
Abstract
Oxidative stress has an important role in the pathogenesis of many muscle diseases. The major contributors to oxidative stress in muscle tissue are reactive oxygen species such as oxygen ions, free radicals, and peroxides. Insulin-like growth factor I (IGF-I) has been shown to increase muscle mass and promote muscle cell proliferation, differentiation, and survival. We, therefore, hypothesized that IGF-I might also be cytoprotective for muscle cells during oxidative stress. Exogenous hydrogen peroxide (H2O2) was used to induce oxidative stress/damage in two types of skeletal muscle cells. Apoptotic pathways were assessed after the oxidative damage and the effects of IGF-I on oxidative stress in muscle cells were examined. Different IGF-I sub-pathways were analyzed with measurement of the expression of pro-and anti-apoptotic proteins. It was found that H2O2 diminishes muscle cell viability and induces a caspase-independent apoptotic cell death. Pretreatment with IGF-I protects muscle cells from H2O2-induced cell death and enhances muscle cells survival. This effect appears to result from the promotion of the anti-apoptotic protein, Bcl2. Further investigation shows that protection is via an IGF-I sub-pathway: PI3K/Akt and ERK1/2 MAPK pathways. Protecting muscle cells from oxidative damage presents a potential application in the treatment of the muscle wasting, which appears in many muscle pathologies including Duchenne muscle dystrophy and sarcopenia.
Similar content being viewed by others


Main
Many pathological muscle conditions, such as Duchenne muscle dystrophy (DMD) and sarcopenia, have been found to be associated with an increase in oxidative stress.1 DMD is a severe progressive X-linked muscle disease with the absence of dystrophin, a protein providing structural integrity on the muscle cell membrane. Owing to the fragility of the DMD muscle membrane, normal muscular contraction in DMD destabilizes the myocyte membrane, causing intracellular accumulation of calcium. This stimulates oxidative metabolism, which generates free radicals and triggers the key pathological processes.2 Sarcopenia is a condition with degenerative loss of skeletal muscle mass and strength associated with ageing. Mitochondrial (Mt) production of reactive oxygen species (ROS) has been shown to increase in skeletal muscle over the course of ageing,3 which in turn reduces bioenergetic efficiency and leads to muscle fiber atrophy/loss.4 Although DMD and sarcopenia have different pathological properties, they share a common syndrome, that is, muscle wasting. Abundant evidence implicates oxidative stress as a potential regulator of proteolytic pathways leading to muscle wasting.5
Oxidative stress results from the activities of the reactive compounds called ROS. ROS are natural by-products in the normal metabolism of oxygen and have important roles in cell signalling. ROS including oxygen ions, free radicals, and peroxides usually have highly reactive properties because of the presence of unpaired valence shell electrons. During physiological homeostasis overall oxidative balance is maintained by the removal of ROS via a variety of antioxidants to match the production of ROS from a variety of sources.6 Overproduction of ROS or lack of antioxidants leads to oxidative stress and cause oxidative damage such as deleterious cellular effects, cell death, and diverse pathological conditions. One major contributor to oxidative damage is hydrogen peroxide (H2O2), which is produced from superoxide that leaks from the mitochondria. Exogenous H2O2 has been used to induce myotube oxidative stress7 and atrophy8 in previous studies.
Preventing oxidative stress or protection against oxidative damage offers a potential strategy for cure or delaying muscle wasting.1, 4 Insulin-like growth factor I (IGF-I) has been found to contribute to oxidative balance and has a protective effect against iron-induced-lipid oxidative stress.9 We, therefore, hypothesized that IGF-I might be able to protect muscle cells from oxidative stress. To test this hypothesis exogenous H2O2 was used to induce skeletal muscle cell oxidative stress/damage. The effects of IGF-I on oxidative stress in muscle cells was examined and different IGF-I pathways were analyzed. It was found that IGF-I has a protective effect on muscle cells after oxidative stress. Protecting muscle cells from oxidative damage by IGF-I offers a potential application in a variety of muscle pathologies.
MATERIALS AND METHODS
Reagents
Recombinant human IGF-I was purchased from Peprotech EC (UK). Caspase 3/7, 8, and 9 activity assay kits and CellTiter-Blue cell viability assay kits were purchased from Promega (Madison, USA). Yo Pro-1 iodide was purchased from Invitrogen (Paisley, UK). Polyclonal anti-Akt and Phospho-Akt (Ser473) antibodies were purchased from New England Bio-labs (Hitchin, UK). Polyclonal anti-human Bax and Bcl2 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Anti-rabbit IgG antibody was purchased from GE Healthcare (Amersham, UK). LY294002, SB 203580, and MEK1/2 inhibitor were purchased from Merck (UK). TUNEL assay kit was purchased from Calbiochem (UK). Apoptotic DNA ladder detection kit was purchased from Millipore (USA). All cell culture media, serums, and antibiotics were purchased from GIBCO (UK).
Cell Lines and Culture Conditions
C2C12(mouse) and L6E9 (rat) skeletal muscle cell lines were purchased from European Collection of Cell Cultures (ECACC). Cells were cultured and maintained at 37 °C in 5% CO2/95% air using DMEM medium containing 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (PenStrep). All cells were used at passages 10–30 after their receipt from the supplier.
Cell Treatments
Pretreatment with IGF-I
C2C12 or L6E9 cells were seeded in 96-well plates at a density of 2 × 104 cells/well in DMEM medium containing 10% FBS, 1% PenStrep, and maintained for 24 h. After seeding, cells were pretreated with different concentrations (0, 50, and 100 ng/ml) of IGF-I (Peprotech EC, UK) in DMEM medium containing 10% FBS and 1% PenStrep at 37 °C in 5% CO2/95% air for 24 h. After pretreatments, cells were challenged with different concentrations of H2O2.
H2O2 induce cell oxidative stress
After seeding or pretreatment with IGF-I, cells were challenged by the addition of H2O2 solution at the desired final concentrations (0.25, 0.5 and 1 mM) in DMEM medium containing 10% FBS and 1% PenStrep at 37 °C in 5% CO2/95% air for 2 h. After treatments and oxidative challenge, cells were subjected to cell viability or cell death assays.
Inhibition of PI3K/Akt, p38, and ERK1/2 MAPK pathways
To determine which pathway IGF-I executes its cell protection from oxidative injury, a cell permeable phosphatidylinositol 3 kinase (PI3K) inhibitor (LY294002), p38 mitogen-activation protein kinase (MAPK) inhibitor (SB 203580), or MEK1/2 inhibitor (cat no. 444939, Merck) were used to inhibit PI3K/Akt, p38MAPK, or MEK1/2 MAPK pathways. Briefly, C2C12 and L6E9 cells were seeded in 96-well plates at a density of 1 × 104 cells/well in DMEM medium containing 10% FBS and 1% PenStrep and maintained for 24 h. After seeding, cells were treated with IGF-I at a concentration of 100 ng/ml plus LY294002 (20 μM), SB203580 (20 μM), or MEK1/2 inhibitor (20 μM) in DMEM medium containing 10% FBS and 1% PenStrep at 37 °C in 5% CO2/95% air for 24 h before they were challenged with different concentrations of H2O2. After treatments and oxidative challenge, cells were subjected to cell viability or cell death assays.
Cell Viability Assay
After 24 h of pretreatment with different concentration of IGF-I and 2 h of oxidative challenge, cell viability was assessed using a previously reported method.10, 11 Briefly, after treatment with IGF-I and oxidative stress, 20 μl of CellTiter-Blue reagent (Promega) was added to each well without the removal of the medium. The plate was rocked on an orbital shaker gently for 2 min to help mixing of the CellTiter-Blue reagent with culture medium. Cells were then further incubated at 37 °C in 5% CO2/95% air for 5 h. Fluorescence was measured with excitation at 530 and emission at 620 nm using a fluorescent plate reader (Fluoroskan Ascent FL; Labsystems, Helsinki, Finland). The experiments were performed in triplicate and repeated on two separately initiated cultures.
Cell Death Assay
After pretreatment by IGF-I and oxidative stress by H2O2, dead cells were assessed using a previously reported method.11, 12 Briefly, cells were seeded in 96-well plates with DMEM containing 10% FBS and 1% PenStrep at a density of 2 × 104 cells/well. After overnight culture, cells were pretreated with multiple IGF-1 concentrations for 24 h and oxidatively stressed with different concentrations of H2O2. After treatment, YOPRO-iodide (Invitrogen) was added to each well at a final concentration of 4.0 μM/l and mixed for 2 min on an orbital shaker. Cells were incubated at 37 °C in 5% CO2/95% air for 5 h. Fluorescence was measured with excitation at 485 nm and emission at 510 nm using a fluorescent plate reader (Fluoroskan Ascent FL; Labsystems). For comparison purposes results were expressed as the percentage of dead cells to viable cells, which was calculated using the following formula:

where FD represents fluorescence reading for dead cells, FV represent fluorescence reading for viable cells determined in the cell viability assay. The experiments were performed in triplicate and repeated on two separately initiated cultures.
TUNEL Assay
To examine whether H2O2 induces apoptotic cell death, C2C12 and L6E9 cells were seeded in chambers mounted on a glass slide with DMEM media containing 10% FBS and 1% PenStrep at a density of 2 × 105 cells/slide. After overnight incubation, cells were treated by H2O2 (C2C12 and L6E9 cells at concentrations of 1 mM and 0.25 mM, respectively) for 1 and 2 h. One group of cells was treated by camptothecin (20 μM) for 2 h as a positive control. After treatment, cells were fixed with formalin (10%), and apoptotic cells were detected using a terminal deoxynucleotidyl transferase mediated dUTP nick-end-labeling (TUNEL) kit (Calbiochem).
Detection of DNA Fragmentation
DNA fragmentation, the hallmark of apoptosis, was examined using DNA ladder detection kit (Millipore). Briefly, C2C12 and L6E9 cells were seeded in a 5-ml flask at a density of 2 × 106 cells/flask with DMEM media containing 10% FBS and 1% PenStrep. After overnight incubation, cells were treated with H2O2 (C2C12 and L6E9 cells at concentrations of 1 and 0.25 mM, respectively) for 2 h. After the treatment, cells were washed three times with PBS and pelletted by centrifugation. The DNA fragments were isolated and purified according to the manufactory instruction and visualized by electrophoresis.
Caspase Activity Assay
After oxidative stress, cells were subjected to caspase 3/7, 8, and 9 activity measurements with Caspase-Glo assay kit (Promega). Briefly, the plates containing cells were removed from the incubator and allowed to equilibrate to room temperature for 30 min. A volume of 100 μl of Caspase-Glo reagent was added to each well, the contents of wells were gently mixed with a plate shaker at 300–500 r.p.m. for 30 s. The plate was then incubated at room temperature for 2 h. The luminescence of each sample was measured in a plate-reading luminometer (Thermo Labsystems) with parameters of 1-min lag time and 0.5 s/well read time. The experiments were performed in triplicate and repeated on two separately initiated cultures. To assess whether the Caspase-Glo assay kit is suitable for measurement of caspase activity in muscle cells, both muscle cell lines were treated by cycloheximide at 0.1 mM for 24 h (cycloheximide is a protein translation inhibitor, which has been proven to cause both muscle cell apoptosis), caspase 3/7, 8, and 9 activity was measured with Caspase-Glo assay kit using the above protocol.
Protein Extraction and Western Blotting
C2C12 and L6E9 cells were seeded in a six-well plate at a density of 18 × 105 cells/well with DMEM medium containing 10% FBS and 1% PenStrep and incubated at 37 °C in 5% CO2/95% air for 24 h. After seeding, cells were washed with serum-free medium (99% DMEM, 1% PenStrep) three times and then cultured in serum-free medium for 24 h. The volume of medium in each well was 3 ml. After serum deprivation, the cells were divided into four groups with each group containing two wells. For the first group, cells were incubated with serum-free medium, the second group were incubated with serum-free medium containing IGF-I (100 ng/ml), the third group were incubated with serum-free medium containing IGF-I (100 ng/ml) and LY294002 (20 μM), and the fourth group were incubated with serum-free medium containing IGF-I (100 ng/ml) and SB203580 (20 μM). All treatments lasted 24 h. After treatment, cells were washed three times with PBS, lysed by the addition of RIPA buffer containing protease inhibitor cocktail (Roche), and harvested with a cell scraper. To assist cell lysis, cell suspensions were repeatedly frozen and thawed. Total protein concentration in the samples was determined using a modified Lowry protein assay kit (Pierce Biotechnology, USA). In total, 60 μg of total protein was mixed with the same volume of the laemmli sample buffer containing 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.004% bromphenol blue and 0.125 M tris HCl, pH approximately 6.8 (Sigma, UK). The samples were then denatured by heating at 95 °C for 5 min. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) was used to separate the proteins, which were then electro-blotted on to polyvinylidene fluoride (PVDF) membrane (Bio-Rad, USA). The membrane was blocked with 5% milk in PBS (Marvel semi-skimmed milk powder) and incubated with primary antibodies at 4 °C overnight followed by incubation with anti-rabbit IgG antibody conjugated with horse radish peroxidase (Dako, UK) at 1/2000 dilution. The primary antibody dilution for Bax, Bcl2 and Akt was 1/200, for Phospho-Akt was 1/1000. After secondary antibody incubation, the membrane was washed three times with PBS and incubated with Super Signal West Dura extended duration Substrate (Pierce, USA) for 5 min and the protein bands were illuminated with a chemi-doc system (Bio-Rad). The density of protein bands was measured using densitometry software (Molecular Analyst, windows software for Bio-Rad image analysis system version 1.5, USA).
Statistical Analysis
All data have been examined and shown a normal distribution. All results were expressed as mean±s.e.m. One-way ANOVA (Prism version 4 2004 edition, USA) with multiple comparison tests were used. Statistical analysis was performed on n=6 samples and Bonferroni's Multiple Comparison Test was used after ANOVA. P<0.05 was considered as significant and indicated as *P<0.01 was considered as higher significance and indicated as **P>0.05 was considered as not significant and marked as NS.
RESULTS
H2O2-Induced Muscle Cell Oxidative Stress
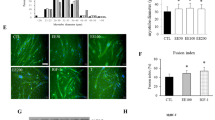
Two mammalian skeletal muscle cell lines (C2C12 and L6E9) were used in the experiment. After 2 h of exposure to different concentrations of H2O2, survival of both C2C12 and L6E9 cells decreased in a dose-dependent manner (Figures 1a and b); the higher the concentration of H2O2, the less the survival of cells. When the ratios of dead cells to survival cells was examined, it revealed that exposure to H2O2 also increased the number of dead cells in a dose-dependent manner (Figures 1c and d). This indicates exogenous H2O2-induced muscle cell oxidative stress, which not only reduced cell viability, but also caused cell death. It is interesting to note that L6E9 cells are more susceptible to H2O2 than C2C12 cells as it is observed that all three concentrations (0.25, 0.5, and 1 mM) of H2O2 decrease L6E9 cell viability significantly compared with the untreated control cells (Figure 1b), while only the highest concentration (1 mM) of H2O2 reduces the C2C12 cell viability significantly (Figure 1a). Similar to cell survival, the ratio of dead cells is also significantly higher in L6E9 (Figure 1d) cells than C2C12 cells (Figure 1c).

C2C12 (a, c) and L6E9 (b, d) skeletal muscle cells were challenged by H2O2 at concentrations of 0.25, 0.5, and 1 mM for 2 h. Cell viability was determined with CellTiter-Blue reagent (Promega), cell survival (%) (a, b) was expressed as percentage of viable cell in treated groups to the untreated control group (0 mM). Dead cell was assessed with YOPRO-iodide (Invitrogen); cell death/survival ratio (c, d) was calculated according to the method mentioned in the text. All data were represented as mean±s.e.m. of six samples (n=6). P<0.05 was considered as significant and indicated as *P<0.01 as higher significance and indicated as **P>0.05 as not significant and marked as NS. C2C12 and L6E9 cells were seeded in chambers mounted on glass slide for 24 h and treated by H2O2 (C2C12 and L6E9 cells at concentrations of 1 and 0.25 mM, respectively) for 1 and 2 h. One group of cells was treated by camptothecin (20 μM) for 2 h as a positive control; one group was untreated as negative control. After treatment, apoptotic cells were detected by TUNEL assay. Dark blue stained cells are apoptotic cells (e). C2C12 and L6E9 cells were seeded in a 5-ml flask at a density of 2 × 106cells/flask overnight and then treated with H2O2 (C2C12 and L6E9 cells at concentrations of 1 and 0.25 mM, respectively) for 2 h. Each cell line included one group that was untreated as negative control (N). The DNA fragments were detected by apoptotic DNA ladder detection kit (Millipore), and DNA fragmentation occurs in both cell line (f). It seems that H2O2 caused more severe DNA destruction in C2C12 cells than in L6E9 cells.
To analyze whether H2O2-induced cell death is a type of apoptosis, TUNEL assay was used to examine dead cells after H2O2 exposure. The results showed that H2O2-induced apoptosis in both C2C12 and L6E9 cells after 1- and 2-h exposure (Figure 1e). It seems that 2-h exposure caused more apoptosis than 1-h exposure. To validate the method, camptothecin that has been proven to be able to induce muscle cell apoptosis previously,13 was included in the experiment as a positive control. It was found that exposure to camptothecin for 2 h also caused apoptosis in both cell lines.
To further prove H2O2-induced cell death is a type of apoptosis, DNA fragmentation was examined. The results showed that H2O2-induced DNA fragmentation in both cell lines after 2-h exposure (Figure 1f). It seems that H2O2 caused more severe DNA destruction in C2C12 cells than in L6E9 cells; this may be due to the different concentration of H2O2 used in the experiment, for C2C12 cells 1 mM of H2O2 was used, while for L6E9 cell 0.25 mM of H2O2 was used.
H2O2-Induced Muscle Cell Death is a Type of Caspase-Independent Apoptosis
To analyze by which apoptotic pathway H2O2-induced muscle cell death occurs, caspase activity was assessed after cells had been exposed to H2O2 at 1 mM for 2 h. It was found that H2O2 significantly induced cell apoptotic death (Figures 1b and d–f), however, it does not affect caspase 3/7, 8, and 9 activity (Figures 2a and b) in either cell lines. It was also shown that cycloheximide treatments (which has been proven to be able to cause both cell lines apoptotic death) can significantly increase the activity of caspase 3/7 and 9 in both cell lines and caspase 8 in C2C12 but not in L6E9 cells (Figures 2c and d) measured by Caspase-Glo assay kit indicating the kit used in experiments is suitable for the measurement of caspase activity in both muscle cell lines. All together these data suggest that H2O2-induced muscle cell death is a type of caspase-independent apoptosis.

C2C12 (a) and L6E9 (b) skeletal muscle cells were challenged by H2O2 at concentrations of 1 mM for 2 h. Cellular caspase 3/7, 8, and 9 activity was determined by Caspase-Glo assay kit (Promega) and expressed as percentage of the treated cell to the untreated control cell. A positive control group (c, d) which received cycloheximide treatment (0.1 mM) was included in each cell line to assess whether the Caspase-Glo assay kit is suitable for the measurement of caspase activity in muscle cells. Caspase 3/7, 8, and 9 activity in control groups (c, d) was measured using the same method as (a, b) used. Caspase activity was represented as the percentage (%) to untreated control group, and data were represented as mean±s.e.m. of six samples (n=6). P<0.05 was considered as significant and indicated as *P<0.01 as higher significance and indicated as **P>0.05 as not significant and marked as NS.
IGF-I Increase Muscle Cell Survival After H2O2-Induced Oxidative Stress
To determine whether IGF-I has a protective effect on muscle cells from H2O2-induced oxidative injury, muscle cells were pretreated with different concentrations of IGF-I for 24 h before they were exposed to a variety concentrations of H2O2. It was found that the viability of both muscle cell lines was higher in the IGF-I pretreated groups than the untreated control group (Figures 3a and b). The increase in the viability of the pretreated cells was in a dose-dependent manner in both cell lines indicating that IGF-I can increase muscle cell survival after oxidative stress. It is interesting to note that the response to oxidative challenge and IGF-I treatment were different in the two cell lines. The greater viable cell loss in L6E9 cells (Figure 3b) compared with C2C12 cells (Figure 3a), after H2O2 treatment, indicates that L6E9 cells are more sensitive to oxidative challenge than the C2C12 cells. Although IGF-I has a protective effect in both cell lines in a dose-dependent manner, the extent of protection is different. In C2C12 cells, the highest concentration of IGF-I (100 ng/ml) always brings the cell survival level to the level of the untreated normal cells no matter what concentration of H2O2 is used in oxidative stress (Figure 3a). In the L6E9 cells, the highest concentration of IGF-I (100 ng/ml) only brings the cell survival level to the level of the untreated normal cells in the lowest level of H2O2 treatment (0.25 mM), while in the higher levels of H2O2 treatments (0.5 and 1 mM), the highest concentration of IGF-I only brings cell survival to the level of 65 and 45% of the untreated normal cells, respectively (Figure 3b). The difference may be due to the fact that L6E9 cells are more vulnerable to the higher level of H2O2 treatments (0.5 and 1 mM), which decrease cell viability to the 45 and 25% of the untreated normal cells, respectively (Figure 3b) and IGF-I protection may not be influential enough to recover these severely damaged cells.

C2C12 (a) and L6E9 (b) skeletal muscle cells were pretreated with different concentrations (0, 50, and 100 ng/ml) of IGF-I for 24 h and then challenged by H2O2 at concentrations of (0, 0.25, 0.5, and 1 mM) for 2 h. Cell viability was determined with CellTiter-Blue reagent (Promega); cell survival (%) was expressed as percentage of viable cell in treated groups to the untreated control group (0). All data were represented as mean±s.e.m. of six samples (n=6). P<0.05 was considered as significant and indicated as *P<0.01 as higher significance and indicated as **P>0.05 as not significant and marked as NS.
IGF-I Reduced Muscle Cell Death After H2O2-Induced Oxidative Stress
After pretreatment with IGF-I and challenge by H2O2, cell death was assessed by a previously reported method.11, 12 The results show that a higher concentration of H2O2 increases cells death in both cell lines (Figures 4a and b). In respect of cell death, the response of two cell lines to oxidative stress is different. In C2C12 cells, only the highest concentration (1 mM) of H2O2 causes significant cell death compared with the untreated cells (Figure 4a), while in L6E9 cells both concentrations of H2O2 (0.5 and 1 mM) cause significantly more cell death compared with untreated cells (Figure 4b). This may also be due to the fact that L6E9 cells are more vulnerable to H2O2-induced oxidative stress. When cells were pretreated by IGF-I, the trends of cell death appear to be neutralized in both cell lines (Figures 4a and b) indicating IGF-I can prevent cells from death induced by oxidative stress.

C2C12 (a) and L6E9 (b) skeletal muscle cells were pretreated with different concentrations (0, 50, and 100 ng/ml) of IGF-I for 24 h and then challenged by H2O2 at concentrations of (0, 0.25, 0.5, and 1 mM) for 2 h. Dead cell was assessed with YOPRO-iodide (Invitrogen); cell death/survival ratio was calculated according to the method mentioned in the text. All data were represented as mean±s.e.m. of six samples (n=6). P<0.05 was considered as significant and indicated as *P<0.01 as higher significance and indicated as **P>0.05 as not significant and marked as NS.
IGF-I Execute Its Cell Protection from H2O2-Induced Oxidative Injury Likely via PI3K and ERK1/2 MAPK Pathways, but not via p38 MAPK Pathway
It has been established that the effects of IGF-I are mediated through the PI3K/Akt pathway or MAPK pathway. To determine by which pathway IGF-I executes its protective effect from H2O2-induced oxidative stress, a cell permeable, potent and specific PI3K inhibitor (LY294002) was used to examine the relation between the PI3K/Akt pathway and IGF-I protection in muscle cells undergoing oxidative stress. It was found that 1 mM H2O2 treatment reduced cell viability significantly and IGF-1 reversed the H2O2 effects. However, when cells were pretreated with both IGF-I and LY294002, the protection of IGF-I vanished in both cell lines (Figures 5a and c). Similar to cell survival, IGF-I preventing cells from death induced by H2O2 is also mediated by the PI3K/Akt pathway. It can be observed that IGF-I prevents cell from death caused by H2O2, while pretreatment by both IGF-I and LY294002 have neutralized IGF-I protection in both cell lines (Figures 5b and d). As previous studies have shown that the PI3K/Akt pathway has conflicting roles under different conditions, to make sure the PI3K/Akt pathway was activated by IGF-I in muscle cells and LY294002 inhibits PI3K activation at the same conditions used in the experiment, both cell lines were treated with IGF-I or IGF-I plus LY294002 for 24 h. The level of the total Akt and phosphor-Akt was determined by western blotting. The results show that the PI3K/Akt pathway can be activated by IGF-I and LY294002 can inhibit PI3K activation in both muscle cell lines under the same conditions in all experiments (Figures 6a–d).

C2C12 (a, b, e, f) and L6E9 (c, d, g, h) muscle cells were pretreated with IGF-I (100 ng/ml) alone, IGF-I (100 ng/ml) plus LY294002 (20 μM), IGF-I (100 ng/ml) plus SB203580 (20 μM), IGF-I (100 ng/ml) plus MEK1/2 (20 μM) for 24 h and challenged by H2O2 (1 mM) for 2 h. Cell viability was determined with CellTiter-Blue reagent (Promega), cell survival (%) (a, c, e, g) was expressed as percentage of viable cell in treated groups to the untreated control group. Dead cell was assessed with YOPRO-iodide (Invitrogen); cell death/survival ratio (b, d, f, h) was calculated according to the method mentioned in the text. All data were represented as mean±s.e.m. of six samples (n=6). P<0.05 was considered as significant and indicated as *P<0.01 as higher significance and indicated as **P>0.05 as not significant and marked as NS.

C2C12 (a, b) and L6E9 (c, d) cells cells were pretreated by IGF-I (100 ng/ml) alone, IGF-I (100 ng/ml) plus LY294002 (20 μM) or IGF-I (100 ng/ml) plus SB203580 (20 μM) for 24 h, cellular Akt and phosphor-Akt proteins were detected by western blotting (b, d) and quantized (a, c) by measuring the density of protein bands. All data were represented as mean±s.e.m. of four samples (n=4). The result was represented as the ratio of phosphorylated Akt/Akt (a, c).
The MAPK pathway is another major signal transduction cascade, which ultimately triggers multiple biological cell responses of IGF-I.14 To evaluate the role of the MAPK pathways in IGF-I protection cells from oxidative stress, two inhibitors of the MAPK pathway were used. One is SB203580, which specifically inhibits p38 MAPK and has been used to identify the physiological roles and targets of p38MAPK;15 another one is the MEK1/2 inhibitor, which selectively inhibits MEK1 and MEK2. Interestingly, the MEK1/2 inhibitor not only markedly neutralized IGF-I protection in both cell lines (Figures 5e–h), but also significantly induced cell death (Figures 5f and h) and decreased cell survival (Figures 5e and g) in both cell lines even if cells have not been challenged by H2O2. This confirmed that MEK1 and 2 are major downstream signalling molecules of the IGF pathway and is involved with IGF-I protection for oxidative stress. In contrast to LY294002 and MEK1/2 inhibitors, the addition of SB203580 appears not to inhibit IGF-I protection cell from oxidative stress (Figures 5e–h). Together all these data suggest that IGF-I protection of muscle cells from oxidative damage appears to proceed via the PI3K/Akt and ERK1/2 MAPK pathways, but not proceed via p38 MAPK pathway.
IGF-I Affect Pro- and Pre- Apoptotic Proteins Expression in Muscle Cells
To test whether pretreatment with IGF-I has any effect on apoptotic regulation proteins, the expression level of Bax (a pro-apoptotic protein) and Bcl2 (an anti-apoptotic protein) were examined by western blotting after muscle cells were incubated either with IGF-I alone, or with IGF-I plus PI3K inhibitor or with the p38 MAPK inhibitor. It was found that the ratio of Bcl2 to Bax in IGF-I-treated cells was significantly higher compared with untreated cells (Figures 7a and b) indicating that pretreatment with IGF-I can reduce muscle cell apoptosis. When cells were pretreated with IGF-I and PI3K inhibitor (LY294002) the effect of IGF-I on the ratio of Bcl2 to Bax was neutralized (Figures 7a and b) confirming that IGF-I prevents cell from apoptosis via the PI3K/Akt pathway. If cells were pretreated with IGF-I plus a p38 MAPK inhibitor (SB203580), the effect of IGF-I on the ratio of Bcl2 to Bax was not significantly influenced compared with LY294002 suggesting that the p38 MAPK pathway seems not involved in the protective effect executed by IGF-I.

C2C12 cells were pretreated by IGF-I (100 ng/ml) alone, IGF-I (100 ng/ml) plus LY294002 (20 μM) or IGF-I (100 ng/ml) plus SB203580 (20 μM) for 24 h. Cellular Bcl2 and Bax proteins were detected by western blotting (b) and quantized (a) by measuring the protein band density. All data were represented as mean±s.e.m. of two samples (n=2). The result was represented as the ratio of Bcl2/Bax.
DISCUSSION
Oxidative stress has a major role in the pathogenesis of many muscle diseases. The main contributors to oxidative stress in muscle tissue are ROS such as oxygen ions, free radicals, and peroxides. We used exogenous H2O2 to induce two types of muscle cell oxidative stress and found that exogenous H2O2 decreased muscle cell viability with an increase in caspase-independent apoptosis. IGF-I is able to prevent muscle cells from H2O2-induced cell death and recovers cell viability. After further investigation, it was found that it is likely IGF-I executes its cell protection via the PI3K/Akt and ERK1/2 MAPK, but not p38 MAPK pathways.
It has been known that caspases have a central role in most apoptotic cell death, however, in addition to the classic caspase-mediated apoptosis, mammalian cells can also undergo caspase-independent apoptosis,16 that is mediated by the dissipation of the inner Mt membrane potential and the release of apoptosis factors. The caspase-independent apoptosis has been documented in various death models including exposure to some agents that affect protein redox status.17 Oxidation of redox-sensing inner membrane proteins was found to cause caspase-independent apoptosis in hybridoma T cells.18 Although H2O2 has been found to cause muscle myotube apoptosis,7 it is not clear that H2O2-induced muscle cell death is what type of apoptosis. As far as we know this is the first report that H2O2 induce muscle cell death is a type of caspase-independent apoptosis. It is interesting to note that two types of muscle cells used in the experiment have different sensitivity against H2O2 challenge and L6E9 cells are more susceptible to H2O2 than C2C12 cells. The reason for diverse responses to oxidative challenge in the two cell lines are not yet known. It is known that the level of IGF-I expression is very low or nonexistent in L6E9 cells but the IGF-I receptor is present, while in C2C12 cells both IGF-I and its receptor are highly expressed.19 The fact that L6E9 cells produce a very low level (if any) of IGF-I could be a reason why L6E9 cells are more vulnerable to H2O2-induced oxidative stress than C2C12 cells.
IGF-I is a single-chain peptide growth factor produced primarily by the liver and executes its action mainly through its receptor- insulin-like growth factor I receptor (IGF-IR). It can act as a systemic endocrine hormone or in a paracrine/autocrine manner in target tissues. A recent in vivo study has found that IGF-IR is a mediator of cardiac hypertrophy, which involved oxidative stress in cardiac muscle.20 In skeletal muscle IGF-I can increase muscle mass, promote muscle cell proliferation, differentiation, and survival.21 Systemic administration of IGF-I or its analog has been shown to improve muscle function and reduce muscle susceptibility to contraction-mediated damage in dystrophic mdx mice.22, 23 The mechanism by which IGF-I and its analog confer protection from contraction-mediated damage in dystrophic skeletal muscle is not clear.23 It was found that prolonged IGF-I treatment in mdx mice can induce muscle fibers from a fast-twitch phenotype toward the slow-twitch phenotype without a change in fiber cross areas.22 Compared with fast-twitch muscle, slow-twitch muscle fibers are rich in mitochondria,24 have higher oxidative capacities,25 and therefore use ATP in a more efficient way and produce less ROS, which would cause less or no damage to muscle cells. Our finding that IGF-I is able to prevent muscle cell from H2O2-induced cell death and recover cell viability may offer a rational explanation for IGF-I conferring protection on dystrophic muscle from contraction-induced damage. It is also interesting to note that this protective effect exists only when cells are pretreated by IGF-I for at least 24 h; if cells are treated with IGF-I and H2O2 simultaneously, there is no protection (unpublished data). This indicates that IGF-I executes its protection action through an indirect way.
The effects of IGF-I are mediated mainly through IGF-IR, which has tyrosine kinase activity and signals through the PI3K/Akt pathway and MAPK pathway. There have been a number of studies highlighting the contradictory roles of the PI3K/Akt pathway in cell survival and apoptosis. For instance, it has been previously found that the PI3K/Akt pathway is involved in cell survival and the prevention of apoptosis26 while another study reported that PI3K/Akt pathway activation by IGF-I is not required for cell survival,27 and not involved in the IGF-I anti-apoptotic function against IFN/TNF-induced apoptosis.28, 29 These contradictory findings raise the possibility of conflicting roles of PI3K/Akt under different conditions. To make sure the PI3K/Akt pathway is activated by IGF-I in muscle cells under the same conditions used in the experiment, both cell lines were treated with IGF-I alone or IGF-I plus LY294002 for 24 h, the level of total Akt and phosphor-Akt was determined by western blotting. It was found that PI3K/Akt pathway was activated by IGF-I in muscle cells under the same conditions used in all experiments and LY294002 can inhibit PI3K activation under the same conditions. Further examination of cell viability and cell death after incubation with LY294002 revealed that IGF-I protects muscle cells from oxidative damage through the activation of PI3K/Akt pathway. Similarly, MAPK pathway is another major signal transduction cascade responding to the IGF-I stimulation. Using MEK1/2 inhibitor and SB203580 to block MAPK pathways, we confirmed MEK1 and 2 are major downstream signalling molecules of IGF pathway and involved with IGF-I protection for oxidative stress. P38 MAPK seems not to be involved in the muscle cell protection shown by IGF-I. It is interesting to note a different response between cell survival and cell death in IGF-I and p38 MAPK inhibitor (SB203580) treatment groups (Figures 5e and f). The difference may be due to the role that p38 has in the myoblast cell cycle. A previous report30 has shown that p38 has a critical role in the myoblast cell cycle. As p38 MAPK pathway is more implicated with the cell cycle, when it was inhibited, cell may stop proliferation and stay in the arrest state but not undergo apoptosis. As a result, the cell proliferation/survival will be severely disturbed, but cell death would not be affected.
It has been reported that caspase-independent apoptosis results from the protein damage caused by the unbalance of redox status16 and is linked with Mt membrane permeability.31 A recent study using IGF-I to treat old rats has found that IGF-I treatment has led to a reduction of free radical production in the liver and that this action is through a Mt protection.32 Bax and Bcl-2 are two main proteins to mediate Mt membrane permeability.33 Bax is a pro-apoptotic protein, which forms a trans-membrane pore across the outer Mt membrane, leading to loss of membrane potential and enhances apoptosis. Bcl-2 is an anti-apoptotic protein, which prevents the pore formation and cell death. The ratio of Bcl2 to Bax can be a marker to indicate the level of apoptosis.33 Our study shows that IGF-I reduces the muscle cell apoptotic level significantly and the PI3K inhibitor neutralise IGF-I action on apoptosis but p38 MAPK inhibitor has not affected apoptotic level. All these data suggest that IGF-I prevents cell from apoptosis through the PI3K/Akt and ERK1/2 MAPK pathways, but not via the p38 MAPK pathway.
Recombinant IGF-I has been produced on a large scale and has been in clinical trials for a variety of pathological conditions including growth failure,34 type I35 and type II diabetes.36 Although there are many studies using IGF-I as a therapeutic agent to treat dystrophic muscle wasting in an animal model, the mechanism by which IGF-I protects cells from oxidative stress is still not clear. This study has offered a rational mechanism by which IGF-I prevents muscle from contract-mediated damage in a dystrophic animal model.
In summary, exogenous H2O2 can induce oxidative damage in skeletal muscle cells and cause muscle cell caspase-independent apoptotic death. Pretreatment by IGF-I can protect muscle cells from oxidative stress by increasing the ratio of the anti-apoptotic protein (Bcl 2) to the pro-apoptotic protein (Bax). The protection of IGF-1 seems to be via the PI3K/Akt and ERK1/2 MAPK pathways. Protecting muscle cells from oxidative damage presents a potential application in the treatment of muscle wasting, which occurs in many pathological muscle conditions including DMD and sarcopenia.
References
Arthur PG, Grounds MD, Shavlakadze T . Oxidative stress as a therapeutic target during muscle wasting: considering the complex interactions. Curr Opin Clin Nutr Metab Care 2008;11:408–416.
Wagner KR . Approaching a new age in Duchenne muscular dystrophy treatment. Neurotherapeutics 2008;5:583–591.
Chabi B, Ljubicic V, Menzies KJ, et al. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008;7:2–12.
Marzetti E, Anne LH, Eva WS, et al. Sarcopenia of aging: underlying cellular mechanisms and protection by calorie restriction. Biofactors 2009;35:28–35.
Powers SK, Kavazis AN, DeRuisseau KC . Mechanisms of disuse muscle atrophy: role of oxidative stress. Am J Physiol Regul Integr Comp Physiol 2005;288:R337–R344.
Bonekamp NA, Volkl A, Fahimi HD, et al. Reactive oxygen species and peroxisomes: struggling for balance. Biofactors 2009;35:346–355.
Siu PM, Wang Y, Alway SE . Apoptotic signaling induced by H2O2-mediated oxidative stress in differentiated C2C12 myotubes. Life Sci 2009;84:468–481.
McClung JM, Judge AR, Talbert EE, et al. Calpain-1 is required for hydrogen peroxide-induced myotube atrophy. Am J Physiol Cell Physiol 2009;296:C363–C371.
Kokoszko A, Dabrowski J, Lewinski A, et al. Protective effects of GH and IGF-I against iron-induced lipid peroxidation in vivo. Exp Toxicol Pathol 2008;60:453–458.
Yang SY, Sales KM, Fuller BJ, et al. Inducing apoptosis of human colon cancer cells by an IGF-I D domain analogue peptide. Mol Cancer 2008;7:17.
Yang SY, Bolvin C, Sales KM, et al. IGF-I activates caspases 3/7; 8 and 9 but does not induce cell death in colorectal cancer cells. BMC Cancer 2009;9:158.
Remacle-Bonnet M, Garrouste F, Baillat G, et al. Membrane rafts segregate pro- from anti-apoptotic insulin-like growth factor-I receptor signaling in colon carcinoma cells stimulated by members of the tumor necrosis factor superfamily. Am J Pathol 2005;167:761–773.
Cottle DL, McGrath MJ, Wilding BR, et al. Slimmer (FHl1B/KYOT3) interacts with the pro-apoptotic protein siva-1 (CD27BP) and delays skeletal myoblast apoptosis. J Biol Chem 2009;284:26964–26977.
Baserga R, Peruzzi F, Reiss K . The IGF-1 receptor in cancer biology. Int J Cancer 2003;107:873–877.
Cuenda A, Rouse J, Doza YN, et al. SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett 1995;364:229–233.
Liang H, Salinas RA, Leal BZ, et al. Caspase-mediated apoptosis and caspase-independent cell death induced by irofulven in prostate cancer cells. Mol Cancer Ther 2004;3:1385–1396.
Cande C, Vahsen N, Garrido C, et al. Apoptosis-inducing factor (AIF): caspase-independent after all. Cell Death Differ 2004;11:591–595.
Costantini P, Belzacq AS, Vieira HL, et al. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2-independent permeability transition pore opening and apoptosis. Oncogene 2000;19:307–314.
Engert JC, Berglund EB, Rosenthal N . Proliferation precedes differentiation in IGF-I-stimulated myogenesis. J Cell Biol 1996;135:431–440.
Araujo AS, Enzveiler AT, Schenkel P, et al. Oxidative stress activates insulin-like growth factor I receptor protein expression, mediating cardiac hypertrophy induced by thyroxine. Mol Cell Biochem 2007;303:89–95.
Velloso CP . Regulation of muscle mass by growth hormone and IGF-I. Br J Pharmacol 2008;154:557–568.
Gehrig SM, Ryall JG, Schertzer JD, et al. Insulin-like growth factor-I analogue protects muscles of dystrophic mdx mice from contraction-mediated damage. Exp Physiol 2008;93:1190–1198.
Schertzer JD, Ryall JG, Lynch GS . Systemic administration of IGF-I enhances oxidative status and reduces contraction-induced injury in skeletal muscles of mdx dystrophic mice. Am J Physiol Endocrinol Metab 2006;291:E499–E505.
Isaeva EV, Shkryl VM, Shirokova N . Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J Physiol 2005;565:855–872.
Negredo P, Rivero JL, Gonzalez B, et al. Slow- and fast-twitch rat hind limb skeletal muscle phenotypes 8 months after spinal cord transection and olfactory ensheathing glia transplantation. J Physiol 2008;586:2593–2610.
Franke TF, Hornik CP, Segev L, et al. PI3K/Akt and apoptosis: size matters. Oncogene 2003;22:8983–8998.
Galvan V, Logvinova A, Sperandio S, et al. Type 1 insulin-like growth factor receptor (IGF-IR) signaling inhibits apoptosis signal-regulating kinase 1 (ASK1). J Biol Chem 2003;278:13325–13332.
Garrouste F, Remacle-Bonnet M, Fauriat C, et al. Prevention of cytokine-induced apoptosis by insulin-like growth factor-I is independent of cell adhesion molecules in HT29-D4 colon carcinoma cells—evidence for a NF-kappa B-dependent survival mechanism. Cell Death Differ 2002;9:768–779.
Remacle-Bonnet MM, Garrouste FL, Heller S, et al. Insulin-like growth factor-I protects colon cancer cells from death factor-induced apoptosis by potentiating tumor necrosis factor alpha-induced mitogen-activated protein kinase and nuclear factor kappa B signaling pathways. Cancer Res 2000;60:2007–2017.
Perdiguero E, Ruiz-Bonilla V, Serrano AL, et al. Genetic deficiency of p38alpha reveals its critical role in myoblast cell cycle exit: the p38alpha-JNK connection. Cell Cycle 2007;6:1298–1303.
Herzig MC, Trevino AV, Liang H, et al. Apoptosis induction by the dual-action DNA- and protein-reactive antitumor drug irofulven is largely Bcl-2-independent. Biochem Pharmacol 2003;65:503–513.
Puche JE, Garcia-Fernandez M, Muntane J, et al. Low doses of insulin-like growth factor-I induce mitochondrial protection in aging rats. Endocrinology 2008;149:2620–2627.
Durai R, Yang SY, Sales KM, et al. Insulin-like growth factor binding protein-4 gene therapy increases apoptosis by altering Bcl-2 and Bax proteins and decreases angiogenesis in colorectal cancer. Int J Oncol 2007;30:883–888.
Chernausek SD, Backeljauw PF, Frane J, et al. Long-term treatment with recombinant insulin-like growth factor (IGF)-I in children with severe IGF-I deficiency due to growth hormone insensitivity. J Clin Endocrinol Metab 2007;92:902–910.
Carroll PV, Christ ER, Umpleby AM, et al. IGF-I treatment in adults with type 1 diabetes: effects on glucose and protein metabolism in the fasting state and during a hyperinsulinemic-euglycemic amino acid clamp. Diabetes 2000;49:789–796.
Clemmons DR, Moses AC, Sommer A, et al. Rh/IGF-I/rhIGFBP-3 administration to patients with type 2 diabetes mellitus reduces insulin requirements while also lowering fasting glucose. Growth Horm IGF Res 2005;15:265–274.
Acknowledgements
SYY has received financial support from Motor Neurone Disease Association-UK and IOC World Anti-Doping Agency.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article
Cite this article
Yang, S., Hoy, M., Fuller, B. et al. Pretreatment with insulin-like growth factor I protects skeletal muscle cells against oxidative damage via PI3K/Akt and ERK1/2 MAPK pathways. Lab Invest 90, 391–401 (2010). https://doi.org/10.1038/labinvest.2009.139
Received:
Revised:
Accepted:
Published:
Issue date:
DOI: https://doi.org/10.1038/labinvest.2009.139
Keywords
This article is cited by
-
Direct exposure to mild heat stress stimulates cell viability and heat shock protein expression in primary cultured broiler fibroblasts
Cell Stress and Chaperones (2020)
-
Mechanical stress-induced apoptosis of nucleus pulposus cells: an in vitro and in vivo rat model
Journal of Orthopaedic Science (2014)
-
Independent and additive effects of atenolol and methionine restriction on lowering rat heart mitochondria oxidative stress
Journal of Bioenergetics and Biomembranes (2014)
-
Sodium fluoride suppress proliferation and induce apoptosis through decreased insulin-like growth factor-I expression and oxidative stress in primary cultured mouse osteoblasts
Archives of Toxicology (2011)
-
An evolutionary comparative scan for longevity-related oxidative stress resistance mechanisms in homeotherms
Biogerontology (2011)
