
Abstract
Viral infections can trigger increased reactive oxygen species (ROS) production and a reduced antioxidant response in the host, leading to redox stress, inflammation, apoptosis, and ultimately, cell and tissue damage, which contribute to disease development. A better understanding of how ROS contributes to viral pathogenesis is critical for the development of novel therapeutic interventions. In this review, we discuss the current knowledge on ROS production and its effects across various viral infections, including severe acute respiratory syndrome-coronavirus-2, influenza A virus, dengue virus, Zika virus, hepatitis B virus, hepatitis C virus, and human immunodeficiency virus infections, to improve future therapeutic and preventive strategies for these infections.
Similar content being viewed by others


Introduction
Reactive oxygen species (ROS), which are byproducts of cellular metabolism, are generated during viral infections and play important roles in cell signaling, transcription, apoptosis, ion transport, inflammation, immunomodulation, and neuromodulation1,2,3. Any imbalance that leads to increased ROS production and decreased antioxidant defenses results in oxidative stress4,5,6. ROS exert cellular toxicity by modifying DNA, lipids, and proteins through DNA base oxidation, lipid peroxidation, and protein carbonylation (protein oxidation), respectively7,8,9.
ROS are highly reactive radical and non-radical forms of oxygen comprising superoxide anion (O2−), hydrogen peroxide (H2O2), and highly reactive hydroxyl (OH) groups that are produced during oxygen metabolism during photosynthesis, oxidative phosphorylation, and peroxisomal activity10,11. These oxidative entities have dual effects on nuclear factor (NF)-κB signaling, either activating or inhibiting it to produce anti- or pro-oxidative responses12. Antioxidants, such as thiol donors, inhibit NF-κB-induced gene transcription13. Proinflammatory cytokines, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta, and interferon-gamma (IFN-γ), promote ROS production, while ROS scavengers such as pyrrolidine dithiocarbamate and N-acetyl-cysteine (NAC) can reduce the ROS levels induced by these cytokines14.
ROS are involved in the regulation of a wide range of physiological and pathological processes15,16. ROS generation is controlled under physiological conditions but increases under pathological conditions, with high ROS levels potentially inducing carcinogenesis and oxidative stress-stimulated diseases. Optimal ROS levels are essential for cellular functions such as cell proliferation, differentiation, and apoptosis15,16,17. ROS also play important roles in the activation and regulation of innate and adaptive immunity, including respiratory bursts and inflammasome activation18,19. Excessive mitochondrial ROS (mtROS) production can suppress immunoglobulin production via T-cell-dependent antigens20.
ROS generation occurs via multiple metabolic pathways that are tightly controlled by oxidant and antioxidant signaling and plays an important role in modulating inflammation during viral infections21,22. Although ROS are involved in the initiation, progression, and metastasis of cancer, they also exert beneficial activities, such as apoptosis by chemotherapeutic agent activation23. Nuclear factor-erythroid 2-related factor 2 (Nrf2) is a key transcription factor upregulating the antioxidant response element (ARE)-mediated expression of antioxidant and detoxifying enzymes, and the Nrf2-antioxidant pathway is critical in host defense against acute lung injury (ALI)24.
Nitrative stress induced by reactive nitrogen species similarly plays a significant role in viral pathogenesis25. However, here we mainly focus on viral infection-induced ROS production and its effects on viral pathogenesis. Different viruses, including the severe acute respiratory syndrome-coronavirus-2 (SARS-CoV-2)26, influenza A virus (IAV)21,27,28,29, herpes simplex virus (HSV)22, hepatitis C virus (HCV)30,31,32, human immunodeficiency virus-1 (HIV-1)33, respiratory syncytial virus (RSV)34, and enterovirus (EV)-7135, induce ROS generation during infection. Mitochondria are the primary source of intracellular ROS, and ROS modulator 1 is one of the key mitochondrial proteins involved in ROS production36. Cytosolic ROS produced by NADPH oxidase 2 (NOX2) in innate immune cells damage pathogens via lipid and DNA oxidation37. During autooxidation of glucose, enzymatic pathways, such as the NOX pathway, become the main sources of ROS38,39. Potential ROS induction pathways include the NF-κB, mitogen-activated protein kinase, phosphoinositide 3-kinase–Akt, and Keap1–Nrf2–ARE signaling pathways39.
NOXs are present in most eukaryotes, including amoeba, fungi, plants, nematodes, echinoderms, urochordates, insects, fish, reptiles, and mammals40,41. They perform a variety of roles in eukaryotes but share the common function of ROS generation by innate immune cells in response to pathogens. However, the specific effects of ROS on viral infections remain unknown42. To et al. showed that NOX2 oxidase, the primary enzymatic source of ROS, is activated by single-stranded RNA and DNA viruses in endocytic compartments, inducing endosomal H2O2 generation, thereby suppressing the antiviral and humoral signaling networks by modifying the unique highly conserved cysteine residue on Toll-like receptor (TLR)-742. Viral infections activate endosomal NOX2 oxidase and restrict TLR7 signaling, and inhibition of endosomal NOX2 decreases viral pathogenicity42.
Many studies have reported the roles of ROS in the pathogeneses of various infections, and a better understanding of ROS involvement in viral infections should help in the development of new therapeutic interventions. In this review, we discuss ROS generation and highlight their roles in the pathogeneses of various viral infections, with an emphasis on SARS-CoV-2, IAV, HCV, hepatitis B virus (HBV), HIV, dengue virus (DENV), and Zika virus (ZIKV) infections.
ROS production during viral infection
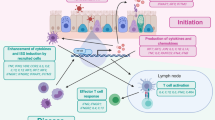
ROS production occurs with numerous viral infections (Fig. 1). ROS can be either beneficial or harmful to the host, depending on the infectious agent, by either providing protection or enabling pathogen attack (Table 1). Low ROS levels activate cell survival signaling pathways, such as the unfolded protein response and Nrf2 pathways, whereas high ROS levels activate cell death signaling pathways, such as apoptosis and necroptosis pathways43. ROS play an important role in microbial decontamination either directly by inducing oxidative stress or indirectly by promoting pathogen clearance through a number of non-oxidative mechanisms, including autophagy, T-cell responses, and pattern recognition receptor signaling44. ROS also exert potent antiviral effects on DENV-infected human monocyte-derived dendritic cells by activating the interferon regulatory factor (IRF)-3/IRF7/signal transducer and activator of transcription 1 and NF-κB signaling45.

Viral infections enhance the activities of oxidative enzymes, such as NADPH oxidase (NOX) and suppress those of antioxidant enzymes, such as reduced glutathione (GSH) and nuclear factor-erythroid 2-related factor 2 (Nrf2), thereby increasing ROS levels. Increased ROS production modulates viral replication and pathogenesis. IAV Influenza A virus, HCV Hepatitis C virus, DENV Dengue virus, ZIKV Zika virus, HIV human immunodeficiency virus, HBV hepatitis B virus. This figure was originally designed with a graphical abstract generation service by Editage Co. under our instruction.
High ROS levels generated via endogenous (mitochondria, endoplasmic reticulum [ER], and NOXs) and exogenous sources (radiation, drugs, foods, cigarette smoking, and pollution) induce oxidative stress that is harmful to the host46,47. Viruses interact with mitochondrial membranes and other mitochondria-associated components, leading to increased ROS production48. During viral infection, the antioxidant defense of the host is perturbed, resulting in oxidative stress48. A number of viruses, such as HCV, HBV, IAV, and ZIKV, modulate the antioxidant defense system to produce ROS32,49,50,51,52. Viruses control ROS-generating machinery of the host, thereby altering ROS metabolism and enhancing viral replication during infection53,54.
Mechanisms that commonly induce ROS generation in viral infections include the deregulation of mitochondrial function, activation and upregulation of NOX production, and modification of the Nrf2/ARE pathway, with superoxide being the most commonly produced ROS55. HCV core protein inhibits the mitochondrial electron transport complex I and increases ROS production32. We previously reported significantly increased ROS levels in the sera and livers of HCV-infected tree shrew, which is an emerging small animal model of the Tupaiidae family susceptible to HCV infection56,57. ROS production is commonly observed during viral infections and modulates the host responses (Table 1).
ROS-induced promotion of viral pathogenesis
Moderate ROS levels are necessary for numerous cellular functions, including gene expression58. Generally, ROS protect cells against invading bacteria and viruses; however, some viruses readily replicate despite ROS induction, suggesting that ROS can exert bidirectional effects on viral replication59. ROS trigger oxidative stress via various biological processes, such as apoptosis, necrosis, and autophagy, playing critical roles as signaling molecules in the cell death pathway60. Oxidative stress is associated with the pathogenesis of many viral infections, including those caused by the influenza virus, SARS-CoV-2, HIV, and HCV. However, the specific effects of ROS vary depending on their origin61.
During host cell infection, CoVs trigger an imbalance involving increased ROS production and reduced antioxidant host responses, thereby leading to increased redox stress, reduced antiviral host responses, and increased virus-induced inflammation and apoptosis, ultimately causing cell and tissue damage and end-organ disease3. ROS are directly or indirectly associated with cytokine storm induction in SARS-CoV-2 infection, and ROS can activate NF-κB and NLRP3 inflammasomes, thereby leading to the production of inflammatory cytokines and the development of acute respiratory distress syndrome and multiple organ dysfunction62,63,64. SARS-CoV-2 infection induces oxidative stress and is associated with lower plasma reduced glutathione (GSH) levels. Lower GSH levels have been reported in the frontal grey matter of the brain of post-acute sequelae of COVID-19 (PASC) participants, suggesting ongoing oxidative stress in the brains of individuals with PASC65. Nrf2 has been shown to negatively regulate stimulator of interferon genes (STING) expression by decreasing STING mRNA stability, thus indicating an association in antiviral sensing66. SARS-CoV-2 infection has been shown to suppress Nrf2 expression in patients with COVID-19, thereby enhancing the infection severity67. Recent studies show that SARS-CoV-2 nonstructural viral protein NSP14 inhibits Nrf2/HMOX-168. SARS-CoV-2 ORF6 protein has also been shown to dysregulate redox homeostasis by decreasing Nrf2 expression, which can enhance viral replication69.
RSV infection affects cellular mitochondrial membrane potential by enhancing mtROS generation, which facilitates viral infection34. HSV infection, however, induces early ROS production and NF-κB and IRF-3 pathways, resulting in type I IFN and interferon-stimulated gene production22.
The pathogenesis of IAV infection has been associated with oxidative stress27. IAV infection induces oxidative stress by decreasing the intracellular content of GSH and increasing ROS levels in the host cells. IAV infection has also been shown to suppress glucose-6-phosphate dehydrogenase expression, leading to decreased Nrf2 expression, induction of oxidative stress, and viral replication70. IAV has been shown to suppress SIRT2, thereby enhancing viral replication70. IAV can induce glutathionylation, and glutaredoxin-1-mediated deglutathionylation exerts an inhibitory effect on viral replication71. Although NOX2 has been shown to enhance the pathogenesis in IAV infection through endosomal NOX2 oxidase-dependent ROS production, NOX4 oxidase exerts a protective effect against IAV infection72. IAV infection increases the levels of xanthine oxidase, which is an enzyme responsible for oxygen-free radical generation in mouse serum and lung tissues, thus promoting disease pathogenesis73. Uncontrolled production of ROS during influenza virus infection can induce cell injury74. Lin et al. reported that H5N1 IAV infection in lung epithelial cells (A549 cells) decreased Cu/Zn superoxide dismutase (SOD1) expression, which could enhance ROS production, viral replication, and the inflammatory response27. Similarly, SOD1 overexpression has been reported to reduce ROS generation and inhibit viral replication and the inflammatory response27.
Protein disulfide isomerases (PDIs) constitute a family of redox chaperones that interact with IAV- hemagglutinin (HA) and IAV- neuraminidase (NA)75,76. Inhibition of PDIA3 can reduce IAV HA and NA activity, resulting in a decreased viral burden and thus indicating a role in viral pathogenesis75,76. IAV infection can induce MAPK phosphatase-5 expression, which has been shown to inhibit IRF3 activation and type I IFN response, thereby enhancing viral replication77.
IAV infection activates NF-κB binding and induces the expression of antioxidant genes, including manganese SOD and indoleamine 2,3-dioxygenase, in human airway epithelial cells78,79. Association between ROS generation and IFN-λ secretion to control IAV infection has been reported in human nasal epithelial cells49. Inhibition of mtROS generation and knockdown of dual oxidase 2 enhance IAV replication and decrease IFN-λ secretion49.
Imai et al. have shown that TLR4–TIR-domain-containing adapter-inducing IFN-β (TRIF)–TNF receptor-associated factor 6 signaling is a key pathway controlling the severity of acid-induced ALI80. They found that microbial infections, such as SARS and H5N1 virus infections in the lungs, trigger NADPH-dependent ROS production, thereby generating oxidized phospholipids, such as oxidized 1-palmitoyl-2-arachidonoyl-sn-glycerol-3-phosphatidylcholine, to induce lung injury and cytokine production by lung macrophages via TLR4–TRIF80. Inhibition of TLR4 or TRIF expression protects mice from H5N1-induced ALI80. A recent study has shown that the influenza virus matrix 1 protein can induce the production of proinflammatory cytokines and cellular ROS as well as trigger cell death through the activation of TLR4 signaling28.
Oxidative stress also occurs with chronic viral infections, such as viral hepatitis and acquired immunodeficiency syndrome81. Oxidative stress induced by hepatitis viruses, such as HBV and HCV, contributes to the development of hepatocellular carcinoma82. HCV infection can induce ROS-mediated oxidative stress, leading to the formation of 8-hydroxydeoxyguanosine (8-OHdG), which is an indicator of oxidative DNA damage83. Using a tree shrew model, we also observed a significantly increased level of 8-OHdG in all HCV-infected tree shrews compared to that in control tree shrews56.
Significant ROS induction has been reported during HBV infection, whereby the viral protein HBx plays a key role in the induction of oxidative stress51. The C-terminal region of HBx is essential for mitochondrial DNA damage84. Oxidative stress supports HBV replication, whereas ROS inhibitors, such as NAC, inhibit HBV replication51. HBV capsid assembly has been reported to be facilitated by ROS with heat shock protein 90, enhancing viral replication85. Hepatitis delta virus (HDV) also induces ROS generation by the expression of NOX1 and 4, cytochrome P450 2E1, and ER oxidoreductin 1α86. Large HDV antigens activate the antioxidant Nrf2/ARE pathway86. Yoon et al. reported that H2O2 inhibited HBV replication in human hepatoma cell lines in a p53-dependent manner87. Moreover, the use of NAC and knockdown of either p53 or seven-in-absentia homolog 1 prevented the inhibitory effects of H2O287.
Although almost all HCV proteins induce ROS generation, HCV core protein and NS5 are major inducers30,88,89,90. HCV-induced ROS play a significant role in HCV pathogenesis, as Kupffer cell activation contributes to pro-fibrotic cytokine and ROS production, leading to hepatic damage91,92. Damaged hepatocytes release ROS into the extracellular environment, thereby activating ROS-generating hepatic stellate cells. Activation of NOX enzymes in Kupffer and hepatic stellate cells induces ROS generation, thereby promoting liver fibrosis and hepatocarcinogenesis93,94,95,96. ROS generation induced by HCV infection triggers anti-3β-hydroxysterol Δ24-reductase (DHCR24) antibody production, suggesting that DHCR24 may be used as a prognostic biomarker for hepatitis C progression to hepatocellular carcinoma56,97.
HCV induces ER stress and alters ER calcium homeostasis, leading to oxidative stress98. Additionally, a positive correlation between HCV viral load and oxidative stress has been reported99. Dynamic thiol/disulfide homeostasis plays an important role in oxidative stress, and an imbalance in this homeostasis may be indicative of oxidative stress100. HCV infection also induces oxidative stress by impairing the thiol/disulfide balance, while direct-acting antiviral therapy restores this balance and decreases the pro-fibrotic process in patients with chronic hepatitis C101. Recently, Yoon et al. showed that H2O2 inhibits HCV replication in a p53-dependent manner by downregulating HCV core levels102.
Impairment of Nrf2/ARE signaling has been observed in HCV infection, leading to elevated ROS levels that can activate autophagy and facilitate HCV maturation and release103,104. HCV core protein induces ROS generation and antioxidant gene expression52,105. Mitochondrial dysfunction has been reported both in vitro and in vivo due to the direct effect of HCV core protein on mitochondria52. Although the direct association between HCV and apoptosis remains controversial, HCV-induced oxidative stress has been reported to enhance apoptosis106,107. However, HCV NS5A-mediated inhibition of apoptosis has also been reported108. Chronic HCV infection has been associated with higher expression of 8-OHdG, an indicator of oxidative DNA damage, which may lead to carcinogenesis83,109. NS5A and core proteins trigger oxidative stress-mediated Ca2+ homeostasis alterations in human hepatocyte-derived cells, thereby enhancing HCV-induced pathogenesis110. HCV infection-induced oxidative stress enhances fibrosis progression in chronic hepatitis C94.
HIV infection induces oxidative stress, which plays an important role in the development of various virus-associated pathologies111,112,113. Alteration of the redox balance is a key factor in HIV-1 pathogenicity, including neurotoxicity and dementia, depletion of CD4 + /CD8 + T cells, susceptibility to lung infections, and side effects of antiretroviral therapy111. HIV-1 proteins, including Tat, Nef, Vpr, protease, and gp120, modulate ROS-generating systems and induce oxidative stress, thereby promoting the expression of viral genes and proinflammatory lymphokines114,115,116. An association between ROS levels and IFN-γ response in HIV-1 infection has been reported, whereby HIV-1 reverse transcriptase enzyme-induced increased ROS levels induce stronger IFN-γ responses115. Oxidative stress enhances NF-κB-dependent activation of HIV transcription, thereby facilitating viral replication117. Vpr induces oxidative stress by stabilizing p53 protein, increasing transforming growth factor-beta protein levels and ROS production, and suppressing antioxidant enzymes118,119,120.
Increased oxidative stress and decreased antioxidant defense responses have been reported in HIV-positive patients121. HIV-1 protein Vpr increases H2O2 production and activates hypoxia-inducible factor-1, thereby stimulating HIV-1 gene transcription via hypoxia-inducible factor-1 association with the HIV-1 long terminal repeats122. The HIV-1 Tat protein induces oxidative stress in B cells via mtROS production. Tat-induced oxidative stress, DNA damage, and chromosomal aberrations are novel oncogenic factors promoting B-cell lymphomas in HIV-1 infection123. Both HCV and HIV infections enhance the induction of oxidative stress; however, HCV–HIV co-infection increases oxidative stress more potently than HIV mono-infection124,125.
Wang et al. have shown that DENV-2 infection alters oxidative stress by increasing the levels of malondialdehyde (MDA), the end product of lipid peroxidation, and the oxidized glutathione/GSH ratio126. Increased MDA levels and decreased serum SOD levels enhance the serum levels of TNF-α and IL-6 during DENV-2 infection126. Moreover, a positive correlation between viremia and MDA levels indicates an association between DENV-2 infection and oxidative stress126.
ZIKV induces oxidative stress, which further promotes ZIKV replication. Downregulation of Nrf2 and GSH significantly enhances viral replication127. ZIKV infection has been shown to increase the expression of oxidative stress-controlling proteins, including thioredoxin reductase 1, SOD, and biliverdin reductase B128. Piperlongumine (PL), an oxidative stress inducer, inhibits ZIKV replication in various cells, including the human brain microvascular, Vero, and human umbilical vein endothelial cells129. Moreover, NAC mitigates the inhibitory effect of PL on ZIKV replication129.
Almeida et al. reported that ZIKV induces ROS production and suppresses the activities of antioxidant enzymes, including SOD and catalase, in U87-MG (human glioblastoma) and HepG2 (human liver carcinoma) cell lines50. Additionally, ZIKV inhibits Nrf2 activation in both cell lines50. In contrast, a recent study reported that ROS levels are not altered by ZIKV infection in undifferentiated SH-SY5Y (human neuroblastoma) cells130. Moreover, extensive death of SH-SY5Y cells induced by ZIKV infection cannot be prevented by ROS scavenger NAC130. West Nile virus infection induces both ROS and antioxidant responses, with virus-induced changes in cellular redox status supporting viral replication131.
Enteroviral 2B protein has been shown to interact with mitochondrial voltage-dependent anion channel 3 (VDAC3) to induce ROS production, and knockdown of VDAC3 significantly reduces EV71-induced mtROS generation and viral replication132. ROS-induced impairment of the IFN response occurs in murine herpes virus infection, with the inhibition taking place downstream of cytoplasmic DNA sensing59. The suppression of the type I IFN response by ROS is due to oxidation of cysteine 147 of murine stimulator of IFN, an ER membrane protein that mediates the IFN response after cytoplasmic DNA sensing59,133,134. Therefore, ROS-induced immunomodulatory effects may facilitate viral replication.
Beck et al. showed that nutritionally induced oxidative stress in mice fed an antioxidant-deficient diet enhances the pathogenesis of coxsackievirus B3 infection and the development of myocarditis135. In a recent study, Hu et al. demonstrated that rabbit hemorrhagic disease virus (RHDV) infection induces NF-κB activation, and Keap1 interaction reduces Nrf2-related antioxidant function136. Notably, increased Nrf2 activity delays death in RHDV-infected rabbits, indicating oxidative damage during RHDV infection136.
Therapeutic potential of ROS modulators in viral pathogenesis
Numerous studies have investigated the therapeutic potential of inhibiting viral replication and pathogenesis by ROS inhibitors in vitro and in vivo in animal models55,137. Antioxidants and agents that downregulate pro-inflammatory cytokine and lipid mediator levels can be used as therapeutic complements to specific antiviral drugs33. SIRTs play important roles by stimulating antioxidant expression, repairing cells damaged by oxidative stress, and preventing cell dysfunction. Specifically, SIRT1, SIRT3, and SIRT5 protect the cell from ROS, and SIRT2, SIRT6, and SIRT7 modulate oxidative stress138,139. SIRT2 has been reported to induce Nrf2 activation and GSH production140, and pharmacological activation of SIRT2 has been shown to inhibit IAV replication70. Injection of the specific superoxide radical scavenger SOD 5–8 d after infection in conjugation with a pyran copolymer protects mice against lethal influenza virus infection73, thus suggesting that it has therapeutic potential against influenza virus infection. NAC, a precursor of L-cysteine, is a powerful antioxidant that acts as a scavenger of free radicals, particularly oxygen-derived radicals141. NAC inhibits TLR4 expression and protects mice against lethal H9N2 swine influenza virus infection142.
To et al. demonstrated that IAV infection induces mtROS generation, thereby causing innate immune inflammation and exacerbating viral pathogenesis in a mouse model137. Inhibition of mtROS by Mito-TEMPO attenuates the pathogenic effects of IAV (Hkx31 and H3N2) in mice137. The mtROS scavenger MitoQ strongly suppresses RSV production in both primary human bronchial epithelial cells and mouse models34, suggesting ROS inhibitors as potential therapeutic tools against RSV.
Decreased GSH levels increase cellular oxidative stress and are involved in many diseases and conditions, such as cancer, viral infections, and immune dysfunctions. Low GSH levels have been reported to increase susceptibility to viral infections, including SARS-CoV-2 infection143. GSH has been shown to exert an anti-influenza activity in vitro and in vivo, and depletion of GSH may have a fundamental role in COVID-19 and IAV infection susceptibility and disease severity144,145. SARS-CoV-2 has been reported to markedly decrease the level of GSH146. NAC has been reported to prevent the depletion of GSH in vitro146. Antioxidants, such as GSH and NAC, can hence be used as therapeutic tools to decrease the viral load and treat lung injuries caused by ROS overproduction during viral infections143. Olagnier et al. demonstrated that the Nrf2 agonists 4-octyl-itaconate and clinically approved dimethyl fumarate can inhibit the replication of several types of virus, including SARS-CoV-268 and ZIKV by an IFN-independent pathway67. However, future investigations are warranted for its clinical use.
Nadhan et al. proposed the use of ROS inducers as therapeutic agents against RNA viruses to induce the degradation of nascent viral RNA within host cells, as RNA is more susceptible to ROS-mediated degradation than DNA or proteins147. It has been reported that the highly potent ROS inducer plumbagin148 can be used against RNA viruses, including SARS-CoV-2147. SIRT3 overexpression has been reported to inhibit HBV replication by reducing cellular ROS levels51. Another study reported the interplay between ROS and SIRT1 in cell death during EV71 infection, and inhibition of ROS by NAC was shown to reduce EV71 propagation149. Overall, ROS modulators potentially reduce viral pathogenesis in the host, thus warranting further investigation.
Discussion
Oxidative stress occurs due to an imbalance between ROS generation and antioxidant capacity. ROS can exert multiple effects on the immune system, including the activation and interaction of immune cells, host defense, and immune suppression150. Additionally, ROS may act as signal molecules with regulatory activity on viral replication, potentially providing new insights into the treatment approaches151.
Although numerous studies have demonstrated ROS production during viral infections, the mechanisms of its production can vary between viruses3,33. Depending on the virus-host interaction, virus-induced ROS production can either enhance or inhibit the replication of viruses45,152,153,154. For example, IAV or SARS-CoV-2-induced NOX2 activity may suppress the TLR7-dependent antiviral response, thereby enhancing viral replication and pathogenicity42,155. Similarly, DENV replication has been reported to be either enhanced or inhibited by NOX modulation45,152, suggesting a critical role of ROS in viral replication.
Inhibition of ROS by GSH or its analogs has been reported to block HIV replication, whereas increased intracellular GSH levels have been shown to enhance African swine fever virus replication156,157, indicating that the effect of GSH on viral replication varies according to the virus type.
Conclusions
ROS production, mainly via mitochondrial dysfunction, is a common feature of various viral infections. While ROS production can be either beneficial or harmful to the host, excessive ROS generation is widely recognized for its harmful effects, enhancing viral pathogenesis. Therefore, the overall effect of ROS on the host depends on the context. Oxidative stress is associated with the pathology of various viral infections, including SARS-CoV-2, IAV, HCV, ZIKV, and HIV infections. Further exploration of ROS production mechanism(s) in viral infections and their interactions within the host can provide insights for new antiviral development.
Data availability
Data is provided within the manuscript.
References
Gloire, G., Legrand-Poels, S. & Piette, J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem. Pharmacol. 72, 1493–1505 (2006).
Lander, H. M. An essential role for free radicals and derived species in signal transduction. FASEB J. 11, 118–124 (1997).
Gain, C., Song, S., Angtuaco, T., Satta, S. & Kelesidis, T. The role of oxidative stress in the pathogenesis of infections with coronaviruses. Front Microbiol. 13, 1111930 (2022).
Betteridge, D. J. What is oxidative stress? Metabolism 49, 3–8 (2000).
Ryter, S. W. et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 9, 49–89 (2007).
Schieber, M. & Chandel, N. S. ROS function in redox signaling and oxidative stress. Curr. Biol. 24, R453–R462 (2014).
Yan, L. L. & Zaher, H. S. How do cells cope with RNA damage and its consequences? J. Biol. Chem. 294, 15158–15171 (2019).
Ito, F., Sono, Y. & Ito, T. Measurement and clinical significance of lipid peroxidation as a biomarker of oxidative stress: oxidative stress in diabetes, atherosclerosis, and chronic inflammation. Antioxidants (Basel) 8, 72 (2019).
Hawkins, C. L. & Davies, M. J. Detection, identification, and quantification of oxidative protein modifications. J. Biol. Chem. 294, 19683–19708 (2019).
Schwarz, K. B. Oxidative stress during viral infection: a review. Free Radic Biol. Med. 21, 641–649 (1996).
Checa, J. & Aran, J. M. Reactive oxygen species: drivers of physiological and pathological processes. J. Inflamm. Res. 13, 1057–1073 (2020).
Lingappan, K. NF-kappaB in Oxidative Stress. Curr. Opin. Toxicol. 7, 81–86 (2018).
Schreck, R., Meier, B., Mannel, D. N., Droge, W. & Baeuerle, P. A. Dithiocarbamates as potent inhibitors of nuclear factor kappa B activation in intact cells. J. Exp. Med. 175, 1181–1194 (1992).
Yang, D. et al. Pro-inflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp. Eye Res. 85, 462–472 (2007).
Droge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 82, 47–95 (2002).
Martindale, J. L. & Holbrook, N. J. Cellular response to oxidative stress: signaling for suicide and survival. J. Cell Physiol. 192, 1–15 (2002).
Juan, C. A., Perez de la Lastra, J. M., Plou, F. J. & Perez-Lebena, E. The chemistry of reactive oxygen species (ROS) revisited: outlining their role in biological macromolecules (DNA, Lipids and Proteins) and induced pathologies. Int. J. Mol. Sci. 22, 4642 (2021).
Andres, C. M. C., Perez de la Lastra, J. M., Juan, C. A., Plou, F. J. & Perez-Lebena, E. The role of reactive species on innate immunity. Vaccines (Basel) 10, 1735 (2022).
Bassoy, E. Y., Walch, M. & Martinvalet, D. Reactive oxygen species: do they play a role in adaptive immunity?. Front Immunol. 12, 755856 (2021).
Ogura, M. et al. Mitochondrial reactive oxygen species suppress humoral immune response through reduction of CD19 expression in B cells in mice. Eur. J. Immunol. 47, 406–418 (2017).
Wang, L., Cao, Z., Wang, Z., Guo, J. & Wen, J. Reactive oxygen species associated immunoregulation post influenza virus infection. Front Immunol. 13, 927593 (2022).
Gonzalez-Dosal, R. et al. HSV infection induces production of ROS, which potentiate signaling from pattern recognition receptors: role for S-glutathionylation of TRAF3 and 6. PLoS Pathog 7, e1002250 (2011).
Pizzimenti, S., Toaldo, C., Pettazzoni, P., Dianzani, M. U. & Barrera, G. The “two-faced” effects of reactive oxygen species and the lipid peroxidation product 4-hydroxynonenal in the hallmarks of cancer. Cancers (Basel) 2, 338–363 (2010).
Zhao, H., Eguchi, S., Alam, A. & Ma, D. The role of nuclear factor-erythroid 2 related factor 2 (Nrf-2) in the protection against lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 312, L155–L162 (2017).
Akaike, T. et al. 8-nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc. Natl. Acad. Sci. USA 100, 685–690 (2003).
Cecchini, R. & Cecchini, A. L. SARS-CoV-2 infection pathogenesis is related to oxidative stress as a response to aggression. Med. Hypotheses 143, 110102 (2020).
Lin, X. et al. The influenza virus H5N1 infection can induce ROS production for viral replication and host cell death in A549 cells modulated by human Cu/Zn superoxide dismutase (SOD1) overexpression. Viruses 8, 13 (2016).
Kim, C. U., Lim, D., Kim, Y. S., Ku, B. & Kim, D. J. Influenza viral matrix 1 protein aggravates viral pathogenicity by inducing TLR4-mediated reactive oxygen species production and apoptotic cell death. Cell Death Dis. 14, 228 (2023).
He, G. et al. Oxygen free radical involvement in acute lung injury induced by H5N1 virus in mice. Influenza. Other Respir. Viruses 7, 945–953 (2013).
Ivanov, A. V., Bartosch, B., Smirnova, O. A., Isaguliants, M. G. & Kochetkov, S. N. HCV and oxidative stress in the liver. Viruses 5, 439–469 (2013).
Rebbani, K. & Tsukiyama-Kohara, K. HCV-Induced oxidative stress: battlefield-winning strategy. Oxid. Med. Cell Longev. 2016, 7425628 (2016).
Korenaga, M. et al. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ROS) production. J. Biol. Chem. 280, 37481–37488 (2005).
Peterhans, E. Oxidants and antioxidants in viral diseases: disease mechanisms and metabolic regulation. J. Nutr. 127, 962S–965S (1997).
Hu, M. et al. Respiratory syncytial virus co-opts host mitochondrial function to favour infectious virus production. Elife 8, e42448 (2019).
Cheng, M. L., Weng, S. F., Kuo, C. H. & Ho, H. Y. Enterovirus 71 induces mitochondrial reactive oxygen species generation that is required for efficient replication. PLoS ONE9, e113234 (2014).
Amini, M. A., Karimi, J., Talebi, S. S. & Piri, H. The association of COVID-19 and reactive oxygen species modulator 1 (ROMO1) with oxidative stress. Chonnam Med. J. 58, 1–5 (2022).
Marques, E., Kramer, R. & Ryan, D. G. Multifaceted mitochondria in innate immunity. NPJ Metab Health Dis. 2, 6 (2024).
Araki, E. & Nishikawa, T. Oxidative stress: A cause and therapeutic target of diabetic complications. J. Diabetes Investig. 1, 90–96 (2010).
Rauf, A. et al. Reactive oxygen species in biological systems: pathways, associated diseases, and potential inhibitors-a review. Food Sci. Nutr. 12, 675–693 (2024).
Kawahara, T., Quinn, M. T. & Lambeth, J. D. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol. Biol. 7, 109 (2007).
Aguirre, J. & Lambeth, J. D. Nox enzymes from fungus to fly to fish and what they tell us about Nox function in mammals. Free Radic. Biol. Med. 49, 1342–1353 (2010).
To, E. E. et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 8, 69 (2017).
Redza-Dutordoir, M. & Averill-Bates, D. A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta. 1863, 2977–2992 (2016).
Kaushik, N. et al. The inactivation and destruction of viruses by reactive oxygen species generated through physical and cold atmospheric plasma techniques: current status and perspectives. J. Adv. Res. 43, 59–71 (2023).
Olagnier, D. et al. Cellular oxidative stress response controls the antiviral and apoptotic programs in dengue virus-infected dendritic cells. PLoS Pathog 10, e1004566 (2014).
Jomova, K. et al. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: chronic diseases and aging. Arch Toxicol. 97, 2499–2574 (2023).
Wu, Z., Wang, H., Fang, S. & Xu, C. Roles of endoplasmic reticulum stress and autophagy on H2O2‑induced oxidative stress injury in HepG2 cells. Mol. Med. Rep. 18, 4163–4174 (2018).
Foo, J., Bellot, G., Pervaiz, S. & Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 30, 679–692 (2022).
Kim, H. J. et al. Reactive oxygen species induce antiviral innate immune response through IFN-lambda regulation in human nasal epithelial cells. Am. J. Respir. Cell Mol. Biol. 49, 855–865 (2013).
Almeida, L. T. et al. Zika virus induces oxidative stress and decreases antioxidant enzyme activities in vitro and in vivo. Virus Res. 286, 198084 (2020).
Ren, J. H. et al. Protective role of sirtuin3 (SIRT3) in oxidative stress mediated by hepatitis B virus X protein expression. PLoS ONE 11, e0150961 (2016).
Okuda, M. et al. Mitochondrial injury, oxidative stress, and antioxidant gene expression are induced by hepatitis C virus core protein. Gastroenterology 122, 366–375 (2002).
Hyodo, K., Hashimoto, K., Kuchitsu, K., Suzuki, N. & Okuno, T. Harnessing host ROS-generating machinery for the robust genome replication of a plant RNA virus. Proc. Natl Acad. Sci. USA 114, E1282–E1290 (2017).
Sheyn, U., Rosenwasser, S., Ben-Dor, S., Porat, Z. & Vardi, A. Modulation of host ROS metabolism is essential for viral infection of a bloom-forming coccolithophore in the ocean. ISME J. 10, 1742–1754 (2016).
Sander, W. J. et al. Reactive oxygen species as potential antiviral targets. Rev. Med. Virol. 32, e2240 (2022).
Kayesh, M. E. H. et al. Oxidative stress and immune responses during hepatitis C virus infection in Tupaia belangeri. Sci. Rep. 7, 9848 (2017).
Kayesh, M. E. H., Sanada, T., Kohara, M. & Tsukiyama-Kohara, K. Tree shrew as an emerging small animal model for human viral infection: a recent overview. Viruses 13, 1641 (2021).
Perillo, B. et al. ROS in cancer therapy: the bright side of the moon. Exp. Mol. Med. 52, 192–203 (2020).
Tao, L. et al. Reactive oxygen species oxidize STING and suppress interferon production. Elife 9, e57837 (2020).
He, L. et al. Antioxidants Maintain Cellular Redox Homeostasis by Elimination of Reactive Oxygen Species. Cell Physiol. Biochem. 44, 532–553 (2017).
DiNicolantonio, J. J. & McCarty, M. Thrombotic complications of COVID-19 may reflect an upregulation of endothelial tissue factor expression that is contingent on activation of endosomal NADPH oxidase. Open Heart 7, e001337 (2020).
Mehta, P. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet 395, 1033–1034 (2020).
Xu, Z. et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 8, 420–422 (2020).
Huang, C. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 395, 497–506 (2020).
Saleh, M. G. et al. Ongoing oxidative stress in individuals with post-acute sequelae of COVID-19. NeuroImmune Pharm. Ther. 2, 89–94 (2023).
Olagnier, D. et al. Nrf2 negatively regulates STING indicating a link between antiviral sensing and metabolic reprogramming. Nat. Commun. 9, 3506 (2018).
Olagnier, D. et al. Author Correction: SARS-CoV2-mediated suppression of NRF2-signaling reveals potent antiviral and anti-inflammatory activity of 4-octyl-itaconate and dimethyl fumarate. Nat. Commun.11, 5419 (2020).
Zhang, S., Wang, J., Wang, L., Aliyari, S. & Cheng, G. SARS-CoV-2 virus NSP14 Impairs NRF2/HMOX1 activation by targeting Sirtuin 1. Cell Mol. Immunol. 19, 872–882 (2022).
De Angelis, M., Anichini, G., Palamara, A. T., Nencioni, L. & Gori Savellini, G. Dysregulation of intracellular redox homeostasis by the SARS-CoV-2 ORF6 protein. Virol. J. 20, 239 (2023).
De Angelis, M. et al. Influenza virusdDown-modulates G6PD expression and activity to induce oxidative stress and promote its replication. Front Cell Infect. Microbiol. 11, 804976 (2021).
Checconi, P. et al. Influenza virus replication is affected by glutaredoxin1-mediated protein deglutathionylation. FASEB J. 37, e22729 (2023).
Hendricks, K. S. et al. Endothelial NOX4 oxidase negatively regulates inflammation and improves morbidity during influenza a virus lung infection in mice. Front Cell Infect. Microbiol. 12, 883448 (2022).
Oda, T. et al. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 244, 974–976 (1989).
Liu, M. et al. The role of oxidative stress in influenza virus infection. Microbes Infect. 19, 580–586 (2017).
Chamberlain, N. et al. Lung epithelial protein disulfide isomerase A3 (PDIA3) plays an important role in influenza infection, inflammation, and airway mechanics. Redox Biol. 22, 101129 (2019).
Chamberlain, N. et al. Protein disulfide isomerase A3 regulates influenza neuraminidase activity and influenza burden in the lung. Int. J. Mol. Sci. 23, 1078 (2022).
James, S. J. et al. MAPK phosphatase 5 expression induced by influenza and other RNA virus infection negatively regulates IRF3 activation and type I interferon response. Cell Rep 10, 1722–1734 (2015).
Jacoby, D. B. & Choi, A. M. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic Biol. Med. 16, 821–824 (1994).
Knobil, K., Choi, A. M., Weigand, G. W. & Jacoby, D. B. Role of oxidants in influenza virus-induced gene expression. Am. J. Physiol. 274, L134–L142 (1998).
Imai, Y. et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 133, 235–249 (2008).
Peterhans, E. Reactive oxygen species and nitric oxide in viral diseases. Biol. Trace. Elem. Res. 56, 107–116 (1997).
Ivanov, A. V. et al. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 8, 3895–3932 (2017).
Valavanidis, A., Vlachogianni, T. & Fiotakis, C. 8-hydroxy-2’ -deoxyguanosine (8-OHdG): a critical biomarker of oxidative stress and carcinogenesis. J. Environ. Sci. Health C. Environ. Carcinog Ecotoxicol. Rev. 27, 120–139 (2009).
Jung, S. Y. & Kim, Y. J. C-terminal region of HBx is crucial for mitochondrial DNA damage. Cancer Lett. 331, 76–83 (2013).
Kim, Y. S., Seo, H. W. & Jung, G. Reactive oxygen species promote heat shock protein 90-mediated HBV capsid assembly. Biochem. Biophys. Res. Commun. 457, 328–333 (2015).
Smirnova, O. A. et al. Hepatitis delta virus antigens trigger oxidative stress, activate antioxidant Nrf2/ARE pathway, and induce unfolded protein response. Antioxidants 12, 974 (2023).
Yoon, H., Lee, H. K. & Jang, K. L. Hydrogen peroxide inhibits hepatitis B virus replication by downregulating HBx levels via siah-1-mediated proteasomal degradation in human hepatoma cells. Int. J. Mol. Sci. 24, 13354 (2023).
Ivanov, A. V. et al. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 6, e24957 (2011).
Pal, S. et al. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 25, 627–634 (2010).
Garcia-Mediavilla, M. V. et al. Differential contribution of hepatitis C virus NS5A and core proteins to the induction of oxidative and nitrosative stress in human hepatocyte-derived cells. J. Hepatol. 43, 606–613 (2005).
Vasallo, C. & Gastaminza, P. Cellular stress responses in hepatitis C virus infection: mastering a two-edged sword. Virus Res. 209, 100–117 (2015).
Zampino, R. et al. Chronic HCV infection and inflammation: Clinical impact on hepatic and extra-hepatic manifestations. World J. Hepatol. 5, 528–540 (2013).
Sanchez-Valle, V., Chavez-Tapia, N. C., Uribe, M. & Mendez-Sanchez, N. Role of oxidative stress and molecular changes in liver fibrosis: a review. Curr. Med. Chem. 19, 4850–4860 (2012).
Fierbinteanu-Braticevici, C. et al. Role of oxidative stress in the pathogenesis of chronic hepatitis C (CHC). Rom. J. Morphol. Embryol. 50, 407–412 (2009).
Higgs, M. R., Chouteau, P. & Lerat, H. Liver let die’: oxidative DNA damage and hepatotropic viruses. J. Gen. Virol. 95, 991–1004 (2014).
Choi, J., Corder, N. L., Koduru, B. & Wang, Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic. Biol. Med. 72, 267–284 (2014).
Ezzikouri, S. et al. Serum DHCR24 Auto-antibody as a new Biomarker for Progression of Hepatitis C. EBioMed. 2, 604–612 (2015).
Tardif, K. D., Waris, G. & Siddiqui, A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 13, 159–163 (2005).
Awady, B. A. M. E., Hussein, B., El-Seidi, E. & Abou-Hamed, Y. Viral load and oxidative stress in Egyptian chronic hepatitis C patients. Int. J. Infect. Dis. 53, 106-107 (2016).
Damar Cakirca, T., Ceylan, M. R., Koyuncu, I. & Cakirca, G. Thiol-disulphide balance and total oxidant-antioxidant status in patients with chronic hepatitis C. Int. J. Clin. Pract. 75, e13988 (2021).
Bal, T., Doğan, S., Özcan, O., Çabalak, M. & Çirkin, B. Direct-acting antiviral therapy may help restore HCV-induced impaired redox balance and liver fibrosis process. Tourkish J. Biochem. 48, 44–50 (2023).
Yoon, H. & Jang, K. L. Hydrogen peroxide inhibits hepatitis C virus replication by downregulating hepatitis C virus core levels through E6-associated protein-mediated proteasomal degradation. Cells 13, 62 (2023).
Medvedev, R. et al. HCV-induced oxidative stress by inhibition of Nrf2 triggers autophagy and favors release of viral particles. Free Radic. Biol. Med. 110, 300–315 (2017).
Chu, J. Y. K. & Ou, J. J. Autophagy in HCV replication and protein trafficking. Int. J. Mol. Sci. 22, 1089 (2021).
Tang, W. et al. Responses of nontransformed human hepatocytes to conditional expression of full-length hepatitis C virus open reading frame. Am. J. Pathol. 171, 1831–1846 (2007).
Walters, K. A. et al. Host-specific response to HCV infection in the chimeric SCID-beige/Alb-uPA mouse model: role of the innate antiviral immune response. PLoS Pathog 2, e59 (2006).
Lau, D. T., Luxon, B. A., Xiao, S. Y., Beard, M. R. & Lemon, S. M. Intrahepatic gene expression profiles and alpha-smooth muscle actin patterns in hepatitis C virus induced fibrosis. Hepatology 42, 273–281 (2005).
Gong, G. Z. et al. HCV NS5A abrogates p53 protein function by interfering with p53-DNA binding. World J. Gastroenterol.10, 2223–2227 (2004).
Fujita, N. et al. Comparison of hepatic oxidative DNA damage in patients with chronic hepatitis B and C. J. Viral. Hepatol. 15, 498–507 (2008).
Dionisio, N. et al. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 50, 872–882 (2009).
Ivanov, A. V. et al. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell Longev. 2016, 8910396 (2016).
Rajopadhye, S., Marita, A. R., Ansari, M., Chowdhary, A. & Dandekar, S. Assessment of oxidative stress parameters in HIV infection. BMC Infect. Dis. 14, 8910396 (2014).
Buckley, S. et al. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain Behav. Immun. Health 13, 100235 (2021).
Margaritis, M. Endothelial dysfunction in HIV infection: experimental and clinical evidence on the role of oxidative stress. Ann. Res. Hosp. 3, 185 (2019).
Isaguliants, M. et al. Oxidative stress induced by HIV-1 reverse transcriptase modulates the enzyme’s performance in gene immunization. Hum. Vaccin. Immunother. 9, 2111–2119 (2013).
Perl, A. & Banki, K. Genetic and metabolic control of the mitochondrial transmembrane potential and reactive oxygen intermediate production in HIV disease. Antioxid Redox Signal 2, 551–573 (2000).
Israel, N. & Gougerot-Pocidalo, M. A. Oxidative stress in human immunodeficiency virus infection. Cell Mol. Life Sci. 53, 864–870 (1997).
Liu, R. M. & Desai, L. P. Reciprocal regulation of TGF-beta and reactive oxygen species: a perverse cycle for fibrosis. Redox Biol. 6, 565–577 (2015).
Niwa-Kawakita, M. et al. PML is a ROS sensor activating p53 upon oxidative stress. J. Exp. Med. 214, 3197–3206 (2017).
Verma, S., Ali, A., Arora, S. & Banerjea, A. C. Inhibition of beta-TrcP-dependent ubiquitination of p53 by HIV-1 Vpu promotes p53-mediated apoptosis in human T cells. Blood 117, 6600–6607 (2011).
Allard, J. P., Aghdassi, E., Chau, J., Salit, I. & Walmsley, S. Oxidative stress and plasma antioxidant micronutrients in humans with HIV infection. Am. J. Clin. Nutr. 67, 143–147 (1998).
Deshmane, S. L. et al. Activation of the oxidative stress pathway by HIV-1 Vpr leads to induction of hypoxia-inducible factor 1alpha expression. J. Biol. Chem. 284, 11364–11373 (2009).
El-Amine, R. et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 15, 97–108 (2018).
Gravier-Hernandez, R. et al. Oxidative stress in hepatitis C virus-human immunodeficiency virus co-infected patients. Ann. Hepatol. 19, 92–98 (2020).
Baum, M. K. et al. Coinfection with hepatitis C virus, oxidative stress and antioxidant status in HIV-positive drug users in Miami. HIV Med. 12, 78–86 (2011).
Wang, J. et al. Inhibitory effect of glutathione on oxidative liver injury induced by dengue virus serotype 2 infections in mice. PLoS One 8, e55407 (2013).
Sahoo, B. R. et al. Redox regulation and metabolic dependency of zika virus replication: inhibition by Nrf2-antioxidant response and NAD(H) antimetabolites. J. Virol. 97, e0136322 (2023).
Denolly, S. et al. Zika virus remodelled ER membranes contain proviral factors involved in redox and methylation pathways. Nat. Commun. 14, 8045 (2023).
Lu, W. et al. Piperlongumine inhibits zika virus replication In vitro and promotes up-regulation of HO-1 expression, suggesting an implication of oxidative stress. Virol. Sin. 36, 510–520 (2021).
Mendonca-Vieira, L. R. et al. Reactive oxygen s/pecies (ROS) are not a key determinant for zika virus-induced apoptosis in SH-SY5Y neuroblastoma cells. Viruses 13, 2111 (2021).
Basu, M., Courtney, S. C. & Brinton, M. A. Arsenite-induced stress granule formation is inhibited by elevated levels of reduced glutathione in west Nile virus-infected cells. PLoS Pathog 13, e1006240 (2017).
Cheng, M. L. et al. Enteroviral 2B interacts with VDAC3 to regulate reactive oxygen species generation that is essential to viral replication. Viruses 14, 1717 (2022).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Ishikawa, H. & Barber, G. N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678 (2008).
Beck, M. A., Handy, J. & Levander, O. A. The role of oxidative stress in viral infections. Ann. N. Y. Acad. Sci. 917, 906–912 (2000).
Hu, B. et al. NF-kappaB and Keap1 interaction represses Nrf2-mediated antioxidant response in rabbit hemorrhagic disease virus infection. J. Virol. 94, e00016–20 (2020).
To, E. E. et al. Mitochondrial reactive oxygen species contribute to pathological inflammation during influenza A virus infection in mice. Antioxid Redox Signal 32, 929–942 (2020).
Singh, C. K. et al. The role of sirtuins in antioxidant and redox signaling. Antioxid. Redox. Signal 28, 643–661 (2018).
Alam, F. et al. Interplay between oxidative stress, SIRT1, reproductive and metabolic functions. Curr. Res. Physiol. 4, 119–124 (2021).
Cao, W. et al. SIRT2 mediates NADH-induced increases in Nrf2, GCL, and glutathione by modulating Akt phosphorylation in PC12 cells. FEBS Lett. 590, 2241–2255 (2016).
Mokhtari, V., Afsharian, P., Shahhoseini, M., Kalantar, S. M. & Moini, A. A review on various uses of N-acetyl cysteine. Cell J. 19, 11–17 (2017).
Zhang, R. H. et al. N-acetyl-l-cystine (NAC) protects against H9N2 swine influenza virus-induced acute lung injury. Int. Immunopharmacol. 22, 1–8 (2014).
Fraternale, A. et al. Antiviral and immunomodulatory properties of new pro-glutathione (GSH) molecules. Curr. Med. Chem. 13, 1749–1755 (2006).
Cai, J. et al. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 34, 928–936 (2003).
Labarrere, C. A. & Kassab, G. S. Glutathione deficiency in the pathogenesis of SARS-CoV-2 infection and its effects upon the host immune response in severe COVID-19 disease. Front Microbiol. 13, 979719 (2022).
Bartolini, D. et al. SARS-CoV2 infection impairs the metabolism and redox function of cellular glutathione. Redox. Biol.45, 102041 (2021).
Nadhan, R. et al. Perspectives on mechanistic implications of ROS inducers for targeting viral infections. Eur. J. Pharmacol. 890, 173621 (2021).
Srinivas, P., Gopinath, G., Banerji, A., Dinakar, A. & Srinivas, G. Plumbagin induces reactive oxygen species, which mediate apoptosis in human cervical cancer cells. Mol. Carcinog. 40, 201–211 (2004).
Li, H., Bai, Z., Li, C., Sheng, C. & Zhao, X. EV71 infection induces cell apoptosis through ROS generation and SIRT1 activation. J. Cell Biochem. 121, 4321–4331 (2020).
Li, X. et al. Precise modulation and use of reactive oxygen species for immunotherapy. Sci. Adv. 10, eadl0479 (2024).
Reshi, M. L., Su, Y. C. & Hong, J. R. RNA viruses: ROS-mediated cell death. Int. J. Cell Biol. 2014, 467452 (2014).
Meuren, L. M. et al. Infection of endothelial cells by dengue virus induces ROS production by different sources affecting virus replication, cellular activation, death and vascular permeability. Front Immunol. 13, 810376 (2022).
Hu, M., Bogoyevitch, M. A. & Jans, D. A. Subversion of host cell mitochondria by RSV to favor virus production is dependent on inhibition of mitochondrial complex I and ROS generation. Cells 8, 1417 (2019).
Khan, N. A. et al. Oxidative stress specifically inhibits replication of dengue virus. J. Gen. Virol. 102, 10.1099/jgv.0.001596 (2021).
Violi, F. et al. Nox2 activation in covid-19. Redox Biol. 36, 101655 (2020).
Gao, H. et al. African swine fever virus enhances viral replication by increasing intracellular reduced glutathione levels, which suppresses stress granule formation. Vet. Res. 55, 172 (2024).
Garaci, E. et al. Intracellular GSH content and HIV replication in human macrophages. J. Leukoc. Biol. 62, 54–59 (1997).
Vlahos, R. et al. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog 7, e1001271 (2011).
Chen, K. K. et al. Redox control in the pathophysiology of influenza virus infection. BMC Microbiol. 20, 214 (2020).
Amatore, D. et al. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell Microbiol. 17, 131–145 (2015).
Gil, L. et al. Contribution to characterization of oxidative stress in HIV/AIDS patients. Pharmacol. Res. 47, 217–224 (2003).
Allard, J. P. et al. Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects. AIDS 12, 1653–1659 (1998).
Couret, J. & Chang, T. L. Reactive oxygen species in HIV infection. EC Microbiol. 3, 597–604 (2016).
Acknowledgements
This study was supported by grants from the Tokyo Metropolitan Government and the Japan Agency for Medical Research and Development.
Author information
Authors and Affiliations
Contributions
Conceptualization: M.E.H.K., M.K., and K.T-K.; Writing—Original draft preparation: M.E.H.K. and K.T.-K.; Writing—Review and editing: M.E.H.K., M.K., and K.T.-K. All the authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Kayesh, M.E.H., Kohara, M. & Tsukiyama-Kohara, K. Effects of oxidative stress on viral infections: an overview. npj Viruses 3, 27 (2025). https://doi.org/10.1038/s44298-025-00110-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s44298-025-00110-3
