
Abstract
Alternative splicing significantly contributes to gene expression heterogeneity and disease progression, yet analyzing its dynamics in short genetic regions such as microexons remains challenging. Here we identified notable variations in ribosomal protein S24 (RPS24) splicing patterns across breast cancer subtypes and investigated this novel regulatory mechanism. To overcome the complexity of analyzing three consecutive microexons (3 bp, 18 bp and 22 bp), we developed a specialized approach combining splice junction read analysis with fragment analysis for accurate isoform quantification. We observed distinct isoform compositions across breast cancer cell lines. The 3-bp exon-containing isoform (ex4:3 bp) of RPS24 showed significantly higher expression in estrogen receptor-positive (ER+) cells, demonstrating the strongest association with estrogen receptor signaling among all analyzed genes. This isoform functions as a molecular sensor for therapeutic interventions, being consistently upregulated following mTOR or CDK4/6 inhibition but consistently reduced across diverse drug-resistant cell lines, regardless of resistance mechanism. Through systematic RNA-binding protein screening and crosslinking immunoprecipitation followed by high-throughput sequencing analysis, we identified PTBP1 as a critical upstream regulator mediating microexon skipping. Analysis of multiple patient cohorts demonstrated that decreased ex4:3 bp expression strongly correlated with poor epithelial differentiation and metastatic progression specifically in ER+ breast cancer. Our findings suggest that RPS24 alternative splicing is associated with a multilayered regulatory network integrating ER signaling, cell cycle and PTBP1-mediated splicing. The ex4:3 bp isoform serves as a potential biomarker for drug resistance and treatment response in ER+ breast cancer.
Similar content being viewed by others


Introduction
Alternative splicing (AS) allows a single gene to produce multiple mRNA isoforms, contributing considerably to gene expression heterogeneity and phenotypic diversity1. This mechanism plays crucial roles in development, differentiation and disease progression. Despite the importance of AS in gene regulation, understanding AS dynamics in large-scale transcriptome data remains challenging. While many tools support AS analysis in RNA-sequencing (RNA-seq) data, their results often differ owing to varying algorithms and models2,3. Analysis accuracy further decreases when splicing regulation is complex or when the genetic regions regulated by AS are too short.
We previously developed a database called AS-CMC, designed to facilitate exploration of AS changes in cancer subtypes recorded in The Cancer Genome Atlas (TCGA)4. Within this database, subtype-specific AS patterns were commonly found in the ribosomal protein S24 (RPS24) gene in 13 out of 24 cancer types, suggesting its potential role in cancer-specific regulation. The RPS24 gene has three microexons, each less than 30 nucleotides in length. The short length of these microexons renders their detection difficult as they can be missed or misaligned during read mapping. In addition, the combination of multiple short exons makes it challenging to identify and quantify all possible AS events. Therefore, many studies have focused on the inclusion and skipping of the 22 bp exon in the RPS24 gene, which is the longest among the three5,6,7. Olivieri et al. conducted the first comprehensive analysis of all three microexons in the RPS24 gene across 12 tissues using single-cell analysis, revealing its remarkable tissue specificity8. Our group has further analyzed RPS24 AS isoform composition across cancer types and reported its relevance in epithelial–mesenchymal transition and response to KRAS-targeted therapy in cancer9,10.
Breast cancer is a highly heterogeneous disease with varied molecular and clinical characteristics11,12. Eukaryotic ribosomes, once perceived as uniform cellular machinery for protein synthesis, are now recognized for their regulatory roles in gene expression. As the roles of ribosomal proteins in breast cancer progression and treatment response continue to emerge13,14, understanding RPS24 AS behavior in breast cancer could provide valuable insights into disease mechanisms.
Here, we aimed to examine RPS24 AS patterns across breast cancer cell lines and associated isoforms, components and regulatory mechanisms in estrogen receptor (ER)-positive (ER+) breast cancer. The objective was to investigate the clinical significance of RPS24 AS patterns by exploring its potential as a predictive biomarker for drug resistance and epithelial differentiation status in ER+ breast cancer.
Materials and methods
Data source for cell lines
Raw RNA-seq data in fastq format from the Cancer Cell Line Encyclopedia (CCLE) were downloaded from the Sequence Read Archive (SRA) under project PRJNA523380 (https://www.ncbi.nlm.nih.gov/sra). Cell line annotation, gene dependency and other processed genomic data were obtained using the depmap R package from Bioconductor (https://bioconductor.org). For RNA-seq datasets from non-CCLE cell lines, fastq files were acquired from SRA through searches on GEO (https://www.ncbi.nlm.nih.gov/geo) or ArrayExpress (https://www.ebi.ac.uk/biostudies/arrayexpress) websites. Initial data in SRA format were converted to fastq files using the SRA Toolkit (https://www.ncbi.nlm.nih.gov/home/tools).
Sources for patient data
For the cohort analysis of patients with breast cancer, we obtained transcriptome profiling data from the TCGA (n = 1,098 patients)15, Clinical Proteomic Tumor Analysis Consortium (CPTAC; n = 134 patients)16 and Metastatic Breast Cancer (MBC; n = 200 patients) project17 through the Genomic Data Commons (GDC) portal (https://portal.gdc.cancer.gov). We downloaded splice junction quantification files generated by the GDC bioinformatics pipeline after dbGaP approval. Additional TCGA sample data were accessed using the TCGAbiolinks R package in Bioconductor18, and CPTAC sample data were obtained using the CPTAC Python package (https://paynelab.github.io/cptac). For the MBC project, corresponding clinical data were acquired from cBioPortal (https://www.cbioportal.org).
RPS24 AS analysis using splicing junction reads
The fastq files were mapped to the reference genome (hg38) using the STAR aligner in two-pass mode19. We extracted information from the SJ.out.tab files, which summarize splice junctions and serve as output files from the STAR alignment process. RPS24 AS isoform analysis was performed following the protocol outlined in a recently published paper10. For each sample, we extracted splice junction reads at the 5′ splice site (ss) of exon 4 and categorized them on the basis of the 3′ ss. As exon 4 is constitutive, we considered the total number of reads with the exon 4 5′ ss as RPS24 gene expression and reads corresponding to the 3′ ss as expression of each isoform. To assess isoform proportions, we calculated the ratio of junctional reads representing each isoform to the total number of junctional reads starting from exon 4.
Crosslinking immunoprecipitation followed by high-throughput sequencing data analysis
We downloaded raw fastq files from a crosslinking immunoprecipitation followed by high-throughput sequencing (CLIP-seq) dataset (GSE220184). The sequencing reads were aligned to the human reference genome (hg38) using the Burrows–Wheeler Aligner (version 0.7.17) with default parameters. The resulting SAM files were converted to BAM format, sorted by genomic coordinates and indexed using Samtools (version 1.13). To generate genome coverage files suitable for visualization, we processed the sorted BAM files using bamCoverage from the deepTools package (version 3.5.5). The generated bigWig (bw) files were loaded into the Integrative Genomics Viewer (version 2.19.1) for visualization and qualitative analysis. The visualization in Integrative Genomics Viewer allowed us to validate peaks on motifs identified by RBPmap (version 1.2), including a database of 274 RNA-binding motifs20. The PTBP1 binding sites were identified using the PTBP3 motif model in RBPmap, as PTBP1 and PTBP3 share highly similar RNA recognition motifs and sequence specificity21.
Developmental lineage and stemness score analysis
Ectoderm, mesoderm, endoderm and stemness scores for TCGA breast cancer samples from patients were obtained using the TCGAanalyze_Stemness function from the TCGAbiolinks R package. The analysis workflow involved several steps: first, TCGA RNA-seq data were downloaded and prepared using the GDCquery and GDCdownload functions from TCGAbiolinks. The downloaded data were then normalized and filtered using TCGAanalyze_Normalization and TCGAanalyze_Filtering functions to remove low-expression genes and ensure data quality. Finally, stemness and lineage scores were calculated using specific stem cell signatures, including SC_PCBC_stemSig for stemness characteristics and lineage-specific signatures ECTO_PCBC_stemSig, MESO_PCBC_stemSig and ENDO_PCBC_stemSig for ectoderm, mesoderm and endoderm scores, respectively22. The stemness scores reflect the degree of oncogenic dedifferentiation, while the lineage scores indicate the developmental origin characteristics of tumor samples.
Experimental validation of RPS24 AS in breast cancer cell lines
We studied 11 breast cancer cell lines categorized by ER expression. ER+ lines included MCF7, T47D, HCC1428 and ZR751, whereas ER-negative (ER−) lines included HCC2218, HCC1954, MDA-MB-453, HCC1143, HCC1187, MDA-MB-231 and HCC38. Cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium (HyClone) supplemented with 10% fetal bovine serum (HyClone) and 1% penicillin–streptomycin (Thermo Fisher Scientific). For mTOR inhibition studies, MCF7 cells were treated with 500 nM everolimus (Sigma-Aldrich) for 48 h. For drug resistance studies, tamoxifen-resistant MCF7 cells were obtained from the American Type Culture Collection (CRL-3435) and cultured according to the manufacturer’s instructions in DMEM medium (WELGENE) supplemented with 10% fetal bovine serum, 10 μg/ml of human insulin (Sigma-Aldrich) and 1 μM 4-hydroxytamoxifen (Sigma-Aldrich).
For fragment analysis of RPS24 AS, cells were lysed using TRIzol (Thermo Fisher Scientific) followed by the TRIzol–chloroform extraction protocol. The AS region was amplified using previously published primers23, with the forward primer modified with FAM fluorescent dye for capillary gel electrophoresis detection. Primer sequences were as follows: RPS24 ex4F:ATGGCCTGTATGAGAAGAAA and RPS24 ex6R: CACAGCTAACATCATTGCAG. The fluorescently labeled PCR products were diluted to 1:100 and analyzed using an ABI 3130xl Genetic Analyzer (Thermo Fisher Scientific).
Through the Peak Scanner program (Thermo Fisher Scientific), fragment sizes were determined and peak areas were analyzed by measuring fluorescence intensity. Each peak represented an RPS24 AS isoform, and the relative expression of RPS24 AS isoforms was determined by calculating the ratio of peak areas.
Results
Characterizing RPS24 AS isoforms in breast cancer cell lines
To assess the diversity among breast cancer cell lines, we first investigated the distribution of RPS24 AS isoforms in 51 breast cancer cell lines from CCLE24 and observed profound heterogeneity in isoform composition between cell lines (Fig. 1a). Both the ex4:22 bp and ex4:ex6 junctions were present in 49 out of 51 cell lines (96% of total cell lines), with expressions exceeding 5%. However, the ex4:3 bp junction was detected in only two-thirds of the investigated cell lines (34 out of 51 cell lines). The ex4:18 bp junction was not detected, and when RNA-seq profiles were visualized, the 18-bp exon showed no expression (Supplementary Fig. 1).

a Heterogeneity of RPS24 AS isoform composition in 51 CCLE breast cancer cell lines. Top: the RPS24 gene structure between exons 4 and 6. In RNA-seq data, junction reads derived from exon 4 (marked by colored lines) were used to determine the expression of each isoform. The stacked bar graphs below show the proportion of each isoform. Cell lines are ordered according to the ex4:3 bp isoform ratio. Cell lines highlighted in red were used for experimental validation (Fig.1d). b Fragment analysis by capillary gel electrophoresis for experimental validation. From 51 breast cancer cell lines in CCLE, 11 representative cell lines were selected for experimental validation. PCR amplification was performed with fluorescent primers, followed by fragment analysis by capillary gel electrophoresis to detect and quantify RPS24 AS isoforms. c Representative electrograms for MCF7 and MDA-MB-231 cell lines are shown. The fragment types corresponding to each peak and their proportions are indicated below. Through fragment size analysis, we discovered that all transcripts containing the ex4:3 bp junction lacked the 18-bp exon but invariably included the 22-bp exon, with a 3-bp size difference between the two peaks. The area of each peak was quantified as the expression of each isoform, and the proportion of total expression accounted for by each isoform was calculated. d Correlation analysis between experimental results and CCLE RNA-seq data for the 11 cell lines. The x-axis represents values calculated from RNA-seq analysis, while the y-axis shows values obtained from fragment analysis. Scatter plots display the proportion of each RPS24 AS isoform with corresponding Pearson correlation coefficients (r = 0.90 for ex4:3 bp, r = 0.95 for ex4:22 bp and r = 0.82 for ex4:ex6). e Protein sequences resulting from the three RPS24 AS isoforms present in breast cancer. The ex4:3 bp and ex4:22 bp isoforms differ by just one lysine (K) residue, while the ex4:ex6 isoform generates a distinct C-terminal sequence with three additional amino acids (PKE). The red lollipop symbol indicates the stop codon position in each isoform. f Correlation between ex4:3 bp isoform proportion and RPS24 protein levels. Analysis of both CCLE breast cancer cell lines (n = 30, left) and CPTAC data from patients with breast cancer (n = 107, right) shows negative correlations (r = −0.38 and r = −0.51, respectively).
To ensure the reliability of our RPS24 AS isoform measurement, we performed two methods of validation. First, we compared the CCLE results with four independent RNA-seq datasets of breast cancer cell lines from GEO (GSE48213, GSE58135, GSE73526 and PRJEB30617) (Supplementary Fig. 2). For pairwise comparison, we utilized cell lines that overlapped between CCLE and the four datasets. The correlation was particularly strong for ex4:3 bp (median r = 0.92), whereas ex4:22 bp (median r = 0.81) and ex4:ex6 (median r = 0.81) also showed good correlation between CCLE and GEO.
Next, we experimentally determined the composition of RPS24 microexons in 11 breast cancer cell lines (cell lines marked in red in Fig. 1a) using fragment analysis (Fig. 1b). In each cell line, we observed a maximum of three peaks, showing the types of isoforms generated by AS between exon 4 and exon 6. Through fragment size analysis, we discovered that all transcripts containing the ex4:3 bp junction lacked the 18-bp exon but invariably included the 22 bp exon. Therefore, the ex4:3 bp and ex4:22 bp junctions differ by only 3 bp, which the fragment analysis successfully separated and quantified (Fig. 1c). On the basis of these findings, we named the isoforms as follows: ex4:3 bp for the isoform containing both 3-bp and 22-bp exons, ex4:22 bp for the isoform containing only the 22-bp exon and ex4:ex6 for the isoform lacking all microexons. When comparing the proportion of each isoform calculated from RNA-seq data with experimental validation data from the same cell lines, both showed high concordance (r > 0.8) across all three isoforms (Fig. 1d). These results indicate that our bioinformatic method is a reliable approach for analyzing the composition of RPS24 AS isoforms in RNA-seq data.
The three RPS24 isoforms present in breast cancer exhibited variations in their C-terminal amino acid composition (Fig. 1e). The difference between ex4:3 bp and ex4:22 bp lies in the presence of one lysine (K), and translation terminates at the stop codon found in the 22-bp exon. By contrast, ex4:ex6 generates a protein ending with three amino acids: proline (P), lysine (K) and glutamic acid (E). To determine whether these differences in C-terminal amino acid composition affect protein abundance, we examined transcriptomics and proteomics data from CCLE breast cancer cell lines (n = 30) and CPTAC data from patients with breast cancer (n = 107). We found that transcripts containing the ex4:3 bp junction showed no correlation with RPS24 RNA levels but exhibited a negative correlation with the protein levels in both datasets (Fig. 1f and Supplementary Fig. 3). These findings suggest that the ex4:3 bp isoform may undergo post-transcriptional regulation (for example, nonsense-mediated decay) or post-translational mechanisms (for example, altered protein stability).
RPS24 AS isoform composition is regulated by ER in breast cancer
We investigated the differences in RPS24 AS isoform expression across molecular subtypes in the CCLE breast cancer dataset. Analysis based on ER and human epidermal growth factor receptor 2 (HER2) status (ER+/HER2+, n = 6; ER+/HER2−, n = 12; ER−/HER2+, n = 8; ER−/HER2−, n = 24; unclassified, n = 1) revealed contrasting regulatory patterns between the ex4:3 bp and ex4:ex6 isoforms (P < 0.01), whereas the ex4:22 bp isoform showed minimal variation (P = 0.09) (Fig. 2a). Accordingly, further classification by molecular subtypes (luminal, n = 11; HER2_amp, n = 13; luminal_HER2_amp, n = 2; basal_A, n = 15; and basal_B, n = 9) demonstrated higher expression of the ex4:3 bp isoform in luminal subtypes and lower expression in basal subtypes (Supplementary Fig. 4). This finding suggests a potential association between ex4:3 bp expression and breast cancer subtypes, particularly relating to ER status.

a Comparison of RPS24 AS isoform expression between breast cancer cell line molecular subtypes. Cell lines are categorized into four types on the basis of ER and HER2 expression. The y-axis represents the proportion of each isoform. P values derived from analysis of variance tests are shown for each plot. b Association of the ex4:3 bp isoform with the ESR1 gene. Columns sort cell lines by ex4:3 bp expression (green). Lower rows show ESR1 mRNA expression and two gene dependency scores (KO by RNAi or CRISPR) in the corresponding cell lines. Correlation with ex4:3 bp is displayed on the right. c Changes in ex4:3 bp expression due to ESR1 gene mutations. GSE108304: T47D cell line with Y537S and D538G mutations, treated with ethanol or 10 nM estrogen (estradiol, E2) for 4 h or 24 h. GSE112243: MCF7 with E380Q, Y537S and D538G mutations, with or without 24 h of estrogen (E2) treatment. Differences between ESR1 wild type and mutants were analyzed using a t-test (***P < 0.0005, **P < 0.005, *P < 0.05). d ER-alpha interactions with RPS24 at multiple levels (GSE173631, MCF7 cells). Left: proteins interacting with ESR1 in the cytoplasm, identified by immunoprecipitation followed by mass spectrometry. The x-axis shows median log2 fold change (ER-alpha/immunoglobulin G (IgG)) in protein abundance between ER-alpha immunoprecipitation and IgG control, with higher values indicating stronger interactions with ER-alpha. The y-axis shows −log10(Best.Expect.Val), with higher values indicating greater statistical confidence of protein identification. Right: a heat map showing that RPS24 (highlighted at bottom) is among ESR1-bound mRNAs essential for breast cancer cell fitness, as determined by CRISPR interference of ESR1-binding sites. The top 15 genes were selected on the basis of Mann–Whitney P value, and each cell value represents the average phenotype of the three strongest single-guide RNAs on cell growth across day 5, 9 and 14 time points compared with day 0. Negative values indicate stronger growth inhibition.
To further elucidate the relationship between ex4:3 bp and ER expression, we examined the association between ESR1 expression and its dependency on the RPS24 ex4:3 bp isoform. Our analysis revealed a strong positive correlation between ESR1 messenger RNA (mRNA) expression and ex4:3 bp expression (r = 0.7) (Fig. 2b). Furthermore, ex4:3 bp expression showed negative correlations with ESR1 gene dependency scores, as determined by both RNA interference (RNAi) and CRISPR–Cas9 knockout (KO) methodologies (r < −0.6). These negative correlations indicate that cells expressing high levels of ex4:3 bp are more dependent on ESR1 for survival. Notably, the correlation between ex4:3 bp and both ESR1 mRNA expression and gene dependency scores ranked among the highest when compared with all other genes (Supplementary Fig. 5a), emphasizing the strong association between ex4:3 bp expression and ESR1 gene function in breast cancer cells.
To further investigate the relationship between ESR1 and RPS24 AS, we analyzed transcriptome data from two luminal A-type breast cancer cell lines (MCF7 and T47D) harboring mutations in the ligand-binding domain of the ESR1 gene, a common occurrence in metastatic breast cancer (Fig. 2c). Analysis of the GSE108304 dataset (T47D) revealed a significant decrease in the proportion of ex4:3 bp isoform in ESR1 mutant cells (Y537S and D538G) compared with that in wild-type cells (P < 0.0005). Similarly, the GSE112243 dataset (MCF7) demonstrated consistent decreases in ex4:3 bp expression across various ESR1 mutations (E380Q, Y537S and D538G); however, it depicted some variation depending on the mutation type (P < 0.0005). Estrogen treatment did not significantly alter the ex4:3 bp proportions within each genotype, suggesting that the effect of ESR1 mutations on RPS24 splicing occurs independently of estrogen stimulation (Fig. 2c). These findings were consistently observed across additional datasets, and we also noted a decrease in ex4:3 bp expression in ESR1 fusion (Supplementary Fig. 5b–d), further confirming the relationship between ER signaling and RPS24 splicing.
Our analysis of ESR1-related datasets (GSE173631) revealed multiple levels of ER-alpha interaction with RPS24 (ref. 25). ESR1 immunoprecipitation followed by quantitative mass spectrometry analysis demonstrated that ER-alpha physically interacts with RPS24 protein in the cytoplasm (Fig. 2d, left). CLIP-seq data showed that ER-alpha directly binds to RPS24 transcripts, specifically to the intronic regions between exon 4 and exon 6 and the 3′UTR (Fig. 2d, middle). When ER-alpha binding sites were repressed using CRISPR interference, RPS24 emerged as one of the top 15 genes of which the loss significantly compromised breast cancer cell fitness (Fig. 2d, right). These data indicate that ER-alpha interacts with RPS24 at both protein and RNA levels and that this interaction is critical for breast cancer cell viability.
Differential regulation of ex4:3 bp isoform by cell cycle/mTOR inhibition and in endocrine therapy resistance
Having established the regulatory relationship between ER-alpha and RPS24 AS, we next aimed to understand how this splicing event responds to therapeutic interventions and contributes to treatment resistance, which are critical aspects of breast cancer management. We investigated the regulation of the RPS24 ex4:3 bp isoform in response to two key elements of breast cancer treatment: pathway inhibition and drug resistance. First, we examined the effects of inhibiting critical signaling pathways. In MCF7 cells, mTOR inhibition through INK128, rapamycin or starvation significantly increased the ex4:3 bp isoform (GSE246889, P < 0.0005) (Fig. 3a). Similarly, CDK4/6 inhibition using either abemaciclib (GSE222984) or palbociclib (GSE140758) led to increased ex4:3 bp isoform levels (P < 0.005) (Fig. 3b). These changes occurred only in retinoblastoma (RB)-positive cells (GSE182631, P < 0.05) (Fig. 3c), suggesting that the ex4:3 bp isoform may serve as an indicator of cell cycle pathway activity. The upregulation of ex4:3 bp following mTOR and CDK4/6 inhibition indicates that this isoform may be involved in cellular adaptation to growth-inhibitory conditions, potentially contributing to treatment response.

a The effect of mTOR inhibition in MCF7 cells (GSE246889). The ex4:3 bp isoform proportion was compared among control and three mTOR inhibition conditions: starvation, INK128 and rapamycin. b The response to CDK4/6 inhibition. Left: MCF7 cells treated with abemaciclib (500 nM, 96 h) (GSE222984). Right: MCF7 cells treated with palbociclib (500 nM, 48 h) (GSE140758). c Comparison of the mTOR and CDK4/6 inhibition effects in RB-positive (left) and RB-negative (right) MCF7 cells (GSE182631). Cells were treated with everolimus (500 nM) or palbociclib (250 nM) for 48 h. d Effect of CDK4/6 inhibitor treatment conditions on ex4:3 bp isoform. MCF7 cells expressing wild-type ESR1 (ESR1 WT) or ESR1 mutations (ESR1 D538G and ESR1 Y537S) were treated with vehicle control (DMSO), palbociclib (200 nM), palbociclib combined with estrogen-deprived media or palbociclib combined with fulvestrant (1 nM). Statistical significance was determined using a t-test. Gray brackets indicate statistical comparisons between control and palbociclib groups. e Fragment analysis validation of the mTOR inhibition effect. Left: representative electropherograms showing three RPS24 isoforms in control and everolimus-treated (500 nM, 48 h) MCF7 cells. Right: quantification of isoform proportions. f Analysis of ex4:3 bp isoform in drug-resistant models. Comparison between parental and resistant cells for tamoxifen and fulvestrant resistance (GSE164529 and GSE104985) (tamoR and fulvR) and palbociclib resistance (palboR) (GSE130437). g Fragment analysis comparison between tamoxifen-sensitive (tamoS) and tamoxifen-resistant (tamoR) MCF7 cells. Left: representative electropherograms. Right: quantification of isoform proportions. ***P ≤ 0.0005, ** P≤ 0.005 and * P ≤ 0.05.
We further examined the effect of ESR1 mutations on the ex4:3 bp isoform in response to CDK4/6 inhibition alone or in combination therapy. In MCF7 cells expressing wild-type ESR1 (WT), D538G or Y537S mutations, we observed significant changes in ex4:3 bp isoform across all treatment conditions (GSE281302) (Fig. 3d). Treatment with palbociclib alone, palbociclib combined with estrogen deprivation (palbo+E2 dep) or palbociclib combined with fulvestrant (palbo+fulv) significantly increased the proportion of ex4:3 bp splicing compared with that in control (P < 0.001) in all ESR1 variants. However, both ESR1 mutants (D538G and Y537S) showed distinct splicing patterns compared with wild-type ESR1, suggesting that ESR1 mutation status may influence the splicing response to CDK4/6 inhibition and combination treatments. To validate these findings, we performed fragment analysis in MCF7 cells, with results shown as the average of three independent experiments. These experiments demonstrated a 1.5-fold increase in the proportion of ex4:3 bp following mTOR inhibition with everolimus treatment compared with that in the control (Fig. 3e).
We then examined the behavior of the isoform in drug resistance models. We observed a consistent decrease in the isoform across various drug-resistant cell lines (Fig. 3f). This reduction was evident in cells resistant to different therapeutic agents: tamoxifen (GSE164529, P < 0.0005), fulvestrant (GSE164529 and GSE104985, P < 0.005) and palbociclib (GSE130437, P < 0.0005). These results suggest that downregulation of the RPS24 ex4:3 bp isoform represents a common feature in the development of drug resistance in breast cancer cells, independent of the mechanism of action of the drug. We experimentally confirmed a 1.7-fold decrease in tamoxifen-resistant cells relative to tamoxifen-sensitive cells through fragment analysis (Fig. 3g).
Identification of PTBP1 as an upstream regulator of RPS24 AS
We sought to identify the splicing factors that regulate RPS24 AS patterns. To this end, we leveraged a systematic splicing factor knockdown dataset (305 factors in HeLa cells)26. Since HeLa cells do not express the ex4:3 bp isoform, we focused on splicing factors that substantially altered the proportion of the ex4:22 bp isoform. The screening revealed that the knockdown of U2AF1 and PTBP1 led to a substantial increase in the proportion of ex4:22 bp, yielding proportions of 0.79 and 0.65, respectively, ranking as the top two most potent expression enhancers (Fig. 4a and Supplementary Table 1). By contrast, SF3B1 knockdown replicate experiments (SF3B1_r1 and SF3B1_r2) showed the lowest proportion of this isoform (0.05 and 0.03, respectively), as shown by the blue dots.

a The effect of systematic knockdown of 305 splicing factors on ex4:22 bp in HeLa cells (E-MTAB-11202). The y-axis shows the proportion of the ex4:22 bp isoform after each factor knockdown. U2AF1 and PTBP1 knockdown (red dots) markedly increased the ex4:22 bp proportion, while SF3B1 knockdown (blue dot) dramatically decreased it. b PTBP1 binding site analysis. Top: in silico prediction using RBPmap identifies multiple PTBP1 binding motifs (CUUUCU) in the polypyrimidine tracts upstream of all three microexons, with z-scores greater than 3 (red lines). Note: RBPmap uses the PTBP3 motif model, which is functionally equivalent to PTBP1 owing to shared binding specificity. High stringency (P value(significant) < 0.005 and Pvalue(suboptimal) < 0.01) with a conservation filter was applied. Bottom: experimental validation using CLIP-seq data (GSE220184) from HeLa cells confirming PTBP1 binding at the predicted sites, with binding signals also observed for other splicing factors (SF3B1, SF1, FUBP1, U2AF1 and U2AF2). c The effect of PTBP1 knockdown on RPS24 AS isoform composition in MCF7 breast cancer cells (GSE206142). The stacked bar graph shows notable increases in both ex4:3 bp (2.2-fold) and ex4:22 bp (1.7-fold) isoforms following PTBP1 depletion. d A proposed model of PTBP1-mediated regulation of RPS24 microexon splicing in breast cancer. PTBP1 binds to upstream intronic regions flanking the microexons and functions as a repressor of microexon inclusion. Under low PTBP1 conditions, microexon-containing isoforms (ex4:3 bp and ex4:22 bp) show increased expression, while high PTBP1 levels promote microexon skipping.
In the RBPmap database, we identified PTBP1 binding motifs with z-scores greater than 3 in the polypyrimidine tract upstream of the 3′ ss of all three microexons, using a high stringency filter option (Pvalue(significant) < 0.005 and Pvalue(suboptimal) < 0.01) with conservation filter (Fig. 4b, top). To confirm these in silico predictions, we examined independent CLIP-seq data from HeLa cells (GSE220184). The CLIP-seq data showed clear enrichment of PTBP1 binding at all three microexon regions (particularly strong at the 3-bp and 22-bp positions), with robust signals at positions matching the predicted motifs (Fig. 4b, bottom). This observation was consistent across all replicates. Furthermore, the CLIP-seq data revealed that U2AF2 exhibited similar binding patterns.
To validate the role of these factors in breast cancer models, we observed notable increases in both ex4:3 bp and ex4:22 bp isoforms following PTBP1 depletion (Fig. 4c). We observed a 2.2-fold increase in ex4:3 bp expression (from approximately 0.2 to 0.45) and a 1.7-fold increase in ex4:22 bp expression (from approximately 0.1 to 0.17) in the MCF7 (GSE206142) cell line, with a corresponding decrease in the ex4:ex6 isoform. These findings suggest that PTBP1 acts as a repressor of microexon inclusion in RPS24 (Fig. 4d) in breast cancer. In conditions with low PTBP1 protein expression, the microexon-containing isoforms appear to be derepressed, leading to their increased expression.
Ex4:3 bp as a potential clinical biomarker in breast cancer
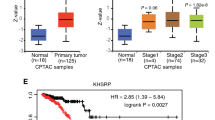
We next investigated the clinical relevance of RPS24 ex4:3 bp isoform expression patterns in cohorts of patients with breast cancer. Analysis of TCGA data revealed significantly higher ex4:3 bp expression in luminal subtypes (LumA and LumB) than in other molecular subtypes (Fig. 5a). This observation aligns with our cell line findings, as luminal subtypes are characteristically ER+. The molecular subtype-specific patterns observed in TCGA were also independently validated in the CPTAC dataset (Fig. 5c).

a The distribution of ex4:3 bp isoform proportions across molecular subtypes in TCGA breast cancer data, showing significantly higher expression in luminal subtypes (LumA and LumB) compared with HER2 and basal subtypes. n indicates the number of samples in each group. Molecular subtypes: LumA (luminal A: ER+/PR+/HER2−, low proliferation), LumB (luminal B: ER+ with high proliferation or HER2+), HER2 (HER2 enriched: ER−/PR−/HER2+), basal (basal-like: triple-negative, ER−/PR−/HER2−). b Correlation between ex4:3 bp expression and developmental lineage scores in TCGA. Ectoderm score shows the strongest positive correlation with ex4:3 bp expression, particularly in ER+ samples (r = 0.44) than in the ER− samples (r = 0.32). By contrast, mesoderm and endoderm scores showed weaker or negative correlations, whereas the stemness score showed weak correlations in opposite directions for ER+ and ER− samples. c Independent validation using CPTAC breast cancer data. Similar pattern of ex4:3 bp isoform distribution across molecular subtypes as observed in TCGA. d Analysis of MBC project data. Left: higher ex4:3 bp expression in ER+ samples than in ER− samples (P = 4 × 10−5). Right: significantly lower ex4:3 bp expression in ER+ samples with distant lymph node metastasis (P = 4 × 10−2), suggesting that decreased expression of this isoform correlates with metastatic progression.
We also examined correlations between ex4:3 bp expression and various developmental lineage scores in TCGA data, including ectoderm, mesoderm, endoderm and stemness scores (Fig. 5b). Among these, the ectoderm score showed a positive correlation with ex4:3 bp expression, particularly pronounced in ER+ samples (r = 0.44) compared with ER− samples (r = 0.32). By contrast, mesoderm and endoderm scores showed weaker or negative correlations in both ER+ and ER− samples. This specific association with ectoderm score suggests that the ex4:3 bp isoform may be linked to the maintenance of epithelial differentiation status in breast cancer, which is consistent with the epithelial origin of mammary tissue.
Considering that, typically, ER+ breast cancer has a more favorable prognosis than ER− breast cancer does, we examined whether patient survival differed on the basis of the ex4:3 bp expression status using the TCGA data. However, no significant correlation was observed with overall survival (P = 0.1) or cancer stages (P = 0.7) (Supplementary Fig. 6). Interestingly, while the ex4:3 bp isoform did not show prognostic value in terms of overall survival or staging in the primary tumor setting, analysis of the MBC project data revealed some clinical associations of potential interest (Fig. 5d). The MBC data confirmed higher ex4:3 bp expression in ER+ samples compared with ER− samples (P < 0.05) and showed an association between lower ex4:3 bp expression and distant lymph node metastasis status in ER+ samples (P < 0.05).
Discussion
Our comprehensive investigation into RPS24 AS in breast cancer reveals a complex regulatory network centered on the ex4:3 bp microexon isoform (Fig. 6). This study highlights three major findings: (1) the ex4:3 bp isoform serves as a molecular sensor for ER signaling and cell cycle regulation, (2) PTBP1 functions as a key upstream regulator controlling microexon inclusion and (3) the ex4:3 bp isoform exhibits potential as a predictive biomarker for drug resistance and epithelial differentiation status.

The schematic summarizes regulatory mechanisms and clinical implications of the RPS24 ex4:3 bp isoform in ER+ breast cancer. Inclusion of the 3-bp microexon (ex4:3 bp isoform) is repressed by PTBP1 binding to intronic regions, while ER signaling and cell cycle/growth pathway inhibition (via mTOR and CDK4/6 inhibitors) promote its expression. The ex4:3 bp isoform is downregulated in drug-resistant and metastatic cells but upregulated in response to targeted therapy, acting as a dynamic biomarker. High ex4:3 bp expression is associated with ER+ status, increased epithelial differentiation and sensitivity to treatment, while reduced expression correlates with drug resistance and metastatic potential. This isoform therefore serves as a predictive marker for therapeutic response and disease progression in breast cancer.
Our data demonstrate a strong dependency of the ex4:3 bp isoform on the ESR1 gene, supported by multiple lines of evidence, including ESR1 gene expression correlation, its elevated expression in cells where ESR1 is essential for survival and the effects of ESR1 gene mutations (Fig. 2). Building on recent reports that ER-alpha (encoded by ESR1) possesses post-transcriptional regulatory functions25, we discovered that ER-alpha interacts with RPS24 at multiple levels: through protein–protein interactions, RNA binding and its impact on cell fitness. This sophisticated interplay reveals a key regulatory mechanism where RPS24 AS actively participates in maintaining ER+ breast cancer cell identity and survival, extending beyond merely being a downstream target of ER signaling.
Our systematic analysis identified PTBP1 as a critical repressor of RPS24 microexon inclusion (Fig. 4). Computational predictions, CLIP-seq validation and functional studies collectively establish PTBP1 as a key regulatory node. PTBP1’s binding to polypyrimidine tracts upstream of all three microexons, coupled with its role in promoting exon skipping during cellular differentiation27,28, suggests RPS24 splicing is integrated into broader developmental programs. Incomplete rescue after PTBP1 depletion indicates the involvement of additional regulatory factors, consistent with models of collaborative RNA-binding protein control over microexon splicing29,30,31. The RPS24 microexons modify the C-terminal region, creating isoforms with distinct amino acid compositions. The negative correlation between ex4:3 bp transcript levels and RPS24 protein abundance suggests complex post-translational regulation. Upregulation of ex4:3 bp after mTOR and CDK4/6 inhibition highlights this C-terminal region as a signaling convergence point for growth control pathways. This parallels findings for RPS26, where the C-terminal domain integrates energy metabolism and AMPK–mTOR signaling32, indicating that ribosomal protein C termini may act as regulatory hubs linking RPS24 AS to ER signaling, growth and cell cycle pathways.
Consistent downregulation of ex4:3 bp across diverse drug resistance models suggests its involvement in a common resistance pathway. Although ex4:3 bp isoform shows potential as a biomarker for monitoring treatment response, large-scale prospective clinical validation using an objective measuring method will be required for clinical implementation. For this, our high-resolution detection method would be helpful.
A correlation between ex4:3 bp expression and ectoderm scores (especially in ER+ samples) suggests a role in maintaining epithelial differentiation. Reduced ex4:3 bp expression being associated with metastatic progression in ER+ breast cancers further provides a mechanistic link to epithelial–mesenchymal transition. However, its lack of correlation with overall survival or primary tumor staging indicates that ex4:3 bp is a dynamic biomarker primarily predicting treatment response rather than universal prognosis.
In summary, our findings reveal RPS24 AS as a sophisticated regulatory mechanism integrating multiple signaling pathways critical to breast cancer biology. The ex4:3 bp isoform acts as a molecular sensor responsive to ER signaling, cell cycle control and therapeutic interventions, regulated by both transcriptional (ER-alpha) and post-transcriptional (PTBP1) factors. C-terminal modifications from RPS24 AS create functional diversity, with distinct isoforms correlating with protein abundance, supporting the emerging concept of ‘specialized ribosomes’ that fine-tune translation on the basis of cellular context.
Key limitations include the need for mechanistic details of ER-alpha-mediated RPS24 splicing regulation, full characterization of the regulatory network beyond PTBP1 and functional consequences of different RPS24 protein isoforms. Future studies should focus on: (1) defining direct ER-alpha–RPS24 RNA interactions, (2) identifying additional PTBP1-cooperating regulatory factors, (3) characterizing functional differences between RPS24 protein isoforms and (4) validating clinical utility in prospective trials.
Data availability
The data generated and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Wright, C. J., Smith, C. W. J. & Jiggins, C. D. Alternative splicing as a source of phenotypic diversity. Nat. Rev. Genet. 23, 697–710 (2022).
Mehmood, A. et al. Systematic evaluation of differential splicing tools for RNA-seq studies. Brief. Bioinform. 21, 2052–2065 (2020).
Jiang, M., Zhang, S., Yin, H., Zhuo, Z. & Meng, G. A comprehensive benchmarking of differential splicing tools for RNA-seq analysis at the event level. Brief. Bioinform. https://doi.org/10.1093/bib/bbad121 (2023).
Park, J., Lee, J. O., Lee, M. & Chung, Y. J. AS-CMC: a pan-cancer database of alternative splicing for molecular classification of cancer. Sci. Rep. 12, 21074 (2022).
Brumwell, A., Fell, L., Obress, L. & Uniacke, J. Hypoxia influences polysome distribution of human ribosomal protein S12 and alternative splicing of ribosomal protein mRNAs. RNA 26, 361–371 (2020).
Wang, Y. et al. RPS24c isoform facilitates tumor angiogenesis via promoting the stability of MVIH in colorectal cancer. Curr. Mol. Med. 20, 388–395 (2020).
Kerry, J. et al. Autophagy-dependent alternative splicing of ribosomal protein S24 produces a more stable isoform that aids in hypoxic cell survival. FEBS Lett. 598, 503–520 (2024).
Olivieri, J. E. et al. RNA splicing programs define tissue compartments and cell types at single-cell resolution. eLife https://doi.org/10.7554/eLife.70692 (2021).
Park, J., Nam, D. H., Kim, D. & Chung, Y. J. RPS24 alternative splicing is a marker of cancer progression and epithelial-mesenchymal transition. Sci. Rep. 14, 13246 (2024).
Nam, D., Tian, B., Park, J. & Chung, Y. J. High-resolution detection of RPS24 microexon variations reveals novel splicing patterns in response to KRAS-targeted therapy in lung adenocarcinoma. BMB Rep. 58, 244–249 (2025).
Zardavas, D., Irrthum, A., Swanton, C. & Piccart, M. Clinical management of breast cancer heterogeneity. Nat. Rev. Clin. Oncol. 12, 381–394 (2015).
Nolan, E., Lindeman, G. J. & Visvader, J. E. Deciphering breast cancer: from biology to the clinic. Cell 186, 1708–1728 (2023).
Ban, Y. et al. Targeting ribosome biogenesis as a novel therapeutic approach to overcome EMT-related chemoresistance in breast cancer. eLife https://doi.org/10.7554/eLife.89486 (2024).
Tsoi, H., You, C. P., Leung, M. H., Man, E. P. S. & Khoo, U. S. Targeting ribosome biogenesis to combat tamoxifen resistance in ER+ve breast cancer. Cancers https://doi.org/10.3390/cancers14051251 (2022).
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 490, 61–70 (2012).
Krug, K. et al. Proteogenomic landscape of breast cancer tumorigenesis and targeted therapy. Cell 183, 1436–1456 (2020).
Parry, M. Introducing the Metastatic Breast Cancer Project: a novel patient-partnered initiative to accelerate understanding of MBC. ESMO Open 3, e000452 (2018).
Colaprico, A. et al. TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71 (2016).
Dobin, A. & Gingeras, T. R. Mapping RNA-seq reads with STAR. Curr. Protoc. Bioinform. https://doi.org/10.1002/0471250953.bi1114s51 (2015).
Paz, I., Kosti, I., Ares, M. Jr., Cline, M. & Mandel-Gutfreund, Y. RBPmap: a web server for mapping binding sites of RNA-binding proteins. Nucleic Acids Res. 42, W361–W367 (2014).
Hu, J., Qian, H., Xue, Y. & Fu, X. D. PTB/nPTB: master regulators of neuronal fate in mammals. Biophys. Rep. 4, 204–214 (2018).
Malta, T. M. et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173, 338–354 (2018).
Jun, Y. et al. Comprehensive analysis of alternative splicing in gastric cancer identifies epithelial–mesenchymal transition subtypes associated with survival. Cancer Res. 82, 543–555 (2022).
Ghandi, M. et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019).
Xu, Y. et al. ERα is an RNA-binding protein sustaining tumor cell survival and drug resistance. Cell 184, 5215–5229 (2021).
Rogalska, M. E. et al. Transcriptome-wide splicing network reveals specialized regulatory functions of the core spliceosome. Science 386, 551–560 (2024).
Linares, A. J. et al. The splicing regulator PTBP1 controls the activity of the transcription factor Pbx1 during neuronal differentiation. eLife 4, e09268 (2015).
Wang, K. et al. PTBP1 knockdown promotes neural differentiation of glioblastoma cells through UNC5B receptor. Theranostics 12, 3847–3861 (2022).
Mochizuki, Y. et al. Alternative microexon splicing by RBFOX2 and PTBP1 is associated with metastasis in colorectal cancer. Int. J. Cancer 149, 1787–1800 (2021).
Li, Y. I., Sanchez-Pulido, L., Haerty, W. & Ponting, C. P. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res. 25, 1–13 (2015).
Ruan, X. et al. Cell-type-specific splicing of transcription regulators and Ptbp1 by Rbfox1/2/3 in the developing neocortex. J. Neurosci. https://doi.org/10.1523/JNEUROSCI.0822-24.2024 (2025).
Havkin-Solomon, T. et al. Translation regulation of specific mRNAs by RPS26 C-terminal RNA-binding tail integrates energy metabolism and AMPK–mTOR signaling. Nucleic Acids Res. 51, 4415–4428 (2023).
Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (grant nos. RS-2022-NR075158, RS-2022-00165497 and 2019R1A5A2027588). This research was also supported by KREONET (Korea Research Environment Open NETwork), which is managed and operated by the Korea Institute of Science and Technology Information (KISTI). In addition, this study was supported by the Basic Medical Science Facilitation Program through the Catholic Medical Center of the Catholic University of Korea, funded by the Catholic Education Foundation.
Author information
Authors and Affiliations
Contributions
J.P. conceived the study, collected data and performed computational analyses. D.N. and S.J. conducted experimental validations. B.T. provided analytical guidance and critical interpretation of data. Y.C. supervised the overall study and coordinated the project. All authors reviewed and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Park, J., Nam, D., Jung, SH. et al. RPS24 microexon isoform as a novel biomarker for estrogen receptor-positive breast cancer progression and therapeutic resistance. Exp Mol Med 57, 2608–2618 (2025). https://doi.org/10.1038/s12276-025-01578-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-025-01578-y
