Biomed. Data Sci. 4, 279–311 (2021).
Zhao, S. et al. Discovery of new enzymes and metabolic pathways by using structure and genome context. Nature 502, 698–702 (2013).
Durairaj, J. et al. Uncovering new families and folds in the natural protein universe. Nature 622, 646–653 (2023).
Barrio-Hernandez, I. et al. Clustering predicted structures at the scale of the known protein universe. Nature 622, 637–645 (2023).
Jia, B., Han, X., Kim, K. H. & Jeon, C. O. Discovery and mining of enzymes from the human gut microbiome. Trends Biotechnol. 40, 240–254 (2022).
Vogel, C. & Marcotte, E. M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 13, 227–232 (2012).
Franzosa, E. A. et al. Sequencing and beyond: integrating molecular ‘omics’ for microbial community profiling. Nat. Rev. Microbiol. 13, 360–372 (2015).
Cravatt, B. F., Wright, A. T. & Kozarich, J. W. Activity-based protein profiling: from enzyme chemistry to proteomic chemistry. Annu. Rev. Biochem. 77, 383–414 (2008).
Li, N., Overkleeft, H. S. & Florea, B. I. Activity-based protein profiling: an enabling technology in chemical biology research. Curr. Opin. Chem. Biol. 16, 227–233 (2012).
Barglow, K. T. & Cravatt, B. F. Activity-based protein profiling for the functional annotation of enzymes. Nat. Methods 4, 822–827 (2007).
Simon, G. M. & Cravatt, B. F. Activity-based proteomics of enzyme superfamilies: serine hydrolases as a case study. J. Biol. Chem. 285, 11051–11055 (2010).
Han, L. & Chang, P. V. Activity-based protein profiling in microbes and the gut microbiome. Curr. Opin. Chem. Biol. 76, 102351 (2023).
Wright, A. T., Hudson, L. A. & Garcia, W. L. Activity-based protein profiling—enabling phenotyping of host-associated and environmental microbiomes. Isr. J. Chem. 63, e202200099 (2023).
Keller, L. J., Babin, B. M., Lakemeyer, M. & Bogyo, M. Activity-based protein profiling in bacteria: applications for identification of therapeutic targets and characterization of microbial communities. Curr. Opin. Chem. Biol. 54, 45–53 (2020).
Hatzios, S. K. et al. Chemoproteomic profiling of host and pathogen enzymes active in cholera. Nat. Chem. Biol. 12, 268–274 (2016).
Parasar, B. et al. Chemoproteomic profiling of gut microbiota-associated bile salt hydrolase activity. ACS Cent. Sci. 5, 867–873 (2019).
Brandvold, K. R. et al. Activity-based protein profiling of bile salt hydrolysis in the human gut microbiome with beta-lactam or acrylamide-based probes. ChemBioChem 22, 1448–1455 (2021).
Whidbey, C. et al. A probe-enabled approach for the selective isolation and characterization of functionally active subpopulations in the gut microbiome. J. Am. Chem. Soc. 141, 42–47 (2019).
Jariwala, P. B. et al. Discovering the microbial enzymes driving drug toxicity with activity-based protein profiling. ACS Chem. Biol. 15, 217–225 (2020).
Simpson, J. B. et al. Diverse but desolate landscape of gut microbial azoreductases: a rationale for idiopathic IBD drug response. Gut Microbes 15, 2203963 (2023).
Thuy-Boun, P. S. et al. Quantitative metaproteomics and activity-based protein profiling of patient fecal microbiome identifies host and microbial serine-type endopeptidase activity associated with ulcerative colitis. Mol. Cell. Proteom. 21, 100197 (2022).
Van Den Bossche, T. et al. The Metaproteomics Initiative: a coordinated approach for propelling the functional characterization of microbiomes. Microbiome 9, 243 (2021).
Wang, Y., Zhou, Y., Xiao, X., Zheng, J. & Zhou, H. Metaproteomics: a strategy to study the taxonomy and functionality of the gut microbiota. J. Proteom. 219, 103737 (2020).
Mayers, M. D., Moon, C., Stupp, G. S., Su, A. I. & Wolan, D. W. Quantitative metaproteomics and activity-based probe enrichment reveals significant alterations in protein expression from a mouse model of inflammatory bowel disease. J. Proteome Res. 16, 1014–1026 (2017).
Rhee, K. Y., Jansen, R. S. & Grundner, C. Activity-based annotation: the emergence of systems biochemistry. Trends Biochem. Sci. 47, 785–794 (2022).
Bhatia, S., Singh, A., Batra, N. & Singh, J. Microbial production and biotechnological applications of alpha-galactosidase. Int. J. Biol. Macromol. 150, 1294–1313 (2020).
Katrolia, P., Rajashekhara, E., Yan, Q. J. & Jiang, Z. Q. Biotechnological potential of microbial α-galactosidases. Crit. Rev. Biotechnol. 34, 307–317 (2014).
Lenders, M. & Brand, E. Fabry disease: the current treatment landscape. Drugs 81, 635–645 (2021).
Kwan, D. H. et al. Toward efficient enzymes for the generation of universal blood through structure-guided directed evolution. J. Am. Chem. Soc. 137, 5695–5705 (2015).
Rahfeld, P. et al. An enzymatic pathway in the human gut microbiome that converts A to universal O type blood. Nat. Microbiol. 4, 1475–1485 (2019).
Anisha, G. S. Molecular advances in microbial α-galactosidases: challenges and prospects. World J. Microbiol. Biotechnol. 38, 148 (2022).
Elango, D. et al. Raffinose Family Oligosaccharides: friend or foe for human and plant health? Front. Plant Sci. 13, 829118 (2022).
Willems, L. I. et al. Potent and selective activity-based probes for GH27 human retaining alpha-galactosidases. J. Am. Chem. Soc. 136, 11622–11625 (2014).
Kytidou, K. et al. Nicotiana benthamiana α-galactosidase A1.1 can functionally complement human α-galactosidase A deficiency associated with Fabry disease. J. Biol. Chem. 293, 10042–10058 (2018).
Merceron, R. et al. The molecular mechanism of thermostable alpha-galactosidases AgaA and AgaB explained by x-ray crystallography and mutational studies. J. Biol. Chem. 287, 39642–39652 (2012).
Jones, D. T., Taylor, W. R. & Thornton, J. M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282 (1992).
Kumar, S., Stecher, G., Li, M., Knyaz, C. & Tamura, K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549 (2018).
Wang, H. et al. Insights into the substrate specificity and synergy with mannanase of family 27 α-galactosidases from Neosartorya fischeri P1. Appl. Microbiol. Biotechnol. 99, 1261–1272 (2015).
Wang, J. et al. Characterization of a protease-resistant α-galactosidase from Aspergillus oryzae YZ1 and its application in hydrolysis of raffinose family oligosaccharides from soymilk. Int. J. Biol. Macromol. 158, 708–720 (2020).
Liu, Y., Yang, S., Yan, Q., Liu, J. & Jiang, Z. High-level expression of a novel protease-resistant α-galactosidase from Thielavia terrestris. Process Biochem. 71, 82–91 (2018).
Rios, S., Pedregosa, A. M., Fernandez Monistrol, I. & Laborda, F. Purification and molecular properties of an alpha-galactosidase synthesized and secreted by Aspergillus nidulans. FEMS Microbiol. Lett. 112, 35–41 (1993).
E, S., Potumarthi, R., A, N. & Mangamoori, L. N. Purification and characterisation of intracellular alpha-galactosidases from Acinetobacter sp. 3 Biotech 5, 925–932 (2015).
Schröder, C., Janzer, V. A., Schirrmacher, G., Claren, J. & Antranikian, G. Characterization of two novel heat-active α-galactosidases from thermophilic bacteria. Extremophiles 21, 85–94 (2017).
Bangoria, P., Patel, A. & Shah, A. R. Characterization of a fungal α-galactosidase and its synergistic effect with β-mannanase for hydrolysis of galactomannan. Carbohyd. Res. 531, 108893 (2023).
Fredslund, F. et al. Crystal structure of alpha-galactosidase from Lactobacillus acidophilus NCFM: insight into tetramer formation and substrate binding. J. Mol. Biol. 412, 466–480 (2011).
Bruel, L. et al. alpha-Galactosidase/sucrose kinase (AgaSK), a novel bifunctional enzyme from the human microbiome coupling galactosidase and kinase activities. J. Biol. Chem. 286, 40814–40823 (2011).
Laskowski, R. A. & Swindells, M. B. LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 (2011).
Ren, W. et al. Revealing the mechanism for covalent inhibition of glycoside hydrolases by carbasugars at an atomic level. Nat. Commun. 9, 3243 (2018).
Adamson, C. et al. Structural snapshots for mechanism-based inactivation of a glycoside hydrolase by cyclopropyl carbasugars. Angew. Chem. Int. Ed. 55, 14978–14982 (2016).
Nguyen, T. L. A., Vieira-Silva, S., Liston, A. & Raes, J. How informative is the mouse for human gut microbiota research? Dis. Model. Mech. 8, 1–16 (2015).
Simpson, J. B. et al. Gut microbial β-glucuronidases influence endobiotic homeostasis and are modulated by diverse therapeutics. Cell Host Microbe 32, 925–944.e910 (2024).
Killinger, B. J. et al. Activity-based protein profiling identifies alternating activation of enzymes involved in the bifidobacterium shunt pathway or mucin degradation in the gut microbiome response to soluble dietary fiber. NPJ Biofilms Microbi. 8, 60 (2022).
Cargile, B. J., Bundy, J. L. & Stephenson, J. L. Jr Potential for false positive identifications from large databases through tandem mass spectrometry. J. Proteome Res. 3, 1082–1085 (2004).
Jagtap, P. et al. A two-step database search method improves sensitivity in peptide sequence matches for metaproteomics and proteogenomics studies. Proteomics 13, 1352–1357 (2013).
Sticker, A., Martens, L. & Clement, L. Mass spectrometrists should search for all peptides, but assess only the ones they care about. Nat. Methods 14, 643–644 (2017).
Noble, W. S. & Keich, U. Response to “Mass spectrometrists should search for all peptides, but assess only the ones they care about”. Nat. Methods 14, 644 (2017).
Sun, Z., Ning, Z. & Figeys, D. The landscape and perspectives of the human gut metaproteomics. Mol. Cell Proteom. 23, 100763 (2024).
Lombard, V., Ramulu, H. G., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495 (2014).
Anisha, G. S. Microbial α-galactosidases: efficient biocatalysts for bioprocess technology. Bioresour. Technol. 344, 126293 (2022).
Davies, G. J., Planas, A. & Rovira, C. Conformational analyses of the reaction coordinate of glycosidases. Acc. Chem. Res. 45, 308–316 (2012).
Koshland, D. E. Jr. Stereochemistry and the mechanism of enzymatic reactions. Biol. Rev. 28, 416–436 (1953).
Liu, Q. P. et al. Bacterial glycosidases for the production of universal red blood cells. Nat. Biotechnol. 25, 454–464 (2007).
Li, N. et al. Relative quantification of proteasome activity by activity-based protein profiling and LC-MS/MS. Nat. Protoc. 8, 1155–1168 (2013).
Cheng, K. et al. MetaLab: an automated pipeline for metaproteomic data analysis. Microbiome 5, 157 (2017).
Tatusova, T. et al. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624 (2016).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301–2319 (2016).
Katoh, K., Rozewicki, J. & Yamada, K. D. MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform 20, 1160–1166 (2019).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Zheng, S. Q. et al. MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332 (2017).
Rohou, A. & Grigorieff, N. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 192, 216–221 (2015).
Medlar, A. & Holm, L. TOPAZ: asymmetric suffix array neighbourhood search for massive protein databases. BMC Bioinforma. 19, 278 (2018).
Scheres, S. H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 (2012).
Emsley, P. & Cowtan, K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D. 60, 2126–2132 (2004).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D. 66, 213–221 (2010).
Acknowledgements
We thank Prof. Hermen S. Overkleeft and Dr. Bogdan I. Florea of Leiden University for providing the specific probes and all the insightful discussions. The Cryo-EM Facility of Southern University of Science and Technology (SUSTech) for the technical assistance on cryo-EM data collection. This work was supported by funding from the National Key Research and Development Program of China (2023YFA0913900), the National Natural Science Foundation of China (32271501, 31971354, 21807106), the Natural Science Foundation of Guangdong Province (2018A030310035), the Shenzhen Science and Technology Innovation Committee (JCYJ20180302145723601, ZDSYS20220303153551001), Shenzhen Peacock Talent Launch Funding (RC00324), Exit Postdoctoral Fellow for Shenzhen Research Funding (K22227502).
Author information
Authors and Affiliations
Contributions
N.L., J.J., and Y.L. conceived the study. J.J., D.C., Y.R., H.P., W.Z., and T.W. designed and performed experiments with supervision from N.L., Y.L., S.S., and W.C. All authors participated in data analysis and interpretation. J.J., N.L., and Y.L. wrote the draft manuscript, with input from all authors.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jiang, J., Czuchry, D., Ru, Y. et al. Activity-based metaproteomics driven discovery and enzymological characterization of potential α-galactosidases in the mouse gut microbiome. Commun Chem 7, 184 (2024). https://doi.org/10.1038/s42004-024-01273-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-024-01273-5
