
Abstract
Background
Acquired resistance (AR) is a major limitation of immune checkpoint inhibitor (ICI) therapy when treating renal cell carcinoma (RCC). Understanding who will get AR is currently unknown. We hypothesized the T-cell-inhibitory glycoprotein non-metastatic melanoma protein B (GPNMB) to be a prognostic marker for patients with AR.
Methods
Alongside other markers, GPNMB was measured in the blood of RCC patients (n = 39) several times after starting ICI treatment and analyzed for association with Response Evaluation Criteria in Solid Tumors (RECIST) tumor response. To better understand the role of GPNMB in AR, we created an ICI-resistant RenCa mouse kidney cancer model by repeatedly selecting the largest tumors that grew in ICI-treated mice.
Results
Here we show that among patients who positively respond to ICI, a subset of patients (n = 9) acquire resistance within 2 years that coincides with an increase in serum GPNMB. Our mouse model recapitulates this elevation in GPNMB at the onset of AR which is triggered by cytoplasmic motif signaling via the Programmed cell death ligand 1 (PDL1) receptor that is known to protect tumors from Interferon-gamma (IFN-γ) cytotoxicity. This PDL1-induced signal leads to upregulation of the SRY-box transcription factor 10 (SOX10), dysregulation of the microphthalmia-associated transcription factor (MITF) pathway, and overexpression of GPNMB. Indeed, activation of SOX10-MITF signaling is present in plasma cell-free RNA from RCC patients who develop AR.
Conclusions
Elevation of the SOX10-MITF-GPNMB signal cascade via the PDL1 receptor leads to AR. Therefore, GPNMB can be both a prognosticator of and a potential treatment target for overcoming AR to ICI treatment in RCC.
Plain language summary
Immune checkpoint inhibitors (ICI) are a type of cancer treatment that helps the immune system kill cancer cells. However, over half of people with kidney cancer who initially see a benefit following ICI treatment find the drug stops working for them, with their cancer acquiring resistance to this treatment. In this study, we investigated if a protein known as glycoprotein non-metastatic melanoma protein B (GPNMB) increases in cancers that do not respond to ICI therapy. We find that elevated GPNMB levels coincide with the onset of treatment resistance to ICI therapy in both humans and a mouse model of kidney cancer. Therefore, measuring GPNMB levels during ICI therapy can inform if a patient will become resistant to treatment. Additionally, GPNMB may serve as a potential target for overcoming such resistance.
Similar content being viewed by others

Introduction
Immune checkpoint inhibitors (ICI) have revolutionized the management of many metastatic cancers including renal cell carcinoma (RCC), but continue to suffer from a high incidence of resistance1,2,3, of which there are two types: innate resistance that occurs at the onset of treatment, and acquired resistance that develops after an initial positive response4,5,6. Incidence of innate resistance can be reduced by combining ICI with other treatments7, but even so, over half of initial positive responders succumb to acquired resistance within 3–5 years of sustained treatment6,8. These outcomes underscore the importance of finding markers that reliably prognosticate resistance to ICI and of developing better-targeted therapy for metastatic disease.
Markers for predicting innate resistance include soluble factors; C-reactive protein, Iterleukin-6 (IL-6), Lactate Dehydrogenase, New York esophageal squamous cell carcinoma 1 (NY-ESO-1) Ab, soluble CD25, Vascular endothelial growth factor (VEGF9) and particular cells; myeloid-derived suppressor cells (MDSC), programmed cell death ligand 1 (PDL1)+ tumor cells, tumor CD8 T-lymphocytes (T-cells)10,11. By contrast, there are no established markers for acquired resistance, though postulated mechanisms have been proposed including (a) deficient tumor antigen (Ag) presentation12; (b) defective Interferon-gamma (IFN-γ) signaling12; (c) depleted neoantigens13; and (d) various immunosuppressive pathways (Lymphocyte activation gene 3 (LAG3), T-cell immunoglobulin and mucin domain 3 (TIM3), V-domain Ig suppressor of T-cell activation (VISTA))14. These possibilities are considered wanting since they are based on studies that compared whole exome sequences from specimens taken at baseline vs. onset of acquired resistance12 and thus contaminated by the influence of innate resistance. Moreover, these studies were limited to very few cases of melanoma and lung cancer, muddled by an inconsistent definition of acquired resistance15,16.
Glycoprotein non-metastatic melanoma protein B (GPNMB) is expressed as a receptor and its extracellular domain is released in the circulation by A disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) sheddase17. Both receptor and soluble versions inhibit T-cell activation and induce angiogenesis by binding to the ligand syndecan-4 (SDC4) on T-cells and on endothelial cells18,19,20,21, respectively. GPNMB-bound SDC4 utilizes membrane protein tyrosine phosphatase CD148 to inhibit T-cells, a mechanism distinct from that of PDL1 and other immune checkpoints22. In non-pathologic (normal) conditions, GPNMB is expressed at low levels on osteoblasts in bone endosteum and on macrophages23. By contrast, GPNMB is expressed at high levels by melanoma and breast cancer, and at lower levels by RCC and non-small cell lung carcinoma (NSCLC). GPNMB is also expressed highly by blood MDSC in patients with various types of metastatic cancer and it mediates the T-cell suppressor function24. Thus, GPNMB is an immune regulator that promotes tumor progression.
In a mouse model of B16 melanoma, we showed GPNMB to mediate resistance to treatment with anti-PDL1 Ab25, even as treatment with combined anti-GPNMB and anti-PDL1 Ab did not produce added value. The lack of the additive effect was also true in our studies using MC38 colon and RenCa kidney cancers. In human studies, we showed high blood levels of soluble GPNMB receptor (sGPNMB) to predict poor responses to ICI treatment of NSCLC, thereby highlighting its potential as a marker for innate resistance25.
These findings led us to ask whether blood sGPNMB can prognosticate ICI response in RCC patients. Our study of ICI-treated RCC patients showed blood sGPNMB to be a poor predictor of innate resistance, but an accurate anticipator of acquired resistance since sGPNMB rose during the transition to progressive disease (PD). To understand the reasons for sGPNMB’s upregulated expression, we created an anti- Programmed cell death protein 1 (PD1) Ab-resistant RenCa mouse model that recapitulated the concurrent onset of AR and rise in blood sGPNMB in ICI-treated RCC patients. In this model, we found the SRY-box transcription factor 10 (SOX10)-Microphthalmia-associated transcription factor (MITF) axis to upregulate GPNMB linked to acquired resistance. We also noted the axis to be dysregulated by intrinsic signaling of the PDL1 receptor activated by ICI treatment. Human relevance was corroborated by a rise in gene expression of SOX10-MITF in plasma cell-free RNA (cfRNA) of RCC patients who developed acquired resistance. These findings indicate blood GPNMB as a prognosticator of risk for acquired resistance and a potential treatment target for overcoming this form of ICI resistance. Underlying these clinical benefits is the discovery of the SOX10-MITF-GPNMB signal cascade as a cause of acquired resistance to anti-PDL1 therapy in RCC.
Methods
RCC patients
RCC patients (III/IV stages, n = 45, Supplementary Table S1) received ICI treatment between January 2018 and January 2022 at Harold C. Simons Comprehensive Cancer Center at The University of Texas Southwestern Medical Center. Patients were treated with standard of care ICI monotherapy (atezolizumab, nivolumab, or pembrolizumab) every 2–3 weeks26,27, with radiographic assessment of disease status every 12 weeks28. Response to treatment was documented every 12 weeks by RECIST version 1.129. Each patient provided informed consent before enrollment. The Institutional Review Board of UT Southwestern approved this study (STU-2020-0757). Peripheral blood samples were collected from all patients before treatment (Week 0) and at follow-up visits extending through 55 weeks post-treatment. The study was conducted in accordance with the amended Declaration of Helsinki and the International Conference on Harmonization Guidelines.
Reagents, animals, and cell culture
Leukocyte marker Ab and anti-mouse PD1 mAb (J43 clone) were purchased from eBioscience and Bio-X-Cell, respectively (All antibodies used are listed in Supplementary Table S2). We generated mAb rabbit anti-mouse Gpnmb mAb (IgG1, UTX103 clone) and genetically engineered to replace the rabbit C-regions with those of mouse IgG130. This chimeric IgG was produced by transient DNA transfection of suspension cultures with the IgG genes using ExpiCHO™ systems in serum-free media (Thermo-Fisher) and purified by Protein A-agarose (Invitrogen). Purified preparations had very low endotoxin level (<0.05 EU/ml), determined by Pierce™ Chromogenic Endotoxin Quant Kit. Female BALB/c mice (~8-week-old) were purchased from Harlan Breeders. Animals were housed in the pathogen-free facility of the Institutional Animal Care Use Center of UT Southwestern Medical Center. All animal protocols were approved by the Center. RenCa, B16F10, 293T and human RCC Caki-2 cells were purchased from the American Type Culture Collection and maintained in DMEM supplemented with 10% fetal bovine serum. All cell lines are negative for mycoplasma using Universal Mycoplasma Detection Kit (ATCC).
Blood samples and flow cytometry
Within 24 h after collecting blood, samples were centrifuged to separate plasma from cells. Cells were mixed with 10 ml cold phosphate buffer saline (PBS), and 10 ml of 6% dextran/0.9% NaCl and incubated for 90 min at room temperature. The supernatant was recovered, centrifuged, and cell pellets were incubated with ACK Lysing buffer (Gibco) for 5 min. After centrifugation, cells were resuspended in PBS buffer; this fraction is referred to as peripheral blood leukocytes (PBL). PBL were further isolated by Ficoll-Paque (1500 rpm for 20 min centrifugation) and applied to the Dead Cell Removal Kit (Miltenyi Biotec); 99% of dead cells are normally removed. The cells were treated with FcR blocking reagent (Militenyi Biotec), and fluorescently stained with marker Ab and 3D5 anti-human GPNMB or the isotype control anti-KLH mAb24 with fluorochrome-labeled antibody and analyzed for cell-bound fluorescence using FACSVerse (BD Bioscience) and FlowJo v10.10.0. Polymorphonuclear (PMN) and peripheral blood mononuclear cells (PBMC) were gated separately in size-scatter plots: M-MDSC in PBMC fraction was identified by the phenotype of CD14+HLA-DRno/lo, and granular MDSC (G-MDSC) in PMN fraction by CD14negCD15+HLA-DRneg. Both MDSC populations were positive for CD33 and CD11b31.
ELISA
An aliquot (100 μl) of 5x PBS-diluted plasma samples was applied to ELISA wells in triplicate for quantification of GPNMB, PDL1, and VEGF (all from R&D Systems). For measuring mouse sGpnmb levels in blood and culture supernatant, 1:100 diluted or undiluted samples were used. To allow reliable and unbiased method comparison, all tests were done in a single-blind manner by an independent laboratory technician.
Tumor growth assays and Ab treatment
RenCa cells were harvested from the growing cultures by EDTA, washed extensively with PBS, and injected subcutaneously (s.c.) into female BALB/c mice (1 × 106 cells/mouse). We prefer to use females because they generally have a more robust immune system than males. Three to six days post-injection, all mice were injected i.p. with Ab (200 μg/mouse) twice weekly, for a total of 5–6 injections. Tumor volume was measured every 2–3 days32. One or two days after the last injection, tumors were excised, weighted, and for some experiments, subjected to histological examination or FACS analysis of leukocyte populations. Blood was also collected from tail veins and serum was prepared using BD Microtainer. For generating ICI-resistant RenCa cells, day 6 post-implantation of parental tumor cells, mice (n = 4) were treated with control or anti-PD1 Ab. A day after the fifth injection, the largest tumors from PD1 Ab-treated mice were chosen, excised, prepared for single cells, and finally cultured in vitro for 2 weeks; these cells were termed P1 cells, which were implanted into new mice and repeated the procedures. After four repeats, the resulting cells were termed P4 cells. Cells used for molecular and biological assays were obtained from cultures with less than 6 time-passages. For experiments measuring survival rate, all mice were injected s.c. with P4 cells (1 × 105 cells/mouse); on day 6 they were randomly sorted into 4 groups (n = 10) and given different Ab every 3 days until day 35; thereafter schedule was changed to longer intervals, weekly until day 60.
Analysis of tumor microenvironment
For cytometric analysis, tumors were excised from treated mice and dissociated into single cells by tumor dissociation kit (Miltenyi Biotec). Isolated cells were FcR-blocked and stained with rabbit anti-mouse Gpnmb UTX103 mAb or control IgG2b and 5 μg/ml PE-anti-mouse IgG [F(ab′)2 fragment]. After washing, cells were also stained with PE-cyanine7-anti-CD45 (BD Biosciences) and FITC-Ab to cell surface markers including CD4, CD8, CD11c, and F4/80 (eBioscience) and analyzed by flow cytometry using FACSVerse (BD BioSciences). For immunohistochemistry (IHC) staining, serial sections of formalin-fixed tissues were deparaffinized, rehydrated, immersed in citrate buffer (pH 6.0), and microwaved for 15 min to retrieve antigens. Slides were incubated with Bloxall Blocking solution for 10 min, with primary Ab in 2% goat serum overnight at 4 °C; including rabbit anti-mCD4 (1:1,000 dilution, Abcam), rabbit anti-mCD8 (1:500, Abcam), mouse anti-mCD11b (1:200, Invitrogen), rabbit anti-mCD31 (1:50, Invitrogen), and mouse anti-mast cell tryptase Ab (1:500, Abcam). After washing, slides were incubated with biotin-secondary Ab and avidin-peroxidase solution (Vector), followed by color development (brown) by 3,3 diaminobenzidine and counterstaining with hematoxylin. These IHC-stained sections were image-analyzed by Nano Zoomer S60 (Hamamatsu) and Aperio ImageScope or ImageJ program for determining tumor density in a blinded fashion; positively stained cells are calculated as stained areas and expressed as the average of 6 separate microscopic views from three different mice per group (2 views/mouse × 3 mice = 9).
Tet-off system
RenCa cells were transfected with the vector driving expression of sGpnmb-V5 under the control of Tet-off system33. All mice were given 2 mg/ml Doxycycline (Dox)-water from day −3 to day 6. Day 0, all mice were implanted s.c. with Tet-off-sGpnmb-V5-RenCa cells (2 × 105 cells/ mouse) and day 6 randomly sorted into two groups (n = 10); one group was continuously supplied with Dox (2 µg/mouse i.p. injection every 5 days) and the other discontinued (PBS injection). From day 7, mice were further divided into 2 groups (n = 5) and treated with i.p. injection of anti-PD1 or control IgG every 3–4 days. Tumor volume was measured. Next day after the last injection, tumors were excised, weighed, and subjected to cytometric flow-analysis.
WES analysis
Genome DNA was extracted from growing P0 and P4 cells using Blood & Cell DNA mini kit (Qiagen); 1.5 μg of DNA per cell was sheared to make small fragments and hybridized with biotinylated RNA library baits (Psomagen). Agilent uses ultra-long 120-mer RNA baits for the highest specificity. An average of 120 Gb of deep sequencing data (~1000×) was generated per mouse (GENEWIZ). Raw reads were processed with fastqc (Version 0.11.9) and trim-galore (Version 0.6.5) to remove reads with low quality and trim adapters34. Then processed reads were mapped to Mouse GRCm38/mm10 with BWA-mem (v0.7.15)70. Picard (v1.119) was used to mark duplicated reads. Variant calling and allele frequency calculation were performed using a customized GATK pipeline [BD, V.d.A.G.O.C. (2020)]. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media). For the calculation of TMB, there are 8658 variants and 9173 variants in the coding domains in P0 and P4 samples respectively. TMB was calculated by the number of the variants divided by 49.6 Mb (mouse exome size); 174.6 and 184.9 for P0 and P4, respectively. Lacking matching normal samples to remove polymorphism, these TMB values were over-estimated.
RNA-seq analysis
Total RNA was extracted from P0 and P4 cells or tumors in mice treated five times with control or PD1 Ab using Aurum Total RNA mini kit (Bio-Rad). Spectrophotometric and electrophoretical verification (Agilent Tapestation 4200, Agilent Technologies) was performed to identify the quality and quantity of total RNA at The McDermott Center Next Generation Sequencing Core in UT Southwestern. Only high-quality RNA was used (RIN Score 8 or higher). RNA (1 μg) was used to construct Illumina-compatible libraries using TruSeq Stranded mRNA Library Prep Kit (Illumina, San Diego) and quantified by Qubit then run on the Illumina NextSeq 2000-P2-100 kit with the run configuration of 100 × 8 × 8. The short ds-cDNA fragment was ligated to the sequencing adapters, and A HiSeqTM2000 platform (Illumina) was used for performing RNA-Seq.
RNA-seq data analysis
Adapters were trimmed using Trim Galore (v0.6.4). The trimmed reads were then aligned to the mouse genome (mm10) using STAR (2.7.3a). The subsequent mapped reads were quantified by feature Counts of the Subread (v1.6.3) package. Low-expressed genes (genes with the expression value of 0 in more than 30% of samples) were removed before any subsequent analysis. Gene expression was normalized using the Voom method from the R package limma (v3.50.3). Differentially expressed genes (DEGs) were then identified using the same package. Gene Set Enrichment Analysis for the DEGs was performed using the R package clusterProfiler (v4.2.2). The Benjamini–Hochberg algorithm was used to align the p values to limit the false discovery rate. R4.1.2 (www.r-project.org) was employed to analyze all data and visualize the DEGs.
MITF isoforms
Transcript_tpm file was created from RNA-seq data and mapped to Transcript ID; Mitf-201 (Isoform-M), ENSMUST00000043628.13; Mitf-202 (Isoform-A), ENSMUST00000043637.14; Mitf-203 (Isoform-H), ENSMUST00000101123.10; or Mitf-204 (Isoform-B), ENSMUST00000113339.8. Isoform quantification was performed using Salmon (v1.2.1). Reads were aligned to the mouse genome (mm10) using DNAstar Lasergene 16. Transcripts per million (TPM) were calculated for the isoforms of each gene. Composition of different isoforms in P0 and P4 cells expressed as percentage of each specific TPM in total MITF TPM.
Luciferase reporter assays
pGL3-basic and pGL4.75 vectors were purchased from Promega. Gpnmb promoter region (−196 nt from the RNA start site) was extracted from genomic DNA of RenCa cells by PCR and ligated to pGL-b (basic promoter-Luc) vector. Either or both E-boxes (CACGAG and TCACATGA) were deleted in the promoter fragment using Quick Change Site-directed mutagenesis kit (Stratagene). P0 or P4 cells were seeded on 96 well-plate (2 × 104 cells/well) and transfected with 25 ng pGL4.75 (Renilla expressing vector) and increasing doses of pGL3-Gpnmb-p (Gpnmb promoter-linked Luc) or pGL3b, using FuGENE® HD (Promega). Two days later, cell extracts were prepared and assayed for Luc (OD560) and Renilla (OD480) using Synergy HT Microplate Reader (Bio Tek) and expressed as OD560/OD480 x 100.
shRNA experiments
We constructed a small interfering RNA (siRNA)-expressing lentivector consisting of gene-targeted short hairpin RNA (oligonucleotides are shown in Supplementary Table S3), with BamHI and EcoRI sites at 5′- and 3′-end, respectively; 4 different shRNAs for Mitf and 3 for Sox10 gene were made. These shRNA sequences are ligated to the downstream of H1 RNA polymerase III promoter in pSIF1-H1-copGFP shRNA lentivector (System Biosciences) that contains a cytomegalovirus-driven fluorescent copepod green fluorescent protein (copGFP). pSIF1-H1-copGFP without a shRNA insert served as a control lentivector. Packaging of pSIF constructs in pseudoviral particles and their titration were performed according to established protocols35. P4 cells (2 × 106) were infected in triplicate with mixed shRNA-lentiviruses at MOI of 1 for 1 day, washed with fresh media, and incubated for another 1 day. GFP+ cells (25–35% positive) were FACS-sorted and subjected to qRT-PCR analysis.
Deletion mutant analysis of PDL1 signaling
Mouse PDL1 full-length sequence was extracted by PCR amplification from cDNA of RenCa cells with the 5′-primer coding V5-tag sequence and inserted into pHR-SIN-ub-Em lentiviral vector (termed pHR-SIN-V5-mPDL1). This vector DNA was denatured and annealed with forward and reverse primers (Supplementary Table S4) for site-directed mutagenesis and PCR-amplified for 12 or 18 cycles, digested with DpnI restriction enzyme, and transformed into E. coli. Lentiviruses encoding mutants were infected into 293T cells (6 × 105 cells) in triplicate; next day cells were harvested (60–75% was GFP+ cells) and re-seeded onto culture wells pre-coated with anti-V5 or control Ab (10 μg/ml). After being cultured for 2 days, RNA was extracted and assayed by qRT-PCR for gene expression with gene-specific primers (Supplementary Table S5).
cfRNA purification
Total RNA purification was performed by Qucik cfRNA Serum/Plasma Kit (ZYMO Research) from 2 ml of plasma samples. To digest trace amounts of contaminating DNA, RNA samples were treated with DNase I, purified, and further concentrated using an oligo clean and concentrator (ZYMO Research), and stored immediately at −80 °C36. cfRNA yielded an average of 90.2 ng with a range of 36 and 134 ng from patients and 32.3 ng (24–36 ng) from HD (Supplementary Table S6).
Nested real-time PCR
Entire amounts of cfRNA were converted into cDNA using an iScript cDNA Synthesis kit (Bio-Rad) and 5% of the cDNA (set up in duplicate) was PCR-amplified for 15 cycles with primers specific to Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), beta-actin (ACTB), GPNMB, MITF or SOX10. After the purification of PCR product, the whole amount was subjected to PCR amplification with nested specific primers. mRNA expression in each sample was expressed as the expression level relative to GAPDH using the formula 2−ΔΔCq37. Standard curves plotting Cq values against starting copy numbers were created: Template DNA was prepared by PCR amplification of total RNA extracted from Caki-2 cells with the same primer sets. The purified PCR product was used for template DNA, diluted to 23 μM, 10-fold serially diluted, and an aliquot (1 μl) was subjected to the initial PCR amplification for 15 cycles and nested qPCR with the same primers (Supplementary Table S6). Cq values are plotted against template DNA dilution.
Statistics and reproducibility
Mann–Whitney U test, Student’s t-test, or two-way ANOVA were used for comparison, using GraphPad Prism 10.4.1. Survival rate and association were evaluated by log-rank (Mantel-Cox test) and Pearson’s correlation coefficient, respectively. p ≤ 0.05 was considered significant. All data shown are presentative of at least two independent experiments.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Results
Blood soluble GPNMB receptor (sGPNMB) correlated significantly with tumor progression during ICI treatment
As was shown for NSCLC, we posited GPNMB expression in RCC to be associated with innate resistance to ICI therapy25. Since GPNMB is expressed as a receptor on MDSC and a secreted form in the blood of cancer patients, we assayed frequencies of GPNMB+ MDSC by flow cytometry and plasma GPNMB by ELISA. We determined baseline MDSC frequencies (see demographic characteristics at baseline, Supplementary Table S1) and classified patients at 12 weeks after treatment onset into positive responders (partial response (PR) or stable disease (SD)) and negative responders (PD). Unexpectedly, we found baseline MDSC frequencies and sGPNMB levels not correlated to tumor response at 12 weeks, inconsistent with data for NSCLC patients24 (Supplementary Figs. S1 and S2). We also noted that neither baseline sPDL138 nor sVEGF39,40,41 correlated with treatment response.
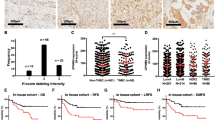
By contrast, GPNMB-expressing monocytic (M)- but not granular (G)- MDSC frequencies at 12 weeks after treatment onset were significantly higher in negative (vs. positive) responders (p = 0.01 vs. p ≥ 0.02) (Fig. 1a, b). % total M-MDSC and G-MDSC also associated significantly. Among blood factors, sGPNMB level was more impressively associated with negative responders (p = 0.0001) compared to sPDL1 (p = 0.09) or sVEGF (p = 0.01) (Fig. 1c–e). sGPNMB level did not correlate with tumor size (Fig. 1f). Thus, while baseline sGPNMB level failed to predict initial response, it was higher in negative (vs. positive) responders 12 weeks after starting treatment.

RCC patients (n = 42) were evaluated for tumor response at 12 weeks post-therapy and determined for sGPNMB levels and myeloid-derived suppressor cells (MDSC) frequencies in blood samples drawn at the same time point. Patients were sorted into responders (stable disease (SD)/partial response (PR)) and non-responders (progressive disease (PD)). % total population or GPNMB+ subpopulation of monocytic (M)-MDSC (a) or granular (G)-MDSC (b) in responders vs. non-responders. Blood levels of sGPNMB (c), sPDL1 (d), and sVEGF (e) between responders vs. non-responders. f Correlation between tumor size and blood sGPNMB levels at 12 weeks. p values are shown, and analyzed by Pearson Correlation Coefficient or Mann–Whitney U test.
Blood sGPNMB level closely paralleled the course of tumor response to ICI treatment
We assayed sGPNMB blood levels at each follow-up visit from baseline and plotted this against RECIST tumor evaluation (PR, SD or PD) (Fig. 2). Examined RCC patients (n = 39) were sorted into positive (n = 32, 82%) and negative responders (n = 7, 18%) in terms of clinical benefit (Fig. 2a). Among initial positive responders, 23 patients (72%) maintained a good response over 55 weeks of treatment, whereas 9 (28%) became ICI-resistant. The rate varied depending on the length of follow-up (mean follow-up of 32 weeks; range of 7–55 weeks). Blood sGPNMB gradually fell in all cases that responded positively (Fig. 2b), while it rose rapidly in all cases of acquired resistance. Among negative responders at the start of treatment, 3 (43%) continued to respond poorly, whereas 4 (57%) eventually responded positively (acquired sensitivity). In all cases of the former group, sGPNMB continued to rise, whereas all cases of the latter group showed reduced sGPNMB. With one exception (Pat #7), all cases showed strong concurrence in sGPNMB rise and PD phenotype. By contrast, GPNMB expression by MDSC did not parallel changes in tumor response (Supplementary Fig. S3), suggesting MDSC is not the major source of sGPNMB. This high association with PD phenotype suggests that blood sGPNMB during ICI treatment is an indicator of acquired resistance.

a RCC patients (n = 39) were categorized into responders (Res) and non-responders (NR) based on 12-week evaluations after starting ICI. While most Res maintained a positive response (persistent Res or pRes), some Res acquired resistance (AR) to ICI. NR were sorted into those who never responded positively (pNR) and those who displayed a delayed positive response (acquired sensitivity or AS). Patient number in each category and frequency among total RCC cases are indicated. b Chronological changes (weeks after onset) in sGPNMB in each individual patient are shown, with red-dashed lines indicating the median sGPNMB value (11.3 ng/ml) of the stable disease (SD)/partial response (PR) group. Red and blue lines indicate a rise in transition to progressive disease (PD) and to PR, respectively. Note: pRes who displayed PR did not take many treatment cycles so that blood samples were not available at >36 weeks.
Gpnmb expression rose in ICI-resistant RenCa cells during ICI treatment
To identify reasons for upregulated GPNMB in negative responders, we established a mouse model of RCC in which RenCa cells were implanted s.c. into BALB/c mice that were then treated with anti-PD1 Ab. Among treated mice, the largest tumors were chosen and re-implanted into new mice, a process repeated four times to generate ICI-resistant RenCa cells (termed P4) that were completely unresponsive to PD1 Ab. Parental RenCa (P0) cells exhibited very positive responses to anti-PD1 Ab (tumor volume of 182 ± 33 for Ab-treated group on day 22 post-implantation vs. 542 ± 40 mm3 for control) (Fig. 3a). In quantifying Gpnmb and PDL1 RNA for each generation of RenCa cells (Fig. 3b), we found Gpnmb RNA to decline from P0 to P1 cells but increased progressively thereafter through P4. PDL1 RNA was similarly reduced but with no difference between P0 and P4 cells. Cell surface expression of Gpnmb also increased in P4 cells, but PDL1 was slightly reduced (Fig. 3c, Supplementary Fig. S4). Confocal analysis of RenCa cells showed much higher Gpnmb protein in P4 vs. P0 cells, on both cell surfaces and intracellularly (Supplementary Fig. S5). sGpnmb secreted into cell culture supernatant also increased for P4 cells (Fig. 3d). This was true in mice treated with PD1 Ab (Fig. 3e, f). IHC staining of untreated P0 and P4 tumors showed reduced CD4 and CD8 T-cell densities from P0 to P4, in the face of increments of 3.5- to 4-fold in CD11b myeloid and CD31 endothelial cells, respectively (Fig. 3g, h). Tryptase+ mast cell density (a better prognostic marker for tumor response42) decreased during this progression, suggesting that ICI-resistant RenCa tumors are more immunosuppressive and angiogenic. Akin to tumor microenvironment (TME), P4 tumor-bearing mice exhibited: altered immunological phenotypes in their bone marrow (BM); upregulated expression of Gpnmb but not PDL1 RNA (Fig. 3i); and increments in %CD4 and CD8 T-cells, CD11b+Gr1+ MDSC, CD11c+ dendritic cells, and F4/80+ macrophages among CD45+ cells by flow cytometry (Fig. 3j). Intrinsic tumor growth capacity in in vitro culture and in immunodeficient mice was similar for P0 and P4 cells (Supplementary Fig. S6). These findings are consistent with those of our human studies43, thereby buttressing the validity of ICI-resistant RenCa as a mouse model of ICI resistance.

a Generation of ICI-resistant RenCa cells. RenCa P0 (parental cells) were implanted subcutaneously into BALB/c mice (n = 4) and treated with anti-PD1 or control Ab (shown by blue reverse arrows). After five shots, the largest tumors (shown by red lines) were excised, cultured in vitro, and implanted into new mice and similarly treated (termed P0 to P4). This enrichment was repeated four times. Tumor volume is shown. b P0 to P4 cells were assayed by qRT-PCR for RNA expression of Gpnmb and PDL1 to Gapdh (mean ± SEM, n = 3). c Dot plots of flow-cytometric analysis of P0 and P4 cells fluorescently stained for expression of Gpnmb or PDL1, with mean fluorescence intensity (MFI). d Kinetics of sGpnmb secretion by in vitro cultured P0 and P4 cells. e Growth curves of P0 and P4 subcutaneous tumors in mice (n = 4) treated with anti-PD1 Ab (by closed arrows on the top). Blue arrows show the date of blood drawn. f Blood sGpnmb levels in mice treated with Ab (e) are plotted at varying time points post-implantation of P0 or P4 cells. −3 means 3 days prior to tumor implantation. Untreated P0 and P4 tumors (~1 cm size) are IHC-stained with control IgG (Ctrl), or Ab to CD4, CD8, CD11b, CD31, or tryptase (scale bar, 100 μm, g) and assayed for tumor density of each cell population (h) (mean ± SEM, n = 5); (Ab-stained area μm2/mm2 microscopic view), average of nine different views ± SD, from five different tumors. Bone marrows of the same mice were analyzed by qRT-PCR for Gpnmb or PDL1 RNA (i) (mean ± SEM, n = 3). and by flow cytometry for frequencies of different leukocyte populations (j) (mean ± SEM, n = 4). Representative of three separate experiments except (a–c). p values are shown, using Student’s t-test comparing two conditions.
ICI-resistant RenCa tumors became highly sensitive to anti-Gpnmb Ab treatment
We posited that upregulated Gpnmb expression conferred on RenCa cells an alternative pathway for escaping ICI-enhanced cancer immunity. We asked whether P4 tumor growth could be inhibited by anti-Gpnmb Ab that prevents Gpnmb from binding to its ligand, acting as an antagonist that blocks Gpnmb-signaling30. We treated P0 (ICI-sensitive RenCa) tumor-bearing mice with the Ab and monitored tumor growth (Fig. 4a). Reproducibly, P0 tumors responded very well to PD1 Ab but only moderately to Gpnmb Ab (153 ± 18 vs. 351 ± 32 mm3). By contrast, P4 tumors were unresponsive to PD1 Ab but highly responsive to Gpnmb Ab (Fig. 4b, c). We measured blood sGpnmb levels in P4 tumor-bearing mice during Ab treatment (Fig. 4d). Mice treated with control or PD1 Ab produced blood sGpnmb of 65 ng/ml on day 6 and proportionally increased, whereas treatment reduced sGpnmb expression almost completely by days 6 and 12 (<5 ng/ml), indicating that sGpnmb is a critical factor in resistance to PD1 Ab. We next asked whether Gpnmb Ab can improve the survival of P4-bearing mice (Fig. 4e). All control mice died at 31 days and PD1 Ab prolonged their survival by 6 days. Gpnmb Ab improved survival by over 60 days (50% mortality on day 42). We also examined changes in the immune phenotypes of P4 tumors after treatment with PD1 vs. Gpnmb Ab. Flow cytometric analysis of tumors showed no significant difference between PD1 and control Ab treatment groups, in frequencies of tumor-infiltrating CD4 and CD8 T-cells, MDSC, CD11c, and F4/80 myeloid cells (Fig. 4f, g; Supplementary Fig. S7). By contrast, Gpnmb Ab treatment increased CD4 and CD8 T-cell densities, while reducing MDSC, CD11b, and F4/80 cell densities.

a–d P0 (a) or P4 cells (b, c) were implanted subcutaneously (s.c.) into mice (n = 5) and treated with control, anti-Gpnmb (αGpnmb) or anti-PD1 (αPD1) mAb. Tumor volume is measured. Blue arrows show the date of blood drawn. P4 tumor weights in each treatment group were measured at the endpoint (c) (mean ± SEM, n = 5). d Blood samples collected from mice (b) were determined for sGPNMB and plotted against days after implantation. e Survival rate (%) of P4 tumor-bearing mice (n = 10) treated with control, anti-PD1 or anti-Gpnmb Ab, until day 60; p value compared to PD1 treatment group. f, g Total cells were prepared from Ab-treated tumors and sorted into CD45+ fraction in flow cytometry, followed by determination of % leukocyte subtype among CD45+ cells. Representative dot plots are shown, with % positivity (f). Data are summarized in a graph (g) (mean ± SEM, n = 5). h–j RenCa parental cells transfected with Tet-Off-controlled sGpnmb gene were implanted subcutaneously (s.c.) into mice treated with doxycycline (Dox). On day 6, all mice were sorted into two groups. Dox-continued and Dox-discontinued (PBS-injected) and treated with anti-PD1 or control (Ctrl) Ab (h). Tumor volume is measured (i). On day 24, tumors were excised and measured (j) (mean ± SEM, n = 5). Data shown are representative of at least two independent experiments. P values are compared to control group using two-way ANOVA.
To determine whether sGpnmb plays an important role in generating resistance to PD1 Ab in RenCa, we introduced a Tet-off-sGpnmb expression system in parental RenCa (P0) cells and examined tumor response to PD1 Ab with or without doxycycline (Dox). With Dox present (sGpnmb is shut off), the transfected P0 tumors displayed a good response to PD1 Ab, but without Dox (sGpnmb is expressed), the response was dampened (Fig. 4h– j). Immunological analysis of TME in treated mice revealed similar changes after Gpnmb Ab treatment (Supplementary Fig. S8). These data indicate P4 tumors utilize the Gpnmb (rather than PDL1/PD1) pathway to suppress T-cell immunity.
ICI-resistant RenCa acquired genetic alterations while converting to resistance phenotype
To discover genetic alterations associated with ICI resistance, we performed whole exome sequencing (WES)44 in which we identified 8658 and 9173 variants, respectively, in exons of P0 and P4 samples (Supplementary Data 1). Among all unique P4 variants (total of 1358 absent in P0 exomes), missense mutations were the most common (87%) (Supplementary Fig. S9a). Gene ontology term enrichment analysis of P4 variants showed great impact on cell-cell adhesion and molecular assembly (Supplementary Fig. S9b). P4 variants were found in genes for H-2 histocompatibility class I (MHC-I) antigens including H2-D1, H2-Q4, H2-T23, and H2-M10.5, but only 4 acquired mutations of total 29 MHC-I genes in the genome45 may have limited influences on presentation of tumor-associated antigens (Supplementary Fig. S9c). Genome instability of P4 cells was suggested by increased expression of Apobec 1 and 3 genes (DNA mutators) and decreased expression of Atm and Atr genes (DNA repairing kinases)46,47 (Supplementary Fig. S9d). These results suggest PD1 Ab-resistant phenotype may be obtained by genetic alterations and genome instability.
We also analyzed transcriptome of P0 and P4 cells (Supplementary Fig. S10a). P4 cells display a more active transcriptome than P0 cells. All top 9 gene ontology terms were related to nuclear structures and processing (Supplementary Fig. S10b), again suggesting that P4 cells are genomically unstable. We next examined gene expression profiles of tumors in mice treated with PD1 vs. control Ab (Fig. 5a; Supplementary Data 2). In P0 tumors, 18,301 genes were examined and showed PD1 Ab (vs. control Ab) to produce significant changes in 218 genes, in which 199 and 19 genes were up- and down-regulated, respectively, with the extent of upregulation in the range of 3-fold log change. By contrast, P4 tumors showed many more genes positively responsive to PD1 Ab and of higher significance and greater magnitude (7-fold log change); in total of 991 genes, 920 upregulated and 71 down-regulated. Intriguingly, top 40 DEGs revealed contrasting RNA expression profiles of P0 vs. P4 tumors (Fig. 5b): P0 tumors had 31 genes down-regulated by PD1 Ab with the remaining nine genes upregulated, whereas P4 tumors upregulated 39 genes with only one gene down-regulated. However, gene ontology enrichment analysis showed that PD1 Ab exerted similar biological effects on both P0 and P4 tumors (Supplementary Fig. S11). These results indicate that TME in response to PD1 Ab was more dynamically altered for P4 than P0. However, genome analyses did not identify specific molecular alterations responsible for Gpnmb upregulation.

a A total of 18,301 genes were analyzed between control- vs. PD1 Ab-treated tumors and expressed as Volcano Plots shown with statistical significance (Log10P from 2 or 3 data batches, y-axis) vs. Log2FC x-axis. Each dot (gene expression) shows values in PD1 Ab samples relative to control Ab. Blue, gray, and red dots show up-, non-significant, and down-regulation, respectively. b Heatmap analyses of top 40 differentially expressed genes (DEGs) in P0 or P4 tumors treated with PD1 vs. control Ab are shown.
Sox10-Mitf axis was dysregulated in P4 cells
Since Gpnmb expression is regulated by Mitf48, we compared the expression of Mitf and its three family members (Tfe3, Tfeb, and Tfec)49 in P0 vs. P4 cells. All genes are listed as DEGs in RNA-seq analysis of the two cell lines (Supplementary Data 3) and qRT-PCR showed Mitf gene expression to be higher in P4 cells, with all Tfe genes down-regulated (Fig. 6a), consistent with the RNA-seq data. We next searched for possible Mitf-activators50 in the DEG list and found Creb3/4 and Sox10 to be significantly upregulated in P4 cells (p < 0.04) (Supplementary Table S7). Although Creb3/4 is a member of the Creb family51, it has not been reported to regulate the Mitf gene. Upregulated expression of Sox10 in P4 cells was confirmed by qRT-PCR (Fig. 6b). Because of the absence of information regarding the relationship between Sox10 and Tfe genes52,53, we assumed Sox10 activation to be responsible for upregulated expression of the Mitf gene.

qRT-PCR analysis of Mit family (Mitf, Tfe3, Tfeb, and Tfec) (a) and Sox10 (b) in P0 vs. P4 cells (mean ± SEM, n = 3). c Proportion of Mitf isoform transcripts in P0 vs. P4 cells is calculated and expressed in Pie chart. d RT-PCR analysis of Mitf isoforms in varying cell lines (P0, P4, B16 melanoma, and Raw macrophages), run on 1.5% agarose gel/ethidium bromide. The bp size of PCR products is shown at the right of the figure. e P0 or P4 cells were transfected with increasing doses of pGL3b (basic promoter-Luc), pG-Gpnmb-p (Gpnmb promoter-linked pGL3b), pG-Gpnmb-p-ΔA (MITF-A site-deleted Gpnmb promoter), pG-Gpnmb-p-ΔΒ (MITF-B-deleted), or pG-Gpnmb-p-ΔAB (both A and B sites-deleted). Luciferase (Luc) activities were measured and expressed as Luc/Renilla × 100 (n = 3, average ± SD). Representative of three experiments. f Heatmap analyses of expression of 43 Mitf-target genes in P0 vs. P4 cells (three different batches per sample) are shown using RNA-seq data (Supplementary Data 2 and 3). p values in (a, b) are shown using Student’s t-test. Adjusted p value is indicated in parenthesis, using the Benjamini–Hochberg procedure.
From transcript seq information of the RNA-seq data, we extracted all Mitf transcripts, determined the RNA start site, and sorted these into separate isoforms: Mitf-A (ubiquitous), Mitf-M (melanocyte-associated), Mitf-B (rare), and Mitf-H (heart-specific)54,55,56. P4 cells generated aberrant Mitf isoforms (Mitf-B and Mitf-H) whereas P0 cells expressed Mitf-A and Mitf-M (Fig. 6c). These findings were confirmed by isoform-specific RT-PCR (Fig. 6d; Supplementary Fig. S12).
Since the Gpnmb promoter region (−196 nt from the RNA start site) contains two E-box sequences (referred to as MITF-A distal site and MITF-B proximal site) for Mitf-binding48, we measured the Gpnmb promoter activity in P0 and P4 cells, using luciferase (Luc) reporter assays. Net Luc expression by Gpnmb promoter in P4 cells, over that of basic promoter-Luc vector, was 15-fold-higher than in P0 (Fig. 6e). Deletion of MITF-A site greatly reduced Luc activities of Gpnmb promoter, and deletion of MITF-B showed moderate reduction. Double deletions resulted in a complete loss of the activities, indicating that upregulated Gpnmb expression in P4 was due to elevated Mitf expression.
Our RNA-seq data of P4 vs. P0 cells showed 43 Mitf-target gene57,58 expression altered (Fig. 6f), not only for Gpnmb but also for other genes not known to be direct T-cell regulators; Note that Naip6 (the highest Log2FC 4.2) is involved in innate immunity59. We concluded that the dysregulated Sox10-Mitf axis in P4 cells led to elevated Gpnmb expression.
Sox10 regulates the Mitf-Gpnmb axis
To determine linkage between Sox10 and Mitf-Gpnmb axes, we examined impact of each component gene expression by shRNA-mediated knockdown of Sox10. Transfection of Sox10-targeted shRNA into P4 cells led to substantial reduction of its gene expression alongside an 80% decrement in Mitf and Gpnmb gene expression (Fig. 7a). Mitf-shRNA reduced gene expression of Mitf and Gpnmb without affecting the Sox10 gene. Also, these shRNAs did not alter PDL1 expression. These results indicate that Sox10-regulated Mitf and Gpnmb genes in a unidirectional manner.

a Sox10- or Mitf-targeted or control shRNA was transfected into P4 cells and determined by qRT-PCR for expression of indicated genes. Mitf gene expression is shown as transcripts of Mitf common regions. Expression is shown as % of control gene expression in control shRNA-transfected cells (mean ± SEM, n = 3). b RenCa or Caki-2 cells were cultured for 0 (immediately harvested), 1, and 2 days with immobilized anti-PDL1 Ab and quantified for SOX10 gene expression (mean ± SEM, n = 3). c Deletion mutant analysis of PDL1 signaling. The cytoplasmic amino acid sequences of all mutants with deletion or amino acid substitution are schematically presented. The entire cytoplasmic region is shown at the top. The deleted region is shown by gray-filled bars, with amino acid residues of the junction between both ends. Lysine-to-arginine substitution at 171 aa (R171) or 280 aa (R280) is also constructed. These genes were separately transfected into 293T cells and assayed for upregulated SOX10 gene expression by PDL1-crosslinking and expressed as % of WT-induced full rise (mean ± SEM, n = 3). Data are representative of at least two experiments. p values are shown, compared to control using Student’s t-test.
PDL1-induced signaling activates the Sox10 gene
We assumed interaction of anti-PD1/PDL1 Ab with the PDL1 receptor on tumor cells would activate the Sox10-directed Mitf-Gpnmb axis. RenCa and the human RCC Caki-2 cells were crosslinked with anti-PDL1 Ab, incubated at different times, and measured for RNA expression of Sox10 gene. Compared to results on day 0 post-crosslinking, Sox10 expression rose 4-fold for RenCa and 8-fold for Caki-2 (Fig. 7b). To probe involvement of PDL1 signaling, we employed deletion mutant analysis. Gene for the full-length of PDL1 tagged with the N-terminal V5 epitope was transfected into 293T cells (as model that express PDL1 highly and Sox10 lowly), crosslinked with anti-V5 Ab, and assayed for RNA expression of endogenous Sox10 gene. Crosslinking of WT-PDL1-trasfected 293T cells induced SOX10 RNA expression that was 16-fold greater than control Ab. We next constructed mutants that lack previously reported signal motifs60. Sox10-upregulating ability of PDL1 was abrogated significantly (68%) by deleting the RMLDVEKC motif (previously determined for inhibiting IFN-γ-induced cytotoxicity), but only slightly by deleting the DTSSK motif (for regulating the above activity) (Fig. 7c). Double deletions reduced Sox10 upregulation by 82%. The cytoplasmic tail-lacking mutant or lysine-to-arginine substitution (for ubiquitination)61 produced a small reduction. All mutant-transfected cells expressed similar levels of PDL1 on the cell surface (Supplementary Fig. S13). Thus, the two motifs are required to fully upregulate the Sox10 gene.
Plasma cell-free RNA (cfRNA) from acquired resistance patients showed increased expression of SOX10-MITF RNA
To determine the human relevance of SOX10-MITF activation, we quantified RNA expression levels in plasma cfRNA: Plasma samples were taken from three patients who exhibited acquired resistance (Pat #9, 19, and 20) during transition from SD to PD (Fig. 2b). cfRNA was purified from plasma samples62 at all follow-up time points available and PCR-amplified with specific primers for GAPDH, GPNMB, MITF (specific to the common region of all MITF isoforms), SOX10, or β-ACTIN. Because of the minute quantity of mRNA in cfRNA fraction, we employed nested qPCR assay (Fig. 8a) known to have 10-fold higher sensitivity than regular qPCR63,64. Standard curves plotting Cq values against starting all template DNA doses showed a linear relationship up to 108 or 109 dilutions (0.23–0.023 pM) of the initial dose (23 μM) (Fig. 8b); 10−3–10−6 dilutions for β-ACTIN. cfRNA samples (including three healthy donors or HD) were subjected to nested qPCR assays. GPNMB RNA was detected at very low levels in HD. By contrast, Pat #9 expressed high levels of GPNMB RNA at baseline, and it gradually declined to the level of 35-week sample and increased in 55-week sample beyond the baseline level (Fig. 8c), consistent with blood sGPNMB protein levels (Fig. 2b). The other two patients also displayed similar kinetics of GPNMB RNA expression, indicating positive correlation of sGPNMB protein and GPNMB cfRNA levels. Both transcription factors MITF and SOX10 RNA had basically identical expression patterns as GPNMB RNA. After transition to PD, all patients displayed an increase in both SOX10 and MITF cfRNA, with fold increase range of 1.7 and 3.6. β-ACTIN cfRNA expression was similar at all time points in all patients and healthy donors. SOX10, MITF, and GPNMB RNA expression was confirmed in RNA-seq data of ccRCC cancer tissues65 and Entrez ID10457, NCBI (Supplementary Fig. S14). Thus, plasma cfRNA analysis suggests robust activation of the SOX10-MITF axis in ICI-treated patients that transitioned from SD to PD.

a Schematic presentation of nested real-time PCR analysis. b Nested qPCR standard curves: target cDNA template (23 μM) was 10-fold serially diluted, PCR-amplified for 15 cycles, and an aliquot (5% of total products) was then subjected to nested qPCR. Dilution factors are plotted against Cq value. c Plasma cfRNA at varying time points of acquired resistance patients (Pat #9, 19, and 20, Fig. 2b) and from three healthy donors (HD #1, 2, and 3) were quantified by nested qPCR for expression of GPNMB, MITF, SOX10, and β-ACTIN relative to GAPDH, and expressed as fold change to Week 0 of each patient (average, n = 2). Expression levels in HD are indicated by relative expression to the value at week 0 of Pat #9. Gray-filled bars represent time points for stable disease (SD) just before progressive disease PD (shown in black-filled bars). p values are shown, using Student’s t-test comparing gray- and black-filled bars in patients or #1 and #3 β-ACTIN in HD.
Discussion
Of the mechanisms cited as possible causes of acquired resistance to ICI6, none applied to our anti-PD1 Ab-resistant mouse model. P4 cells were not genetically mutated in the β2 microglobulin (B2M) gene responsible for tumor Ag presentation nor in Janus kinases (JAK) genes for IFN-γ signaling. Genes for WNT, PTEN, and the immune checkpoints Lag3, Tim3, and VISTA, were not altered. We also found no evidence for neoantigen depletion mechanism since the tumor-associated Ag for RenCa has yet to be identified.
By using the RenCa syngeneic mouse model, however, we discovered that PDL1 signal-linked dysregulation of the SOX10-MITF pathway accounted for the acquired resistance phenotype. The RenCa model enabled us to study cellular and molecular interactions of TME with ICI since tumor cells spontaneously grew within a TME with genetically identical tumor stromal cells that included T-cells and other immune cells66. To be sure, some may argue this model falls short of true human ccRCC biology given an inconsistent genetic landscape without stem cells and intratumoral heterogeneity. Nonetheless, the high association of upregulated GPNMB and ICI resistance in both our human and animal studies indicates remarkable consistency across species.
Engagement of PD1 on T-cells to the PDL1 receptor on cancer cells has long been known to inhibit T-cell functions but only recently has the nature of the signaling responsible for this process been elucidated. Skepticism initially surrounded the notion of intrinsic signaling since the cytoplasmic domain of the PDL1 receptor is very short, consisting of only 31 amino acids and without a tyrosine kinase motif67. More recently, the cytoplasmic domain was shown to contain two conserved non-classical signal motifs (RMLDVEKC and DTSSK) that protected cancers from IFN-γ cytotoxicity due to signal transducer and activator of transcription 3 (STAT3) activation mediated via the IFN-γ receptor60. Thus, binding of PD1 to the PDL1 receptor not only led to inhibiting T-cell activation but also protected tumor cells from T-cell attack.
Taking advantage of the two known signal motifs, we performed deletion mutant analysis of each and found the PDL1 receptor to signal a completely different pathway, SOX10/MITF/GPNMB, that in turn endowed RCC cells with resistance to destruction by ICI therapy. This new finding indicates that cancer can exploit PDL1 signaling by unleashing two pathways, the first during its regular confrontation with host T-cells and the second at the onset of acquired resistance to ICI treatment.
P4 tumor-bearing mice displayed comparable immunosuppressive phenotypes in TME and BM, unlike P0 tumors. These alterations may be due to the varied functions of both soluble and receptor Gpnmb that regulate the T-cell response, angiogenesis, and macrophage differentiation, via diverse ligand receptors of Gpnmb expressed on activated T-cells, endothelial cells, and osteoblasts68. In particular, sGpnmb is responsible for contrasting reduced T-cell and increased macrophage density in BM. Thus, upregulated Gpnmb expression shifts TME to an immunosuppressive phenotype.
There is accumulating evidence that elevated GPNMB expression is a negative prognosticator for various malignancies including extensive bone metastasis in RCC69, invasive triple-negative breast cancer64, and colorectal liver metastasis70 in human patients; and MC38 colon cancer in mouse models71. Increased methylation of the GPNMB gene resulting in its reduced expression was also shown for colorectal adenomas with high carcinogenic potential72. Not until our present study, however, has GPNMB been associated with acquired resistance to ICI treatment.
The MITF/MIT family consists of four transcription factors that contain basic helix-loop-helix leucine zipper structures that activate target gene expression by binding to E-boxes73. Their expression is highly regulated by similar mechanisms, including alternative splicing, differential usage of promoter, and post-translational modifications. In RCC, TFE members dominate over MITF in regulating the expression of E-box-targeted genes, and RCC of the translocation type display overexpressed TFE3 and GPNMB (while MITF is low)73. We found PD1 Ab-resistant P4 cells with high Gpnmb expression to have upregulated Mitf in the face of down-regulated other TFE members, even as we note all are potent activators of the Gpnmb gene. We showed ICI treatment of RCC to change the equilibrium between TFE and MITF in favor of MITF with concomitant dysregulation of the MITF gene, as documented by emergence of unusual Mitf-B and Mitf-H isoforms in P4 cells (not expressed in P0 cells nor in normal kidney tissue). Mitf gene generates diverse isoforms by differential use of Exon 1 promoters, some utilized in a lineage-specific manner. Melanoma predominantly expresses Mitf-M, whereas RCC expresses mostly Mitf-A74,75. Thus it is possible that Sox10 activates the Exon 1B promoter (generating Mitf-B) since we found the Sox10 consensus DNA-binding site (ACAAG) in the promoter region76. Alternatively, genome instability in P4 cells (Supplementary Fig. S9d) may unblock repressed promoter activity.
Tumor cells survive through the acquisition and selection of molecular modifications via genetic and/or non-genetic mechanisms77. Our preliminary data showed that acquired resistance phenotype of P4 cells persists after tumor passage in mice without ICI-immune pressure, suggesting this phenotype is inheritable. We compared the epigenetic state of P0 versus P4 cells and discovered that P4 cells display open chromatin in many regions of the Mitf genome, compared to P0 cells; this was not true for other Mitf members (unpublished data). Having shown no genetic alterations that account for acquired resistance development, ICI resistance in our mouse model may be due to non-genetic mechanisms.
SOX10 and MITF were reported to be involved independently in regulating resistance of human melanoma to targeted therapy against mitogen-activated protein kinases. SOX10 gene expression confers AR to BRAF inhibitors (BRAFi)78. Likewise, MITF is thought to cause BRAF resistance in melanoma6,67. Unlike these reports, our studies indicate SOX10 and MITF collaborate in producing a dysregulated MITF-GPNMB axis that leads to ICI resistance.
Plasma cfRNA isolated before and after the development of acquired resistance revealed upregulated expression of SOX10 and MITF RNA during transition to PD. We recognize that such plasma cfRNA may derive from tumor cells and/or normal cells. Plasma samples from cancer donors tend to have a higher cfDNA yield than healthy donors (cfDNA-quantification-tech-note-1170-2020-001, Illumina). We noted a similar trend for plasma cfRNA from cancer patients vs. healthy individuals, and before vs. after acquired resistance development (Supplementary Table S7), leading us to assume the major source of cfRNA is cancer tissue and/or circulating tumor cells. However, we cannot exclude involvement of GPNMB+ MDSC to cfRNA expression. If so, then upregulated SOX10-MITF gene expression in plasma may reflect tumor relapse.
We believe the mechanisms underlying innate and acquired resistance are different. Studies of innate resistance point to a predetermined outcome based on genetic alterations in tumor cells. By contrast, acquired resistance mechanisms appear more complex, akin to chronic inflammatory disease with elevated expression of various genes, including negative regulators. This concept is supported by our RNA-seq data of P4 vs. P0 tumors’ responses to anti-PD1 Ab, in which treatment-resistant (vs. susceptible) tumors had considerably more dynamic transcriptomes, supporting the notion that ICI resistance is inducible (not constitutive), arising from mechanisms that counteract the hyper-activated immunity resulting from sustained ICI treatment.
We conclude that rising blood sGPNMB is a useful real-time indicator of ICI resistance. PDL1-triggered dysregulation of the oncogenic SOX10-MITF pathway causes resistance, making elements of this pathway potential targets for overcoming this limitation to optimal treatment efficacy.
Data availability
Anonymized data and supporting documents are available upon request via Jin-Sung.Chung@utsouthwestern.edu. WES analysis data are provided in Supplementary Data 1. RNA-seq data for tumor RNA and cell lines (P0 vs. P4) are available in Supplementary Data 2 and Supplementary Data 3, respectively. The numerical data (source data) plotted in all figures in the text and in Supplementary figures are available in Supplementary Data 4 and Supplementary Data 5, respectively. These sequence data sets (Supplementary Fig. S14) are also available at NCBI: accession number PRJNA1199538 for RNA-seq data; and PRJNA1200710 for Wes seq data.
References
Hammers, H. Immunotherapy in kidney cancer: the past, present, and future. Curr. Opin. Urol. 26, 543–547 (2016).
Hammers, H. J. et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 study. J. Clin. Oncol. 35, 3851–3858 (2017).
Motzer, R. J. et al. Nivolumab for metastatic renal cell carcinoma: results of a randomized phase II trial. J. Clin. Oncol. 33, 1430–1437 (2015).
Moreira, M. et al. Resistance to cancer immunotherapy in metastatic renal cell carcinoma. Cancer Drug Resist. 3, 454–471 (2020).
Jenkins, R. W., Barbie, D. A. & Flaherty, K. T. Mechanisms of resistance to immune checkpoint inhibitors. Br. J. Cancer 118, 9–16 (2018).
Schoenfeld, A. J. & Hellmann, M. D. Acquired resistance to immune checkpoint inhibitors. Cancer Cell 37, 443–455 (2020).
Spranger, S. & Gajewski, T. Rational combinations of immunotherapeutics that target discrete pathways. J. Immunother. Cancer 1, 16 (2013).
Draghi, A., Chamberlain, C. A., Furness, A. & Donia, M. Acquired resistance to cancer immunotherapy. Semin. Immunopathol. 41, 31–40 (2019).
Li, H., van der Merwe, P. A. & Sivakumar, S. Biomarkers of response to PD-1 pathway blockade. Br. J. Cancer 126, 1663–1675 (2022).
McKean, W.B., Moser, J.C., Rimm, D. & Hu-Lieskovan, S. Biomarkers in precision cancer immunotherapy: promise and challenges. in American Society of Clinical Oncology Educational Book e275–e291 (Lippincott Williams & Wilkins, 2020).
Pender, A. et al. Genome and transcriptome biomarkers of response to immune checkpoint inhibitors in advanced solid tumors. Clin. Cancer Res. 27, 202–212 (2021).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375, 819–829 (2016).
Zhou, B., Gao, Y., Zhang, P. & Chu, Q. Acquired resistance to immune checkpoint blockades: the underlying mechanisms and potential strategies. Front. Immunol. 12, 693609 (2021).
Gettinger, S. et al. Impaired HLA class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discov. 7, 1420–1435 (2017).
Kluger, H. M. et al. Defining tumor resistance to PD-1 pathway blockade: recommendations from the first meeting of the SITC Immunotherapy Resistance Taskforce. J. Immunother. Cancer 8, e000398 (2020).
Jackman, D. et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J. Clin. Oncol. 28, 357–360 (2010).
Rose, A. A. et al. ADAM10 releases a soluble form of the GPNMB/Osteoactivin extracellular domain with angiogenic properties. PLoS ONE 5, e12093 (2010).
Kuan, C. T. et al. Glycoprotein nonmetastatic melanoma protein B, a potential molecular therapeutic target in patients with glioblastoma multiforme. Clin. Cancer Res. 12, 1970–1982 (2006).
Shikano, S., Bonkobara, M., Zukas, P. K. & Ariizumi, K. Molecular cloning of a dendritic cell-associated transmembrane protein, DC-HIL, that promotes RGD-dependent adhesion of endothelial cells through recognition of heparan sulfate proteoglycans. J. Biol. Chem. 276, 8125–8134 (2001).
Chung, J. S., Dougherty, I., Cruz, P. D. Jr. & Ariizumi, K. Syndecan-4 mediates the coinhibitory function of DC-HIL on T cell activation. J. Immunol. 179, 5778–5784 (2007).
Chung, J. S., Sato, K., Dougherty, I. I., Cruz, P. D. Jr. & Ariizumi, K. DC-HIL is a negative regulator of T lymphocyte activation. Blood 109, 4320–4327 (2007).
Chung, J. S., Cruz, P. D. Jr. & Ariizumi, K. Inhibition of T-cell activation by syndecan-4 is mediated by CD148 through protein tyrosine phosphatase activity. Eur. J. Immunol. 41, 1794–1799 (2011).
Chung, J. S., Tamura, K., Cruz, P. D. Jr. & Ariizumi, K. DC-HIL-expressing myelomonocytic cells are critical promoters of melanoma growth. J. Investig. Dermatol. 134, 2784–2794 (2014).
Kobayashi, M. et al. Blocking monocytic myeloid-derived suppressor cell function via anti-DC-HIL/GPNMB antibody restores the in vitro integrity of T cells from cancer patients. Clin. Cancer Res. 25, 828–838 (2019).
Chung, J. S. et al. DC-HIL/Gpnmb is a negative regulator of tumor response to immune checkpoint inhibitors. Clin. Cancer Res. 26, 1449–1459 (2020).
Bailey, A. & McDermott, D. F. Immune checkpoint inhibitors as novel targets for renal cell carcinoma therapeutics. Cancer J. 19, 348–352 (2013).
Choueiri, T. K. et al. Immunomodulatory activity of nivolumab in metastatic renal cell carcinoma. Clin. Cancer Res. 22, 5461–5471 (2016).
Bergerot, P. et al. Advances in the treatment of metastatic renal cell carcinoma. Cancer Treat. Res 175, 127–137 (2018).
Eisenhauer, E. A. et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009).
Chung, J. S., Bonkobara, M., Tomihari, M., Cruz, P. D. Jr. & Ariizumi, K. The DC-HIL/syndecan-4 pathway inhibits human allogeneic T-cell responses. Eur. J. Immunol. 39, 2009 (2009).
Turrentine, J. et al. DC-HIL+ CD14+ HLA-DRno/low cells are a potential blood marker and therapeutic target for melanoma The. J. Investig. Dermatol. 134, 2839–2842 (2014).
Tomihari, M., Chung, J. S., Akiyoshi, H., Cruz, P. D. Jr. & Ariizumi, K. DC-HIL/glycoprotein Nmb promotes growth of melanoma in mice by inhibiting the activation of tumor-reactive T cells. Cancer Res. 70, 5778–5787 (2010).
Ramani, V. et al. Melanoma-derived soluble DC-HIL/GPNMB promotes metastasis by excluding T-lymphocytes from the pre-metastatic niches. J. Investig. Dermatol. 138, 2443–2451 (2018).
Jin, S. et al. Genome-wide specificity of prime editors in plants. Nat. Biotechnol. 39, 1292–1299 (2021).
Zufferey, R., Nagy, D., Mandel, R. J., Naldini, L. & Trono, D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15, 871–875 (1997).
Albitar, M. et al. Combining cell-free RNA (cfRNA) with cell-free total nucleic acid (cfTNA) as a new paradigm for liquid biopsy. J. Clin. Oncol. 40, 3048–3048 (2022).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408 (2001).
Yao, J. et al. Checkpoint molecule PD-1-assisted CD8(+) T lymphocyte count in tumor microenvironment predicts overall survival of patients with metastatic renal cell carcinoma treated with tyrosine kinase inhibitors. Cancer Manag. Res. 10, 3419–3431 (2018).
Shen, H., Liu, Q., Li, M. & Yang, P. The prognostic value of vascular endothelial growth factor in patients with renal cell carcinoma: a systematic review of the literature and meta-analysis. Clin. Investig. Med. 40, E40–E48 (2017).
Jacobsen, J., Rasmuson, T., Grankvist, K. & Ljungberg, B. Vascular endothelial growth factor as prognostic factor in renal cell carcinoma. J. Urol. 163, 343–347 (2000).
Negrier, S. et al. Interleukin-6, interleukin-10, and vascular endothelial growth factor in metastatic renal cell carcinoma: prognostic value of interleukin-6–from the Groupe Francais d’Immunotherapie. J. Clin. Oncol. 22, 2371–2378 (2004).
Yao, J. et al. Mast cell density in metastatic renal cell carcinoma: association with prognosis and tumour-infiltrating lymphocytes. Scand. J. Immunol. 93, e13006 (2021).
Su, S., Akbarinejad, S. & Shahriyari, L. Immune classification of clear cell renal cell carcinoma. Sci. Rep. 11, 4338 (2021).
Retterer, K. et al. Clinical application of whole-exome sequencing across clinical indications. Genet. Med. 18, 696–704 (2016).
Shiina, T., Blancher, A., Inoko, H. & Kulski, J. K. Comparative genomics of the human, macaque and mouse major histocompatibility complex. Immunology 150, 127–138 (2017).
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability—an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010).
Salter, J. D., Bennett, R. P. & Smith, H. C. The APOBEC protein family: united by structure, divergent in function. Trends Biochem. Sci. 41, 578–594 (2016).
Loftus, S. K. et al. Gpnmb is a melanoblast-expressed, MITF-dependent gene. Pigment Cell Melanoma Res. 22, 99–110 (2009).
La Spina, M. et al. MiT/TFE family of transcription factors: an evolutionary perspective. Front. Cell Dev. Biol. 8, 609683 (2021).
Ferguson, J., Smith, M., Zudaire, I., Wellbrock, C. & Arozarena, I. Glucose availability controls ATF4-mediated MITF suppression to drive melanoma cell growth. Oncotarget 8, 32946–32959 (2017).
Ito, T., Saito, A., Kamikawa, Y., Nakazawa, N. & Imaizumi, K. AIbZIP/CREB3L4 promotes cell proliferation via the SKP2-p27 axis in luminal androgen receptor subtype triple-negative breast cancer. Mol. Cancer Res. 22, 373–385 (2024).
Fufa, T. D. et al. Genomic analysis reveals distinct mechanisms and functional classes of SOX10-regulated genes in melanocytes. Hum. Mol. Genet. 24, 5433–5450 (2015).
Lee, M., Goodall, J., Verastegui, C., Ballotti, R. & Goding, C. R. Direct regulation of the Microphthalmia promoter by Sox10 links Waardenburg-Shah Syndrome (WS4)-associated hypopigmentation and deafness to WS2*. J. Biol. Chem. 275, 37978–37983 (2000).
Shibahara, S. et al. Microphthalmia-Associated Transcription Factor (MITF): multiplicity in structure, function, and regulation. J. Investig. Dermatol. Symp. Proc. 6, 99–104 (2001).
Zarei, M. et al. MITF is a driver oncogene and potential therapeutic target in kidney angiomyolipoma tumors through transcriptional regulation of CYR61. Oncogene 40, 112–126 (2021).
Flesher, J. L. et al. Delineating the role of MITF isoforms in pigmentation and tissue homeostasis. Pigment Cell Melanoma Res. 33, 279–292 (2020).
Vu, H. N., Dilshat, R., Fock, V. & Steingrímsson, E. User guide to MiT-TFE isoforms and post-translational modifications. Pigment Cell Melanoma Res. 34, 13–27 (2021).
Ji, Z. et al. MITF modulates therapeutic resistance through EGFR signaling. J. Investig. Dermatol. 135, 1863–1872 (2015).
Kofoed, E. M. & Vance, R. E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Gato-Cañas, M. et al. PDL1 signals through conserved sequence motifs to overcome interferon-mediated cytotoxicity. Cell Rep. 20, 1818–1829 (2017).
Lim, S.-O. et al. Deubiquitination and stabilization of PD-L1 by CSN5. Cancer Cell 30, 925–939 (2016).
Schwarzenbach, H., Hoon, D. S. B. & Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 11, 426–437 (2011).
Tran, T. M. et al. A nested real-time PCR assay for the quantification of Plasmodium falciparum DNA extracted from dried blood spots. Malar. J. 13, 393 (2014).
Neuberger, E. W. I. et al. Establishment of two quantitative nested qPCR assays targeting the human EPO transgene. Gene Ther. 23, 330–339 (2016).
Braun, D. A. et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat. Med. 26, 909–918 (2020).
Sobczuk, P. et al. Choosing the right animal model for renal cancer research. Transl. Oncol. 13, 100745 (2020).
Escors, D. et al. The intracellular signalosome of PD-L1 in cancer cells. Signal Transduct. Target Ther. 3, 26 (2018).
Lazaratos, A. M., Annis, M. G. & Siegel, P. M. GPNMB: a potent inducer of immunosuppression in cancer. Oncogene 41, 4573–4590 (2022).
Zhai, J. P., Liu, Z. H., Wang, H. D., Huang, G. L. & Man, L. B. GPNMB overexpression is associated with extensive bone metastasis and poor prognosis in renal cell carcinoma. Oncol. Lett. 23, 36 (2022).
Cortese, N. et al. High-resolution analysis of mononuclear phagocytes reveals GPNMB as a prognostic marker in human colorectal liver metastasis. Cancer Immunol. Res. 11, 405–420 (2023).
Xu, X. et al. Adaptive resistance in tumors to anti-PD-1 therapy through re-immunosuppression by upregulation of GPNMB expression. Int. Immunopharmacol. 101, 108199 (2021).
Ashktorab, H. et al. GPNMB methylation: a new marker of potentially carcinogenic colon lesions. BMC Cancer 18, 1068 (2018).
Davis, I. J. et al. Oncogenic MITF dysregulation in clear cell sarcoma: defining the MiT family of human cancers. Cancer Cell 9, 473–484 (2006).
Lang, M. et al. Clinical and molecular characterization of microphthalmia-associated transcription factor (MITF)-related renal cell carcinoma. Urology 149, 89–97 (2021).
Goding, C. R. & Arnheiter, H. MITF-the first 25 years. Genes Dev. 33, 983–1007 (2019).
Schlierf, B., Ludwig, A., Klenovsek, K. & Wegner, M. Cooperative binding of Sox10 to DNA: requirements and consequences. Nucleic Acids Res. 30, 5509–5516 (2002).
Marine, J.-C., Dawson, S.-J. & Dawson, M. A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer 20, 743–756 (2020).
Sun, C. et al. Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014).
Acknowledgements
We thank I. Dougherty and S. Ung for technical assistance and T. Byrd for administrative support and the assistance of the University of Texas Southwestern Medical Center Tissue Resource, a shared resource of the Harold C. Simmons Comprehensive Cancer Center, which is supported in part by the National Cancer Institute under award number 5P30 CA 142543. This study was primarily supported by the Department of Defense Kidney Cancer Research Program (W81XWH-20-1-0905) and partially by the VA Merit Award (1 I01 BX004069-01).
Author information
Authors and Affiliations
Contributions
K. Ariizumi and H. Hammers conceptualized the study. J.-S. Chung, V. Ramani, L. Guo, and V. Popat were responsible for the methodology. L. Guo and L. Xu were responsible for the software. J.-S. Chung, V. Ramani, L. Guo, and V. Popat performed the formal analysis and performed investigations. H. Hammers and V. Popat were responsible for access to patient samples and clinical data. K. Ariizumi and J.-S. Chung wrote the original draft of the manuscript. K. Ariizumi, H. Hammers, and P.D. Cruz Jr reviewed and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chung, JS., Ramani, V., Guo, L. et al. Acquired resistance to immune checkpoint therapy is caused by glycoprotein non-metastatic melanoma protein B signal cascade. Commun Med 5, 79 (2025). https://doi.org/10.1038/s43856-025-00786-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s43856-025-00786-x
