
Abstract
Background:
Glyoxalase I (GI) is a cellular defence enzyme involved in the detoxification of methylglyoxal (MG), a cytotoxic byproduct of glycolysis, and MG-derived advanced glycation end products (AGEs). Argpyrimidine (AP), one of the major AGEs coming from MG modifications of proteins arginines, is a pro-apoptotic agent. Radiotherapy is an important modality widely used in cancer treatment. Exposure of cells to ionising radiation (IR) results in a number of complex biological responses, including apoptosis. The present study was aimed at investigating whether, and through which mechanism, GI was involved in IR-induced apoptosis.
Methods:
Apoptosis, by TUNEL assay, transcript and protein levels or enzymatic activity, by RT–PCR, western blot and spectrophotometric methods, respectively, were evaluated in irradiated MCF-7 breast cancer cells, also in experiments with appropriate inhibitors or using small interfering RNA.
Results:
Ionising radiation induced a dramatic reactive oxygen species (ROS)-mediated inhibition of GI, leading to AP-modified Hsp27 protein accumulation that, in a mechanism involving p53 and NF-κB, triggered an apoptotic mitochondrial pathway. Inhibition of GI occurred at both functional and transcriptional levels, the latter occurring via ERK1/2 MAPK and ERα modulation.
Conclusions:
Glyoxalase I is involved in the IR-induced MCF-7 cell mitochondrial apoptotic pathway via a novel mechanism involving Hsp27, p53 and NF-κB.
Similar content being viewed by others

Main
Glyoxalase I (GI) is a key enzyme in the pathway leading to glutathione (GSH)-mediated detoxification of the side product of glycolysis methylglyoxal (MG) that, reacting with DNA and proteins, forms stable products called advanced glycation end products (AGEs) (Rabbani and Thornalley, 2012). Among them, argpyrimidine (AP) represents one of the major products deriving from MG modifications of protein arginine residues (Kim et al, 2012a), recently shown to induce apoptosis to cultured cells (Kim et al, 2012b; Antognelli et al, 2013). Radiotherapy (RT) is an important modality widely used in cancer treatment, including breast cancer (BC). In early-stage breast cancer, whole breast RT is usually applied after conserving surgery; nevertheless, most tumour recurrences occur near the tumour bed, thus indicating that tolerable doses of external RT are often insufficient to achieve control of locally advanced BC. Therefore, intraoperative RT (IORT), a procedure in which a high dose of IR is precisely applied to the tumour bed during surgery (Calvo et al, 2013), has been clinically and epidemiologically suggested as a valid alternative to whole breast RT. However, in vitro research in such a field has been scarcely performed. In such a therapeutic tool ambit (IORT), the Italian intraoperative radiotherapy with electrons (ELIOT) trial appeared a promising feature in early BC, treated with breast-conserving surgery (Veronesi et al, 2010).
Exposure of cells to ionising radiation (IR) results in a number of complex biological responses, including programmed cell death (Chen et al, 2013). Radiation-induced apoptosis is a phenomenon dependent on cell type, dose and time after irradiation (Urashima et al, 2006), and can occur through different mechanisms (Golden et al, 2012; Kwon et al, 2013). Certainly, reactive oxygen species (ROS) are crucial mediators of the apoptosis by IR in a variety of cells (Morales et al, 1998; Van Laethem et al, 2006). Reactive oxygen species are very transient species owing to their high reactivity that gradually leads to impose oxidative damage on biomolecules ultimately compromising cell viability, leading to activation of the programmed cell death machinery (Sinha et al, 2013). In particular, ROS are known to damage cells by peroxidising lipids, disrupting DNA and oxidising amino acids, thus inactivating specific enzymes directly or through oxidation of cofactors (Bolisetty and Jaimes, 2013). Moreover, they act as important signalling molecules regulating many signal-transduction pathways and modulating gene transcription (Forman and Torres, 2001). The ROS-induced apoptosis can occur through a wide variety of mechanisms and signalling pathway often depending on the considered cell model and different cell contexts (Soga et al, 2012; Zhang et al, 2012; Zhu et al, 2012). Biological systems have evolved an effective and complicated network of defence mechanisms that enable cells to cope with lethal oxidative environments. These defence mechanisms involve the tripeptide GSH and some antioxidant enzymes, as superoxide dismutase (SOD) that catalyses the dismutation of O2.– to H2O2 and O2 (McCord and Fridovich, 1969), catalase and peroxidases that remove hydrogen peroxide and hydroperoxides (Chance et al, 1979).
The present study was aimed at investigating whether, and through which molecular mechanism, GI was involved in IR-induced apoptosis, possibly by the involvement of ROS, in human MCF-7 oestrogen-responsive early-stage BC cell line, exposed to a single dose of 21 Gy, as in the ELIOT trial (Veronesi et al, 2010). We demonstrated that IR-induced ROS-mediated GI inhibition, leading to AP accumulation, triggered a mitochondrial apoptotic pathway via a novel mechanism involving heat shock protein 27 (Hsp27), p53 and nuclear factor-κB (NF-κB), and that GI inhibition occurred at both functional and transcriptional levels, the latter via p44/42 mitogen-activated protein kinase (MAPK; extracellular signal-regulated kinase 1/2 (ERK1/2)) and oestrogen receptor-α (ERα) involvement.
Materials and Methods
Irradiation
The same dose used in the partial breast irradiation ELIOT trial, 21 Gy (Veronesi et al, 2010), was administered at a dose rate of 200 cGy min−1 at room temperature using 6 MV photons produced by a linear accelerator (Clinac DBX, Varian Medical Systems, Palo Alto, CA, USA). A 5-mm-thick plexiglass spoiler was placed on the flasks or plates.
Cell culture and exposure
The MCF-7 human breast cancer cell line was obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA) and cultured as per the manufacturer’s recommendations at 37 °C and 5% CO2. Analyses were performed immediately (time T0) and at 0.5, 24, 48 or 72 h post irradiation. During these latter periods, cells were incubated at 37 °C. As no significant biological effects were observed at T0 time compared with unexposed cells, serving as control, results were reported only for all the rest of the time course. Untreated cells were incubated for the same time periods. Independent experiments were also performed by pretreating cells with N-acetyl-L-cysteine (NAC, 10 mM, for 0.5 h), a known antioxidant that increases cellular GSH levels, aminoguanidine bicarbonate (AG, 1 mM, for 2 h), a scavenger of free MG (Fleming et al, 2013) and MG-derived AGEs (Thornalley et al, 2000), the specific wild-type p53 inhibitor, pifithrin-α (PFT-α, 20 μ M in DMSO, for 2 h), the specific NF-κB inhibitor BAY 11-7082 (10 μ M in DMSO, for 0.5 h, Santa Cruz Biotechnology, Inc., Heidelberg, Germany), the ERα anti-oestrogen ICI 182,780 (100 nM in DMSO, for 4 h), ERK-1/2 inhibitor U-0126 (10 μ M in DMSO, for 1 h), and then irradiated in the conditions above described. Unless otherwise stated, these reagents were from Sigma-Aldrich (Milan, Italy) and used at concentrations producing no significant toxicity to MCF-7 cells. Control cells for the experiments with DMSO-dissolved agents did not show any significant difference with respect to control cells in RPMI-1640 medium, and therefore all the relative treatments were compared with these latter controls.
Cell viability assays
Cell viability was evaluated by assessment of reduction of 3-(4,5-dimethylthiazol-2-yl)2,5-diphenyl-tetrazolium bromide (MTT) (Kanagasabai et al, 2010) or Cell Counting Kit-8 (CCK8) (Yi et al, 2013) and lactate dehydrogenase (LDH) protein amount in cell culture medium by LDH ELISA kit (Antibodies-online, GmbH, Aachen, Germany). As to MTT and CCK8 assays, absorbance of cell survival was calculated relative to control cells that were set to 100%; for LDH protein amount in cell culture medium, basic values were provided.
Apoptosis detection
Apoptosis was detected by evaluating DNA fragmentation by TdT-mediated dUTP nick-end labelling (TUNEL) assay (ApoAlert DNA Fragmentation Assay, Clontech Laboratories, Inc., Santa Clara, CA, USA) and agarose gel electrophoresis (Antognelli et al, 2013).
Measurement of oxidative stress: ROS, GSH, malondialdheyde (MDA) and enzymatic antioxidant defences ROS detection
Intracellular ROS production was detected using 2′-7′-dichlorodihydrofluorescein diacetate (DCFH-DA) (Sigma-Aldrich) or dihydroethidium (DHE, Sigma-Aldrich) (Park et al, 2011).
GSH assay
Control and irradiated cells were suspended in 50 mM phosphate buffer containing 1 mM EDTA, pH 6.8, at the concentration of 107 cells per ml. Suspensions were then homogenised and centrifuged at 10 000 g for 15 min at 4 °C. Supernatants were collected, deproteinised with 2.5% metaphosphoric acid in a 1 : 1 ratio (v/v) and centrifuged at 2000 g for 5 min. The resulting extracts were used for glutathione assay. Reduced and oxidised glutathione intracellular concentrations were measured using Cayman’s GSH Assay kit (Cayman Chemical, Ann Arbor, MI, USA) according to the manufacturer’s instructions.
MDA detection
MDA was measured using OxiSelect TBARS Assay Kit (MDA Quantitation) (Cell Biolabs, San Diego, CA, USA).
Enzymatic activity assays for antioxidant defences
Control cells and IR-exposed cells were harvested and suspended at the concentration of 107 cells per ml in 10 mM phosphate buffer, pH 7.0, containing 1 mM dithiothreitol (for GI and GSH-Px) or 1 mM Triton X-100 (for CuZn-SOD) and 0.1 mM phenylmethanesulphonylfluoride (PMSF). Cell suspensions were then homogenised with a Potter-Helvehjem homogeniser, centrifuged at 13 000 g for 30 min and the resulting cell extracts used for enzymatic activities and protein content measurements. The GI activity was assayed according to Mannervik et al (1981, 297–301). The assay solution contained 0.1 M sodium-phosphate buffer, pH 7.2, 2 mM MG and 1 mM GSH. The reaction was monitored spectrophotometrically by following the increase of absorbance at 240 nm and 25 °C. One unit activity is defined as 1 μmol of S-D-lactoylglutathione produced per min. The Se-dependent GSH-Px activity was measured according to Paglia and Valentine (1967). The oxidation of NADPH was followed spectrophotometrically at 340 nm and 25 °C. One unit activity was defined as 1 μmol NADPH oxidised per min. Catalase activity was measured spectrophotometrically at 240 nm at 25 °C, according to Aebi (1974, 673–684), by following the rate of reduction of H2O2 (10 mM). One unit activity is defined as 1 μmol H2O2 reduced per min. The CuZn-SOD activity was assayed spectrophotometrically at 480 nm and 25 °C by its capability to inhibit the autoxidation of adrenaline according to Sun and Zigman (1978). One unit activity is defined as the amount of enzyme required to halve the rate of substrate oxidation.
Knockdown of GI with specific siRNA
Subconfluent MCF-7 cells were transiently transfected with a 100 nmol pool of four siRNAs targeting GI (si-GI) and nonspecific siRNA (si-C) (Dharmacon, Milan, Italy), using Lipofectamine 2000 (Invitrogen, Milan, Italy). After culturing for 72 h (maximum silencing effect in preliminary experiments), the culture medium was removed and cells were added with a fresh one containing new transfection complexes and then irradiated. The effect of silencing was evaluated analysing the specific protein knockdown by western blot using the specific Ab, and β-actin as loading control, and enzymatic activity, either 72 h before or after irradiation. In addition, a mock transfection was performed with all vehicles used except for the siRNA. In all transfection experiments, mock transfection and transfection with si-C did not affect all the studied parameters, providing results comparable with those obtained from untreated cells (data not shown).
Whole-cell protein extraction and western blot
For extraction of total proteins, a total of 106 cells were lysed in precooled radioimmunoprecipitation assay (RIPA) lysis buffer (Fettucciari et al, 2006). For subcellular fractionation, cells were resuspended in Mitobuffer (Fettucciari et al, 2006). For western blot, samples of equal protein concentration (40 μg) were treated with Laemmli buffer (Invitrogen), boiled for 5 min, resolved on 10, 12 or 15% SDS–PAGE and then blotted onto a nitrocellulose membrane using iBlot Dry Blotting System (Invitrogen). Nonspecific binding sites were blocked in Roti-Block (Roth, Bavaria, Germany) for 1 h at room temperature, incubated overnight at 4 °C with an appropriate dilution of the primary specific Abs (mouse anti-GI mAb, BioMac, GmbH, Leipzig, Germany; mouse anti-AP mAb, Antibodies-online, GmbH, Aachen, Germany; rabbit anti-Bcl-XL polyclonal Ab, rabbit anti-Bax (N20) polyclonal Ab, Santa Cruz Biotechnology; rabbit anti-phospho-I-κ-B-α (Ser32) (14D4), anti-I-κ-B-α (44D4) mAbs, phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) rabbit mAb, phospho-oestrogen receptor α (Ser118) (16J4) mouse mAb, oestrogen receptor α (D8H8) rabbit mAb, caspase-7 (D2Q3L) rabbit mAb, Cell Signaling Technology, Milan, Italy; mouse anti-Bcl-2 mAb, DAKO, Milan, Italy; mouse anti-cytochrome c (Cyt c) mAb, BD Pharmingen, Milan, Italy; mouse anti-Cyt c oxidase subunit IV (Cox IV) mAb, Molecular Probes, Monza, Italy). After washing with TBST, antigen–antibody complexes were detected by incubation of the membranes for 1 h at room temperature with the appropriated HRP-conjugated secondary Ab and revealed using ECL system (Amersham Pharmacia, Milan, Italy). As internal loading controls, all membranes were subsequently stripped of the first Ab in a stripping buffer (100 mM 2-ME, 2% SDS and 62.5 mM Tris-HCl, pH 6.8) and reprobed with anti-β-actin mAb (mouse anti-β-actin mAb, Santa Cruz Biotechnology).
Protein assay
Protein concentration was determined by BCA kit (Pierce, Rockford, IL, USA) (Shibuya et al, 1989), using bovine serum albumin as a standard.
Affinity purification of the 27 kDa AGE AP and amino acid sequence analysis
Purification and identification of AGE AP was performed according to Sakamoto et al (2002).
RNA isolation and cDNA synthesis
Total cellular RNA was isolated using TRIzol Reagent (Invitrogen). The cDNA was then synthesised from 1 μg of RNA using the RevertAid H Minus First Strand cDNA Synthesis Kit (M-Medical, Milan, Italy).
Real-time TaqMan PCR analysis
Glyoxalase I expression vs β-actin was evaluated by quantitative real-time TaqMan PCR analysis (qRT–PCR) on a MX3000P Real-Time PCR System (Agilent Technology). The sequences of oligonucleotide primers and TaqMan probes used for qRT–PCR were as follows: GI: 5′-CTCTCCAGAAAAGCTACACTTTGAG-3′ (sense, 400 nM), 5′-CGAGGGTCTGAATTGCCATTG-3′ (antisense, 400 nM), 5′-FAM-TGGGTCGCATCATCTTCAGTGCCC-TAMRA-3′ (TaqMan Probe, 200 nM); β-actin: 5′-CACTCTTCCAGCCTTCCTTCC-3′ (sense, 600 nM), 5′-ACAGCACTGTGTTGGCGTAC-3′ (antisense, 600 nM), 5′-TEXASRED-TGCGGATGTCCACGTCACACTTCA-BHQ-3′ (TaqMan Probe, 200 nM). All PCR primers and probes were designed using Beacon Designer 4 software (Stratagene, Santa Clara, CA, USA), starting from published sequence data supplied by the NCBI database. The PCR reactions were performed in a total volume of 25 μl, containing 250 ng of cDNA, 1 × Brilliant QPCR master mix (Agilent Technology), 0.5 μl of ROX Reference Dye (Agilent Technology) and a concentration of specific primers and probes. The thermal cycling conditions were as follows: 1 cycle at 95 °C for 10 min, followed by 45 cycles at 95 °C for 20 s and 55 °C for 1 min. Data for comparative analysis of gene expression were obtained using the ΔΔCt method (Livak and Schmittgen, 2001).
Statistical analysis
Results were expressed as the percentage of irradiated cells (control) and are representative of at least four independent experiments performed in quadruplicate. The data were expressed as mean±s.d. and compared by Student’s t-test. The null hypothesis was rejected at the 5% significance level (P<0.05).
Results
IR-induced cytotoxicity occurs via apoptosis in MCF-7 cells
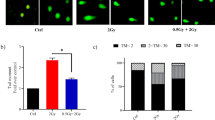
To study IR-induced cytotoxicity, viability was examined at various post-irradiation time points in MCF-7 cells exposed to a single dose (21 Gy) of IR. We found that IR treatment resulted in a significant time-dependent decrease of cell viability since 24 h post irradiation, compared with nonirradiated control cells, as shown by the decrease in viable cell percentage by MTT or CCK8 tests and increase in LDH protein release into the medium (Figure 1A). It is known that IR-induced cell death has been identified in many cases as apoptosis (Urashima et al, 2006; Chen et al, 2013; Kwon et al, 2013). Therefore, to investigate the mechanism of cell death induced by IR in our study, apoptosis was investigated by DNA fragmentation assays. The TUNEL assay showed that the percentage of apoptotic cells measured after IR exposure, calculated by the ratio between the values of green fluorescence (apoptotic cells) and red fluorescence (total amount of cells) by flow cytometry (FITC or PI channel), significantly increased in a time-dependent manner compared with untreated cells (Figure 1B). The DNA fragmentation into oligonucleosomes confirmed the apoptotic responses as evidenced by the typical DNA laddering (Figure 1C).

Effect of irradiation on MCF-7 cell viability and apoptosis. Cell viability (A), measured by MTT- or CCK8-based assays and LDH leakage, and apoptosis (B), measured by TUNEL assay or DNA fragmentation electrophoresis (C), were measured in nonexposed or irradiated cells at the different indicated post-irradiation time points. Results are presented as percentage of control. Basic values for LDH amount of enzyme in cell culture supernatant were provided. Data report the means of four separate experiments performed in quadruplicate and error bars represent the s.d. of the mean. Electrophoresis is representative of three independent experiments providing the same result. ***P<0.001.
Effect of IR on GI and the AGE AP
We recently reported that GI can trigger apoptosis through the control of MG-derived AP intracellular levels in human prostate cancer cell lines (Antognelli et al, 2013). To reveal the potential involvement of GI in the apoptotic cell death caused by treatment with IR in MCF-7 cells, we first studied the levels of GI enzymatic activity by spectrophotometric method. A significant inhibitory time-dependent effect was observed for GI after IR exposure, compared with control cells (Figure 2A). Mean values of GI activity, expressed as mmol min−1 per mg protein were (mean±s.d.) 0.75±0.07 for control cells and 0.51±0.04, 0.41±0.01, 0.30±0.02 or 0.17±0.01 for cells post irradiation. As the biological activity of a protein can be regulated at post-translational level, changes in protein functional level might not be necessarily reflected by similar changes in protein abundance. We, therefore, measured GI protein level in MCF-7 cells after IR treatment by immunoblotting detection. Western blot revealed an equally pronounced decrease of GI at such a level compared with untreated cells (Figure 2B). Next, to determine whether the decrease in GI protein was paralleled by a similar trend at GI transcript level, GI mRNA was examined. The qRT–PCR revealed a significant downregulation of GI mRNA levels occurring in a time-dependent manner after irradiation compared with unirradiated cells (Figure 2C). We then examined the hypothesis that IR-induced GI inhibition was paralleled by increased GI-dependent MG-derived AP intracellular levels. To this end, immunodetection of AP, the AGE derived from spontaneous MG adduction of arginine residues (Kim et al, 2012a), was performed in total protein extracts from both control and irradiated MCF-7 cells. Western blot analysis, using a monoclonal antibody directed against AP, identified a single band of 27 kDa molecular weight. Densitometry analysis revealed that AP cellular levels were significantly increased in IR-treated as compared with control cells (Figure 2D).

Glyoxalase I (GI) expression status and argpyrimidine (AP) levels in MCF-7 cells after irradiation. Histograms show (A) GI enzymatic-specific activity, measured by spectrophotometric methods, (B) GI gene expression at protein level, assessed by densitometric analysis from western blot detection, (C) mRNA level by qRT–PCR and (D) AP intracellular levels assessed by densitometric analysis from western blot detection. Western blots were obtained by using the appropriate mAbs. The blots were stripped of the bound Abs and reprobed with anti-β-actin to confirm equal loading. The western blots shown are representative of three separate experiments. All histograms indicate means±s.d. of three different cultures, and each one was tested in quadruplicate and expressed as percentage of unirradiated cells (control). **P<0.01 and ***P<0.001.
IR induces oxidative stress in MCF-7 cells
It is known that the cytotoxic effects of radiation are mediated primarily through increased formation of hydroxyl radicals and ROS that, in turn, can damage proteins and trigger apoptosis in a variety of cells (Morales et al, 1998; Van Laethem et al, 2006; Na et al, 2012; Soga et al, 2012; Zhu et al, 2012; Zhang et al, 2012; Sinha et al, 2013; Zhang et al, 2013). Therefore, to investigate whether the observed IR-induced GI inhibition, paralleled by induction of apoptosis, was associated with ROS formation, the levels of intracellular H2O2 were initially measured with DCF staining. Compared with control, irradiation caused a significant increase in H2O2 levels starting from 0.5 h post irradiation (Figure 3A). We further confirmed IR-induced ROS generation using another ROS detection dye, DHE for O2.–. Similarly, we found a marked increase in O2.– generation after treatment with IR as compared with untreated cells (Figure 3A). As known, ROS production may result from impairment in the function of the defence mechanisms that enable cells to cope with lethal oxidative environment. Therefore, we analysed the level of GSH as well as some of the major cellular antioxidant enzymes, such as SOD, that catalyse the dismutation of O2− to H2O2 and O2 (McCord and Fridovich, 1969), catalase and GSH-Px that remove H2O2 (Chance et al, 1979), finding that treatment with IR resulted in a significant inhibition of all the studied enzymes with respect to control cells (Figure 3B). As indicative marker of oxidative damage to cells, the occurrence of lipid peroxidation was also evaluated. It is well established that oxidative stress in various cells usually leads to accumulation of potent, cytotoxic lipid peroxides such as MDA (Esterbauer et al, 1991). Exposure to IR significantly increased MDA content compared with unirradiated cells (Figure 3C).

Induction of oxidative stress in irradiated MCF-7 cells. Oxidative stress was evaluated by the detection of (A) ROS, (B) endogenous antioxidant defences (GSH, SOD, catalase, GSH-Px), and (C) lipid peroxidation by MDA cell content. Results are presented as percentage of unirradiated cells (control). Data report the means of four separate experiments performed in quadruplicate and error bars represent the s.d. of the mean. *P<0.05, **P<0.01 and ***P or °P<0.001.
IR-induced apoptosis occurs via ROS-mediated inhibition of GI enzyme and AP accumulation in MCF-7 cells
To determine whether the increase in intracellular ROS levels was directly related to the observed IR-triggered apoptotic cell death and concomitant GI inhibition/AP accumulation, we pretreated MCF-7 cells with the antioxidant NAC before exposing them to IR. A total of 10 mM NAC added to cells 0.5 h before irradiation significantly reduced apoptotic cell death (Figure 4A) and reverted GI inhibition (Figures 4B–D) or AP intracellular content (Figure 4E), caused by IR, to the level of control cells. The NAC did not significantly affect the other studied enzymes playing a protective role against ROS, thus indicating a specific effect to GI (data not shown). The role of GI in triggering AP accumulation and apoptosis was explored by knocking down GI via siRNA methods. We first tested the efficiency of GI siRNA. As shown in Figure 4F MCF-7 cells transfected with GI siRNA showed inhibition of GI protein expression and enzymatic activity, either 72 h before or after irradiation, whereas the control siRNA had no effect. We found that blockade of GI by siRNA additionally increased AP accumulation (Figure 4G) and apoptosis (Figure 4H) but not ROS (Figure 4I), confirming that ROS are upstream signal of GI/AP axis in the observed apoptosis induction by IR. Pretreatment with AG demonstrated the effect of such MG scavenger in directly triggering the apoptosis mediated by ROS (Figure 4J). Aminoguanidine showed no effects on ROS and GI expression level (data not shown), again confirming the proposed mechanism by which IR induces apoptosis in MCF-7 via AP accumulation due to ROS-mediated GI inhibition.

Ionising radiation-induced apoptosis occurs via ROS-mediated inhibition of GI and AP accumulation in MCF-7 cells. Histograms show the effect of 10 mM NAC pretreatment for 0.5 h on (A) MCF-7 cell apoptosis, evaluated by TUNEL assay, (B) GI enzymatic-specific activity, measured by spectrophotometric method, (C) GI mRNA level by quantitative real-time RT–PCR, (D) GI protein and (E) AP levels, assessed by western blot detection. The GI knockdown via small interfering RNA (si-GI) (F) proved GI involvement in triggering (G) AP accumulation and (H) apoptosis, but not ROS content (I). Pretreatment with AG (J) proved AP role in MCF-7 cell apoptosis. The effects of si-GI or AG were evaluated at 72 h post irradiation when their maximum effect was observed in preliminary experiments. Western blots were obtained by using the appropriate mAbs. The blots were stripped of the bound Abs and reprobed with anti-β-actin to confirm equal loading. The western blots shown are representative of three separate experiments. All histograms indicate means±s.d. of three different cultures, and each one was tested in quadruplicate and expressed as percentage of control. (−) Untreated and (+) treated cells; si-C: nonspecific siRNA; *P<0.05, **P<0.01 and ***P<0.001.
IR-induced ROS-mediated GI inhibition, leading to argpyrimidine accumulation, triggers a mitochondrial apoptotic pathway involving NF-κB
It is known that AGEs can act as pro-apoptotic agents (Kim et al, 2012b; Antognelli et al, 2013) by downregulating the anti-apoptotic protein Bcl-2 (Honda et al, 2001; Yamagishi et al, 2002) and upregulating the pro-apoptotic Bax protein (Alikhani et al, 2005) in different cellular models, and that the specific AGE AP drives a mitochondrial apoptotic pathway in prostate cancer cell lines (Antognelli et al, 2013). As our results showed an IR-induced GI inhibition-dependent increase in AP levels, we next investigated whether the observed IR-driven apoptosis occurred via a mitochondrial pathway also in caspase-3-deficient (Santha et al, 2013; Charalambous et al, 2013) MCF-7 cells. To this aim, the levels of the anti-apoptotic Bcl-2 or Bcl-XL or the pro-apoptotic Bax proteins, Cyt c release into the cytosol and the activation of the final executioner caspase-7 (Santha et al, 2013; Charalambous et al, 2013), occurring in a mitochondrial apoptotic pathway (Kaufmann and Hengartner 2001), were analysed. As shown in Figure 5A, we found a significant decrease in the levels of the anti-apoptotic Bcl-2 or Bcl-XL proteins paralleled by a marked increase in the levels of the pro-apoptotic Bax protein in irradiated cells compared with controls. Concomitantly, Cyt c release into the cytosol, as well as the activation of caspase-7, were observed (Figure 5B). We previously found that AP-driven mitochondrial apoptotic pathway occurred via NF-κB downregulation in prostate cancer cell lines (Antognelli et al, 2013). As known, NF-κB is a family of transcription factors present in the cytosol in an inactive state complexed with the inhibitory IκB proteins. Activation occurs via phosphorylation of IκBα at Ser32 and Ser36 followed by proteasome-mediated degradation that results in the release and nuclear translocation of active NF-κB (Baldwin, 2001). It has been described that NF-κB is able to upregulate Bcl-2 and Bcl-XL antiapoptotic genes (Baeuerle and Baltimore, 1988; Gasparian et al, 2002; Suh et al, 2002; Shukla and Gupta, 2004) and is constitutively activated in various human tumours (Bondong et al, 2012; Kim et al, 2012; Ranuncolo et al, 2012) and cancer cell lines (Lindholm et al, 2000; Ludwig et al, 2001; Hwang et al, 2012), including MCF-7 cells (Sovak et al, 1997). Therefore, we hypothesised that AP accumulation following IR-induced ROS-mediated GI inhibition might involve NF-κB signalling pathway via NF-κB desensitisation. In fact, a marked reduction in NF-κB activation was observed in irradiated compared with control MCF-7 cells, as indicated by the decrease in serine 32-phosphorylated IκBα and the increase in total IκBα levels (Figure 5C). The use of the monoclonal antibody that detects endogenous levels of serine 32-phosphorylated IκBα is an excellent marker of NF-κB activation (Baeuerle and Baltimore, 1988), as phosphorylation of IκBα at Ser32 is essential for the release of active NF-κB. The effect of IR was completely abolished by pretreatment with MG-scavenging agent AG, proving the direct involvement of AP in the intrinsic NF-κB-mediated apoptotic pathway (Figures 5A–C). Pretreatment with NAC reverted NF-κB-mediated mitochondrial apoptotic activation pattern induced by IR (data not shown).

Argpyrimidine accumulation triggers a mitochondrial apoptotic pathway involving NF- κ B. (A) Anti-apoptotic proteins Bcl-2 (26 kDa) and Bcl-XL (28 kDa), pro-apoptotic protein Bax (21 kDa), (B) Cyt c (13 kDa) in the cytosolic and mitochondrial subfractions, caspase-7 (intact protein, 35 kDa, solid arrow; active fragment, 20 kDa, open arrow) and (C) phospho-IκBα (40 kDa) or total IκBα (39 kDa) protein expression in unirradiated cells or in 24, 48 and 72 h post-irradiation cells. Aminoguanidine treatments were evaluated at 72 h post irradiation when its maximum effect was observed in preliminary experiments. No significant differences in the analysed proteins were observed in cells 0.5 h post irradiation compared with control cells (data not shown). Whole-cell lysates were subjected to SDS–PAGE and probed with the appropriate Abs. Western blot analysis of β-actin (43 kDa) or Cox IV (17 kDa) expression is provided to show equal loading of the samples. The Cox IV in the cytosolic subfraction was used to check for mitochondrial contamination. Serine 32-phosphorylated IκBα mAb was used as a marker of NF-κB activation. Blots in this figure are representative of three different experiments done independently that gave the same results. (−) Untreated and (+) treated cells.
Involvement of Hsp27 and p53 in the IR-induced ROS-mediated mitochondrial apoptotic pathway
To investigate the mechanism by which AP drove the observed mitochondrium-dependent apoptosis, we identified the detected 27 kDa AP protein in cells 72 h post irradiation, where the maximum AP band intensity was observed. To this aim the 27 kDa AP protein was purified by immunoaffinity chromatography using the anti-AP mAb. Single column fractions, containing the eluted proteins, were individually subjected to SDS–PAGE followed by Coomassie Blue staining. As shown in Figure 6A, one column fraction presented a very marked stained band, containing a 27-kDa polypeptide. Other protein bands were also present in the other fractions of the immunoaffinity elution, showing, however, a by far less staining (data not shown). To determine the identity of the 27 kDa protein, it was digested and resolved as individual peptides by HPLC. As a result, the internal peptides identified with human Hsp27 upon comparison with standard sequencing databases in the public domain (BLAST) (Figure 6B). The Hsp27 is known to accelerate proteasomal degradation of p53 in cancer cells (O'Callaghan-Sunol et al, 2007) and, as demonstrated in a recent research, a negative correlation between Hsp27 and p53 in UV-irradiated MCF-7 cells exists, in a way that Hsp27 potentiates the degradation of p53 post irradiation (Kanagasabai et al, 2010). Moreover, in the same MCF-7 cell line, an inverse correlation has been recently shown between Hsp27, p53 and NF-κB; in particular, the absence of Hsp27 results in accumulation of p53 that, in turn, triggers the NF-κB-dependent signalling pathway (Kanagasabai et al, 2011). Therefore, we assumed that after irradiation, the observed increase in AP-modified Hsp27 protein, along with the concomitant reduction in NF-κB activation, might be paralleled by a decrease in p53 levels. Indeed, we found significantly reduced p53 protein levels starting from 48 h and peaking at 72 h post irradiation compared with unirradiated cells (Figure 6C). Pretreatment with AG completely reversed IR-induced p53 downregulation, indicating a direct action by AP-modified Hsp27 on p53 modulation (Figure 6C). The role of p53 in inducing NF-κB desensitisation was verified via a pharmacological approach by using a specific wild-type p53 inhibitor, PFT-α. Pifithrin-α is a small molecule that binds to the DNA-binding domain of p53, thereby inhibiting its transcriptional activity (Wang and Sun, 2010). Western blot analysis revealed that pretreatment with PFT-α significantly potentiated IR-induced NF-κB desensitisation as shown by p-IkBα and IκBα expression level that resulted undetectable or enhanced, respectively (Figure 6D). In parallel, pretreatment with PFT-α significantly increased the number of apoptotic cells (Figure 6E) but did not affect AP levels (data not shown). Finally, to prove the involvement of NF-κB in triggering IR-induced apoptosis, the specific NF-κB inhibitor BAY 11-7082 that diminishes the activation of NF-κB by preventing phosphorylation of its inhibitory IkB-α protein was used. Figure 6E shows that NF-κB complete inactivation again significantly potentiated apoptosis (Figure 6E) but had no effect on AP or p53 levels (data not shown).

Identification of the 27 kDa AP protein and involvement of p53 in the IR-induced ROS-mediated mitochondrial apoptotic pathway. (A) Proteins eluted from an AP-affinity column from cells 72 h post irradiation were subjected to SDS–PAGE in a gel, followed by Coomassie Blue staining. The molecular weight of molecular size markers (in kDa) are indicated; (B) sequence analysis of the 27 kDa protein is compared with that of the human Hsp27 protein; (C) p53 protein expression by western blot detection in unirradiated cells or 0.5, 24, 48 and 72 h post-irradiation cells or in AG-treated cells. (D) Effect of the specific p53 inhibitor PFT-α on NF-κB signalling; (E) effect of NF-κB inhibitor BAY 11-7082 on MCF-7 cell apoptosis. Whole-cell lysates were subjected to SDS–PAGE and probed with the appropriate Abs. Western blot analysis of β-actin expression is provided to show equal loading of the samples. Blots are representative of three different experiments done independently that gave the same results. Histograms indicate means±s.d. of three different cultures, and each one was tested in quadruplicate and expressed as percentage of unirradiated cells (control). **P<0.01, ***P<0.001.
IR-induced ROS-mediated GI mRNA downregulation occurs through the involvement of ERα and ERK1/2 MAPK
As we found that ROS can even modulate GI gene expression at mRNA level (Figure 4C), we attempted to reveal the molecular mechanism of the observed ROS-mediated GI downregulation by investigating the possible involvement of ERα and ERK1/2 signalling. In fact, it has been shown that ROS can induce post-translational Erk1/2-dependent phosphorylation of ERα at serine 118, leading to ERα downregulation in MCF-7 (Weitsman et al, 2009). Moreover, we previously found that oestrogens positively modulate GI transcript levels in MCF-7 cell, possibly through the interaction of their receptors with degenerated oestrogen response element (ERE) motives, identified in GI promoter by bioinformatics methods (Rulli et al, 2006). Hence, we reasonably assumed that the observed GI transcriptional downregulation could be consequent to the ROS-induced Erk1/2-mediated downregulation of ERα. Indeed, we found that irradiation, associated with ROS production, significantly changed the expression profile of phosphorylated serine 118 (p-ser118) ERα, ERα as well as Erk1/2. In particular, a marked increase in phosphorylation of serine 118 occurred, paralleled by a significant decrease in the level of total ERα and concurrent activation of Erk1/2 over the same period post irradiation (Figure 7A). Pretreatment with NAC abrogated such effects, proving the direct involvement of ROS (Figure 7A). To validate the involvement of ERK1/2 signalling on p-ERα and ERα protein level, or GI mRNA expression, cells were exposed to the specific ERK 1/2 inhibitor, U-0126. As shown in Figure 7B, the effect of IR was completely abolished in the presence of U-0126 (Figure 7B). Western blot analysis of p-Erk1/2 proved the biochemistry evidence of the inhibitory action of U-0126 on ERK1/2 activity (Figure 7B). The inhibitor U-0126 did not affect ROS accumulation (data not shown), confirming that such reactive species act upstream of ERK-1/2 in negatively modulating GI. To prove that ERα was directly involved in the downregulation of GI expression at the mRNA level, ICI 182,780, an ERα-degrading anti-oestrogen (Osborne et al, 2004; Chen et al, 2009), was used. Inhibition of ERα by ICI 182,780 (Figure 7C) potentiated IR-induced GI mRNA inhibition (Figure 7C), indicating its direct effect on GI. The ICI 182,780 did not affect either ROS accumulation or Erk-1/2 activation (data not shown), thus indeed corroborating that ERα receptor acts downstream of ROS/Erk-1/2 axis in downregulating GI mRNA level.

Ionising radiation-induced ROS-mediated GI downregulation occurs through the involvement of ER α and ERK1/2 MAPK. Protein expression of phospho-ERα at Ser118 (p-S118ERα), ERα and phospho-p44/42 MAPK (Erk1/2) ( p-ERK1/2) by western blot detection or GI mRNA level in unirradiated cells or in cells at various time points post irradiation not treated or pretreated with (A) 10 mM NAC, (B) 10 μ M Erk1/2 inhibitor U-0126 or (C) 100 nM ERα anti-oestrogen ICI 182,780. The pretreatment effect of NAC, U-0126 and ICI 182,780 has been shown only for cells after 48 and 72 h of IR treatment, where it yielded a major impact, being similar for the shorter time course points post irradiation. Whole-cell lysates were subjected to SDS–PAGE and probed with the appropriate Abs. Western blot analysis of β-actin expression is provided to show equal loading of the samples. Blots are representative of three different experiments done independently that gave the same results. Histograms indicate means±s.d. of three different cultures, and each one was tested in quadruplicate and expressed as percentage of unirradiated cells (control). ***P<0.001.
Discussion
Glyoxalase I represents the major scavenging enzyme of MG, a potent precursor of AGEs (Rabbani and Thornalley, 2012). Among AGEs, the pro-apoptotic AP represents one of the major products deriving from MG modification of protein arginine residues (Kim et al, 2012a). The present study was aimed at investigating whether, and through which mechanism, GI was involved in IR-induced apoptosis in MCF-7 human early-stage breast cancer cell line exposed to a single dose of 21 Gy, as in the ELIOT trial (Veronesi et al, 2010). In line with published experimental work where IR-induced cell death has been identified in many cases as apoptosis (Urashima et al, 2006; Golden et al, 2012; Chen et al, 2013; Kwon et al, 2013), our results also showed that the IR used in this study was able to activate a programmed cell death pathway in MCF-7 cells. By using si-GI transient transfection and MG/AP scavenging agent, AG, we next demonstrated that IR-induced apoptosis was driven by GI downregulation-dependent AP intracellular accumulation. We recently demonstrated that AP triggers a mitochondrial apoptotic pathway, by modifying the expression of molecules typically involved in such apoptotic mechanism, through NF-κB signalling desensitisation in other human cancer cell lines (Antognelli et al, 2013). In agreement with these observations and as proved by AG pretreatment experiments, the increased level of intracellular AP induced by IR also drove a similar activation pattern in irradiated caspase-3-deficient MCF-7 cells (Santha et al, 2013; Charalambous et al, 2013) compared with untreated controls, as indicated by the decrease in the anti-apoptotic Bcl-2 or Bcl-XL or increase in the pro-apoptotic Bax proteins levels, Cyt c release and procaspase-7 activation, typically involved in the mitochondrial apoptotic pathway (Kaufmann and Hengartner 2001; Charalambous et al, 2013; Santha et al, 2013) and, accordingly, reduced NF-κB activity. Therefore, our initial data indicated that IR induces a NF-κB signalling-mediated mitochondrial apoptotic pathway triggered by GI inhibition-driven AP accumulation. In the attempt to understand how AP accumulation triggered an intrinsic apoptotic pathway moved by NF-κB desensitisation, we identified the AP-modified 27 kDa polypeptide, interestingly finding it to be Hsp27. It is known that HSP family are agents mainly protective against programmed cell death. However, in particular conditions, some of these proteins may promote apoptosis (Kazmierczuk and Kilianska, 2010), as observed for the identified AP (Hsp27) in this study. In addition to AP-modified Hsp27, we observed other polypeptides that formed AP by immunoaffinity purification (data not shown). Therefore, the possibility that other proteins may play some role in the same context cannot be ruled out. The Hsp27 is known to accelerate proteasomal degradation of p53 in cancer cells (O'Callaghan-Sunol et al, 2007) and, as demonstrated in a recent research, a negative correlation between Hsp27 and p53 in UV irradiated MCF-7 cells exists in a way that Hsp27 potentiates the degradation of p53 post irradiation (Kanagasabai et al, 2010). Moreover, in the same MCF-7 cell line, an inverse correlation has been recently shown between Hsp27, p53 and NF-κB; in particular, the absence of Hsp27 resulted in accumulation of p53 that, in turn, activated NF-κB signalling pathway (Kanagasabai et al, 2011). Therefore, we assumed that after irradiation, the observed increase in AP-modified Hsp27 protein might be paralleled by a decrease in p53 levels, leading to the observed concomitant NF-κB desensitisation. Indeed, we found that p53 protein levels were not only significantly reduced in irradiated compared with unirradiated cells, but also that AG completely reverted them, indicating a direct effect of AP on p53 in driving IR-induced apoptosis. It is broadly accepted that p53 is a master tumour suppressor protein promoting apoptosis in many cellular systems. However, it has also been reported that caspase-mediated apoptosis does not always require p53 (Miyake et al, 2012; Yeh et al, 2012; Amir et al, 2013). Here, we showed that IR-induced apoptosis, via GI inhibition and AP-modified Hsp27 protein accumulation, occurred in a mechanism independent of the canonical pro-apoptotic pathway induced by p53 but involving the Hsp27-dependent desensitisation of p53, leading to NF-κB nearly complete suppression, as proved by using the specific p53 inhibitor PFT-α as well as NF-κB inhibitor BAY 11-7082. Therefore, in the present work, we not only propose a novel mechanism in IR-induced apoptosis involving GI, Hsp27, p53 and NF-κB, but also reveal a novel noncanonical role of p53 among apoptotic mechanisms in irradiated MCF-7 cells. It is known that IR induces the production of ROS that play an important causative role in apoptotic cell death (Morales et al, 1998; Van Laethem et al, 2006) and that ROS may arise following antioxidant defence functional depletion. Therefore, after observing an effective reduction in the biological activity of some of the major antioxidant cellular defences, an increase in ROS and in MDA, a marker of lipid oxidative damage, we demonstrated that IR-induced GI inhibition, leading to Hsp27-p53-NF-κB pathway activation, triggering a mitochondrial apoptotic pathway, was mediated by ROS. In fact, NAC pretreatment reversed the IR-induced effect on the above-mentioned proteins as well as on those participating in the mitochondrial apoptotic mechanism. Besides, ROS are very reactive species imposing oxidative damage on indispensable biomolecules such as proteins, ultimately compromising cell viability and leading to activation of the programmed cell death machinery (Sinha et al, 2013). We also found that ROS negatively modulated GI at mRNA and protein levels, thus suggesting that the observed decrease in GI activity might also result from decreased protein expression as evident from western blot analysis for GI protein. In the attempt to reveal the molecular mechanism underlying ROS-dependent GI mRNA downregulation, ERα and ERK1/2 signalling were studied. In fact, it has been shown that ROS can induce post-translational Erk1/2-dependent phosphorylation of ERα at serine 118, leading to ERα downregulation in MCF-7 (Weitsman et al, 2009). Moreover, we previously found that oestrogens positively modulated GI transcript levels in MCF-7 cell, possibly through the interaction of their receptors with degenerated ERE motives, identified in GI promoter by bioinformatics methods (Rulli et al, 2006). We found that irradiation significantly changed the expression profile of phosphorylated serine 118 ERα, ERα as well as Erk1/2. In particular, a marked increase in phosphorylation of serine 118 occurred, paralleled by a significant decrease in the level of total ERα and concurrent activation of Erk1/2 over the same period post irradiation. Pretreatment with NAC, Erk1/2-specific inhibitor U-0126 and ERα anti-oestrogen ICI 182,780 accordingly changed such effects, proving the direct involvement of ROS/Erk1/2/ERα axis in GI mRNA negative control. Therefore, we found that IR-induced ROS-mediated GI downregulation occurred through the involvement of ERα and ERK1/2 MAPK, modulation that, to the best of our knowledge, it was never pointed out before.
In early breast cancer patients, standard treatment is conserving surgery followed by whole breast RT. Such treatment significantly reduces ipsilateral recurrence and increases survival (Veronesi et al, 2010). As most tumour recurrences occur near the tumour bed (Veronesi et al, 2010), studies were designed with the aim to shorten the duration of whole breast RT using partial breast irradiation, that is, irradiation of the excisional cavity plus 1–2 cm margins. Intraoperative RT with electrons is one of the techniques used to administer the treatment. In particular, the Italian ELIOT trial appeared a promising feature in early breast cancer treated with breast-conserving surgery, reducing the exposure of normal tissues to radiations and shortening the radiation course from 6 weeks to one single session (Veronesi et al, 2010). Our in vitro results, demonstrating that the single 21 Gy irradiation dose used in the ELIOT trial efficiently induces apoptosis in human ER-positive MCF-7 cells, appear to support the observed effectiveness of such a therapy in patients with a favourable biopathological characterisation, as hormone receptor expression.
In conclusion, our findings show that IR-induced ROS-mediated GI inhibition results in a mitochondrial apoptotic pathway via Hsp27, p53 and NF-κB, providing a novel mechanism in 21 Gy single dose irradiated MCF-7 cell apoptosis, and significantly contributing to the scarcely investigated in vitro research in such a field.
Change history
15 July 2014
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
30 April 2025
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41416-025-03042-0
References
Aebi HE (1974) Catalase. In Methods in Enzymatic Analysis, Bergmeyer HU (ed) pp 673–684. Academic Press: New York.
Alikhani Z, Alikhani M, Boyd CM, Nagao K, Trackman PC, Graves DT (2005) Advanced glycation end products enhance expression of pro-apoptotic genes and stimulate fibroblast apoptosis through cytoplasmic and mitochondrial pathways. J Biol Chem 280: 12087–12095.
Amir S, Ma AH, Shi XB, Xue L, Kung HJ, Devere White RW (2013) Oncomir miR-125b suppresses p14(ARF) to modulate p53-dependent and p53-independent apoptosis in prostate cancer. PLoS One 8: e61064.
Antognelli C, Mezzasoma L, Fettucciari K, Talesa VN (2013) A novel mechanism of methylglyoxal cytotoxicity in prostate cancer cells. Int J Biochem Cell Biol 45: 836–844.
Antognelli C, Mezzasoma L, Fettucciari K, Mearini E, Talesa VN (2013) Role of glyoxalase I in the proliferation and apoptosis control of human LNCaP and PC3 prostate cancer cells. Prostate 73: 121–132.
Baeuerle PA, Baltimore D (1988) I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 242: 540–546.
Baldwin AS (2001) Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. J Clin Invest 107: 241–246.
Bolisetty S, Jaimes EA (2013) Mitochondria and reactive oxygen species: physiology and pathophysiology. Int J Mol Sci 14: 6306–6344.
Bondong S, Kiefel H, Hielscher T, Zeimet AG, Zeillinger R, Pils D, Schuster E, Castillo-Tong DC, Cadron I, Vergote I, Braicu I, Sehouli J, Mahner S, Fogel M, Altevogt P (2012) Prognostic significance of L1CAM in ovarian cancer and its role in constitutive NF-κB activation. Ann Oncol 23: 1795–17802.
Calvo FA, Gonzalez-Domingo M, Usychin S (2013) Intraoperative irradiation. In Encyclopedia of Radiation Oncology, Brady LW, Yaeger TE (eds) pp 388–399. Springer: Heidelberg.
Chance B, Sies H, Boveris A (1979) Hydroperoxide metabolism in mammalian organs. Physiol Rev 59: 527–605.
Charalambous C, Pitta CA, Constantinou AI (2013) Equol enhances tamoxifen’s anti-tumor activity by induction of caspase-mediated apoptosis in MCF-7 breast cancer cells. BMC Cancer 13: 238.
Chen H, Yang K, Choi S, Fischer JH, Jeong H (2009) Up-regulation of UDP-glucuronosyltransferase (UGT) 1A4 by 17beta-estradiol: a potential mechanism of increased lamotrigine elimination in pregnancy. Drug Metab Dispos 37: 1841–1847.
Chen T, Chen M, Chen J (2013) Ionizing radiation potentiates dihydroartemisinin-induced apoptosis of A549 cells via a caspase-8-dependent pathway. PLoS One 8: e59827.
Esterbauer H, Schaur RJ, Zollner H (1991) Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: 81–128.
Fettucciari K, Fetriconi I, Mannucci R, Nicoletti I, Bartoli A, Coaccioli S, Marconi P (2006) Group B Streptococcus induces macrophage apoptosis by calpain activation. J Immunol 176: 7542–7556.
Fleming TH, Theilen TM, Masania J, Wunderle M, Karimi J, Vittas S, Bernauer R, Bierhaus A, Rabbani N, Thornalley PJ, Kroll J, Tyedmers J, Nawrotzki R, Herzig S, Brownlee M, Nawroth PP (2013) Aging-dependent reduction in glyoxalase 1 delays wound healing. Gerontology 59: 427–437.
Forman HJ, Torres M (2001) Redox signaling in macrophages. Mol Aspects Med 22: 189–216.
Gasparian AV, Yao YJ, Kowalczyk D, Lyakh LA, Karseladze A, Slaga TJ, Budunova IV (2002) The role of IKK in constitutive activation of NF-kappaB transcription factor in prostate carcinoma cells. J Cell Sci 115: 141–151.
Golden EB, Pellicciotta I, Demaria S, Barcellos-Hoff MH, Formenti SC (2012) The convergence of radiation and immunogenic cell death signaling pathways. Front Oncol 2: 88.
Honda S, Farboud B, Hjelmeland L, Handa J (2001) Induction of an aging mRNA retinal pigment epithelial cell phenotype by matrix-containing advanced glycation end products in vitro. Investig Ophthalmol Vis Sci 42: 2419–2425.
Hwang JR, Jo K, Lee Y, Sung BJ, Park YW, Lee JH (2012) Upregulation of CD9 in ovarian cancer is related to the induction of TNF-α gene expression and constitutive NF-κB activation. Carcinogenesis 33: 77–83.
Kanagasabai R, Karthikeyan K, Vedam K, Qien W, Zhu Q, Ilangovan G (2010) Hsp27 protects adenocarcinoma cells from UV-induced apoptosis by Akt and p21-dependent pathways of survival. Mol Cancer Res 8: 1399–1412.
Kanagasabai R, Krishnamurthy K, Druhan LJ, Ilangovan G (2011) Forced expression of heat shock protein 27 (Hsp27) reverses P-Glycoprotein (ABCB1)-mediated drug efflux and MDR1gene expression in adriamycin-resistant human breast cancer cells. J Biol Chem 286: 33289–33300.
Kaufmann SH, Hengartner MO (2001) Programmed cell death: alive and well in the new millennium. Trends Cell Biol 11: 526–534.
Kazmierczuk A, Kilianska ZM (2010) Role of heat shock proteins in cell apoptosis. Postepy Hig Med Dosw 64: 273–283.
Kim A, Lee JE, Jang WS, Lee SJ, Park S, Kang HJ, Lee SS (2012) A combination of methotrexate and irradiation promotes cell death in NK/T-cell lymphoma cells via down-regulation of NF-κB signaling. Leuk Res 36: 350–357.
Kim KM, Kim YS, Jung DH, Lee J, Kim JS (2012a) Increased glyoxalase I levels inhibit accumulation of oxidative stress and an advanced glycation end product in mouse mesangial cells cultured in high glucose. Exp Cell Res 318: 152–159.
Kim J, Kim OS, Kim CS, Sohn E, Jo K, Kim JS (2012b) Accumulation of argpyrimidine, a methylglyoxal-derived advanced glycation end product, increases apoptosis of lens epithelial cells both in vitro and in vivo. Exp Mol Med 44: 167–175.
Kwon JE, Kim BY, Kwak SY, Bae IH, Han YH (2013) Ionizing radiation-inducible microRNA miR-193a-3p induces apoptosis by directly targeting Mcl-1. Apoptosis 18: 896–909.
Lindholm PF, Bub J, Kaul S, Shidham VB, Kajdacsy-Balla A (2000) The role of constitutive NF-kappaB activity in PC-3 human prostate cancer cell invasive behavior. Clin Exp Metastasis 18: 471–479.
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408.
Ludwig L, Kessler H, Wagner M, Hoang-Vu C, Dralle H, Adler G, Böhm BO, Schmid RM (2001) Nuclear factor-kappaB is constitutively active in C-cell carcinoma and required for RET-induced transformation. Cancer Res 61: 4526–4535.
Mannervik B, Aronsson AC, Marmstal E, Tibellin G (1981) Glyoxalase I (rat liver). In Methods in Enzymatic Analysis, Jakoby WB (ed) pp 297–301. Academic Press: New York.
McCord JM, Fridovich I (1969) Superoxide dismutase. an enzymic function for erythrocuprein (hemocuprein). J Biol Chem 244: 6049–6055.
Miyake N, Chikumi H, Takata M, Nakamoto M, Igishi T, Shimizu E (2012) Rapamycin induces p53-independent apoptosis through the mitochondrial pathway in non-small cell lung cancer cells. Oncol Rep 28: 848–854.
Morales A, Miranda M, Sánchez-Reyes A, Biete A, Fernández-Checa JC (1998) Oxidative damage of mitochondrial and nuclear DNA induced by ionizing radiation in human hepatoblastoma cells. Int J Radiat Oncol Biol Phys 42: 191–203.
Na HK, Kim EH, Choi MA, Park JM, Kim DH, Surh YJ (2012) Diallyl trisulfide induces apoptosis in human breast cancer cells through ROS-mediated activation of JNK and AP-1. Biochem Pharmacol 84: 1241–1250.
O'Callaghan-Sunol C, Gabai VL, Sherman MY (2007) Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res 67: 11779–11788.
Osborne CK, Wakeling A, Nicholson RI (2004) Fulvestrant: an oestrogen receptor antagonist with a novel mechanism of action. Br J Cancer 90: S2–S6.
Paglia DE, Valentine WN (1967) Studies on the qualitative and quantitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 70: 158–169.
Park MT, Song MJ, Lee H, Oh ET, Choi BH, Jeong SY, Choi EK, Park HJ (2011) β-lapachone significantly increases the effect of ionizing radiation to cause mitochondrial apoptosis via JNK activation in cancer cells. PLoS One 6: e25976.
Rabbani N, Thornalley PJ (2012) Methylglyoxal, glyoxalase 1 and the dicarbonyl proteome. Amino Acids 42: 1133–1142.
Ranuncolo SM, Pittaluga S, Evbuomwan MO, Jaffe ES, Lewis BA (2012) Hodgkin lymphoma requires stabilized NIK and constitutive RelB expression for survival. Blood 120: 3756–3763.
Rulli A, Antognelli C, Prezzi E, Baldracchini F, Piva F, Giovannini E, Talesa V (2006) A possible regulatory role of 17β-estradiol and tamoxifen on glyoxalase I and glyoxalase II genes expression in MCF7 and BT20 human breast cancer cells. Breast Cancer Res Treat 96: 187–196.
Sakamoto H, Mashima T, Yamamoto K, Tsuruo T (2002) Modulation of heat-shock protein 27 (Hsp27) anti-apoptotic activity by methylglyoxal modification. J Biol Chem 277: 45770–45775.
Santha S, Bommareddy A, Rule B, Guillermo R, Kaushik RS, Young A, Dwivedi C (2013) Antineoplastic effects of α-santalol on estrogen receptor-positive and estrogen receptor-negative breast cancer cells through cell cycle arrest at G2/M phase and induction of apoptosis. PLoS One 8: e56982.
Shibuya T, Watanabe Y, Nalley KA, Fusco A, Salafsky B (1989) The BCA protein determination system: an analysis of several buffers, incubation temperature and protein standards. Tokyo Ika Daigaku Zasshi 47: 677–682.
Shukla S, Gupta S (2004) Suppression of constitutive and tumor necrosis factor alpha-induced nuclear factor (NF)-kappaB activation and induction of apoptosis by apigenin in human prostate carcinoma PC-3 cells: Correlation with downregulation of NF-kappaB-responsive genes. Clin Cancer Res 10: 3169–3178.
Sinha K, Das J, Pal PB, Sil PC (2013) Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol 87: 1157–1180.
Soga M, Matsuzawa A, Ichijo H (2012) Oxidative stress-induced diseases via the ASK1 Signaling pathway. Int J Cell Biol 2012: 439587.
Sovak MA, Bellas RE, Kim DW, Zanieski GJ, Rogers AE, Traish AM, Sonenshein GE (1997) Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 100: 2952–2960.
Suh J, Payvandi F, Edelstein LC, Amenta PS, Zong WX, Gélinas C, Rabson AB (2002) Mechanisms of constitutive NF-kappaB activation in human prostate cancer cells. Prostate 52: 183–200.
Sun M, Zigman S (1978) An improved spectrophotometric assay for superoxide dismutase based on epinephrine autoxidation. Anal Biochem 90: 81–89.
Thornalley PJ, Yurek-George A, Argirov OK (2000) Kinetics and mechanism of the reaction of aminoguanidine with the alpha-oxoaldehydes glyoxal, methylglyoxal, and 3-deoxyglucosone under physiological conditions. Biochem Pharmacol 60: 55–65.
Urashima T, Nagasawa H, Wang K, Adelstein SJ, Little JB, Kassis AI (2006) Induction of apoptosis in human tumor cells after exposure to Auger electrons: comparison with gamma-ray exposure. Nucl Med Biol 33: 1055–1063.
Van Laethem A, Nys K, Van Kelst S, Claerhout S, Ichijo H, Vandenheede JR, Garmyn M, Agostinis P (2006) Apoptosis signal regulating kinase-1 connects reactive oxygen species to p38 MAPK-induced mitochondrial apoptosis in UVB-irradiated human keratinocytes. Free Radic Biol Med 41: 1361–1371.
Veronesi U, Orecchia R, Luini A, Galimberti V, Zurrida S, Intra M, Veronesi P, Arnone P, Leonardi MC, Ciocca M, Lazzari R, Caldarella P, Rotmensz N, Sangalli C, Sances D, Maisonneuve P (2010) Intraoperative radiotherapy during breast conserving surgery: a study on 1,822 cases treated with electrons. Breast Cancer Res Treat 124: 141–151.
Wang Z, Sun Y (2010) Targeting p53 for novel anticancer therapy. Transl Oncol 3: 1–12.
Weitsman GE, Weebadda W, Ung K, Murphy LC (2009) Reactive oxygen species induce phosphorylation of serine 118 and 167 on estrogen receptor alpha. Breast Cancer Res Treat 118: 269–279.
Yamagishi S, Inagaki Y, Amano S, Okamoto T, Takeuchi M, Makita Z (2002) Pigment epithelium-derived factor protects cultured retinal pericytes from advanced glycation end product-induced injury through its antioxidative properties. Biochem Biophys Res Commun 296: 877–882.
Yeh CH, Yang YY, Huang YF, Chow KC, Chen MF (2012) Induction of apoptosis in human Hep3B hepatoma cells by norcantharidin through a p53 independent pathway via TRAIL/DR5 signal transduction. Chin J Integr Med 18: 676–682.
Yi H, Liang B, Jia J, Liang N, Xu H, Ju G, Ma S, Liu X (2013) Differential roles of miR-199a-5p in radiation-induced autophagy in breast cancer cells. FEBS Lett 587: 436–443.
Zhang Y, Luo M, Zu Y, Fu Y, Gu C, Wang W, Yao L, Efferth T (2012) Dryofragin, a phloroglucinol derivative, induces apoptosis in human breast cancer MCF-7 cells through ROS-mediated mitochondrial pathway. Chem Biol Interact 199: 129–136.
Zhang S, He Y, Tong Q, Chen Q, Wu X, Huang W (2013) Deltonin induces apoptosis in MDA-MB-231 human breast cancer cells via reactive oxygen species-mediated mitochondrial dysfunction and ERK/AKT signaling pathways. Mol Med Rep 7: 1038–1044.
Zhu Q, Wang J, Zhang Y, Sun S (2012) Mechanisms of MPP-induced PC12 cell apoptosis via reactive oxygen species. J Huazhong Univ Sci Technolog Med Sci 32: 861–866.
Acknowledgements
We thank Mrs Roberta Frosini for the excellent technical assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License.
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/3.0/ http://creativecommons.org/licenses/by-nc-sa/3.0/
About this article

Cite this article
Antognelli, C., Palumbo, I., Aristei, C. et al. RETRACTED ARTICLE: Glyoxalase I inhibition induces apoptosis in irradiated MCF-7 cells via a novel mechanism involving Hsp27, p53 and NF-κB. Br J Cancer 111, 395–406 (2014). https://doi.org/10.1038/bjc.2014.280
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/bjc.2014.280
Keywords
This article is cited by
-
Is the small heat shock protein HspB1 (Hsp27) a real and predominant target of methylglyoxal modification?
Cell Stress and Chaperones (2019)
-
Accumulation of the advanced glycation end product carboxymethyl lysine in breast cancer is positively associated with estrogen receptor expression and unfavorable prognosis in estrogen receptor-negative cases
Histochemistry and Cell Biology (2017)
