
Abstract
The tumor microenvironment (TME) is often hypoxic. EGLN1, which encodes the oxygen sensor PHD2, plays a crucial role not only in the survival of cancer cells but also in regulating other cell types that reside in the TME. In this Review, we explore the role of this protein in some of the key components of the TME, focusing on the functions of EGLN1/PHD2 in endothelial, stromal and immune cells. So far, the activity of EGLN1/PHD2 has been characterized in different cell types, albeit with controversial outcomes in different cancer settings. This Review aims to discuss the role of EGLN1/PHD2 in the TME and the strategies targeting this protein that might be used to hit tumors.
Similar content being viewed by others


Introduction
Oxygen is an essential element for the survival and growth of all aerobic organisms. However, hypoxic conditions may be experienced in pluricellular organisms during normal life and in pathological conditions. In aerobic cells, hypoxia may have deleterious effects that are prevented by the activation of the hypoxia response pathway.
This pathway is triggered by the oxygen sensors, the prolyl-4-hydroxylase domain proteins (PHD1, PHD2 and PHD3, encoded by the EGLN2, EGLN1, and EGLN3 genes, respectively), and the factor-inhibiting HIF (FIH). The PHD proteins hydroxylate proline residues (Pro402 and Pro564) on the HIF-α subunit in an oxygen-, iron- and α-ketoglutarate- (2-oxoglutarate, 2-OG) dependent manner. HIF-α hydroxylation leads to recognition by the von Hippel-Lindau tumor suppressor (pVHL), which targets HIF-α for proteasome degradation. Conversely, FIH hydroxylates the asparagine 803, leading to decreased affinity with co-activator p300/CBP1,2,3.
The HIF-α subunit is encoded by three genes, HIF1A, HIF2A or HIF3A, and, together with the HIF-β subunit, constitutes the heterodimeric HIF transcription factor. In hypoxic conditions, the PHD hydroxylases are inactive, and the HIF-α subunit is stabilized and translocated into the nucleus, where it dimerizes with HIF-β and binds to hypoxia response elements on the regulatory regions of genes that need to be transcribed in response to hypoxia1,2,3 (Fig. 1).

In normoxia, the PHD enzyme hydroxylates the HIF-α subunit, leading to recognition by pVHL and degradation by the proteasome. In hypoxia, PHD enzyme is inactive and the HIF-α subunit is stabilized, enters the nucleus, dimerizes with HIF-β subunit and activates the transcription of hypoxic response genes. Created in https://BioRender.com.
EGLN1, encoding the PHD2 isoform, is the most abundant and ubiquitously expressed of the three EGLNs. Besides being dependent on oxygen, iron and 2-OG, several regulatory mechanisms converge on PHD2, finely tuning the cellular and systemic response to hypoxia3. Hereafter, EGLN1 will be used to refer to the gene and PHD2 to the protein.
PHD2 is the preferential regulator of HIF-1α, playing a pivotal role during embryonic development and adult life in physiological conditions, as demonstrated by the embryonic lethality of its knockout in mice4.
In cancer, hypoxia and activation of hypoxia response pathways deeply influence both the tumor cells and the tumor microenvironment (TME). PHD2, being the primary oxygen sensor and the main regulator of HIF-1α, has been shown to influence the physiology of different TME cell populations5. In this Review, after discussing the complex relationship between PHD2/HIF and hypoxia in cancer cells, we will focus on the pivotal role of PHD2 in the response to hypoxia in TME cells. Finally, we will report pharmacological targeting strategies of the hypoxia pathway for cancer treatment and discuss open issues in including PHD2 inhibitors among these strategies.
Hypoxia, HIF and EGLN1 in cancer cells: a complex relationship
Hypoxia is highly prevalent in tumors owing to the rapid outgrowth of cancer cells, leading to structural and functional abnormalities in microvessels and increased diffusion distances. Importantly, hypoxia is widely associated with a worse prognosis in different cancer types6.
HIF-1α and HIF-2α have been frequently found aberrantly overexpressed in many different tumor tissues, compared with healthy counterparts, directly linking tumor hypoxia to HIF factor expression7. Meanwhile, different mechanisms besides hypoxia have been shown to induce HIF expression in cancer. In addition, the consequences of HIF activation may be variable as well, depending on HIF isoform and cancer type.
The inactivation of classical tumor suppressors, as well as many pro-oncogenic signals, leads to HIF activation. Loss of pVHL tumor suppressor, which is found in a familial angiomatous syndrome and the majority of sporadic cases of hemangioblastoma and clear-cell renal cell carcinoma (ccRCC), is the typical example that results in HIF-1α subunit stabilization due to lack of Cul2 ubiquitylation complex recognition8. However, the loss of PTEN and p53 has also been shown to promote HIF activity, with p53 directly contributing to HIF-1α degradation9,10. Several oncogenes, including H-RAS, v-src and c-Myc, have been shown to induce activation of HIF and its downstream target genes11,12,13. Many growth factors and intracellular signaling pathways activated during carcinogenesis can also stabilize and activate HIF-1α under normoxic conditions (for example, EGF, FGF2, insulin, IGF1-2, TNF, PDGF, AR, PI3K/AKT and BCR/ABL)8,14,15,16,17,18,19,20. HIF-1α subunits can be phosphorylated by different kinases on different threonine and serine residues with consequent stabilization or destabilization of the protein independently of oxygen tension15,21,22,23,24,25,26. Furthermore, other mechanisms, such as the regulation of mRNA stability and translation rate, may influence HIF levels in cancer cells27,28. Importantly, Rohwer and coworkers reported a poor association between intratumor hypoxic areas and HIF stabilization in colorectal cancer models, supporting a role for noncanonical HIF activation in promoting tumorigenesis29.
Although HIF-1α and HIF-2α have been widely shown to be overexpressed in many cancer types, they often play nonoverlapping and sometimes contrasting roles in cancer development and progression30. In most cases, HIF-1α seems to have a tumor-suppressive role, supported by its capacity to induce metabolic reprogramming, growth arrest and apoptosis, whereas HIF-2α is considered to have pro-oncogenic roles in inducing growth, pluripotency, and angiogenesis30. These differential functions are supported by distinct transcriptional programs regulated by the two factors31. However, the specific function of each factor (pro-oncogenic or tumor suppressor) is largely dependent on tumor type and, in some cases, also on tumor context. For example, in ccRCC, the respective pro-oncogenic role and tumor-suppressive role of HIF-1α and HIF-2α are well established32,33, but the recent development of a mouse model for this disease has questioned this concept. In this mouse model bearing an inducible deletion of VHL, TP53 and RB1 in renal cells, the HIF-1α deletion abrogated tumor growth, whereas HIF-2α had a limited effect. These results suggest a context-dependent effect of the two factors and a possible complementarity of their functions in cancer development34. Meanwhile, the transcriptional targets of the two factors also show a certain overlap, raising the question of whether they can compensate for each other31. Adding complexity to this field, HIF-1α factor expression has been frequently observed in the cytosolic compartment, and nontranscriptional roles of both HIF-1α and HIF-2α are emerging35,36,37.
The genes of the EGLN family have been reported to play variable roles in different cancer contexts. EGLN3 is a tumor suppressor in most cases, whereas EGLN1 and EGLN2 have more controversial roles. EGLN1, in particular, has been described as a tumor suppressor in colorectal and pancreatic cancer38,39, as a pro-oncogenic factor in acute myeloid leukemia and in lung and ovarian carcinoma40,41,42, and as having dual roles depending on the study in breast and hepatocellular carcinomas43,44,45,46. These contradictory roles can be partly explained by the differential enzymatic activity of the PHD proteins on the two main HIF isoforms, with HIF-1α preferentially hydroxylated by PHD2 and HIF-2α by PHD1 and PHD35,47,48. Nevertheless, a growing number of alternative PHD targets are being reported, including several proteins having a pivotal role in cancer development and progression, such as p53, AKT1, BRD4 and DYRK149,50,51,52,53. In addition, all three PHD proteins have been shown to have still poorly characterized enzymatic-independent activities54,55,56. Together, this evidence suggests a widespread function of PHD proteins in sensing oxygen and other nutrient levels that go beyond their canonical role in HIF regulation.
PHDs belong to a wider family of 2-oxoglutarate-dependent dioxygenase (2-OGDD), which comprises approximately 60–70 members, including collagen hydroxylases, Jumonji C (JmjC) domain-containing demethylases, the ten-eleven translocation (TET) DNA demethylases and various RNA demethylases57. Although all these enzymes are dependent on oxygen for their activity, in many cases, it is unclear whether they may play the role of oxygen sensors, due to very high or unknown affinity for oxygen. Interestingly, KDM5A and KDM6A histone demethylases, belonging to the JmjC subfamily, can respond to oxygen level variations and modify histone methylation in a HIF-independent manner, leading to chromatin remodeling and gene expression regulation58,59.
Finally, the cells may sense oxygen also through other mechanisms. For example, Deygas and collaborators demonstrated that epithelial cells migrate toward areas of higher oxygen concentration in a HIF- and PHD-independent manner, mediated by EGFR’s ability to sense reactive oxygen species60.
Taken together, these findings depict a complex regulatory network in which HIF factors are stabilized not only by hypoxia but also by other mechanisms, PHD enzymes have targets beyond HIFs, and the PHD–HIF axis is not the sole cellular oxygen sensor (Fig. 2). This complexity should be taken into consideration when studying hypoxia, both in cancer cells and in the TME.

Hypoxia can regulate other enzymes of the 2-OGDD family, besides the PHDs and FIH. Different mechanisms can regulate the activity of the PHD enzymes, including the presence of binding partners, the metabolic status of the cell and specific post-translational modifications (PTM). The HIF proteins are the main targets of PHDs, but several alternative targets have been described, including p53, AKT1, BRD4 and DYRK1, holding the possibility to influence tumor behaviour. Finally, a number of pro-oncogenic signals have been shown to stabilize the HIF-α subunit in a oxygen-independent way. Created in https://BioRender.com.
EGLN1-dependent response to hypoxia in the TME
Hypoxia affects cancer cells as well as host cell populations residing in the TME, including endothelial cells, fibroblasts and different immune populations. The EGLN1 gene has been shown to have a key role in regulating the response to hypoxia in these populations, supporting various pro-oncogenic or tumor-suppressive functions.
Angiogenesis
Vasculogenesis is classically defined as the de novo blood vessel formation from endothelial progenitor cells during embryonic life, whereas angiogenesis arises from the systemic and local need for nutrition and oxygen supply during adult life. Abnormal vessel formation is often associated with the onset of malignant disorders61,62. However, normal blood vessels distribute oxygen and nutrients evenly, whereas tumor blood vessels have an immature structure and high permeability63. These abnormal blood vessels mainly derive from tumor-induced angiogenesis, but alternative mechanisms may be involved, such as adult vasculogenesis and vascular mimicry exerted by tumor cells64. Increasing tumor volume, hypoxia and downstream activation of the HIF pathway induce angiogenesis to increase blood supply to cancer cells65. VEGF, the key soluble factor involved both in angiogenesis and vascular mimicry, is one of the most widely recognized targets of HIF transcription factors66. Based on these findings, antiangiogenetic drugs were developed and applied for cancer treatment in different settings. However, in most cancers, blood vessels are twisted, deregulated and dysfunctional67,68,69, indicating that vessel normalization might be the best strategy70. Indeed, blocking VEGF expression prunes immature vessels and increases their maturation71 (Fig. 3).

a,b The hypoxic TME shapes blood vessels and induces vasculogenesis (a) to feed the tumor and potentially lead to metastasis (b). c Angiogenesis normalization may lead to hypoxia relief, anticancer drug delivery and immune surveillance reactivation. Created in https://BioRender.com.
PHD2 appears to be the key PHD isoform regulating adult angiogenesis in mice, as its deficiency leads to hyperactive angiogenesis, whereas PHD1- or PHD3-deficient mice do not exhibit angiogenic defects69.
Moreover, in a syngeneic model of cancer growth, EGLN1 haplodeficiency in host cells induces the normalization of the endothelial cell lining, leading to improved tumor oxygenation and drug diffusion, and to decreased metastatization72,73. In addition, inducible PHD2 KO in myeloid cells improves angiogenesis by attenuating the pro-inflammatory phenotype of macrophages74.
Complete PHD2 ablation in mouse endothelial and hematopoietic cells leads to extensive pulmonary vascular remodeling, including vascular occlusion and plexiform-like lesions, recapitulating the pathology of pulmonary arterial hypertension75. Conversely, PHD2 overexpression in endothelial cells suppresses hypoxia-induced cell proliferation76.
PHD2 ablation may enhance both angiogenesis and vasculogenesis, through increased HIF-1α stabilization as well as other collateral pathways. Indeed, PHD2 inhibition induces the release of pro-angiogenic factors, such as IL-8 and angiogenin, in a HIF-independent and NF-kB-dependent manner. These factors mediate tumor angiogenesis and the recruitment of bone marrow-derived cells77.
Overall, all the evidence indicates that PHD2 levels are fundamental for correct angiogenesis in physiological conditions. Conversely, in cancer, where blood vessel formation is aberrantly stimulated, decreasing PHD2 levels or activity may lead to angiogenesis normalization.
Fibroblasts
Cancer-associated fibroblasts (CAFs) are directly or indirectly reprogrammed by cancer cells to be activated and produce extracellular matrix to favor microenvironment remodeling78 in several cancer types79, including pancreatic adenocarcinoma (PDAC)80, gastrointestinal cancer81 and breast cancer79.
Furthermore, the activated CAFs supply high-energy substances, including ketone bodies and lactate, to cancer cells through their autophagic pathways82,83.
The TME is highly oxidative. This activates two pro-autophagic pathways in stromal fibroblasts: one driven by HIF-1α and another orchestrated by NFκB84. As a result, CAFs initiate autophagy and mitophagy, leading to metabolic and proteomic reprogramming85. Autophagy sustains cancer cell growth because CAF debris and nutrients can be recycled. At the same time, HIF-1α-induced mitophagy enhances aerobic glycolysis, and CAFs secrete high-energy nutrients that can further boost oxidative metabolism in cancer cells86.
Nevertheless, autophagy in fibroblasts can also inhibit tumor progression in the early stages because the secreted intermediates might activate immune cells. Conversely, CAF autophagy promotes TME inflammation, hypoxia and the regulation of immune checkpoints at later time points in tumor development87.
In PDAC, myofibroblastic CAFs are characterized by αSMA expression and are considered to have tumor-restraining function, while inflammatory CAFs (iCAF) express lower levels of αSMA, produce inflammatory cytokines and enhance tumor growth88. Hypoxia potentiates the effects of cytokines secreted by PDAC cells, leading to a switch toward an iCAF phenotype, which is associated with worse cancer prognosis and therapy resistance. In this setting, HIF-1α stabilization induces the iCAF phenotype, while the secreted cytokines cooperate to boost HIF-1α transcriptional activity in a self-loop manner. This is mainly supported by IL-1α produced by PDAC cells in hypoxic conditions89.
However, prolonged hypoxia deactivates CAFs and reduces their ability to remodel and invade their surrounding matrix90. Indeed, in vitro treatment with PHD inhibitor dimethyloxalylglycine (DMOG) or PHD2 silencing leads to CAF deactivation and extracellular matrix remodeling in a HIF-1α-dependent manner. These findings are mirrored by results in orthotopic cancer models in which (DMOG) treatment or cotransplant with PHD2-silenced CAFs led to a marked decrease of metastatic load, without altering primary tumor growth90. Another report showed similar results in a spontaneous model of breast tumorigenesis, generated in the EGLN1 haplodeficient background. These mice showed the same primary tumor growth as EGLN1 wild type, but decreased metastases due to reduced CAFs activation and improved blood vessels. Moreover, the authors showed that CAF impairment was not due to PHD2 inhibition in CAFs, but rather in cancer cells. In fact, EGLN1 haplodeficient cancer cells secrete less TGF-β, which in turn leads to reduced CAF activation91.
Thus, inhibiting or reducing PHD2 activity might be crucial to inhibit tumor progression and CAF protumoral support. (Fig. 4).

PHD2 inhibition in CAFs induces their deactivation, resulting in TME matrix remodeling and reduced metastatic potential of cancer cells. Created in https://BioRender.com.
Macrophages
Tumor-associated macrophages (TAMs) can be clustered and characterized on the basis of their receptor expression, effector function and cytokine production. Several microenvironmental signals can induce M1 or M2 polarization in vivo and in vitro. Classically activated M1 macrophages are usually the effector cells that kill microorganisms and tumor cells, while alternatively activated M2 cells dampen inflammatory responses, scavenge debris and promote angiogenesis92. M1 macrophages secrete IL-12 and tumor necrosis factor (TNF), while M2 macrophages typically produce IL-1093.
M1-like macrophages can be found in the TME during the early phase of tumorigenesis when a pro-inflammatory response is still ongoing. They further activate effector immune cells, such as CD8+ cytotoxic T cells and natural killer cells94,95. TNF is a positive regulator of M1 polarization and a negative regulator of M2 polarization when the NF-κB pathway is activated. Furthermore, myeloid differentiation primary response 88 (MyD88) suppresses M2-associated gene expression in TAMs, resulting in an antitumor M1 phenotype polarization96.
An elevated M1/M2 TAM ratio is associated with a better prognosis in non-small cell lung cancer97,98, colorectal cancer99, ovarian cancer100, breast cancer101 and oral squamous cell carcinoma102. High M2 percentages, conversely, are linked to worse outcomes and thrive in a less inflammatory TME, favoring tumor growth103.
Strong hypoxic TME and large numbers of TAMs correlate with a decreased survival rate104. M2 macrophages express low levels of the MHCII complex or HLA-DR (in mice and humans, respectively) and were found in the most hypoxic areas inside human tumors105. Metabolism is tightly linked to TAMs functionality, in particular, M1, with high MHCII expression exhibiting a hampered TCA cycle. Conversely, M2 TAMs showed higher oxidative and glycolytic metabolism compared with M1 TAMs, which show less mitochondrial activity. These M2 TAMs can fuel their activity with lactate, increasing L-arginine metabolism and enhancing their T cell suppressive capacity106. HIF-1α is essential for macrophages activation and infiltration in vivo. HIF1A knockout in these cells results in impaired glycolytic capacity and drastic reduction of ATP production107. HIF-1α expression is also required for myeloid cells to kill pathogenic bacteria107,108.
M2 TAMs are associated with hypoxic regions, whereas M1 TAMs are located preferentially in better-oxygenated areas. EGLN1 haplodeficiency induces vasculature normalization and enhances tumor oxygenation in mice. In this system, oxygen availability does not alter the attraction of monocytes to the tumor site nor the efficiency of monocyte differentiation into M2 or M1 TAMs. Less hypoxia does not influence M2 markers expression, albeit it downregulates the expression of typical genes involved in angiogenesis, metastasis and response to hypoxia. Laoui and colleagues concluded that tumor-infiltrating monocytes do not fail to differentiate because of hypoxia, but are instead influenced by other microenvironmental stimuli. They are attracted to hypoxic areas where they perform their protumoral activity109. In vitro, EGLN1 knockout induces metabolic reprogramming of myeloid cells toward anaerobic glycolysis through increasing pyruvate dehydrogenase kinase 1 (PDK1) protein levels and decreasing pyruvate dehydrogenase enzyme activity, while HIF1A knockout inhibits this glycolytic reprogramming110. The inhibition of the glycolytic shift also suppresses macrophage migration and functionality111. These findings are in line with another report showing that depletion of HIF-1α in myeloid cells dramatically reduces glucose transporters and glycolytic enzyme expressions, resulting in a defective inflammatory response110.
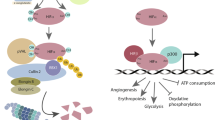
Treatment with PHD2 inhibitor roxadustat in a syngeneic model of lung cancer and melanoma reduces tumor growth by increasing the macrophages’ phagocytic activity and by inducing tumor vessel normalization, through the PHD–HIF axis112 (Fig. 5).

PHD2 genetic or pharmacological inhibition leads to metabolic reprogramming and increased phagocytic activity in TAMs. Created in https://BioRender.com.
T lymphocytes
T cells are one of the most represented immune populations infiltrating solid tumors, being recruited to tumor sites by TME-secreted factors113. There, they can have different phenotypic differentiation and functions. Cytotoxic CD8+ T cells recognize tumor cells’ neo-antigens and kill tumor cells, creating perforin and granzyme pores on their membrane and inducing cell apoptosis through the IFNγ and TNF pathways114. According to their antitumoral function, their numbers are generally associated with a better prognosis115.
CD4+ T cells have more variable roles. In most cases, the anti-inflammatory TME primes them with regulatory functions (Treg). These Treg cells have been characterized in several solid cancers. One of their main activities is to secrete anti-inflammatory cytokines, which hinder other T cell functions and promote tumor growth and metastasis116. CD4+ T cells may also differentiate into T helper cells, recruiting and activating natural killer cells and CD8+ T cells to the tumor site117.
The role of HIF-1α in T cells is rather controversial. Hypoxia induces a metabolic shift in CD8+ T cells, which enhances their antitumor activity118. On the other hand, hypoxic CD8+ T cells have decreased proliferative capacity due to metabolic remodeling119.
Nevertheless, the concomitant deletion of EGLN1 and EGLN3 genes in already primed CD8+ T cells enhanced their effector activity. These cells reduce tumor growth when adoptively transferred into several mouse cancer models. It is also relevant that culturing CD8+ T cells under hypoxic conditions boosted their antitumor activity in vivo through increased glucose metabolism and granzyme/perforin granule production120,121. This effect was independent of IFNγ secretion, even though its mRNA was upregulated.
Genetic deletion of EGLN1 in T lymphocytes increases EG7-OVA tumor-killing activity. These cells have increased intracellular positive staining for IFNγ, TNF and granzyme B compared with wild-type controls122. The stabilization of HIF-1α in CD4+ T cells improves the effect of anti-PD-1 therapy to reduce tumor growth and patient survival. Similarly, VHL deletion in T cells, which leads to HIF-1α accumulation, prevents exhaustion in CD8+ T cells during chronic viral infections. Moreover, PHD protein expression in T cells is needed to induce tolerance against lung metastases due to an increase in Treg cells and a decrease in CD8+ T cells’ effector function. Consequently, T cell-specific deletion of all three PHDs or pharmacological inhibition restrains lung colonization and improves the effect of adoptive transfer immunotherapy123.
Notably, PHD inhibitors activate CD8+ T cells through HIF-1α stabilization and induction of co-stimulatory molecules, leading to increased antitumor efficacy of adoptive T cell therapy124 (Fig. 6).

PHD2 inactivation shifts CD8+ T cell metabolism, resulting in an increased tumor killing potential. Created in https://BioRender.com.
Hypoxia pathway pharmacological targeting for cancer treatment: time to include PHD inhibitors?
Due to the intimate association between hypoxia and tumors, targeting this pathway is regarded as a promising strategy to overcome different types of cancer. Major pharmacological targets in this pathway are the PHD enzymes, the HIF transcription factors and the HIF main effectors, such as VEGF (Fig. 7 and Supplementary Table 1). The latter is a well-established drug target, with specific monoclonal antibodies such as bevacizumab that have been in clinical practice for almost 20 years. Anti-VEGF drugs were originally developed to inhibit hypoxia-induced angiogenesis, aiming to starve tumors and restrain metastatic spreading. However, the efficacy of these treatments was limited in monotherapy and remains circumscribed to some cancer types in combination with other agents. Notably, other mechanisms of action emerged, such as vasculature normalization and immune modulation, partially explaining tumor-specific effects and indicating the possibility of more rational combinations125.

Several PHD enzymes inhibitors such as roxadustat, daprodustat and vadadustat have been developed for the treatment of anemia. Belzutifan is a small molecule specifically inhibiting the dimerization of HIF-2α with HIF-β. Inhibitors of the HIF transcriptional target VEGF have been used for more than 20 years for cancer treatment. Created in https://BioRender.com.
Due to the pivotal role of HIF transcription factors in cancer cell regulation and their multifaceted role in promoting an immune-suppressive microenvironment, they have been considered promising targets for a long time. Several agents have been shown to directly or indirectly target HIF factor expression, protein synthesis, stabilization and dimerization. However, most of these inhibitors regulate HIFs indirectly and/or in addition to other pathways, making it difficult to discriminate the HIF-dependent effects30,126. In 2009, Scheuermann and collaborators discovered a cavity within the PAS-B domain of HIF-2α, which is not present in HIF-1α, leading to the development of specific inhibitors of HIF-2α dimerization with HIF-β subunit127. Belzutifan is the most advanced of these inhibitors and demonstrated efficacy in preclinical models and clinical trials of VHL-related cancers such as ccRCC and hemangioblastoma, receiving US Food and Drug Administration approval for these indications in 2021128,129. Combinations of belzutifan with other anticancer agents, including immunotherapy, are currently under investigation in several clinical trials130.
A relatively recent strategy proposes to alleviate TME hypoxia itself, resulting in simultaneous downregulation of HIF activity and TME reprogramming, including the promotion of immune cell recruitment and activation. These effects may be achieved by vascular normalization, leading to increased oxygen and nutrient concentration but also fostering immune cell infiltration and anticancer drug delivery131,132.
Mazzone and colleagues developed a syngeneic mouse model carrying EGLN1 haplodeficiency in the host animal and showed that PHD2 deficiency leads to vessel normalization, improving tumor perfusion and oxygenation, and restraining metastatic spreading72. In this model, loss of PHD2 also results in better chemotherapy delivery and protection of normal organs from chemotherapy side effects133. In addition, EGLN1 deletion in T cells promotes the differentiation of effector T cells, improving tumor control and response to immune checkpoint blockade treatment122. Notably, PHD2 genetic or pharmacologic inhibition in cancer cells has variable effects depending on cancer type, showing anti-oncogenic activity in melanoma, lung, ovarian and osteosarcoma cancer40,41,134. Taken together, this evidence indicates that PHD2 systemic inhibition through pharmacologic approaches may exert anticancer activity through a double effect on cancer cells and TME, at least in some cancer types.
PHD enzyme inhibitors have been developed for the treatment of other diseases, such as anemia and ischemia135. Clinical trials of different PHD inhibitors showed efficacy in the management of anemia associated with chronic kidney disease with limited side effects, leading to the approval of roxadustat, daprodustat and vadadustat for the treatment of this pathology in different countries136. These results indicate that the use of PHD inhibitors may also be safe in neoplastic disease and can be repurposed as agents capable of influencing both the cancer cells and the TME.
Notably, all the available inhibitors rely on competition with the 2-OG cofactor, which is shared by all three enzymes and also by the other dioxygenases of the superfamily. As the three PHD enzymes play different roles in different cancer contexts, specificity issues may arise. All the tested inhibitors induce erythropoiesis in vivo, while they show some isoform specificity in vitro135. However, due to differential functions of the PHD proteins in cancer, the activity of each inhibitor on the three PHD enzymes and downstream pathways should be carefully evaluated before repurposing in the oncology context. Furthermore, the activity of these inhibitors on other dioxygenases is often poorly investigated and should be taken into consideration. Ideally, the development of isoform-selective inhibitors would be optimal for the precise targeting of these enzymes and for limiting undesired side effects.
Conclusions
In summary, EGLN1/PHD2 is known as a key regulator in the cellular response to hypoxia. Furthermore, it can trigger a plethora of other metabolic and regulatory mechanisms that affect the TME as a whole. PHD2 activities can have different outcomes based on the context, including the tumor subtype or the TME cell population. In most cases, however, PHD2 triggers a cascade of events that promote tumor growth, drive abnormal blood vessel formation, and suppress immune cell activity. For these reasons, inhibiting PHD2 may represent a novel and effective anticancer strategy, as it affects both cancer cells and the TME. Although several PHD inhibitors are already in clinical practice for the treatment of anemia, specificity issues may arise and limit their use in oncology. Therefore, the generation of new, stronger and more selective inhibitors may be beneficial for the optimal targeting of this intriguing protein for anticancer therapy.
References
Kaelin, W. G. J. & Ratcliffe, P. J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 (2008).
Ivan, M. & Kaelin, W. G. J. The EGLN–HIF O2-sensing system: multiple inputs and feedbacks. Mol. Cell 66, 772–779 (2017).
Strocchi, S., Reggiani, F., Gobbi, G., Ciarrocchi, A. & Sancisi, V. The multifaceted role of EGLN family prolyl hydroxylases in cancer: going beyond HIF regulation. Oncogene 41, 3665–3679 (2022).
Takeda, K. et al. Placental but not heart defects are associated with elevated hypoxia-inducible factor alpha levels in mice lacking prolyl hydroxylase domain protein 2. Mol. Cell Biol. 26, 8336–8346 (2006).
Appelhoff, R. J. et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 279, 38458–38465 (2004).
Vaupel, P. & Mayer, A. Hypoxia in cancer: significance and impact on clinical outcome. Cancer Metastasis Rev. 26, 225–239 (2007).
Talks, K. L. et al. The expression and distribution of the hypoxia-inducible factors HIF-1α and HIF-2α in normal human tissues, cancers, and tumor-associated macrophages. Am. J. Pathol. 157, 411–421 (2000).
Maxwell, P. H., Pugh, C. W. & Ratcliffe, P. J. Activation of the HIF pathway in cancer. Curr. Opin. Genet. Dev. 11, 293–299 (2001).
Zundel, W. et al. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 14, 391–396 (2000).
Ravi, R. et al. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1alpha. Genes Dev. 14, 34–44 (2000).
Mazure, N. M., Chen, E. Y., Laderoute, K. R. & Giaccia, A. J. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood 90, 3322–3331 (1997).
Jiang, B. H., Agani, F., Passaniti, A. & Semenza, G. L. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: involvement of HIF-1 in tumor progression. Cancer Res. 57, 5328–5335 (1997).
Shim, H. et al. c-Myc transactivation of LDH-A: implications for tumor metabolism and growth. Proc. Natl Acad. Sci. USA 94, 6658–6663 (1997).
Mabjeesh, N. J., Willard, M. T., Frederickson, C. E., Zhong, H. & Simons, J. W. Androgens stimulate hypoxia-inducible factor 1 activation via autocrine loop of tyrosine kinase receptor/phosphatidylinositol 3′-kinase/protein kinase B in prostate cancer cells. Clin. Cancer Res. 9, 2416–2425 (2003).
Richard, D. E., Berra, E. & Pouyssegur, J. Nonhypoxic pathway mediates the induction of hypoxia-inducible factor 1α in vascular smooth muscle cells. J. Biol. Chem. 275, 26765–26771 (2000).
Zelzer, E. et al. Insulin induces transcription of target genes through the hypoxia-inducible factor HIF-1α/ARNT. EMBO J. 17, 5085–5094 (1998).
Zhong, H. et al. Modulation of hypoxia-inducible factor 1α expression by the epidermal growth factor/phosphatidylinositol 3-kinase/PTEN/AKT/FRAP pathway in human prostate cancer cells: implications for tumor angiogenesis and therapeutics. Cancer Res. 60, 1541–1545 (2000).
Feldser, D. et al. Reciprocal positive regulation of hypoxia-inducible factor 1α and insulin-like growth factor 2. Cancer Res. 59, 3915–3918 (1999).
Mayerhofer, M., Valent, P., Sperr, W. R., Griffin, J. D. & Sillaber, C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood 100, 3767–3775 (2002).
Mayro, B. et al. ABL kinases regulate the stabilization of HIF-1α and MYC through CPSF1. Proc. Natl Acad. Sci. USA 120, e2210418120 (2023).
Flügel, D., Görlach, A., Michiels, C. & Kietzmann, T. Glycogen synthase kinase 3 phosphorylates hypoxia-inducible factor 1alpha and mediates its destabilization in a VHL-independent manner. Mol. Cell Biol. 27, 3253–3265 (2007).
Xu, D., Yao, Y., Lu, L., Costa, M. & Dai, W. Plk3 functions as an essential component of the hypoxia regulatory pathway by direct phosphorylation of HIF-1α. J. Biol. Chem. 285, 38944–38950 (2010).
Cam, H., Easton, J. B., High, A. & Houghton, P. J. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol. Cell 40, 509–520 (2010).
Warfel, N. A., Dolloff, N. G., Dicker, D. T., Malysz, J. & El-Deiry, W. S. CDK1 stabilizes HIF-1α via direct phosphorylation of Ser668 to promote tumor growth. Cell Cycle 12, 3689–3701 (2013).
Bullen, J. W. et al. Protein kinase A-dependent phosphorylation stimulates the transcriptional activity of hypoxia-inducible factor 1. Sci Signal 9, ra56 (2016).
Casillas, A. L. et al. Direct phosphorylation and stabilization of HIF-1α by PIM1 kinase drives angiogenesis in solid tumors. Oncogene 40, 5142–5152 (2021).
El-Naggar, A. M. et al. Translational activation of HIF1α by YB-1 promotes sarcoma metastasis. Cancer Cell 27, 682–697 (2015).
Lu, M. et al. CIRBP is a novel oncogene in human bladder cancer inducing expression of HIF-1α. Cell Death Dis. 9, 1046 (2018).
Rohwer, N. et al. Non-canonical HIF-1 stabilization contributes to intestinal tumorigenesis. Oncogene 38, 5670–5685 (2019).
Cowman, S. J. & Koh, M. Y. Revisiting the HIF switch in the tumor and its immune microenvironment. Trends Cancer 8, 28–42 (2022).
Downes, N. L., Laham-Karam, N., Kaikkonen, M. U. & Ylä-Herttuala, S. Differential but complementary hif1α and hif2α transcriptional regulation. Mol. Ther. 26, 1735–1745 (2018).
Gordan, J. D. et al. HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 14, 435–446 (2008).
Shen, C. et al. Genetic and functional studies implicate HIF1α as a 14q kidney cancer suppressor gene. Cancer Discov. 1, 222–235 (2011).
Hoefflin, R. et al. HIF-1α and HIF-2α differently regulate tumour development and inflammation of clear cell renal cell carcinoma in mice. Nat. Commun. 11, 4111 (2020).
Persson, C. U. et al. ARNT-dependent HIF-2 transcriptional activity is not sufficient to regulate downstream target genes in neuroblastoma. Exp. Cell Res. 388, 111845 (2020).
Uniacke, J. et al. An oxygen-regulated switch in the protein synthesis machinery. Nature 486, 126–129 (2012).
Jn, L. et al. A transcription-independent role for HIF-1α in modulating microprocessor assembly. Nucleic Acids Res. 52, 11806-11821 (2024).
Wang, L. et al. PHD2 exerts anti-cancer and anti-inflammatory effects in colon cancer xenografts mice via attenuating NF-κB activity. Life Sci. 242, 117167 (2020).
Su, Y. et al. Prolyl hydroxylase-2 (PHD2) exerts tumor-suppressive activity in pancreatic cancer. Cancer 118, 960–972 (2012).
Reggiani, F. et al. An integrative functional genomics approach reveals EGLN1 as a novel therapeutic target in KRAS mutated lung adenocarcinoma. Mol. Cancer 20, 63 (2021).
Price, C. et al. Genome-wide interrogation of human cancers identifies EGLN1 dependency in clear cell ovarian cancers. Cancer Res. 79, 2564–2579 (2019).
Lawson, H. et al. The selective prolyl hydroxylase inhibitor IOX5 stabilizes HIF-1α and compromises development and progression of acute myeloid leukemia. Nat. Cancer 5, 916–937 (2024).
Di Conza, G., Trusso Cafarello, S., Zheng, X., Zhang, Q. & Mazzone, M. PHD2 targeting overcomes breast cancer cell death upon glucose starvation in a PP2A/B55α-mediated manner. Cell Rep 18, 2836–2844 (2017).
Zhen, L., Shijie, N. & Shuijun, Z. Tumor PHD2 expression is correlated with clinical features and prognosis of patients with HCC receiving liver resection. Medicine 93, e179 (2014).
Heindryckx, F. et al. Effect of prolyl hydroxylase domain-2 haplodeficiency on the hepatocarcinogenesis in mice. J. Hepatol. 57, 61–68 (2012).
Jiang, W. et al. A mitochondrial EglN1–AMPKα axis drives breast cancer progression by enhancing metabolic adaptation to hypoxic stress. EMBO J 42, e113743 (2023).
Berra, E. et al. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 22, 4082–4090 (2003).
Chowdhury, R. et al. Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases. Nat. Commun. 7, 12673 (2016).
Ullah, H., De Filippis, A., Santarcangelo, C. & Daglia, M. Epigenetic regulation by polyphenols in diabetes and related complications. Med. J. Nutr. Metab. 13, 289–310 (2020).
Rodriguez, J. et al. PHD3 regulates p53 protein stability by hydroxylating proline 359. Cell Rep. 24, 1316–1329 (2018).
Guo, J. et al. pVHL suppresses kinase activity of Akt in a proline-hydroxylation-dependent manner. Science 353, 929–932 (2016).
Erber, L., Luo, A. & Chen, Y. Targeted and interactome proteomics revealed the role of PHD2 in regulating BRD4 proline hydroxylation. Mol. Cell Proteomics 18, 1772–1781 (2019).
Lee, S. B. et al. Proline hydroxylation primes protein kinases for autophosphorylation and activation. Mol. Cell 79, 376–389 (2020).
To, K. K. W. & Huang, L. E. Suppression of hypoxia-inducible factor 1α (HIF-1α) transcriptional activity by the HIF prolyl hydroxylase EGLN1. J Biol. Chem. 280, 38102–38107 (2005).
Garvalov, B. K. et al. PHD3 regulates EGFR internalization and signalling in tumours. Nat. Commun. 5, 5577 (2014).
D’Hulst, G. et al. PHD1 controls muscle mTORC1 in a hydroxylation-independent manner by stabilizing leucyl tRNA synthetase. Nat. Commun. 11, 174 (2020).
Frost, J., Frost, M., Batie, M., Jiang, H. & Rocha, S. Roles of HIF and 2-oxoglutarate-dependent dioxygenases in controlling gene expression in hypoxia. Cancers 13, 350 (2021).
Chakraborty, A. A. et al. Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222 (2019).
Batie, M. et al. Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–1226 (2019).
Deygas, M. et al. Redox regulation of EGFR steers migration of hypoxic mammary cells towards oxygen. Nat. Commun. 9, 4545 (2018).
Chung, M. S. & Han, S. J. Endometriosis-associated angiogenesis and anti-angiogenic therapy for endometriosis. Front. Glob. Womens Health 3, 856316 (2022).
D’Alessio, A., Moccia, F., Li, J.-H., Micera, A. & Kyriakides, T. R. Angiogenesis and vasculogenesis in health and disease. BioMed Res. Int. 2015, 126582 (2015).
Majidpoor, J. & Mortezaee, K. Angiogenesis as a hallmark of solid tumors—clinical perspectives. Cell Oncol. 44, 715–737 (2021).
Lugano, R., Ramachandran, M. & Dimberg, A. Tumor angiogenesis: causes, consequences, challenges and opportunities. Cell Mol. Life Sci. 77, 1745–1770 (2020).
Höckel, M. & Vaupel, P. Tumor hypoxia: definitions and current clinical, biologic, and molecular aspects. J. Natl Cancer Inst. 93, 266–76 (2001).
Asikainen, T. M. et al. Stimulation of HIF-1α, HIF-2α, and VEGF by prolyl 4-hydroxylase inhibition in human lung endothelial and epithelial cells. Free Radic. Biol. Med. 38, 1002–1013 (2005).
Thurston, G. et al. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 286, 2511–2514 (1999).
Semenza, G. L. Hypoxia-inducible factor 1: master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 8, 588–594 (1998).
Takeda, K., Cowan, A. & Fong, G.-H. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 116, 774–781 (2007).
Visconti, R. P., Richardson, C. D. & Sato, T. N. Orchestration of angiogenesis and arteriovenous contribution by angiopoietins and vascular endothelial growth factor (VEGF). Proc. Natl Acad. Sci. USA 99, 8219–8224 (2002).
Abramovitch, R., Dafni, H., Smouha, E., Benjamin, L. E. & Neeman, M. In vivo prediction of vascular susceptibility to vascular susceptibility endothelial growth factor withdrawal: magnetic resonance imaging of C6 rat glioma in nude mice. Cancer Res. 59, 5012–5016 (1999).
Mazzone, M. et al. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell 136, 839–851 (2009).
De Bock, K., Cauwenberghs, S. & Carmeliet, P. Vessel abnormalization: another hallmark of cancer? Molecular mechanisms and therapeutic implications. Curr. Opin. Genet. Dev. 21, 73–79 (2011).
Sluiter, T. J. et al. Myeloid PHD2 conditional knockout improves intraplaque angiogenesis and vascular remodeling in a murine model of venous bypass grafting. J. Am. Heart Assoc. 13, e033109 (2024).
Dai, Z., Li, M., Wharton, J., Zhu, M. M. & Zhao, Y.-Y. Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor-2α. Circulation 133, 2447–2458 (2016).
Takeda, K. & Fong, G.-H. Prolyl hydroxylase domain 2 protein suppresses hypoxia-induced endothelial cell proliferation. Hypertension 49, 178–184 (2007).
Haley, M. J. et al. Hypoxia coordinates the spatial landscape of myeloid cells within glioblastoma to affect survival. Sci. Adv. 10, eadj3301 (2024).
Sahai, E. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 20, 174–186 (2020).
Yang, D., Liu, J., Qian, H. & Zhuang, Q. Cancer-associated fibroblasts: from basic science to anticancer therapy. Exp. Mol. Med. 55, 1322–1332 (2023).
Dominguez, C. X. et al. Single-cell RNA sequencing reveals stromal evolution into LRRC15+ myofibroblasts as a determinant of patient response to cancer immunotherapy. Cancer Discov. 10, 232–253 (2020).
Kobayashi, H. et al. The origin and contribution of cancer-associated fibroblasts in colorectal carcinogenesis. Gastroenterology 162, 890–906 (2022).
Martinez-Outschoorn, U. E. et al. Ketones and lactate increase cancer cell ‘stemness,’ driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle 10, 1271–1286 (2011).
Narita, M. et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science 332, 966–970 (2011).
Martinez-Outschoorn, U. E. et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle 9, 3515–33 (2010).
Sari, D., Gozuacik, D. & Akkoc, Y. Role of autophagy in cancer-associated fibroblast activation, signaling and metabolic reprograming. Front. Cell. Dev. Biol. 11, 1274682 (2024).
Deng, M., Zhang, W., Yuan, L., Tan, J. & Chen, Z. HIF-1a regulates hypoxia-induced autophagy via translocation of ANKRD37 in colon cancer. Exp. Cell Res. 395, 112175 (2020).
Wang, X. et al. Overcoming cancer treatment resistance: unraveling the role of cancer-associated fibroblasts. J. Natl Cancer Center https://doi.org/10.1016/j.jncc.2025.03.002 (2025).
Biffi, G. & Tuveson, D. A. Diversity and biology of cancer-associated fibroblasts. Physiol. Rev. 101, 147–176 (2021).
Schwörer, S. et al. Hypoxia potentiates the inflammatory fibroblast phenotype promoted by pancreatic cancer cell-derived cytokines. Cancer Res. 83, 1596–1610 (2023).
Madsen, C. D. et al. Hypoxia and loss of PHD2 inactivate stromal fibroblasts to decrease tumour stiffness and metastasis. EMBO Rep. 16, 1394–408 (2015).
Kuchnio, A. et al. The cancer cell oxygen sensor PHD2 promotes metastasis via activation of cancer-associated fibroblasts. Cell Rep. 12, 992–1005 (2015).
Mantovani, A., Sozzani, S., Locati, M., Allavena, P. & Sica, A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol. 23, 549–555 (2002).
Strizova, Z. et al. M1/M2 macrophages and their overlaps—myth or reality? Clin. Sci. 137, 1067–1093 (2023).
Boutilier, A. J. & Elsawa, S. F. Macrophage polarization states in the tumor microenvironment. Int. J. Mol. Sci. 22, 6995 (2021).
Susek, K. H., Karvouni, M., Alici, E. & Lundqvist, A. The role of CXC chemokine receptors 1–4 on immune cells in the tumor microenvironment. Front. Immunol. 9, 2159 (2018).
Kratochvill, F. et al. TNF counterbalances the emergence of M2 tumor macrophages. Cell Rep. 12, 1902–1914 (2015).
Ma, J. et al. The M1 form of tumor-associated macrophages in non-small cell lung cancer is positively associated with survival time. BMC Cancer 10, 112 (2010).
Jackute, J. et al. Distribution of M1 and M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC Immunol. 19, 3 (2018).
Edin, S., Wikberg, M. L., Oldenborg, P.-A. & Palmqvist, R. Macrophages: Good guys in colorectal cancer. Oncoimmunology 2, e23038 (2013).
Zhang, M. et al. A high M1/M2 ratio of tumor-associated macrophages is associated with extended survival in ovarian cancer patients. J. Ovarian Res. 7, 19 (2014).
Honkanen, T. J. et al. Prognostic and predictive role of tumour-associated macrophages in HER2 positive breast cancer. Sci. Rep. 9, 10961 (2019).
Alves, A. M., Diel, L. F. & Lamers, M. L. Macrophages and prognosis of oral squamous cell carcinoma: a systematic review. J. Oral Pathol. Med. 47, 460–467 (2018).
Basak, U. et al. Tumor-associated macrophages: an effective player of the tumor microenvironment. Front. Immunol. 14, 1295257 (2023).
Andrejeva, G. & Rathmell, J. C. Similarities and distinctions of cancer and immune metabolism in inflammation and tumors. Cell Metab. 26, 49–70 (2017).
Osinsky, S. et al. Hypoxia, tumour-associated macrophages, microvessel density, VEGF and matrix metalloproteinases in human gastric cancer: interaction and impact on survival. Clin. Transl. Oncol. 13, 133–8 (2011).
Geeraerts, X. et al. Macrophages are metabolically heterogeneous within the tumor microenvironment. Cell Rep. 37, 110171 (2021).
Cramer, T. et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell 112, 645–57 (2003).
Peyssonnaux, C. et al. HIF-1α expression regulates the bactericidal capacity of phagocytes. J. Clin. Invest. 115, 1806–15 (2005).
Laoui, D. et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 74, 24–30 (2014).
Guentsch, A. et al. PHD2 is a regulator for glycolytic reprogramming in macrophages. Mol. Cell Biol. 37, e00236-16 (2017).
Semba, H. et al. HIF-1α–PDK1 axis-induced active glycolysis plays an essential role in macrophage migratory capacity. Nat. Commun. 7, 11635 (2016).
Nishide, S. et al. Controlling the phenotype of tumor-infiltrating macrophages via the PHD–HIF axis inhibits tumor growth in a mouse model. iScience 19, 940–954 (2019).
Kohli, K., Pillarisetty, V. G. & Kim, T. S. Key chemokines direct migration of immune cells in solid tumors. Cancer Gene Ther. 29, 10–21 (2022).
Koh, C.-H., Lee, S., Kwak, M., Kim, B.-S. & Chung, Y. CD8 T-cell subsets: heterogeneity, functions, and therapeutic potential. Exp. Mol. Med. 55, 2287–2299 (2023).
Blessin, N. C. et al. Prognostic role of proliferating CD8+ cytotoxic Tcells in human cancers. Cell Oncol. 44, 793–803 (2021).
Speiser, D. E., Chijioke, O., Schaeuble, K. & Münz, C. CD4+ T cells in cancer. Nat. Cancer 4, 317–329 (2023).
Wang, Z., Chimenti, M. S., Strouse, C. & Weiner, G. J. T cells, particularly activated CD4+ cells, maintain anti-CD20-mediated NK cell viability and antibody dependent cellular cytotoxicity. Cancer Immunol. Immunother. 71, 237–249 (2022).
Zhang, Y. et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 32, 377–391 (2017).
Saragovi, A. et al. Systemic hypoxia inhibits T cell response by limiting mitobiogenesis via matrix substrate-level phosphorylation arrest. eLife 9, e56612 (2020).
Gropper, Y. et al. Culturing CTLs under hypoxic conditions enhances their cytolysis and improves their anti-tumor function. Cell Rep. 20, 2547–2555 (2017).
Dvorakova, T. et al. Enhanced tumor response to adoptive T cell therapy with PHD2/3-deficient CD8 T cells. Nat. Commun. 15, 7789 (2024).
Bisilliat Donnet, C. et al. PHD2 constrains antitumor CD8+ T-cell activity. Cancer Immunol. Res. 11, 339–350 (2023).
Clever, D. et al. Oxygen sensing by T cells establishes an immunologically tolerant metastatic niche. Cell 166, 1117–1131 (2016).
Jackson, W. et al. Pharmacologic HIF stabilization activates costimulatory receptor expression to increase antitumor efficacy of adoptive T cell therapy. Sci. Adv. 10, eadq2366 (2024).
Garcia, J. et al. Bevacizumab (Avastin®) in cancer treatment: a review of 15 years of clinical experience and future outlook. Cancer Treat. Rev. 86, 102017 (2020).
Schönberger, T., Fandrey, J. & Prost-Fingerle, K. Ways into understanding HIF inhibition. Cancers 13, 159 (2021).
Scheuermann, T. H. et al. Artificial ligand binding within the HIF2α PAS-B domain of the HIF2 transcription factor. Proc. Natl Acad. Sci. USA 106, 450–455 (2009).
Cho, H. et al. On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 539, 107–111 (2016).
Wallace, E. M. et al. A small-molecule antagonist of HIF2α is efficacious in preclinical models of renal cell carcinoma. Cancer Res. 76, 5491–5500 (2016).
Rioja, P., Rey-Cardenas, M. & De Velasco, G. Targeting HIF-2α and anemia: a therapeutic breakthrough for clear-cell renal cell carcinoma. Cancer Treat. Rev. 129, 102801 (2024).
Semenza, G. L. Targeting intratumoral hypoxia to enhance anti-tumor immunity. Semin. Cancer Biol. 96, 5–10 (2023).
Fan, P. et al. Alleviating hypoxia to improve cancer immunotherapy. Oncogene 42, 3591–3604 (2023).
Leite de Oliveira, R. et al. Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell 22, 263–277 (2012).
Klotzsche-von Ameln, A. et al. Inhibition of HIF prolyl hydroxylase-2 blocks tumor growth in mice through the antiproliferative activity of TGFβ. Cancer Res. 71, 3306–3316 (2011).
Chan, M. C., Holt-Martyn, J. P., Schofield, C. J. & Ratcliffe, P. J. Pharmacological targeting of the HIF hydroxylases—a new field in medicine development. Mol. Aspects Med. 47–48, 54–75 (2016).
Nakanishi, T. & Kuragano, T. Growing concerns about using hypoxia-inducible factor prolyl hydroxylase inhibitors for the treatment of renal anemia. Clin. Kidney J. 17, sfae051 (2024).
Acknowledgements
Figures were created with BioRender.com. This study was partially supported by Italian Ministry of Health – Ricerca Corrente Annual Program 2025 and by AIRC (Associazione Italiana Ricerca contro il Cancro) IG26377 grant to V.S. G.V. was supported by a fellowship from Fondazione Umberto Veronesi.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Verna, G., Fantini, V., Grieco, A. et al. EGLN1 (PHD2) role in tumor microenvironment: insights for therapeutic targeting. Exp Mol Med 57, 2739–2748 (2025). https://doi.org/10.1038/s12276-025-01602-1
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-025-01602-1
