
Abstract
Immune responses are finely regulated by multiple mechanisms, among which immune regulatory coreceptor family molecules play a central role in both enhancing and suppressing immune responses. Traditionally, T cells have been considered the primary cell type expressing these receptors, through which their responses are modulated. This understanding led to the emergence of the field of ‘immune checkpoint blockade’, which aims to rejuvenate T cells that have become exhausted in the context of chronic infections or the tumor environments. The molecules targeted by such approaches include PD1, CTLA4, Lag3, Tim3 and TIGIT, coinhibitory receptors predominantly expressed on conventional T cells exhibiting functionally impaired, exhausted phenotypes. Interestingly, an expanding array of non-T cell types also express these receptors, although their specific roles remain largely elusive. Here we explore the immune regulatory functions of these coreceptors as expressed on non-conventional T cells, such as myeloid cells and B cells, highlighting their potential contributions to immune regulation.
Similar content being viewed by others


Introduction
Immune responses are regulated by highly complex processes at multiple levels1,2. In case of T cells, T cell-receptor recognition of specific peptides presented by self-MHC molecules on antigen presenting cells ensures the antigen-specific T cell expansion and effector function3. The responses are further shaped by secondary and tertiary signals, known as costimulatory and cytokine stimulation, which enable activated T cells to clonally expand and differentiate into effector T cell subsets with distinct functions, particularly in the case of helper CD4+ T cells. Secondary costimulatory molecules are instrumental in supporting IL2 production and proliferation, with CD28 being the founding member, establishing key principles of clonal expansion, differentiation, as well as nonresponsiveness such as anergy, all of which are critical for effective immune responses and peripheral tolerance4. Tertiary cytokine signals typically derived from T cell-extrinsic sources induce master transcriptional programs capable of orchestrating T cell differentiation processes5. T cells activated in the presence of antigen presenting cell-derived IL12 become IFNγ-producing Th1 type effector cells specialized in activating macrophages and eliminating intracellular pathogens6,7. T cells activated with IL4 possibly provided by basophils and dendritic cells instead become IL4/IL13-producing Th2 type effector T cells that mount anti-helminth immunity8,9. The presence of IL6 plus TGFβ supports effector T cells exhibiting IL17-producing Th17 type lineage phenotypes essential for anti-extracellular pathogens and autoimmune inflammation10,11. Activated CD8+ T cells predominantly become cytotoxic effector cells capable of eliminating virus infected or transformed cells.
Effector immune responses are tightly regulated by counterbalancing mechanisms, and coinhibitory receptors play a central role in preventing excessive activation. A growing number of inhibitory coreceptor families have been identified, and much of the research so far has focused on their ability to suppress T cell activation12. In chronic infections and tumor settings, dysfunctional T cells that highly express these coinhibitory receptors are found13,14. These so-called exhausted T cells display impaired effector functions, contributing to pathogen persistence and tumor immune evasion. To counteract this, immunotherapies targeting coinhibitory receptors have been developed, aiming to reverse T cell exhaustion, restore effector functions and ultimately promote pathogen clearance and tumor eradication.
Although conventional effector T cells are the most well-characterized cell type expressing coinhibitory receptors, a variety of other immune cell types also express these receptors, though their functions remain relatively poorly understood. This review will provide an overview of the biology of key coinhibitory receptors—cytotoxic T-lymphocyte antigen 4 (CTLA4), lymphocyte activation gene 3 (Lag3), programmed cell death protein 1 (PD1), T cell immunoglobulin and mucin domain 3 (Tim3) and T cell immunoreceptor with Ig and ITIM domains (TIGIT)—and examine their roles in non-conventional cell types.
PD1
PD1 was discovered in IL3-deprived Ly9D (a murine hematopoietic progenitor cell line) and activated 2B4-11 (a T cell hybridoma cell line) cells that undergo programmed cell death15. PD1, along with CTLA4, is a prototype coinhibitory receptor that suppresses T cell activation and their effector function16. Upon engagement with its ligands, PDL1 or PDL2, PD1 undergoes phosphorylation at two conserved tyrosine residues located within the cytoplasmic domain: the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the immunoreceptor tyrosine-based switch motif (ITSM)17. The phosphorylated motifs recruit the phosphatases SHP1 and SHP2, which then dephosphorylate key signaling intermediates downstream of the TCR and CD28 costimulatory pathway18. By dephosphorylating kinases such as ZAP70, PI3K and RAS, PD1 inhibits multiple downstream signaling pathways, reducing the activation of key transcription factors, including NFAT, AP1 and NFκB, thereby suppressing immune responses19.
Besides interfering with activation signaling pathways, PD1 signaling also shifts T cell metabolism by inhibiting glycolysis while promoting fatty acid oxidation, limiting the energy supply required for robust T cell responses20. PD1 signaling primarily dampens T cell activation to prevent excessive immune responses that could cause autoimmunity and tissue damage. However, sustained PD1 expression in chronic infections and cancers can lead to T cell exhaustion, characterized by impaired proliferation, cytokine secretion and cytotoxicity21,22. Exhausted T cells fail to control chronic pathogens and eliminate tumor cells, allowing immune evasion. Therefore, the recognition that reversing PD1 function restore T cell function from exhausted states has been instrumental in the development of immune checkpoint blockade therapies.
Unlike its well-characterized roles in T cell activation and exhaustion, the function of PD1 in non-T cell populations remains less understood. PD1 is expressed on both human and mouse dendritic cells (DCs). The impact of PD1 on DCs’ antigen presentation, cytokine production and survival have been examined23. In vitro, PD1-deficient DCs induce enhanced antigen specific CD8+ T cell responses, resulting in increased IFNγ and IL2 secretion compared with PD1-sufficient DCs23. In vivo, hepatocellular carcinoma (HCC) intertumoral transfer of PD1-deficient DCs promote CD8 T cell infiltration as well as their cytotoxicity through the increased expression of perforin and granzyme B, enhancing antitumor immunity23. The roles of PD1 in DCs were also examined in tumor-infiltrating DCs in ovarian cancer24,25. It was found that PD1 blocks NFκB activation via SHP2, which reduces the production of cytokines and prevents proper DC maturation in response to Lipopolysaccharide (LPS) and CpG. It also suppresses antigen presentation through a SHP2 independent mechanism, ultimately weakening T cell activation and promoting tumor immune evasion. The roles of PD1 in DC function are further extended to systemic inflammatory and infectious diseases. In the model of multiple organ dysfunction syndrome induced by zymosan injection, DC surface molecules including CD86, PD1 and PDL1, are highly expressed in splenic DCs, which are accompanied with poor T cell proliferation and IL2 production26. Anti-PDL1 antibody treatment to intervene the PD1–PDL1 pathway increases DC production of IL12p70, while decreasing IL12p40 and IL10 production by DCs, restoring T cell proliferation and IL2 production26. PD1-dependent DC function was also investigated in LPS-induced DC survival in vivo. LPS stimulated wild-type DCs express PD1 and undergo apoptosis, while PD1-deficient DCs are more resistant to apoptosis27. Treating mice with anti-PD1 antibody during DC maturation enhances DC survival. T cell responses generated under these conditions display elevated antigen-specific IFNγ production and proliferation27. Mechanistically, PD1 signaling in DCs limits CD40-40L signaling, contributing DC survival27. PD1 expressed on DCs also interferes with the innate function of DCs to clear bacterial infection28. Chen and colleagues reported that PD1 expression is induced in DCs upon stimulation and that PD1-deficient DCs exhibit the ability to protect mice from lethal Listeria infection28. PD1-dependent superior protection is also observed even without adaptive immunity, where PD1-deficient DCs produce more IL12 and TNF upon Listeria monocytogenes infection28. Therefore, PD1 appears to play an important role in negatively regulating DC function.
Mast cells are tissue resident immune cells best known for their roles in allergic inflammation and immune surveillance, mediating inflammation through the release of granules containing histamine, proteases and cytokines and influencing both innate and adaptive immunity29. PD1 expression on mast cells was first reported by investigating mastocytosis, a condition characterized by dysregulated mast cell proliferation30. Kataoka et al.31 examined patients with cutaneous mastocytosis and found that in approximately one-third of the cases, mast cells express PD131. From analyzing human mastocytosis line LAD2 cells, they found that recombinant PDL1 suppresses the growth of the LAD2 cells, in part by activating SHP1/SHP2 and by inhibiting AKT phosphorylation. Therefore, PD1 may regulate mast cell cell growth31. Bioinformatics analysis of patients with melanoma uncovers that mast cells are associated with resistance to anti-PD1 immunotherapy. Anti-PD1 antibody induces PD1+ mast cell activation, triggering the release of histamine and cytokines32. Importantly, inhibiting the release of histamine and cytokines from mast cells substantially improves the efficacy of PD1 immunotherapy in a subcutaneous melanoma model32. Mast cells also play an immune regulatory role via PD1. PD1+ mast cells directly interact with immature DCs to support their differentiation into tolerogenic subsets. Direct interaction with mast cells downregulates MHCII and increases PDL1 expression in DCs, supporting regulatory T cell development33. The tolerogenic DCs developed from interacting with mast cells express high levels of indoleamine-2,3-deoxigenase (IDO), a rate-limiting enzyme catabolizing tryptophan and inhibiting T cell proliferation33. Treating mast cells with anti-PD1 antibody or DCs with anti-PDL1/PDL2 antibody abrogates the generation of tolerogenic DCs33.
Human and murine B cells express both PD1 and PDL1 upon activation and PD1 inhibits B cell receptor (BCR) signaling and B cell activation34,35,36,37. Coligation of PD1 with BCR phosphorylates tyrosine in PD1, recruiting SHP2. Recruited SHP2 dephosphorylates proximal signaling molecules of the BCR, leading to inactivation of downstream molecules, such as PI3K and ERK, thereby dampening BCR signaling and reducing B cell activation35. Certain B cell subsets are capable of downregulating tumor T cell immunity via PD1. In advanced-stage HCC, TLR4-induced BCL6 induces PD1+ regulatory (protumorigenic) B cell subsets. Upon engaging PDL1, these cells acquire regulatory properties and suppress antitumor T cell responses through IL10 production38. In addition to suppressive function of PD1 in B cell activation, a recent study from Tangye and colleagues reported that PD1 signaling is essential for optimal memory B cell and antibody responses both in human and mouse39.
Mechanistically, PD1- (or PDL1)-deficient B cells express diminished key transcription regulator, cMyc and its target genes necessary for Ig class switch and optimal antibody production39. Supporting this, spleens from B cell-specific PD1-deficient mice show disrupted physiologic accumulation of memory B cells, along with reduced numbers of total and memory B cells in both primary and secondary lymphoid tissues. These findings suggest that PD1 on B cells is essential for maintaining B cell homeostasis.
Macrophages are another cell type expressing PD1. In sepsis, PD1 expression in macrophages is critical in protection from the lethality, in part by suppressing bacterial burden and inflammatory cytokine production40. PD1 is also known to regulate macrophage differentiation process. In the model of spinal cord injury, PD1 expressed on macrophages/microglia is involved in the cellular polarization process, that is, to classically activated proinflammatory M1 phenotype41. PD1 signal suppresses M1 differentiation by diminishing STAT1 activation and supports anti-inflammatory M2 phenotype cell differentiation by enhancing STAT6 activation41. Circulating monocytes that infiltrate tumors and differentiate into macrophages in response to the tumor microenvironment are called tumor-associated macrophages (TAMs). PD1 is highly expressed in TAMs in response to IFNγ, MYD88/IL1R signaling and TLR2/3 stimulation42,43,44, inhibiting the phagocytic capacity of M1 phenotype TAMS and impairing their ability to clear tumor cells45. PD1 signal also drives M2 phenotype TAMs polarization, promoting an immunosuppressive, tumor supportive environment46. PD1 expressed on TAM increases in mouse models of cancer and in primary human cancers, negatively correlated with their phagocytic activity against tumor cells46. Blocking the PD1–PDL1 interaction in vivo enhances their phagocytic function, thereby reducing tumor cell growth and prolonging the survival in a macrophage-dependent manner46,47. More recently, Rathmell and colleagues reported that PD1 also suppresses glycolytic activity of TAMs48. Myeloid cell-specific PD1 deficiency is associated with enhanced TAM glycolysis and antigen presentation ability, leading to enhanced CD8+ T cell activity48.
From analyzing single-cell RNA sequencing data of bone marrow progenitor cells, Yu et al. reported that innate lymphoid cells (ILCs) express PD1, marking ILC progenitors49. PD1 is also an important negative regulator of KLRG1+ ILC2 cell function in both humans and mice, as PD1 deficiency increases ILC2 numbers and reduces worm burden during helminth infection50. In tumor microenvironments, IL33/ST2 signaling induces PD1 expression in ILC2s, limiting their ability to support DC recruitment by suppressing CCL5 production51. PD1 is also constitutively expressed on a subset of ILC3, a key ILC subset important in gut homeostasis maintenance52. Lack of PD1 in ILC3s results in reduced IL22 production, which is essential for intestinal barrier function53. Lastly, PD1 expression is found in ILC1 type cells, controlling their proliferation and function within tumor environment54,55. Interestingly, tumor-derived lactate increases PD1 expression on the ILCs, altering their metabolic programing56. In support, PD1-deficient ILC1 display enhanced IFNγ and granzyme B expression, delaying tumor growth56.
The recognition that PD1 regulates immune function beyond T cells has profound implications for immunotherapy and immune modulation. Although traditionally studied in the context of T cell exhaustion, PD1 expression on DCs, macrophages, B cells, ILCs and mast cells indicates a broader function in immune suppression. This expanded understanding calls for a reassessment of PD1 blockade therapies, as targeting PD1 may not only reinvigorate T cell function but also modulate the functions of other immune cell populations. Further research is required to clarify the context-dependent roles of PD1 across diverse immune cell types, evaluate the impact of its blockade on non-T cell-mediated immunity and refine therapeutic strategies to enhance antitumor efficacy while minimizing immune-related adverse effects.
CTLA4
Cytotoxic T-lymphocyte antigen 4 (CTLA4) was first discovered from searching for a cytotoxic surface molecule from cDNA of activated CD8+ T cells57. Initial screen of CTLA4 mRNA found that its expression is predominantly found in CD28+ T cells58. Surface CTLA4 expression is not found in resting T cells, and intracellular CTLA4 is mainly localized in endocytic compartments that contain other secretory granules such as perforin59. Activation rapidly increases surface CTLA4 expression, which localizes toward the sites of TCR engagements59. The avidity that CTLA4 binds its ligand, B7 molecules on antigen presenting cells, is 20-fold greater than that of CD2860. It was thus initially thought that CTLA4 might be the second CD28-like costimulatory molecule, based on the observation that anti-CTLA4 antibody increases CD28-induced T cell proliferation61,62. However, it was found that crosslinking CTLA4 greatly diminishes T cell proliferation and IL2 production, proposing inhibitory property of CTLA4-derived signals63. Mouse model deficient in CTLA4 develop massive lymphoproliferative and fatal multiorgan inflammation, further supporting the importance of CTLA4-derived coinhibitory signal in T cell tolerance and activation in vivo64.
CTLA4 associates with tyrosine phosphatase SYP or SHP2 that dephosphorylates TCR signaling molecules, such as the TCRζ chain65,66. It was additionally reported that CTLA4 recruits the PIX-GIT2-PAK2 complex via the kinase PKCη, especially in Foxp3+ Treg cells, regulating receptor endocytosis, actin dynamics and T cell-APC interaction67. CTLA4 mutation found in patients with common variable immunodeficiency leads to decreased CTLA4 protein expression in Treg cells68. However, Treg cell numbers remain unaltered in these patients; instead, their suppressive function and their ability to control antigen presenting cell function seem impaired, suggesting that CTLA4 expressed on Treg cells may boost their regulatory functions.
The role of CTLA4 in Treg cells appears to be context dependent as evidenced by several conflicting results. CTLA4 expressed on Treg cells depletes CD80/CD86 from antigen presenting cells by trogocytosis, enabling more PDL1 available to inhibit T cell activity69. On the other hand, Treg cells deficient in CTLA4 fail to control diabetes in an adoptive transfer model70, although CTLA4-deficient Treg cells are fully capable of preventing colitis or autoimmune inflammation71,72. Interestingly, Paterson et al. reported using a Treg cell-specific CTLA4-deficient mouse model that CTLA4 deletion during adulthood does not induce systemic autoimmune inflammation and that these mice instead develop resistance from autoimmune inflammation73. This unexpected resistance is attributed to expansion of functionally competent CTLA4-deficient Treg cells and to the compensatory upregulation of other immune inhibitory molecules in conventional T cells73, concluding that CTLA4 controls Treg cell expansion and function to regulate conventional T cell activity. The precise mechanisms underlying the opposing functions of CTLA4 in Treg cell activity remain unclear and warrant future investigation.
Myeloid cells, including monocytes and DCs also express CTLA4. Earlier studies demonstrated that CD14+ monocytes from human PBMCs constitutively express CTLA4, and similar CTLA4 expression is found in activated myelomonocytic cell line U93774. Treating these cells with anti-CTLA4 antibody inhibits the proliferation and upregulation of surface markers, including CD86 and HLA-DR, induced by IFNγ treatment, suggesting that CTLA4 may regulate monocyte-associated immune responses74. CTLA4 is also expressed on human monocyte-derived DCs75,76. The expression level changes over the maturation process, as the expression is high on freshly isolated monocytes and is downregulated upon differentiation into immature DCs. DCs matured following stimulation with Toll-like receptor (TLR) ligands regain high CTLA4 expression75. Agonistic anti-CTLA4 antibody treatment drastically increases IL10 secretion while diminishing inflammatory IL12 secretion, suggesting that CTLA4 expressed on DCs regulates DC functions75. CTLA4 on DCs also plays an inhibitory role, as crosslinking of CTLA4 inhibits DC maturation and antigen presentation ability76. Moreover, DCs treated with siRNA targeting CTLA4 display enhanced stimulatory activity in vivo77. T cell activation and cytokine production is substantially elevated when DCs do not express CTLA478. Halpert et al.79 reported an additional mechanism by which DCs exploit immune regulatory functions via CTLA4. DCs constitutively secrete microvesicles containing intracellular CTLA4, which competitively bind B7 molecules on bystander DCs to downregulate surface B7 expression, limiting their ability to activate T cells79. Thus, CTLA4 expression on myeloid cells could support tumor immune evasion. Targeting this pathway may improve the efficacy of immune checkpoint blockade therapies. Future studies should focus on elucidating the precise molecular mechanisms of CTLA4 in different myeloid subpopulations and its potential as a therapeutic target in cancer and autoimmune diseases.
Lastly, B cells activated in the presence of IL4 also express CTLA480. CTLA4 engagement in B cells inhibits IgE and IgG1 production. Mechanistically, CTLA4 signaling inhibits germline Cε and Cγ1 gene mRNA expression and transcription factor activation, required for isotype switching80. CTLA4 can also be expressed by CD5+ B-1a B cells, an innate B cell subset that produces autoreactive natural antibodies81. In the absence of CTLA4, B-1a cells exhibit dysregulated immune functions, with transcriptional programs enriched in antigen processing and presentation81. Reconstituting B cell-depleted newborn mice with CTLA4-deficient B-1a cells generates B cell chimeras in which conventional B cells are different from reconstituted self-replenishing B-1a cells. CTLA4-deficient B-1a cell reconstitution induces germinal center B cell and follicular helper T cell responses expressing a highly selected repertoire, which are not observed in CTLA4+ B-1a cell recipients81.
Lag3
A novel Ig superfamily surface molecule expressed in activated T cells and natural killer (NK) cells, termed Lag3, was discovered in the year 199082. Structurally, it resembles CD4 surface antigen and shares its binding ligand, MHCII83. The Lag3 gene is embedded within the CD4 locus, probably generated by gene duplication of the CD4 gene and probably controlled by similar CD4 regulatory elements84. It was initially proposed that Lag3 is associated with Th1 type T cell responses, as surface Lag3 expression correlates with IFNγ-producing T cell subsets85. Lag3 expression on activated T cells is further upregulated by certain cytokines including IL12 but not by IL4 or IL1084, supporting its association with type 1 Immunity. Lag3 indeed contributes to the development of Th1 type T cell responses, by crosslinking MHCII on antigen presenting cells, which stimulates APCs to secrete IL1286.
It was then discovered that Lag3 downregulates phorbol ester-activated T cell proliferation and cytokine production87. Lag3 is now widely recognized as a crucial inhibitory coreceptor expressed on activated T cells88. The observation that Lag3 modulates the activity of CD8 and NK cells, which do not interact with MHCII, suggests the presence of additional ligands. LSECtin, a member of the DC-SIGN family expressed on endothelial cells and melanoma cells, interacts with Lag3, inhibiting tumor specific T cell responses89. Galectin-3 (Gal-3), a lectin with the ability to negatively control T cell responses, binds Lag3, and Lag3 expression is required for Gal-3 to suppress CD8 T cell responses90. In support, mice deficient in Gal-3 also display enhanced CD8+ T cell functions. Fibrinogen-like protein 1 (FGL1) is a liver secreted protein identified as another ligand of Lag3, inhibiting antigen specific T cell responses through binding the Lag3 and blocking the interaction improves antitumor immunity91. However, this notion has recently been challenged, as FGL1 binding may be dispensable for Lag3 to inhibit T cell activation and autoimmune disease development92. Due to the potent action of Lag3 to modulate CD8+ T cell responses, targeting Lag3 is being considered the promising strategy to reverse T cell exhaustion and to restore CD8 immunity. The phase 3 clinical trial treating patients with metastatic melanoma has yielded prolonged patient survival when combined with anti-PD1 therapy93. A phase 1 clinical trial utilizing a bispecific antibody targeting both PD1 and Lag3, tebotelimab, is also being conducted94.
The immune regulatory roles of Lag3 in Treg cells have garnered significant attention. Germline Lag3 deficiency does not exhibit any defects in Treg cell development nor develop systemic autoimmunity95. The first hint of Lag3 in Treg cell function came from an elegant study from Huang et al. that shows Lag3 expression in thymus-derived Treg cells is necessary for their regulatory function and that ectopic Lag3 expression confers the ability to reduce effector T cell proliferation in vitro96. It is also shown that Lag3+ Treg cells preferentially expand in the PBMCs of patients with cancer and express immunosuppressive cytokines and potent suppressive activity97. Lag3-dependent Treg cell function could operate through Lag3-dependent inhibition of DC activation via MHCII98. We also reported that Lag3-deficient Treg cells fail to suppress homeostatic proliferation and T cell-induced colitis development and that Lag3 expression in Treg cells is indispensable for in vivo suppressive function in autoimmune inflammation99. Importantly, Lag3 programs Treg cell metabolic profiles to support oxidative phosphorylation suitable for optimal suppression100. However, some conflicting findings were also reported by Vignali and colleagues, where they demonstrated that Lag3 dampens Treg cell suppressive function especially within the target tissues (islet in this study)101. Although the precise mechanism underlying this discrepancy is not clear, it is possible that Lag3 targeting strategy or genetic background may account for the discrepant results. For example, our study targeted the first three exons of the Lag3 gene, the same exons targeted in germline Lag3−/− mouse model generated by Mathis and colleagues95, whereas Vignali’s group targeted the exon 7 from which surface Lag3 expression is abolished, while continuing to release soluble Lag3101. Also worth noting is their genetic background. Our Lag3-targeted animal model is in C57BL/6 background, while Lag3-targeted model from Vignali and colleagues is in NOD background.
Accumulating evidence suggests that Lag3 can be expressed in various non-T cell populations. The role of Lag3 in DCs has been explored. Lag3 controls the immune stimulatory property of BMDCs102. Bone marrow-derived DCs deficient in Lag3 express four times as much TNF-α than Lag3-sufficient BMDCs at basal level, although there is no difference in cytokine expression upon LPS stimulation102. Lag3-deficient BMDCS are also more glycolytic utilizing fewer fatty acids for mitochondrial respiration102. When it comes to their CD4+ T cell priming abilities, Lag3-deficient BMDCs are more potent inducers of Th1 effector differentiation in ex vivo coculture experiments102. Plasmacytoid DCs (pDCs) are a unique DC subset implicated as the main source of type 1 interferons103. pDCs express a substantial amount of Lag3 mRNA, 10–15 times more than any other T cell population regardless of activation status, suggesting a constitutive expression of Lag3 in pDCs104. Interestingly, the role of Lag3 in pDCs appears to be selective as Lag3-deficient pDCs expand more with CpG stimulation compared with control pDCs, while expressing comparable levels of MHCII, TLR9, IDO, CCR5 and CCR7104.
Activated B cells also express surface Lag3105. B cell Lag3 expression is dependent on T cell-derived soluble factor, suggesting Lag3 as a marker for T cell-induced B cell activation105. Lag3 is found on a subset of natural plasma cells that also express other inhibitory receptors such as PDL1 and PDL2. Following Salmonella infection, Lag3+CD138+ cells are found in the spleen, which also highly express IL10106. The Lag3+ plasma cell subsets display a distinct transcriptome that maintains quiescent state and IL10 expression. Functionally, Lag3+ plasma cells influence host resistance against Salmonella infection through IL10 production. Upon infection, mice with higher levels of Lag3+IL10+CD138+ plasma cells exhibit a weaker immune response, whereas mice with fewer LAG3+ plasma cells show increased levels of memory T cells, suggesting an immunosuppressive role of Lag3 in plasma cells106.
Emerging evidence suggests that microglia, the resident macrophages of the central nervous system, also express Lag3107. Excessive microglial activation is linked to several neurological disorders, and Lag3 may play a key role in controlling microglial activation. Microglial Lag3 expression is further enhanced by IFNγ via STAT1 signaling108. Importantly, Lag3 knockdown in microglia enhances the expression of inducible nitric oxide synthetase by IFNγ, indicating that Lag3 may modulate inflammatory functions of microglia108. Microglial Lag3 expression increases in chronic unpredictable stress-exposed mice, a model for depression, whereas electroconvulsive stimulation, a known antidepressant treatment, reduces microglial Lag3 levels109. Blocking Lag3 in microglia induces antidepressant effects and enhances neurogenesis, suggesting that Lag3 may be a potential target for antidepressant therapeutics109. A separate recent study examining hippocampal sections from post-mortem brains of patients with bipolar disorder who committed suicide uncovers a significant increase of microglial density110. Moreover, the proportion of microglia expressing Lag3 is diminished in patients with bipolar disorder that are suicidal, highlighting a potential role of Lag3 in neuroimmune dysfunction110.
Tim3
Searching for a marker distinguishing Th1 and Th2 type effector cells, Kuchroo and colleagues first discovered the Tim3 (encoded by the Havcr2 gene) as a major surface molecule identifying IFNγ-producing CD4+ T cells111. Tim3 is an inhibitory coreceptor preventing autoimmunity and promoting immunological tolerance112. Functional blockade with antibody or gene deficiency results in exacerbated autoimmune inflammation, whereas Tim3 overexpression ameliorates the disease. Multiple ligands for Tim3 have been identified. Galectin-9, the first identified ligand, binds glycosylated sites on Tim3 and induces apoptosis or dysfunction of T cells113. Carcinoembryonic antigen cell adhesion molecule 1 (CEACAM1) is both a cis and trans ligand important for the tolerance-inducing property of Tim3114. T cells from patients with multiple sclerosis and psoriasis express low expression of Tim3 and are resistant to tolerance induction, suggesting that low Tim3 expression supports greater expansion of inflammatory T cells115,116.
Tim3 is widely expressed across various immune cell types, including DCs, macrophages, NK cells and mast cells. This broad expression suggests that Tim3 functions not only a regulator of T cell responses but also as a multifaceted immune checkpoint involved in both innate and adaptive immunity. Tim3 expression is often dynamically controlled by environmental cues, that is, cytokines or pathogen-derived signals, and influence processes such as cytokine production, phagocytosis, antigen presentation and immune tolerance.
Recent studies emphasized the importance of Tim3 in the immune regulatory functions of DCs. Tim3 expressed on DCs binds the phosphatidylserine (PtdSer) on apoptotic cells not only to trigger phagocytosis of apoptotic cells but also to cross-present antigens117,118,119. In addition, Tim3 expression is particularly high on type 1 conventional DCs (cDCs1), where Tim3 may exert the inhibitory effects in DCs. The high mobility group box 1 (HMGB1), an alarmin that binds nucleic acids from dying cells and facilitates their recognition by endosomal TLRs, is identified as another Tim3 ligand, and the interaction suppresses downstream signaling pathways that would otherwise promote the production of type I interferons and proinflammatory cytokines120,121. This sequestration of HMGB1 by Tim3 effectively reduces DC activation and limits their ability to activate immune responses. In tumor models, Tim3 on BATF3⁺CD103⁺ cDC1s suppresses the production of CXCL9, a chemokine critical for the recruitment of effector CD8⁺ T cells to the tumor microenvironment, and blockade of Tim3 enhances CXCL9 production and improves antitumor immunity122. Tim3 expressed on DCs also modulate antitumor immunity through inflammasome activation123. Loss of Tim3 in DCs increases reactive oxygen species accumulation and NLRP3 inflammasome activation, supporting stem-like CD8+ T cell expansion. Neutralization of IL1 and IL18 abrogates Tim3-deficient DC-derived antitumor immunity123. Tim3-dependent DC function is reported by the Bat3, an adapter protein that binds to the Tim3 cytoplasmic domain and interferes with its inhibitory function124. By examining Bat3-deficient DC function in autoimmune inflammation model, the authors show that Bat3 deficiency decreases encephalitogenic differentiation while enhances Treg cell development124. Thus, Tim3 serves as a checkpoint molecule in DCs, restraining their ability to sense danger signals and limiting their contribution to antitumor immunity. Tim3-dependent DC function is reported by the Bat3, an adapter protein that binds to the Tim3 cytoplasmic domain and interferes with its inhibitory function124. By examining Bat3-deficient DC function in autoimmune inflammation model, the authors show that Bat3 deficiency decreases encephalitogenic differentiation while enhances Treg cell development124. Thus, Tim3 serves as a checkpoint molecule in DCs, restraining their ability to sense danger signals and limiting their contribution to antitumor immunity.
Analogous to DCs, Tim3 plays important regulatory roles in macrophages. Tim3 is constitutively expressed on CD14+ PBMC monocytes. Tim3 knockdown increases TLR-induced IL12 production, demonstrating a regulatory role of Tim3 in innate immunity of myeloid cells125,126. Similarly, Tim3 overexpression in macrophages greatly suppress TLR-mediated inflammatory cytokine production127. Tim3 also plays a role in modulating TAM function in tumor progression. In the model of HCC, Tim3 expression in TAM is greatly increased, and the expression correlates with higher tumor grades and poor survival of patients with HCC128. Tim3 knockdown by siRNA inhibits TAM activation and suppresses HCC cell growth, suggesting a negative role of Tim3 in TAMs. Notably, Tim3–Gal9 interaction could activate Mycobacterium tuberculosis-infected macrophages for better bactericidal activity via IL1β production129,130.
In the brain, microglia express Tim3 and the expression is further increased by LPS stimulation131. Tim3 activation increases microglial production of TGFβ and IL1β, while Tim3 blockade decreases microglial phagocytosis of apoptotic neurons, suggesting that Tim3 controls microglial activity131. Microglial Tim3 expression is increased following intracerebral hemorrhage, and Tim3 knockdown mitigates intracerebral hemorrhage-induced brain damage accompanied with diminished IL1β secretion. Mechanistically, Tim3 regulates microglial polarization process, namely Tim3 knockdown supports microglial cell differentiation into anti-inflammatory M2-like subsets132. Tim3 can also be upregulated in microglia under cerebral hypoxia-ischemia in a HIF1α-dependent manner133. Blocking Tim3 by antibody treatment markedly reduces infarct size, neuronal cell death and neutrophil infiltration, and HIF1α deficiency substantially reduces infarct size and neutrophil infiltration. However, Tim3 overexpression is sufficient to reverse neuronal damage in HIF1α-deficient condition, suggesting a key role of Tim3 in regulating hypoxia-induced brain injury.
TIGIT
TIGIT discovered as immune receptors containing tyrosine-based inhibition motifs is expressed primarily by activated T cells134,135. TIGIT binds poliovirus receptor (PVR, also known as CD155) expressed on DCs136,137 and the interaction induces IL10 and diminishes IL12p40 production by DCs138. T cells from TIGIT-deficient mice display hyperproliferative responses and increased susceptibility to autoimmunity, suggesting a T cell intrinsic inhibitory function139. Moreover, a TIGIT-Fc fusion protein inhibits T cell activation and delayed type hypersensitivity reactions by IL10-dependent mechanism. TIGIT is highly expressed on tumor-infiltrating lymphocytes, and blocking TIGIT together with PD1 markedly enhances CD8 T cell activity140. In a recent randomized combination therapy targeting both TIGIT (tiragolumab) and PDL1 (atezolizumab) in patients with metastatic non-small cell lung cancer results in great clinical benefit compared to atezolizumab alone141. TIGIT is also expressed on NK cells141. NK cell subsets that emerge during cytomegalovirus infection, referred to as ‘adaptive NK cells’, are characterized by the expression of activating markers and inflammatory cytokines, along with low levels of TIGIT. Their expansion following cytomegalovirus reactivation is strongly associated with a reduced leukemia relapse142. Myeloid-derived suppressor cells inhibit ZAP70/ERK pathways and NK cell cytotoxic activity via a TIGIT-dependent mechanism, and adaptive NK cells expressing lower TIGIT are resistant to myeloid-derived suppressor cell-derived suppression143.
TIGIT also directly influences macrophage function. In the bone marrow of patient with acute myeloid leukemia, M2-type macrophage level is increased144. These macrophages exhibit elevated expression of immunosuppressive receptors, including TIGIT, although stimulatory receptor CD226 expression is decreased, suggesting impaired immune activation. TIGIT blockade in M2 macrophages led to macrophage repolarization toward proinflammatory M1 phenotype, an increase in stimulatory receptor CD226 expression, and enhanced CD47-mediated phagocytosis, demonstrating that TIGIT on leukemia-associated macrophages can be targeted for therapeutic purposes144.
TIGIT is also expressed in B cells, especially Tim1+ B cell subsets145. B cells from TIGIT-deficient mice produce less IL10. B cell-specific TIGIT-deficient mice develop severe autoimmune inflammation, suggesting a key role of TIGIT expression for B cell-mediated immune tolerance in the central nervous system145. TIGIT expressed on B cells interacts with CD155+ follicular helper T (TFH) cells to suppress TFH cell proliferation and reduced TIGIT expression on B cells causes CCR6+ TFH cell expansion in patients with multiple sclerosis, demonstrating a negative feedback loop between TIGIT+ B cells and TFH cell in multiple sclerosis pathogenesis146. TIGIT is also expressed on human memory B cell subsets that are crucial for immune regulation147. B cell-derived TIGIT directly controls T cell and DC functions, suppressing T cell responses. A decrease of TIGIT+ memory B cells is associated with elevated donor specific antibody and TFH responses in allograft patients147. TIGIT-dependent regulatory functions of B cells are also observed in memory B cell subsets both TIGIT and Tim1148. Depending on the expression of TIGIT and/or Tim1, B cells display differential capacity to control T cell responses. For example, TIGIT/Tim1-double negative memory B cells strongly induce T cell proliferation and cytokine production, whereas TIGIT/Tim1-double positive memory T cells display strong regulatory ability to reduce T cell production of inflammatory cytokines. Therefore, the expression of TIGIT along with Tim1 defines a bona fide regulatory B cell subset whose function is dysregulated in patients with multiple sclerosis.
Concluding remarks
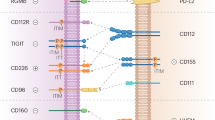
Although significant advances have been made in the development of immunotherapies targeting coinhibitory receptors on dysfunctional T cells in chronic inflammatory diseases and cancer, enhancing therapeutic efficacy and improving patient response rates remain critical challenges. Increasing evidence indicates that coinhibitory receptors are not restricted to T cells but are also expressed by myeloid lineage cells, including monocytes, macrophages, DCs and microglia, across diverse physiological and pathological contexts (Fig. 1 and Table 1). This broader expression suggests that the current T cell-centric perspective on immune checkpoint modulation may underestimate the wider immunological roles and therapeutic potentials of targeting these receptors.

a This schematic illustrates the ligand–receptor interactions and downstream immunoregulatory effects of five inhibitory receptors: PD1, CTLA4, Lag3, Tim3 and TIGIT. Each receptor interacts with one or more ligands and transduces intracellular signals that lead to diverse function outcomes in a variety of cell types. The question marks indicate that the precises signaling mechanisms or intermediates remain unknown. The black text denotes effects in T cells. The gray text denotes effects in non-T cells (DCs, macrophages, B cells, mast cells and ILCs). The asterisk indicates that the effect is shared between both populations. The pound symbol (#) indicates that the effect is specific to regulatory T cells. Ab prod, antibody production; FAO, fatty acid oxidation; ROS, reactive oxygen species. b This diagram illustrates the expression patterns of inhibitory receptors—PD1, CTLA4, Lag3, Tim3 and TIGIT—across immune cell types, T cells, DCs, macrophages, microglia, B cells, mast cells and ILCs.
Current therapeutic antibodies targeting coinhibitory receptors can influence both lymphoid and myeloid compartments, underscoring the importance of elucidating the cell type-specific functions and signaling mechanisms associated with these molecules. A deeper understanding of how coinhibitory receptors regulate immune function in non-T cell populations may reveal novel predictive biomarkers for immunotherapy responsiveness and resistance. Furthermore, dissecting the contributions of myeloid-expressed coinhibitory receptors to immunosuppression, resistance to checkpoint blockade and the emergence of immune-related adverse events could uncover new mechanistic insights and inform the development of more targeted therapeutic strategies.
References
Chaplin, D. D. Overview of the immune response. J. Allergy Clin. Immunol. 125, S3–S23 (2010).
Arneth, B. Molecular mechanisms of immune regulation: a review. Cells 14, 283 (2025).
Hwang, J.-R. et al. Recent insights of T cell receptor-mediated signaling pathways for T cell activation and development. Exp. Mol. Med. 52, 750–761 (2020).
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013).
Luckheeram, R. V. et al. CD4⁺ T cells: differentiation and functions. Clin. Dev. Immunol. 2012, 925135 (2012).
Zhu, J. & Paul, W. E. CD4 T cells: fates, functions, and faults. Blood 112, 1557–1569 (2008).
Trinchieri, G., Pflanz, S. & Kastelein, R. A. The IL-12 family of heterodimeric cytokines: new players in the regulation of T cell responses. Immunity 19, 641–644 (2003).
Licona-Limón, P. et al. TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol. 14, 536–542 (2013).
Zhu, J. et al. Stat6 is necessary and sufficient for IL-4’s role in Th2 differentiation and cell expansion. J. Immunol. 166, 7276–7281 (2001).
Veldhoen, M. et al. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24, 179–189 (2006).
Mangan, P. R. et al. Transforming growth factor-β induces development of the TH17 lineage. Nature 441, 231–234 (2006).
Odorizzi, P. M. & Wherry, E. J. Inhibitory receptors on lymphocytes: insights from infections. J. Immunol. 188, 2957–2965 (2012).
Blank, C. U. et al. Defining ‘T cell exhaustion’. Nat. Rev. Immunol. 19, 665–674 (2019).
Wherry, E. J. T cell exhaustion. Nat. Immunol. 12, 492–499 (2011).
Ishida, Y. et al. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 11, 3887–3895 (1992).
Chamoto, K. et al. Insights from a 30-year journey: function, regulation and therapeutic modulation of PD1. Nat. Rev. Immunol. 23, 682–695 (2023).
Boussiotis, V. A. Molecular and biochemical aspects of the PD-1 checkpoint pathway. N. Engl. J. Med. 375, 1767–1778 (2016).
Sheppard, K.-A. et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 574, 37–41 (2004).
Sharpe, A. H. & Pauken, K. E. The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 (2018).
Patsoukis, N. et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 6, 6692 (2015).
Jubel, J. M. et al. The role of PD-1 in acute and chronic infection. Front Immunol. 11, 487 (2020).
Jiang, Y., Li, Y. & Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 6, e1792–e1792 (2015).
Lim, T. S. et al. PD-1 expression on dendritic cells suppresses CD8+ T cell function and antitumor immunity. Oncoimmunology 5, e1085146 (2016).
Karyampudi, L. et al. PD-1 blunts the function of ovarian tumor-infiltrating dendritic cells by inactivating NF-κB. Cancer Res. 76, 239–250 (2016).
Krempski, J. et al. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J. Immunol. 186, 6905–6913 (2011).
Liu, Q. et al. Changes in the PD-1 and PD-L1 expressions of splenic dendritic cells in multiple-organ dysfunction syndrome mice and their significance. Genet Mol. Res. 13, 7666–7672 (2014).
Park, S. J. et al. Negative role of inducible PD-1 on survival of activated dendritic cells. J. Leukoc. Biol. 95, 621–629 (2014).
Yao, S. et al. PD-1 on dendritic cells impedes innate immunity against bacterial infection. Blood 113, 5811–5818 (2009).
Baran, J. et al. Mast cells as a target-A comprehensive review of recent therapeutic approaches. Cells 12, 1187 (2023).
Metcalfe, D. D. Mast cells and mastocytosis. Blood 112, 946–956 (2008).
Kataoka, T. R. et al. PD-1 regulates the growth of human mastocytosis cells. Allergol. Int. 62, 99–104 (2013).
Li, J. et al. PD-1+ mast cell enhanced by PD-1 blocking therapy associated with resistance to immunotherapy. Cancer Immunol. Immunother. 72, 633–645 (2023).
Rodrigues, C. P. et al. Tolerogenic IDO+ dendritic cells are induced by PD-1-expressing mast cells. Front Immunol. 7, 9 (2016).
Haro, M. A. et al. PD-1 suppresses development of humoral responses that protect against Tn-bearing tumors. Cancer Immunol. Res. 4, 1027–1037 (2016).
Okazaki, T. et al. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc. Natl Acad. Sci. USA 98, 13866–13871 (2001).
Thibult, M. L. et al. PD-1 is a novel regulator of human B-cell activation. Int Immunol. 25, 129–137 (2013).
Wang, X. et al. PD-1-expressing B cells suppress CD4+ and CD8+ T cells via PD-1/PD-L1-dependent pathway. Mol. Immunol. 109, 20–26 (2019).
Xiao, X. et al. PD-1hi Identifies a novel regulatory B-cell population in human hepatoma that promotes disease progression. Cancer Discov. 6, 546–559 (2016).
Ogishi, M. et al. Impaired development of memory B cells and antibody responses in humans and mice deficient in PD-1 signaling. Immunity 57, 2790–2807.e15 (2024).
Huang, X. et al. PD-1 expression by macrophages plays a pathologic role in altering microbial clearance and the innate inflammatory response to sepsis. Proc. Natl Acad. Sci. USA 106, 6303–6308 (2009).
Yao, A. et al. Programmed death 1 deficiency induces the polarization of macrophages/microglia to the M1 phenotype after spinal cord injury in mice. Neurotherapeutics 11, 636–650 (2014).
Bally, A. P. et al. NF-κB regulates PD-1 expression in macrophages. J. Immunol. 194, 4545–4554 (2015).
Cho, H.-Y. et al. Interferon-sensitive response element (ISRE) is mainly responsible for IFN-α-induced upregulation of programmed death-1 (PD-1) in macrophages. Biochimica et. Biophysica Acta 1779, 811–819 (2008).
Tartey, S. et al. A MyD88/IL1R axis regulates PD-1 expression on tumor-associated macrophages and sustains their immunosuppressive function in melanoma. Cancer Res. 81, 2358–2372 (2021).
Kono, Y. et al. Increased PD-1-positive macrophages in the tissue of gastric cancer are closely associated with poor prognosis in gastric cancer patients. BMC Cancer 20, 1–9 (2020).
Gordon, S. R. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 545, 495–499 (2017).
Rao, G. et al. Anti-PD-1 induces M1 polarization in the glioma microenvironment and exerts therapeutic efficacy in the absence of CD8 cytotoxic T cells. Clin. Cancer Res. 26, 4699–4712 (2020).
Bader, J. E. et al. Obesity induces PD-1 on macrophages to suppress anti-tumour immunity. Nature 630, 968–975 (2024).
Yu, Y. et al. Single-cell RNA-seq identifies a PD-1hi ILC progenitor and defines its development pathway. Nature 539, 102–106 (2016).
Taylor, S. et al. PD-1 regulates KLRG1+ group 2 innate lymphoid cells. J. Exp. Med 214, 1663–1678 (2017).
Moral, J. A. et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature 579, 130–135 (2020).
Zeng, B. et al. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 10, 315 (2019).
Jacquelot, N. et al. PD-1 regulates ILC3-driven intestinal immunity and homeostasis. Mucosal Immunol. 17, 371–386 (2024).
Gao, Y. et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 18, 1004–1015 (2017).
Heinrich, B. et al. The tumour microenvironment shapes innate lymphoid cells in patients with hepatocellular carcinoma. Gut 71, 1161–1175 (2022).
Lim, J. X. et al. Programmed cell death-1 receptor-mediated regulation of Tbet+NK1.1− innate lymphoid cells within the tumor microenvironment. Proc. Natl Acad. Sci. USA 120, e2216587120 (2023).
Brunet, J. F. et al. A new member of the immunoglobulin superfamily-CTLA-4. Nature 328, 267–270 (1987).
Lindsten, T. et al. Characterization of CTLA-4 structure and expression on human T cells. J. Immunol. 151, 3489–3499 (1993).
Linsley, P. S. et al. Intracellular trafficking of CTLA-4 and focal localization towards sites of TCR engagement. Immunity 4, 535–543 (1996).
Linsley, P. S. et al. CTLA-4 is a second receptor for the B cell activation antigen B7. J. Exp. Med. 174, 561–569 (1991).
Leach, D. R., Krummel, M. F. & Allison, J. P. Enhancement of antitumor immunity by CTLA-4 blockade. Science 271, 1734–1736 (1996).
Linsley, P. S. et al. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science 257, 792–795 (1992).
Krummel, M. F. & Allison, J. P. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J. Exp. Med. 182, 459–465 (1995).
Tivol, E. A. et al. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity 3, 541–547 (1995).
Lee, K. M. et al. Molecular basis of T cell inactivation by CTLA-4. Science 282, 2263–2266 (1998).
Marengere, L. E. et al. Regulation of T cell receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science 272, 1170–1173 (1996).
Kong, K. F. et al. Protein kinase C-eta controls CTLA-4-mediated regulatory T cell function. Nat. Immunol. 15, 465–472 (2014).
Schubert, D. et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 20, 1410–1416 (2014).
Tekguc, M. et al. Treg-expressed CTLA-4 depletes CD80/CD86 by trogocytosis, releasing free PD-L1 on antigen-presenting cells. Proc. Natl Acad. Sci. USA 118, e2023739118 (2021).
Schmidt, E. M. et al. CTLA-4 controls regulatory T cell peripheral homeostasis and is required for suppression of pancreatic islet autoimmunity. J. Immunol. 182, 274–282 (2009).
Verhagen, J. et al. Enhanced selection of FoxP3+ T-regulatory cells protects CTLA-4-deficient mice from CNS autoimmune disease. Proc. Natl Acad. Sci. USA 106, 3306–3311 (2009).
Read, S. et al. Blockade of CTLA-4 on CD4+CD25+ regulatory T cells abrogates their function in vivo. J. Immunol. 177, 4376–4383 (2006).
Paterson, A. M. et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J. Exp. Med. 212, 1603–1621 (2015).
Wang, X. B. et al. Expression of CTLA-4 by human monocytes. Scand. J. Immunol. 55, 53–60 (2002).
Laurent, S. et al. CTLA-4 is expressed by human monocyte-derived dendritic cells and regulates their functions. Hum. Immunol. 71, 934–941 (2010).
Wang, X. B. et al. CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol. 12, 21 (2011).
Ghorbaninezhad, F. et al. CTLA-4 silencing in dendritic cells loaded with colorectal cancer cell lysate improves autologous T cell responses in vitro. Front Immunol. 13, 931316 (2022).
Bakhshivand, M. et al. Boosting immunotherapy efficacy: empowering the potency of dendritic cells loaded with breast cancer lysates through CTLA-4 suppression. Heliyon 10, e37699 (2024).
Halpert, M. M. et al. Dendritic cell-secreted cytotoxic T-lymphocyte-associated protein-4 regulates the T-cell response by downmodulating bystander surface B7. Stem Cells Dev. 25, 774–787 (2016).
Pioli, C. et al. Inhibition of IgG1 and IgE production by stimulation of the B cell CTLA-4 receptor. J. Immunol. 165, 5530–5536 (2000).
Yang, Y. et al. CTLA-4 expression by B-1a B cells is essential for immune tolerance. Nat. Commun. 12, 525 (2021).
Triebel, F. et al. LAG-3, a novel lymphocyte activation gene closely related to CD4. J. Exp. Med. 171, 1393–1405 (1990).
Baixeras, E. et al. Characterization of the lymphocyte activation gene 3-encoded protein. A new ligand for human leukocyte antigen class II antigens. J. Exp. Med 176, 327–337 (1992).
Bruniquel, D. et al. Regulation of expression of the human lymphocyte activation gene-3 (LAG-3) molecule, a ligand for MHC class II. Immunogenetics 48, 116–124 (1998).
Annunziato, F. et al. Expression and release of LAG-3-encoded protein by human CD4+ T cells are associated with IFN-gamma production. FASEB J. 10, 769–776 (1996).
Avice, M.-N. et al. Lymphocyte activation gene-3, a MHC class II ligand expressed on activated T cells, stimulates TNF-alpha and IL-12 production by monocytes and dendritic cells. J. Immunol. 162, 2748–2753 (1999).
Huard, B. et al. T cell major histocompatibility complex class II molecules down-regulate CD4+ T cell clone responses following LAG-3 binding. Eur. J. Immunol. 26, 1180–1186 (1996).
Aggarwal, V., Workman, C. J. & Vignali, D. A. A. LAG-3 as the third checkpoint inhibitor. Nat. Immunol. 24, 1415–1422 (2023).
Xu, F. et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 74, 3418–3428 (2014).
Kouo, T. et al. Galectin-3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG-3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol. Res. 3, 412–423 (2015).
Wang, J. et al. Fibrinogen-like protein 1 is a major immune inhibitory ligand of LAG-3. Cell 176, 334–347e12 (2019).
Maruhashi, T. et al. Binding of LAG-3 to stable peptide-MHC class II limits T cell function and suppresses autoimmunity and anti-cancer immunity. Immunity 55, 912–924e8 (2022).
Lipson, E. J. et al. Nivolumab plus Relatlimab in advanced melanoma: RELATIVITY-047 4-year update. Eur J Cancer 225, 115547 (2025).
Luke, J. J. et al. The PD-1- and LAG-3-targeting bispecific molecule tebotelimab in solid tumors and hematologic cancers: a phase 1 trial. Nat. Med. 29, 2814–2824 (2023).
Miyazaki, T. et al. Independent modes of natural killing distinguished in mice lacking Lag3. Science 272, 405–408 (1996).
Huang, C. T. et al. Role of LAG-3 in regulatory T cells. Immunity 21, 503–513 (2004).
Camisaschi, C. et al. Alternative activation of human plasmacytoid DCs in vitro and in melanoma lesions: involvement of LAG-3. J. Invest. Dermatol. 134, 1893–1902 (2014).
Liang, B. et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene-3 engagement of MHC class II. J. Immunol. 180, 5916–5926 (2008).
Do, J. S. et al. An IL-27/Lag3 axis enhances Foxp3+ regulatory T cell-suppressive function and therapeutic efficacy. Mucosal Immunol. 9, 137–145 (2016).
Kim, D. et al. Inhibitory co-receptor Lag3 supports Foxp3+ regulatory T cell function by restraining Myc-dependent metabolic programming. Immunity 57, 2634–2650.e5 (2024).
Zhang, Q. et al. LAG3 limits regulatory T cell proliferation and function in autoimmune diabetes. Sci. Immunol. 2, eaah4569 (2017).
Garcia Cruz, D. et al. Lymphocyte activation gene-3 regulates dendritic cell metabolic programing and T cell priming function. J. Immunol. 207, 2374–2384 (2021).
Reizis, B. Plasmacytoid dendritic cells: development, regulation, and function. Immunity 50, 37–50 (2019).
Workman, C. J. et al. LAG-3 regulates plasmacytoid dendritic cell homeostasis. J. Immunol. 182, 1885–1891 (2009).
Kisielow, M. et al. Expression of lymphocyte activation gene 3 (LAG-3) on B cells is induced by T cells. Eur. J. Immunol. 35, 2081–2088 (2005).
Lino, A. C. et al. LAG-3 inhibitory receptor expression identifies immunosuppressive natural regulatory plasma cells. Immunity 49, 120–133.e9 (2018).
Galatro, T. F. et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 20, 1162–1171 (2017).
Morisaki, Y. et al. LAG-3 expression in microglia regulated by IFN-γ/STAT1 pathway and metalloproteases. Front Cell Neurosci. 17, 1308972 (2023).
Rimmerman, N. et al. Microglia and their LAG3 checkpoint underlie the antidepressant and neurogenesis-enhancing effects of electroconvulsive stimulation. Mol. Psychiatry 27, 1120–1135 (2022).
Naggan, L. et al. Suicide in bipolar disorder patients is associated with hippocampal microglia activation and reduction of lymphocytes-activation gene 3 (LAG3) microglial checkpoint expression. Brain Behav. Immun. 110, 185–194 (2023).
Monney, L. et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature 415, 536–541 (2002).
Sanchez-Fueyo, A. et al. Tim-3 inhibits T helper type 1-mediated auto- and alloimmune responses and promotes immunological tolerance. Nat. Immunol. 4, 1093–1101 (2003).
Zhu, C. et al. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat. Immunol. 6, 1245–1252 (2005).
Huang, Y.-H. et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 517, 386–390 (2015).
Kanai, Y. et al. Impaired expression of Tim-3 on Th17 and Th1 cells in psoriasis. Acta Derm. Venereol. 92, 367–371 (2012).
Koguchi, K. et al. Dysregulated T cell expression of TIM3 in multiple sclerosis. J. Exp. Med. 203, 1413–1418 (2006).
Nakayama, M. et al. Tim-3 mediates phagocytosis of apoptotic cells and cross-presentation. Blood 113, 3821–3830 (2009).
DeKruyff, R. H. et al. T cell/transmembrane, Ig, and mucin-3 allelic variants differentially recognize phosphatidylserine and mediate phagocytosis of apoptotic cells. J. Immunol. 184, 1918–1930 (2010).
Santiago, C. et al. Structures of T cell immunoglobulin mucin protein 4 show a metal-Ion-dependent ligand binding site where phosphatidylserine binds. Immunity 27, 941–951 (2007).
Chiba, S. et al. Tumor-infiltrating DCs suppress nucleic acid–mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat. Immunol. 13, 832–842 (2012).
de Mingo Pulido, Á et al. The inhibitory receptor TIM-3 limits activation of the cGAS-STING pathway in intra-tumoral dendritic cells by suppressing extracellular DNA uptake. Immunity 54, 1154–1167.e7 (2021).
de Mingo Pulido, Á et al. TIM-3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer cell 33, 60–74. e6 (2018).
Dixon, K. O. et al. TIM-3 restrains anti-tumour immunity by regulating inflammasome activation. Nature 595, 101–106 (2021).
Tang, R. et al. Tim-3 adapter protein Bat3 acts as an endogenous regulator of tolerogenic dendritic cell function. Sci. Immunol. 7, eabm0631 (2022).
Zhang, Y. et al. Tim-3 regulates pro- and anti-inflammatory cytokine expression in human CD14+ monocytes. J. Leukoc. Biol. 91, 189–196 (2012).
Zhang, Y. et al. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS One 6, e19664 (2011).
Yang, X. et al. T cell Ig mucin-3 promotes homeostasis of sepsis by negatively regulating the TLR response. J. Immunol. 190, 2068–2079 (2013).
Yan, W. et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 64, 1593–1604 (2015).
Sada-Ovalle, I. et al. The Tim3-galectin 9 pathway induces antibacterial activity in human macrophages infected with Mycobacterium tuberculosis. J. Immunol. 189, 5896–5902 (2012).
Jayaraman, P. et al. Tim3 binding to galectin-9 stimulates antimicrobial immunity. J. Exp. Med. 207, 2343–2354 (2010).
Wang, H. W. et al. Microglia activity modulated by T cell Ig and mucin domain protein 3 (Tim-3). Cell Immunol. 293, 49–58 (2015).
Chen, Z. Q. et al. Negative regulation of glial Tim-3 inhibits the secretion of inflammatory factors and modulates microglia to antiinflammatory phenotype after experimental intracerebral hemorrhage in rats. CNS Neurosci. Ther. 25, 674–684 (2019).
Koh, H. S. et al. The HIF-1/glial TIM-3 axis controls inflammation-associated brain damage under hypoxia. Nat. Commun. 6, 6340 (2015).
Yu, X. et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 10, 48–57 (2009).
Chauvin, J. M. & Zarour, H. M. TIGIT in cancer immunotherapy. J. Immunother. Cancer 8, e000957 (2020).
Chiang, E. Y. & Mellman, I. TIGIT-CD226-PVR axis: advancing immune checkpoint blockade for cancer immunotherapy. J. Immunother. Cancer 10, e004711 (2022).
Boles, K. S. et al. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur. J. Immunol. 39, 695–703 (2009).
Yue, C. et al. TIGIT as a promising therapeutic target in autoimmune diseases. Front Immunol. 13, 911919 (2022).
Joller, N. et al. Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 186, 1338–1342 (2011).
Inozume, T. et al. Melanoma cells control antimelanoma CTL responses via interaction between TIGIT and CD155 in the effector phase. J. Invest Dermatol 136, 255–263 (2016).
Wang, F. et al. TIGIT expression levels on human NK cells correlate with functional heterogeneity among healthy individuals. Eur. J. Immunol. 45, 2886–2897 (2015).
Cichocki, F. et al. CD56dimCD57+NKG2C+ NK cell expansion is associated with reduced leukemia relapse after reduced intensity HCT. Leukemia 30, 456–463 (2016).
Sarhan, D. et al. Adaptive NK cells with low TIGIT expression are inherently resistant to myeloid-derived suppressor cells. Cancer Res. 76, 5696–5706 (2016).
Brauneck, F. et al. TIGIT blockade repolarizes AML-associated TIGIT+ M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J. Immunother. Cancer 10, e004794 (2022).
Xiao, S. et al. Checkpoint receptor TIGIT expressed on Tim-1+ B cells regulates tissue inflammation. Cell Rep. 32, 107892 (2020).
Asashima, H. et al. Impaired TIGIT expression on B cells drives circulating follicular helper T cell expansion in multiple sclerosis. J. Clin. Invest 132, e156254 (2022).
Hasan, M. M. et al. Implication of TIGIT+ human memory B cells in immune regulation. Nat. Commun. 12, 1534 (2021).
Varghese, J. F. et al. Human regulatory memory B cells defined by expression of TIM-1 and TIGIT are dysfunctional in multiple sclerosis. Front Immunol. 15, 1360219 (2024).
Funding
Supported by NIH grants, AI125247, AI147498, and AI172135.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Khaled, C., Kim, M. & Min, B. The role of coinhibitory receptor-expressing non-T cells in inflammation and immunity: unsung heroes or peripheral players?. Exp Mol Med 57, 2397–2407 (2025). https://doi.org/10.1038/s12276-025-01562-6
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-025-01562-6
