
Abstract
Hyperpolarized 13C MRI visualizes real-time metabolic processes in vivo. In this study, we achieved high 13C polarization in situ in the bore of an MRI system for precursor molecules of most widely employed hyperpolarized agents: [1-13C]acetate and [1-13C]pyruvate ethyl esters in their perdeuterated forms, enhancing hyperpolarization lifetimes, hyperpolarized to P13C ≈ 28% at 80 mM concentration and P13C ≈ 19% at 10 mM concentration, respectively. Using vinyl esters as unsaturated Parahydrogen-Induced Polarization via Side-Arm Hydrogenation (PHIP-SAH) precursors and our novel polarization setup, we achieved these hyperpolarization levels by fast side-arm hydrogenation in acetone-d6 at elevated temperatures (up to 90°C) and hydrogenation pressures (up to 32 bar). We optimized the hyperpolarization process, reducing it to under 10 s, and employed advanced pulse sequences to enhance the polarization transfer efficiency. The hyperpolarization system has a small footprint, allowing it to be positioned in the same magnet, where 13C MRI is performed. We exemplified the utility of the design with sub-second in situ 13C MRI of ethyl [1-13C]pyruvate-d6. However, challenges remain in side-arm cleavage and purification in the MRI system to extract highly polarized aqueous agent solutions. Our results showcase efficient and rapid 13C hyperpolarization of these metabolite precursors in an MRI system with minimal additional hardware, promising to enhance future throughput and access to hyperpolarized 13C MRI.
Similar content being viewed by others
Introduction
Non-invasive imaging of the human anatomy is an important pillar of diagnostic medicine. Morphological criteria are heavily used to diagnose and grade disease, as exemplified by the RECIST criteria for tumor assessment1. However, they hold limitations in capturing early molecular responses, while changes may not emerge early enough in traditional size-based assessments, even when patients respond favorably2. Hence, the evolving field of oncology calls for non-radioactive methods that provide swift and rich insights into metabolism for cancer staging and grading, treatment selection and response analysis, and survival3,4.
Magnetic Resonance Imaging (MRI) is a powerful and versatile modality with great promise for molecular imaging. Notably, Nuclear Magnetic Resonance (NMR) is a gold standard in analytical chemistry for determining structure and for discriminating between molecules enabled by chemically shifted resonance frequencies and line splitting due to spin-spin interactions between neighboring spins. However, the intrinsically low sensitivity of magnetic resonance has been a persistent challenge, limiting MRI mostly to detect 1H nuclei in water and lipids that are highly abundant in the mammalian body. In the quest to amplify the diagnostic potential of MRI, hyperpolarization techniques have garnered significant attention enabling MRI of selected molecules with over 10,000-fold signal enhancement5,6. The ability to hyperpolarize key metabolites such as pyruvate has propelled this interest by offering real-time insights into metabolic processes3,4. For such applications, non-1H nuclei such as carbon-13 (13C) are typically hyperpolarized (HP), because of their background-free detection in vivo, large chemical shift dispersion, and long lifetimes of HP states in vivo compared to proton (i.e. longitudinal relaxation time T1 of 1 min for [1-13C]pyruvate versus ≈1 s for most protons). This endeavor has been supported by the identification of specific metabolic alterations delivering powerful surrogate markers for disease staging and evaluating progression and therapy response. HP MRI, particularly using HP [1-13C]pyruvate, is currently under investigation in over 50 clinical trials, and interim results have shown significant promise for future patient care5,7,8,9.
While pyruvate remains the most widely applied hyperpolarization agent, acetate probes aberrant fatty acid metabolism10,11,12. For instance, non-HP [1-13C]acetate has been used in brain metabolism studies13,14,15,16,17, and HP [1-13C]acetate has been used in studies of liver and brain metabolism13,18,19. Hence, the efficient hyperpolarization of acetate is as important in the context of biomedical applications.
Biomedical applications, including all clinical studies, have relied on dissolution Dynamic Nuclear Polarization (d-DNP), which is recognized as the most established method for producing a variety of 13C HP metabolites3,4,20. The d-DNP hyperpolarization requires strong magnetic fields (>1 T, typically employing superconducting magnets), cryogenic temperatures (<2 K), and microwave irradiation to polarize agents in the solid phase. This is followed by rapid thawing and sample dissolution, typically using superheated water, to transfer HP metabolites into the liquid aqueous phase. This process often takes about one hour e.g. for producing a bolus of HP [1-13C]pyruvate. The impressive results achieved with HP MRI have sparked a persistent effort within the scientific community to explore other technologies and to enhance d-DNP for a faster, more flexible, and more cost-effective production of HP media21,22,23,24,25,26.
Parahydrogen-based techniques hyperpolarize in the liquid state using the readily available spin order of the singlet isomer of molecular hydrogen (parahydrogen, pH2), often employing inexpensive hardware and non-elaborate sample preparation methods24,25. Hence, hydrogenative pH2-Induced Polarization (PHIP)27,28 and Signal Amplification by Reversible Exchange (SABRE, also known as non-hydrogenative PHIP)25,29 have emerged as promising high-throughput alternatives to d-DNP both capable of producing agents swiftly within few minutes as recently exemplified with safe in vivo demonstrations30,31,32,33,34,35.
Hydrogenative PHIP experiments can be classified into two fundamentally distinct subtypes: (a) Parahydrogen And Synthesis Allows a Dramatically Enhanced Nuclear Alignment (PASADENA), where hydrogenation, optional heteronuclear polarization transfer using pulse sequences, and readout occur at the high magnetic field28,36,37; b) Adiabatic Longitudinal Transport After Dissociation Engenders Net Alignment (ALTADENA), a magnetic field cycling (MFC) scheme, where hydrogenation takes place at low field followed by adiabatic transfer to higher magnetic fields for detection of the in-phase 1H spin order38. Similarly, an MFC can be used for heteronuclear polarization transfer by passaging ultra-low fields39,40. Until recently, hydrogenative PHIP for biomedical applications was severely limited in its choice of agents, primarily due to the scarcity of suitable unsaturated precursors that could undergo hydrogenation to produce metabolically active molecules24,41. However, PHIP by Side Arm Hydrogenation (PHIP-SAH) has made strides in enabling the hyperpolarization of various molecules including pyruvate and acetate that have no direct unsaturated precursor that can be hydrogenated with parahydrogen (pH2)32,40. In PHIP-SAH experiments, in a three-step procedure, unsaturated esters of a metabolite of interest are hydrogenated typically in acetone or chloroform yielding PHIP-SAH precursor molecules of the desired metabolite, followed by side-arm cleavage, and a swift purification to extract the metabolite in water32,42,43.
A variety of advanced instruments for PHIP has been developed, each offering distinct advantages, as detailed in a recent review24. Standalone polarizers were presented, many of the first used polysulfone reactors for hydrogenation in water, allowing for elevated pH2 pressures and temperatures to enhance the hydrogenation rate44,45,46,47. However, water’s limited solubility for H2 and some PHIP-SAH esters, compared to organic solvents, restricts the concentration of agents. Additionally, polysulfone is incompatible with many organic solvents, such as acetone and chloroform. To overcome these limitations, 5-mm NMR tubes have been employed48,49,50,51,52, which can tolerate some overpressure, or steel, brass and aluminum reactors have been used53,54. However, for hyperpolarization at high magnetic fields, metallic reactors are unsuitable because they shield radio frequencies and have much different magnetic susceptibilities compared to liquids, causing significant magnetic field inhomogeneity. Similar to d-DNP, all these hydrogenative PHIP implementations still require an additional device and investment for a “polarizer” to prepare the hyperpolarized (HP) agents for in vivo 13C MRI. The polarizer itself can be complex and require considerable space within the imaging facility in close proximity to the MRI system due to signal decay during sample transfer.
Our team has pushed forward a PASADENA variant that enables the ultra-fast polarization of 13C agents in situ within an MRI system with a minimized footprint: Synthesis Amid the Magnet Bore Allows Dramatically Enhanced Nuclear Alignment (SAMBADENA)55. The primary advantage of this method is the elimination of the need for an external polarizer, as the highly advanced MRI hardware, augmented with a “PHIP upgrade,” to polarize agents directly in the application field. Consequently, SAMBADENA circumvents relaxation losses and the need for transporting agents to the MRI setup. However, the elevated magnetic field restricts the materials that can be used and, unlike external polarizers, the limited space available in preclinical MRI systems poses an additional engineering challenge for successful implementation.
In our previous work, we demonstrated the administration and in vivo imaging within seconds using [1-13C]hydroxyethyl propionate-d3 (HEP), a xenobiotic molecule hyperpolarized in water and administered without purification56. We showed that both the reactor and the mouse can be positioned at the isocenter of the MRI system for hyperpolarization and in vivo imaging, respectively, facilitated by a motorized slider moving the mouse bed and reactor in between the two experimental steps during the tail vein administration. To date, high 13C polarizations of several PHIP agents in water were demonstrated with SAMBADENA using custom polysulfone reactors, namely HEP, [1-13C]succinate-d2, and [1-13C]phospholactate-d255,57,58. Additionally, high 13C-polarization throughput (hyperpolarization every 15 s) has been shown59. However, PHIP-SAH and the hyperpolarization of acetate and pyruvate esters have not been demonstrated with SAMBADENA yet due to challenges in synthetic chemistry, spin physics, and the development of a suitable reaction chamber.
Recent advances in the synthesis of suitable 13C and 2H isotope labeled PHIP-SAH precursors60,61,62,63,64, and polarization transfer methods operating at clinical magnetic fields65,66,67,68 have facilitated investigating this promising technique. PHIP-SAH at Tesla fields was predominantly pioneered by the Glöggler group, demonstrating impressively high 13C polarization levels of up to P13C ≈ 50% for ethyl [1-13C]acetate-d6 and for ethyl [1-13C]pyruvate-d6 inside a 300 MHz NMR system43,69. Recently, it has been suggested to use a low-cost benchtop NMR systems or a standalone polarizer operating in the millitesla regime to avoid necessitating an additional costly NMR system for polarization65,70,71. Still, these approaches require an external polarizer to produce HP metabolites.
In this study, we present a SAMBADENA approach that enabled 13C hyperpolarization up to P13C ≈ 30% for ethyl [1-13C]acetate-d6 and ethyl [1-13C]pyruvate-d6. The key innovation was the development of an efficient hydrogenation process at high field, in acetone-d6, at elevated temperature ≥90°C and pressure >30 bar that resulted in complete hydrogenation of unsaturated precursor under 10 s. This achievement was made possible by a newly designed organic solvent-compatible non-magnetic SAMBADENA setup, Fig. 1. While we omit the side-arm cleavage and purification steps typically used with PHIP-SAH for biomedical applications, we demonstrate the potential of our results by showcasing the rapid 13C MRI of 1 mL of 80 mM of ethyl [1-13C]pyruvate-d6 in situ within seconds following the completion of the hyperpolarization process.

a A fluid control unit with two manometers, four electric solenoid valves (V1-V4), and one hand valve was used to inject the fluids and pH2 at the bottom (labeled as V1 in Supplementary Fig. 1a) of the reactor and to create backpressure with N2 or release pressure on top (labeled as V2 in Supplementary Fig. 1a). The liquid chamber was held in the isocenter of the MRI system. b Drawing of the SAMBADENA setup with magnification to show the new PEEK reactor installed in the MRI system (cross-sectioned). Note that this reactor is designed to be compatible with a mouse bed setup, as utilized in our previous in vivo experiments56.
Materials and methods
Experimental setup
The SAMBADENA setup comprised a custom-made, solvent-compatible, actively-heated reactor for fast pH2 addition, described in the next section. For hyperpolarization and MRI, a preclinical 7-T MRI setup (BioSpec 70/20, Advance III, Bruker, USA) and a 1H-13C volume coil (V-XLS-HL-070, Rapid Biomedical, Germany) were used, Fig. 1.
The fluid control included a set of four magnetic valves (G052 with solvent-compatible perfluoroelastomer (FFKM) sealings, GSR Ventiltechnik, Germany), which were operated via a microcontroller-based relay box from the MRI pulse program that was described previously59, and one hand valve. A non-magnetic, FFKM-sealed PEEK check valve (CV-3320, IDEX, USA) was installed in the reactor inlet at the bottom. A fluid trap was installed right before valve V3 to avoid contaminating the magnetic valves with solvents and catalyst. This fluid trap was additionally used as exhaust when tubes and the reactor were cleaned by flushing acetone at the end of each experiment day. This setup was automated and actuated via the MRI system’s console.
Reactor design
A reactor was custom-designed for this study (Inventor Professional, Autodesk, USA) and made from polyether ether ketone (PEEK) via computer numerical control (CNC), Fig. 2. PEEK was chosen because of good magnetic and mechanical properties making it robust against elevated pressures and temperatures, combined with excellent resistance to many acids, bases and solvents including water, acetone and chloroform. The reactor features two chambers: an inner chamber for the hydrogenation reaction and an outer chamber for active heating. The inner chamber has four inlets, two of which (one at the top, one at the bottom) are utilized in this study. The bottom inlet of the reactor facilitates pH2 bubbling. The top inlet serves three main purposes: pre-pressurizing the reactor with nitrogen to prevent boil-off, depressurizing the reactor after each experiment, and for cleaning the setup by flushing acetone. The remaining inlets, intended for injection of the base for cleavage and extraction of the solution, were sealed for this study.

a Cross-sectional view of the reactor designed using CAD. It illustrates the two-chamber structure, with the inner chamber designated for hosting the hydrogenation reaction and the outer chamber for active heating. b Photograph of the reactor, showing the top lid covering for the reaction chamber. Two inlets, one on each side (black), are connecting the heating chamber of the reactor for the hot water circulation. A one-way PEEK check valve (black) is connected to the bottom inlet of the reaction chamber to direct gas flow into the reactor. The white attachment connected to the reactor was used for positioning the reactor in the MRI system when used without the mouse bed.
The reactor was engineered to withstand high pressures and a finite element method based numerical simulation suggested it can hold up to 162 bar with no permanent deformation (i.e. a pressure up to 50 bar can be applied with a safety factor larger three, Supplementary Fig. 2). Experimentally, we successfully tested the setup operation with up to p = 32 bar at T = 90 °C. Solvent compatible FFKM materials were used to seal the top and bottom lids of the reactor.
The reactor also included a heating chamber and was actively heated using a water-circulation system during the experiments (PC300 immersion circulator, ThermoFisher scientific, USA). At the beginning of an experiment day, the water heating was turned on for approximately 30 min before performing hyperpolarization experiments to ensure an equilibrium temperature for the reactor. After this initial heating, the reactor was used for polarization experiments at a rate of up to one sample per two minutes. The reactor was mountable in the MRI system either directly to the 1H-13C coil using an attachment (see Fig. 2b), or to a mouse bed. For the experiments here, the reactor was attached to a mouse bed (Fig. 1b) and positioned in the isocenter of the magnet using an automated positioning system (AutoPac, Bruker, USA).
Technical drawings of the reactor, cross sections, and a pressure simulation are provided in the SI.
Sample preparation
Parahydrogen was enriched to ≈90%72. Reaction solutions were prepared by mixing vinyl acetate-d6 (VA, ≈1.1% naturally abundant 13C, CAS: 189765-98-8, ID:UN1301, EQ Laboratories, Germany) or custom-synthesized vinyl [1-13C]pyruvate-d6 (VP, ≈98% 13C enriched61 provider: Prof. Rainer Herges, QuantView GmbH, Germany) and a rhodium-based hydrogenation catalyst ([Rh(dppb)(COD)]BF4, CAS: 79255-71-3, ID:341134, Sigma Aldrich, USA) in 1 mL of acetone-d6 (CAS: 666-52-4, ID:151793, Sigma Aldrich, USA). During the experiments, VA was hydrogenated to ethyl-acetate-d6 (EA) and VP formed ethyl [1-13C]pyruvate-d6 (EP), Fig. 3a.

a Upon catalytic, pairwise addition of parahydrogen (pH2) to the precursors (top) and SOT, the 1-13C-hyperpolarized esters were obtained (bottom). b The ESOTHERIC sequence66 was used with three 90° 1H- and one 90° 13C-pulses (single bars, y/-y phase) and two composite refocusing pulses in each free evolution interval (90°x-180°y-90°x) (Supplementary Fig. 5). The intervals ∆1 = 1/(2J12) and ∆2 = 1/(2J23) were calculated using the J-couplings of the three polarization-exchanging nuclei. The J-couplings were taken from the literature: for VA, J12 = 6.94 Hz, J23 = 3.16 Hz; and for VP, J12 = 7.1 Hz and J23 = 3 Hz69. J13 = 0 was assumed for both molecules.
Hyperpolarization process
To commence the experiment, 1 mL of the precursor catalyst solution was filled into the reactor (inside the MRI system), pressurized from the top of the reactor under a 5 bar N2 atmosphere to avoid boil-off, and warmed using the reactor-integrated water heating for 10 s. Then, the hand valve at the reactor outlet was closed to limit the outlet volume filled during the pH2 injection. The pH2 addition was started by opening valve V1 for 7 s to inject pressurized pH2 (≈90% enrichment) from the bottom of the reactor before the spin order transfer (SOT) sequence was started. The ESOTHERIC (efficient spin order transfer to heteronuclei via relayed INEPT (insensitive nuclei enhanced by magnetization transfer) chains) sequence was used with two composite refocusing pulses per time interval (90°x-180°y-90°x, Fig. 3b) for the SOT from pH2-derived protons to 13C71.
In total, the hyperpolarization process for a sample was completed in approximately 7.4 s, consisting of 7 s for hydrogenation followed by 388 ms for SOT. An additional 50 s time was allocated for sample filling and warming. Hence, with our technique, producing one hyperpolarized batch per minute becomes feasible.
Signal detection and quantification
In most cases, the hyperpolarized 13C signal was detected at the end of the SOT by acquiring free induction decay of 13C signal. The polarization level P13C was quantified by comparing the Fourier-transformed 13C signals of the HP sample and an external reference sample at thermal equilibrium (4 M [1-13C]sodium acetate in 1.5 mL H2O, 99% 13C, CAS: 23424-28-4, ID: 279293, Sigma Aldrich, USA; doped with gadolinium (7.5 µL Vasovist), T1 = 832 ms). In contrast to other studies73, the direct detection of thermal-equilibrium 13C signal from our HP samples was not possible due to the relatively long 13C T1 relaxation times and the low detection sensitivity of our volume resonator (see SI, section 8). We calculated 13C polarizations via the formula
where “HP” and “ref” refers to the hyperpolarized and reference signal, respectively. P is the polarization with PB ≈ 6.0 × 10−4% referring to the Boltzmann equilibrium polarization at 7 T at 300K, N is the number of summated MR signal (NHP = 1 in all cases), F is the fraction of 13C isotope, c is the concentration of the molecule, V is the volume of the sample and S is the measured signal. Here, 13C polarization level was quantified assuming 100% hydrogenation yield. The reaction kinetics investigated in Fig. 3d uses a two-handed fitting model to reveal time constants for the hydrogenation, Thydr, and relaxation of the pH2-derived 1H spin order before SOT, Trelax55,68:
Pmax is the maximum polarization value reached if no relaxation is present, t0 is the time offset to consider time needed for the delivery and dissolution of pH2, pressure build-up, and catalyst activation.
13C MRI
An MRI sequence and acquisition started subsequently after application of SOT with an additional 90oy 13C flip-back pulse (single-shot 13C RARE, 128x64x1 matrix size, 40×24 mm2 field of view (FOV), 15-mm slice thickness, 0.31×0.38 mm2 in-plane resolution, 64 echoes measured after one excitation, 438.6 ms acquisition time). For coregistration, an X-ray micro-tomography (µCT) image of the empty reactor was acquired to depict the setup (res.: 20 µm, exposure: 266 ms angular step: 0.8°, Bruker SkyScan 1276). Additional experimental details are presented in the supporting information (SI).
Results and discussion
Hyperpolarization of PHIP-SAH precursors
High 1-13C polarizations of 28% and 19% (Fig. 4a) were obtained for 1 mL EA (80 mM, 5 mM catalyst) and EP (10 mM, 5 mM catalyst), respectively, at T = 90 °C and p = 30 bar pH2 pressure after 7 s total hydrogenation time (i.e., 5 s bubbling plus a 2 s delay). For these results, deuterium-labeled unsaturated metabolite esters, which became available only recently43,61,62 were essential, as in the presence of non-pH2-nascent 1H the SOT efficiency is reduced67. Additionally, these results were preceded by extensive optimization and analysis as described in the following.

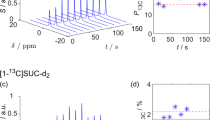
a 13C NMR spectra of a thermally-polarized reference solution (black) and hyperpolarized EA (red) and EP (blue). 13C-HP of EA and EP was quantified to 28% and 19%, respectively. The 13C polarization P13C for EA was optimized as function of temperature T (b), pH2 pressure p (c), and hydrogenation time th (d) using VA (mean, red dots, and stdev, black error bars, of N = 3 experiments each). The reaction kinetics were fitted with a described model (eq.-2). Unless stated otherwise parameters were set constant at T = 90°C, p = 25 bar, cVA = 80 mM, and th = 7 s. The different polarization levels observed in (b)–(d) are due to the typical inter-day reproducibility in SAMBADENA experiments (a factor of 1 ± 0:255,59, which is similar to state-of-the-art dDNP)81. Additionally, in (d) pH2 with a lower enrichment was used.
Maximizing 13C hyperpolarization
The polarization process within PHIP-SAH involves two main stages: hydrogenation and SOT. In our investigation using VA, we examined the impact of various hydrogenation reaction parameters on 13C polarization, including reaction temperature T, pH2 pressure p, and hydrogenation time th (Fig. 4b–d) as well as catalyst concentration ccat (Supplementary Fig. 4), and unsaturated precursor concentration cVA (Supplementary Fig. 6). Unless otherwise specified, the standard parameters are T = 90°C, p = 25 bar, cVA = 80 mM, ccat = 5 mM and th = 7 s.
When we varied the temperature from 30 to 90 °C, we observed a progressive increase in 13C polarization, reaching its peak at temperatures above 70°C (Fig. 4b), with polarization values at 70°C and higher being within the standard deviation. Similarly, as we adjusted the pH2 injection pressure across a range of 10 to 32 bar, we observed an increase in polarization, with the maximum achieved above 25 bar, where values were again equal within the standard deviation (Fig. 4c). Note that the solubility of hydrogen in solutions demonstrates a linear relationship with pressure and is approximately 4.5 mM/bar at 50°C for acetone74. Hence, it was concluded that elevated reaction temperatures and pressures play pivotal roles in achieving high polarization of contrast agents using our setup.
We varied th from 5 to 70 s and discovered that the maximum 13C polarization of 80 mM EA occurred around 7 s (Fig. 4d). Beyond this time, further hydrogenation of the residual unsaturated precursor was dominated by relaxation of the pH2-derived 1H spin order. We examined the data closer by fitting Eq. 2 and obtained the following fit parameters along with their associated errors: Pmax = (14.73 ± 0.17)%, t0 = (1.99 ± 0.03) s, Thydr = (0.44 ± 0.01) s, and Trelax = (68.4 ± 2.7) s, with Trelax the 1H relaxation of pH2-derived double spin order before SOT.
As we increased the concentration of VA from 10 to 160 mM while maintaining an unsaturated precursor-to-catalyst ratio of approximately 10, we observed a gradual decrease of the polarization (Supplementary Fig. 6) which was estimated under the assumption of complete conversion of VA to EA, which was likely not the case, especially at higher concentrations. This incomplete chemical conversion resulted in seemingly lower 13C polarizations for higher concentrations, where actually the chemical conversion was not fully realized; probably because of lack of diluted H2. The influence of the catalyst concentration on 13C polarization was investigated to determine its role in the hyperpolarization process. Notably, within the range of 1 to 20 mM, the concentration of the catalyst demonstrated minimal impact on the achieved 13C polarization indicating that the catalyst is not a limiting factor. This was particularly evident as all values at or exceeding a catalyst concentration of 2 mM were consistent within error intervals (Supplementary Fig. 4). Hence, for highly concentrated unsaturated precursors, optimal th represents a tradeoff between hydrogenation time and relaxation time; in agreement with previous reports55,68,75.
The ESOTHERIC sequence features a remarkable theoretical transfer efficiency, approaching nearly 100%, for converting the 1H two-spin order from the ethyl ester to the 1-13C metabolite site76. In theory, this implies the potential for nearly 100% 13C polarization, provided that pure pH2 is utilized and relaxation effects are negligible. However, practical considerations must also account for imperfections in the MRI system, including static field inhomogeneity and errors in pulse flip angles. A strategy to mitigate losses resulting from these MRI system imperfections is incorporating multiple 180° composite pulses77. In our investigations here, we determined that the maximum 13C polarizations were achieved when two 180° composite pulses were applied during each free evolution interval of the SOT sequence (as depicted in Fig. 4b and detailed in Supplementary Fig. 5). As found in two subsequent HP experiments evaluating 1H and 13C polarization, this sequence allowed for the conversion of the initial 1H polarization P1H ≈ 30% achieved after 7 s of hydrogenation of an 80 mM VA into P13C ≈ 20% 13C polarization (Supplementary Fig. 8) that corresponds to about 66% transfer efficiency.
After the analysis of hydrogenation parameters, it’s worth noting that the highest 13C polarizations achieved for EP were lower, at 19%, compared to those for EA at 28%, although EP had an unsaturated precursor concentration eight times lower than EA. Several factors may contribute to this observation. Firstly, the hydrogenation process was optimized for EA, which is commercially available, but we expect a similar trend of improved polarization with higher temperatures and pH2 pressure for EP. Secondly, prior studies have indicated that the hydrogenation rate for VP may be lower than that of VA under the same reaction conditions, potentially leading to less complete reaction within executed 7 s78. Lastly, we measured the 13C longitudinal relaxation of EP and found a time constant T1 of 22.4 ± 5.28 s (Supplementary Fig. 7), which is shorter than the 67 s reported under similar conditions in a recent paper43,71. Additionally, we observed side-arm cleavage forming [1-13C]pyruvate-d3 over time. Both findings may be attributable to impurities in the samples stemming from the preparation of the reaction solution. In a recent study using VP from the same synthesis, this phenomenon was led back to the presence of water in the reaction solution76. Consequently, we attribute the lower 13C polarizations primarily to a fraction of EP that had cleaved before the SOT had successfully transferred the pH2 spin order to the pyruvate [1-13C]site.
In summary, achieving maximum 13C hyperpolarization relies on a short hydrogenation time, minimized relaxation effects, and a high transfer efficiency. Enhancing the hydrogenation process for faster turnover might involve optimizing pH2 dissolution, potentially by maximizing gas-liquid interfaces. Moreover, the pH2 used had an enrichment fraction of ≈90% right after the ortho-to-para conversion (Supplementary Fig. 3). If 100% pH2 were employed, the 13C polarization levels for EA and EP would increase to approximately 31% and 21%, respectively, i.e. by a factor of 1.11. Furthermore, a comprehensive examination of the polarization transfer, especially under clinical field strengths within the SAMBADENA setup, warrants further investigation, and we intend to present this analysis in future work.
13C imaging
Motivated by the substantial 13C polarization levels achieved at concentrations suitable for in vivo imaging, we embarked on demonstrating the rapid 13C imaging capabilities. Utilizing HP EP prepared less than eight seconds prior, we conducted a sub-second 13C MRI experiment. The results revealed an intense 13C signal (Fig. 5). Coregistration with a µCT image of the reactor provided spatial context, clearly indicating the signal’s origin from the lower region of the reaction chamber and effectively picturing its contours.

HP 13C MRI of 10 mM ethyl [1-13C]pyruvate-d6 (color) superimposed on an X-ray micro-tomography (µCT, gray) of the reactor. For the 13C MRI, the signal was plotted on a red-to-yellow color scheme (Matlab: “autumn”) and red was set transparent. Note that the two images were acquired at different times in different imaging devices and that the reactor and water-heating chamber were drained for µCT.
Outlook: towards obtaining HP metabolites in biocompatible aqueous solutions
In this study, we have not yet addressed the side-arm cleavage and purification steps of the PHIP-SAH procedure required to obtain HP acetate and HP pyruvate in aqueous formulations suitable for preclinical applications. The current acetone solutions with rhodium-based catalyst heated to >60°C are unsuitable for in vivo applications. While elevated temperatures facilitate fast hydrogenation and potentially fast side-arm cleavage reactions, the final agent solution needs to be adapted to a physiological temperature range for safe administration.
We have previously demonstrated the rapid and reliable adaption of temperature during the administration of HP aqueous solution of xenobiotic HEP to (35 ± 1) °C via heat exchange with injection syringe and catheter56. However, the PHIP-SAH experiment is more complex than our previous PHIP experiments, hence posing a significant yet unresolved obstacle.
Methods for rapid side-arm cleavage and purification are available in the literature and they may be adapted for our approach31,53. Techniques such as rapid base addition, solvent evaporation of acetone, and catalyst removal through filtration30,53 seem promising for our approach. Additionally, phase separation using non-polar solvents like dichloromethane or chloroform may enable the separation of an aqueous phase containing the hydrolyzed contrast agents40,48,79—note that our presented setup is compatible with these solvents as well. In the future, a combination of these purification methods, potentially along with additional metal scavenging80, may enable the production of highly pure and safe injection solutions.
During the work-up of the solution, the precious signal enhancement inevitably decreases due to T1 relaxation. For instance, recent publications reported a factor of 0.5 of the starting 13C hyperpolarization remaining after the work-up procedures32,43. Considering these values, our current levels would enable >10% 13C polarization in the final HP agent solutions, which is sufficient for in vivo 13C metabolic imaging40. Nonetheless, improving the 13C polarization levels further is desirable and motivates more thorough future investigations of polarization transfer under MRI setup conditions.
For the SAMBADENA technique, side-arm cleavage, purification, and adaption of the agent solution will need to be incorporated within the magnetic field of the MRI system, while leaving space for the rodent, which particularly is not a trivial task. Purification at high magnetic fields, either at the fringe or inside the magnet, may benefit from extended T1 relaxation times30. However, in the bore of a preclinical small rodent MRI setup as utilized in this study, this appears an engineering challenge. As part of our ongoing efforts, we are also exploring the implementation of SAMBADENA on a clinical MRI system. While this setting provides more room for sample hyperpolarization and purification, it introduces new complexities related to scaling the setup, positioning the reactor and accommodating the purification apparatus, and adapting to varying MRI specifications.
Conclusion
SAMBADENA offers a compact footprint, fast hyperpolarization and administration and hence, holds promise for future applications. Although the implementation of a complete PHIP-SAH procedure including side-arm cleavage and purification presents challenges on both preclinical and clinical MRI systems, they appear to be solvable obstacles that merit exploration, especially now that we have demonstrated the efficient in situ hyperpolarization of pyruvate and acetate precursors within an MRI environment. Considering the low (additional) cost, small footprint55, high sample throughput59, and fast in vivo administration56, SAMBADENA holds great promise to accelerate the translation of PHIP to lower the translational burden and enable widespread use of HP for metabolic MRI.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: Revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Wang, D.-R., Wu, X.-L. & Sun, Y.-L. Therapeutic targets and biomarkers of tumor immunotherapy: response versus non-response. Signal Transduct. Target. Ther. 7, 331 (2022).
Chaumeil, M. M. et al. New horizons in hyperpolarized 13C MRI. Mol. Imaging Biol. https://doi.org/10.1007/s11307-023-01888-5 (2023).
Wodtke, P., Grashei, M. & Schilling, F. Quo Vadis Hyperpolarized 13C MRI? Z. Für Med. Phys. S0939388923001204 (2023) https://doi.org/10.1016/j.zemedi.2023.10.004 (2003).
Eills, J. et al. Spin hyperpolarization in modern magnetic resonance. Chem. Rev. 123, 1417–1551 (2023).
Ardenkjær-Larsen, J. H. et al. Increase in signal-to-noise ratio of > 10,000 times in liquid-state NMR. Proc. Natl Acad. Sci. 100, 10158–10163 (2003).
Chen, H.-Y. et al. Hyperpolarized 13C-pyruvate MRI detects real-time metabolic flux in prostate cancer metastases to bone and liver: a clinical feasibility study. Prostate Cancer Prostatic Dis. 23, 269–276 (2020).
Sinharay, S. & Pagel, M. D. Advances in magnetic resonance imaging contrast agents for biomarker detection. Annu. Rev. Anal. Chem. 9, 95–115 (2016).
Goldman, M., Jóhannesson, H., Axelsson, O. & Karlsson, M. Design and implementation of 13C hyper polarization from para-hydrogen, for new MRI contrast agents. C. R. Chim. 9, 357–363 (2006).
Yoshii, Y., Furukawa, T., Saga, T. & Fujibayashi, Y. Acetate/acetyl-CoA metabolism associated with cancer fatty acid synthesis: overview and application. Cancer Lett. 356, 211–216 (2015).
Koellisch, U. et al. Investigation of metabolic changes in STZ-induced diabetic rats with hyperpolarized [1-13C]acetate. Physiol. Rep. 3, e12474 (2015).
Koellisch, U. et al. Metabolic imaging of hyperpolarized [1‐13C]acetate and [1‐13C]acetylcarnitine ‐ investigation of the influence of dobutamine induced stress. Magn. Reson. Med. 74, 1011–1018 (2015).
Jensen, P. R. et al. Tissue-specific short chain fatty acid metabolism and slow metabolic recovery after ischemia from hyperpolarized NMR in Vivo. J. Biol. Chem. 284, 36077–36082 (2009).
Mikkelsen, E. F. R. et al. Hyperpolarized [1-13C]-acetate renal metabolic clearance rate mapping. Sci. Rep. 7, 16002 (2017).
Bastiaansen, J. A. M. et al. In vivo enzymatic activity of acetylCoA synthetase in skeletal muscle revealed by 13C turnover from hyperpolarized [1-13C]acetate to [1-13C]acetylcarnitine. Biochim. Biophys. Acta BBA—Gen. Subj. 1830, 4171–4178 (2013).
Sailasuta, N., Tran, T. T., Harris, K. C. & Ross, B. D. Swift Acetate Glial Assay (SAGA): An accelerated human 13C MRS brain exam for clinical diagnostic use. J. Magn. Reson. 207, 352–355 (2010).
Blüml, S., Moreno‐Torres, A., Shic, F., Nguy, C. ‐H. & Ross, B. D. Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed. 15, 1–5 (2002).
Le Page, L. M., Guglielmetti, C., Taglang, C. & Chaumeil, M. M. Imaging brain metabolism using hyperpolarized 13C magnetic resonance spectroscopy. Trends Neurosci. 43, 343–354 (2020).
Mishkovsky, M., Comment, A. & Gruetter, R. In vivo detection of brain krebs cycle intermediate by hyperpolarized magnetic resonance. J. Cereb. Blood Flow. Metab. 32, 2108–2113 (2012).
Ardenkjaer-Larsen, J. H. On the present and future of dissolution-DNP. J. Magn. Reson. 264, 3–12 (2016).
Ardenkjær‐Larsen, J. H. et al. Cryogen‐free dissolution dynamic nuclear polarization polarizer operating at 3.35 T, 6.70 T, and 10.1 T. Magn. Reson. Med. 81, 2184–2194 (2019).
Capozzi, A., Cheng, T., Boero, G., Roussel, C. & Comment, A. Thermal annihilation of photo-induced radicals following dynamic nuclear polarization to produce transportable frozen hyperpolarized 13C-substrates. Nat. Commun. 8, 15757 (2017).
Jannin, S., Bornet, A., Melzi, R. & Bodenhausen, G. High field dynamic nuclear polarization at 6.7T: Carbon-13 polarization above 70% within 20 min. Chem. Phys. Lett. 549, 99–102 (2012).
Schmidt, A. B. et al. Instrumentation for hydrogenative parahydrogen-based hyperpolarization techniques. Anal. Chem. 94, 479–502 (2022).
Salnikov, O. G., Burueva, D. B., Skovpin, I. V. & Koptyug, I. V. Parahydrogen-based NMR signal amplification by reversible exchange (SABRE): recent advances and applications. Mendeleev Commun. 33, 583–596 (2023).
Stern, Q. et al. Rapid and simple 13 C-hyperpolarization by 1 H dissolution dynamic nuclear polarization followed by an inline magnetic field inversion. J. Am. Chem. Soc. 145, 27576–27586 (2023).
Reineri, F., Cavallari, E., Carrera, C. & Aime, S. Hydrogenative-PHIP polarized metabolites for biological studies. Magn. Reson. Mater. Phys. Biol. Med. 34, 25–47 (2021).
Bowers, C. R. & Weitekamp, D. P. Parahydrogen and synthesis allow dramatically enhanced nuclear alignment. J. Am. Chem. Soc. 109, 5541–5542 (1987).
Adams, R. W. et al. Reversible interactions with para-hydrogen enhance NMR sensitivity by polarization transfer. Science 323, 1708–1711 (2009).
de Maissin, H. et al. In vivo metabolic imaging of [1‐13C]Pyruvate‐d3 hyperpolarized by reversible exchange with parahydrogen. Angew. Chem. Int. Ed. 62, e202306654 (2023).
Hune, T. et al. Metabolic tumor imaging with rapidly signal‐enhanced 1‐ 13C‐pyruvate‐d3. ChemPhysChem https://doi.org/10.1002/cphc.202200615 (2022).
Nagel, L. et al. Parahydrogen‐polarized [1‐3C]pyruvate for reliable and fast preclinical metabolic magnetic resonance imaging. Adv. Sci. 10, 2303441 (2023).
MacCulloch, K. et al. Facile hyperpolarization chemistry for molecular imaging and metabolic tracking of [1–13C]pyruvate in vivo. J. Magn. Reson. Open 16–17, 100129 (2023).
Petersen, S. et al. In vivo molecular imaging of breast cancer metabolic heterogeneity using [1-13C]pyruvate-d3 hyperpolarized by reversible exchange with parahydrogen. https://doi.org/10.26434/chemrxiv-2024-qk0fh (2024).
Schmidt, A. B. et al. Signal amplification by reversible exchange and its translation to hyperpolarized magnetic resonance imaging in biomedicine. Anal. Sens. e202400039 https://doi.org/10.1002/anse.202400039 (2024).
Bowers, C. R. & Weitekamp, D. P. Transformation of symmetrization order to nuclear-spin magnetization by chemical reaction and nuclear magnetic resonance. Phys. Rev. Lett. 57, 2645–2648 (1986).
Eisenschmid, T. C. et al. Para hydrogen induced polarization in hydrogenation reactions. J. Am. Chem. Soc. 109, 8089–8091 (1987).
Pravica, M. G. & Weitekamp, D. P. Net NMR alignment by adiabatic transport of parahydrogen addition products to high magnetic field. Chem. Phys. Lett. 145, 255–258 (1988).
Jóhannesson, H., Axelsson, O. & Karlsson, M. Transfer of para-hydrogen spin order into polarization by diabatic field cycling. C. R. Phys. 5, 315–324 (2004).
Reineri, F., Boi, T. & Aime, S. Parahydrogen induced polarization of 13C carboxylate resonance in acetate and pyruvate. Nat. Commun. 6, 5858 (2015).
Hövener, J. et al. Parahydrogen‐based hyperpolarization for biomedicine. Angew. Chem. Int. Ed. 57, 11140–11162 (2018).
Cavallari, E. et al. The 13C hyperpolarized pyruvate generated by ParaHydrogen detects the response of the heart to altered metabolism in real time. Sci. Rep. 8, 8366 (2018).
Ding, Y. et al. Rapidly signal‐enhanced metabolites for atomic scale monitoring of living cells with magnetic resonance. Chemistry–Methods 2 (2022).
Hövener, J.-B. et al. PASADENA hyperpolarization of 13C biomolecules: equipment design and installation. Magn. Reson. Mater. Phys. Biol. Med. 22, 111–121 (2009).
Kadlecek, S. et al. A simple and low‐cost device for generating hyperpolarized contrast agents using parahydrogen. NMR Biomed. 24, 933–942 (2011).
Coffey, A. M. et al. A pulse programmable parahydrogen polarizer using a tunable electromagnet and dual channel NMR spectrometer. J. Magn. Reson. 284, 115–124 (2017).
Kovtunov, K. V. et al. Demonstration of heterogeneous parahydrogen induced polarization using hyperpolarized agent migration from dissolved Rh(I) complex to gas phase. Anal. Chem. 86, 6192–6196 (2014).
Carrera, C. et al. Parahydrogen polarized ethyl‐[1‐13C]Pyruvate in Water, A Key Substrate for Fostering the PHIP‐SAH approach to metabolic imaging. ChemPhysChem 22, 1042–1048 (2021).
Eills, J. et al. Polarization transfer via field sweeping in parahydrogen-enhanced nuclear magnetic resonance. J. Chem. Phys. 150, 174202 (2019).
Joalland, B. et al. Pulse-programmable magnetic field sweeping of parahydrogen-induced polarization by side arm hydrogenation. Anal. Chem. 92, 1340–1345 (2020).
Marshall, A. et al. Radio-frequency sweeps at microtesla fields for parahydrogen-induced polarization of biomolecules. J. Phys. Chem. Lett. 14, 2125–2132 (2023).
Nantogma, S. et al. MATRESHCA: microtesla apparatus for transfer of resonance enhancement of spin hyperpolarization via chemical exchange and addition. Anal. Chem. 96, 4171–4179 (2024).
Knecht, S. et al. Rapid hyperpolarization and purification of the metabolite fumarate in aqueous solution. Proc. Natl Acad. Sci. 118, e2025383118 (2021).
Hale, W. G. et al. Toward continuous‐flow hyperpolarisation of metabolites via heterogenous catalysis, side‐arm‐hydrogenation, and membrane dissolution of parahydrogen. ChemPhysChem 22, 822–827 (2021).
Schmidt, A. B. et al. Liquid-state carbon-13 hyperpolarization generated in an MRI system for fast imaging. Nat. Commun. 8, 14535 (2017).
Schmidt, A. B. In vivo 13C-MRI using SAMBADENA (2018).
Berner, S. et al. High field parahydrogen induced polarization of succinate and phospholactate. Phys. Chem. Chem. Phys. 23, 2320–2330 (2021).
Berner, S. et al. SAMBADENA hyperpolarization of 13C‐succinate in an MRI: singlet‐triplet mixing causes polarization loss. ChemistryOpen 8, 728–736 (2019).
Schmidt, A. B. et al. Quasi-continuous production of highly hyperpolarized carbon-13 contrast agents every 15 seconds within an MRI system. Commun. Chem. 5, 21 (2022).
Chukanov, N. V. et al. Synthesis of unsaturated precursors for parahydrogen-induced polarization and molecular imaging of 1-13 C-acetates and 1- 13C-pyruvates via side arm hydrogenation. ACS Omega 3, 6673–6682 (2018).
Brahms, A. et al. Synthesis of 13C and 2H labeled vinyl pyruvate and hyperpolarization of pyruvate. Chem.—Eur. J. 28 (2022).
Brahms, A. et al. Exceptionally mild and high-yielding synthesis of vinyl esters of alpha-ketocarboxylic acids, including vinyl pyruvate, for parahydrogen-enhanced metabolic spectroscopy and imaging. J. Org. Chem. 88, 15018–15028 (2023).
Jagtap, A. P., Mamone, S. & Glöggler, S. Molecular precursors to produce para‐hydrogen enhanced metabolites at any field. Magn. Reson. Chem. mrc.5402 https://doi.org/10.1002/mrc.5402 (2023).
Shchepin, R. V., Barskiy, D. A., Coffey, A. M., Manzanera Esteve, I. V. & Chekmenev, E. Y. Efficient synthesis of molecular precursors for para‐hydrogen‐induced polarization of ethyl acetate‐1‐13C and beyond. Angew. Chem. Int. Ed. 55, 6071–6074 (2016).
Mamone, S. et al. A field‐independent method for the rapid generation of hyperpolarized [1‐3C]pyruvate in clean water solutions for biomedical applications. Angew. Chem. Int. Ed. 61, e202206298 (2022).
Korchak, S., Yang, S., Mamone, S. & Glöggler, S. Pulsed magnetic resonance to signal-enhance metabolites within seconds by utilizing para-hydrogen. ChemistryOpen 7, 344–348 (2018).
Pravdivtsev, A. N., Hövener, J. & Schmidt, A. B. Frequency‐selective manipulations of spins allow effective and robust transfer of spin order from parahydrogen to heteronuclei in weakly‐coupled spin systems. ChemPhysChem 23, e202100721 (2022).
Schmidt, A. B. et al. Selective excitation of hydrogen doubles the yield and improves the robustness of parahydrogen-induced polarization of low-γ nuclei. Phys. Chem. Chem. Phys. 23, 26645–26652 (2021).
Korchak, S., Mamone, S. & Glöggler, S. Over 50% 1H and 13C polarization for generating hyperpolarized metabolites—a para‐hydrogen approach. ChemistryOpen 7, 672–676 (2018).
Ellermann, F. et al. Spying on parahydrogen-induced polarization transfer using a half-tesla benchtop MRI and hyperpolarized imaging enabled by automation. Nat. Commun. 14, 4774 (2023).
Korchak, S., Emondts, M., Mamone, S., Blümich, B. & Glöggler, S. Production of highly concentrated and hyperpolarized metabolites within seconds in high and low magnetic fields. Phys. Chem. Chem. Phys. 21, 22849–22856 (2019).
Hövener, J.-B. et al. A continuous-flow, high-throughput, high-pressure parahydrogen converter for hyperpolarization in a clinical setting: a high-throughput parahydrogen converter for hyperpolarization. NMR Biomed. 26, 124–131 (2013).
Cheng, T. et al. Automated transfer and injection of hyperpolarized molecules with polarization measurement prior to in vivo NMR. NMR Biomed. 26, 1582–1588 (2013).
Purwanto, Deshpande, R. M., Chaudhari, R. V. & Delmas, H. Solubility of hydrogen, carbon monoxide, and 1-octene in various solvents and solvent mixtures. J. Chem. Eng. Data 41, 1414–1417 (1996).
Nelson, C. et al. Parahydrogen‐induced carbon‐13 radiofrequency amplification by stimulated emission of radiation. Angew. Chem. Int. Ed. 62, e202215678 (2023).
Pravdivtsev, A. N. et al. Parahydrogen-induced polarization and spin order transfer in ethyl pyruvate at high magnetic fields. Sci. Rep. 12, 19361 (2022).
Tycko, R., Cho, H. M., Schneider, E. & Pines, A. Composite pulses without phase distortion. J. Magn. Reson. 61, 90–101 (1985).
Dey, A. et al. Hyperpolarized NMR metabolomics at natural 13C abundance. Anal. Chem. 92, 14867–14871 (2020).
Reineri, F. et al. Use of labile precursors for the generation of hyperpolarized molecules from hydrogenation with parahydrogen and aqueous-phase extraction. Angew. Chem. Int. Ed. 50, 7350–7353 (2011).
Barskiy, D. A. et al. Rapid catalyst capture enables metal-free para -hydrogen-based hyperpolarized contrast agents. J. Phys. Chem. Lett. 9, 2721–2724 (2018).
Ferrari, A. et al. Performance and reproducibility of 13C and 15N hyperpolarization using a cryogen-free DNP polarizer. Sci. Rep. 12, 11694 (2022).
Acknowledgements
This work was supported by the German Cancer Consortium (DKTK), the DKTK Joint-Funding Project “HYPERBOLIC”, B.E.S.T. Fluidsysteme GmbH I Swagelok Stuttgart, the Preclinical Imaging Research Center - Core Facility (AMIRCF; DFG-RIsources No RI 00052) with lab and imaging systems (INST 39/1225-1) access, the Research Commission of the University Medical Center Freiburg, the German Research Foundation (SCHM 3694/1-1, SCHM 3694/2-1, SFB1479, HO-4604/2, HO-4604/3, PR 1868/3-1, PR 1868/5-1), and the German Federal Ministry of Education and Research (BMBF) in the funding program “Quantum Technologies-from Basic Research to Market” under the project “QuE-MRT” (contract number: 13^^N16448) and Juniorverbund 01ZX1915C (A.N.P). A.B.S and E.Y.C thank Wayne State University for Postdoctoral Fellow award. E.Y.C thanks the support of NSF CHE-1905341, and NIBIB R21 EB033872.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Investigation: O.M., H.d.M., M.H. Data curation: O.M. Funding acquisition: A.N.P., L.S., E.Y.C., R.H., J.-B.H., D.v.E., A.B.S. Conceptualization: J.-B.H., A.B.S. Methodology: O.M., H.d.M., A.B.S. Resources: A.N.P., A.B., R.H., J.-B.H., M.Z., D.v.E., A.B.S. Supervision: M.Z., D.v.E., A.B.S. Visualization: O.M., H.d.M., A.B.S. Discussion of results: all authors. Writing original draft: O.M. and A.B.S. Review & editing: all authors.
Corresponding author
Ethics declarations
Competing interests
E.Y.C. declares a stake of ownership in XeUS Technologies, LTD. E.Y.C. holds stock of Vizma Life Sciences, and serves on the scientific advisory board (SAB) of VLS. R.H., A.B., A.N.P., and J.-B.H. have filed an international patent application (patent name: Process for the production of ketocarboxylic acid vinyl esters, number: WO2023062106, author: R.H., R.B., A.N.P., and J.-B.H., geographical region: worldwide). R.H. is one of the founders and shareholder of QuantView GmbH, which commercializes the [1-13C]vinyl pyruvate-d6 and other vinyl esters of alpha-ketocarboxylic esters. All other authors declare no competing interest for both financial and non-financial.
Peer review
Peer review information
Communications Chemistry thanks the anonymous reviewers for their contribution to the peer review of this work. Peer review reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mohiuddin, O., de Maissin, H., Pravdivtsev, A.N. et al. Rapid in situ carbon-13 hyperpolarization and imaging of acetate and pyruvate esters without external polarizer. Commun Chem 7, 240 (2024). https://doi.org/10.1038/s42004-024-01316-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s42004-024-01316-x
