
Abstract
Aberrant mRNAs can arise from errors in RNA processing or from various physicochemical insults. Ribosomes translating such faulty mRNAs may stall, producing incomplete and potentially toxic polypeptides. These aberrant translation products are eliminated by the ribosome-associated quality control pathway. Ribosome stalling also leads to ribosome collisions, which can activate signaling pathways that enable cells to adapt to stress or determine cell fate. Here, in this Review, we summarize the molecular mechanisms of ribosome stalling and the associated quality control and signaling pathways, and discuss their implications in disease and therapeutics.
Similar content being viewed by others


Introduction
Misfolded proteins not only lose function but can also form harmful aggregates that disrupt protein homeostasis (proteostasis) by sequestering protein quality control factors1. One potential source of misfolded proteins is incomplete nascent polypeptides generated by prematurely stalled ribosomes. Elongating ribosomes can stall upon encountering RNA damage or specific sequence motifs. If not promptly resolved, trailing ribosomes collide with stalled ribosomes, forming di-ribosome (disome) complexes2,3,4 (Fig. 1a). Collided ribosomes act as markers of translational stress and recruit factors that initiate rescue, quality control and stress response pathways. This Review summarizes the causes of ribosome stalling and cellular responses to ribosome collisions, with a focus on ribosome-associated quality control (RQC), which directs stalled polypeptides to proteasomal degradation5,6. RQC is critical for maintaining cellular proteostasis7,8,9, and defects in this pathway have been implicated in aging10,11 and a range of diseases12,13,14,15,16. A deeper understanding of RQC and other molecular events at stalled ribosomes may ultimately inform the development of new therapeutic strategies.

a Stalled ribosomes collide with trailing ribosomes, forming disome complexes. b RNA damage caused by chemical or physical stress can induce stalling. c mRNAs lacking a stop codon can arise when a near-cognate polyadenylation signal (PAS) is present within the coding sequence. The canonical PAS motif is AAUAAA. The 5′-cap is also shown (m7Gppp). d Specific RNA sequences or nascent peptide features can inherently cause ribosome stalling. m6A modification typically occurs within the consensus sequence DRACH (D = A/G/U, R = A/G, H = A/C/U).
Causes of ribosome stalling
Ribosome stalling during translation elongation can be broadly attributed to two causes: (1) RNA lesions resulting from physicochemical insults (Fig. 1b) and (2) inherent stall-inducing motifs within coding sequences (Fig. 1c,d). Defective mRNAs are typically detected during translation; thus, at least one round of aberrant protein synthesis is often unavoidable17.

The disome interface is recognized by the E3 ligase ZNF598, which ubiquitylates 40S proteins such as eS10 and uS10. This modification recruits the ASCC to mediate ribosome splitting. The resulting 60S subunit, still attached to the stalled nascent polypeptide, is then processed by the RQC pathway, which targets the stalled polypeptide for proteasomal degradation. Persistent stalling stress activates broader cellular responses, including the ISR and RSR. These signaling cascades help mitigate stalling and influence cell fate decisions. The RQC, ISR and RSR pathways exhibit cross-talk, with activation of one often suppressing the others. The dominant pathway is probably determined by the severity and duration of ribosome stalling. Ub, ubiquitin; P, phosphate.
RNA lesions
Because of their single-stranded nature and lack of higher-order packing, mRNAs are particularly susceptible to damage18,19. Their cytosolic localization exposes them to reactive molecules generated during endogenous metabolism, as well as to environmental toxins taken up by the cell (Fig. 1b).
The mitochondria are a major source of cellular reactive species, producing superoxide as a by-product of oxidative metabolism20. Superoxide is subsequently converted into other reactive oxygen species, such as hydrogen peroxide (H₂O₂) and hydroxyl radicals (HO•)20. Although reactive oxygen species serve essential functions in redox signaling21, their high reactivity also damages cellular components, including nucleic acids22. Oxidative reactions can produce modified nucleobases such as 8-oxo-7,8-dihydroguanine, which induce ribosome stalling by disrupting codon–anticodon pairing23. Oxidative damage can also create abasic sites and cause strand breaks24. Notably, RNA oxidation has been associated with neurodegenerative diseases25.
Aldehydes damage nucleic acids by forming covalent protein–RNA crosslinks, which physically obstruct ribosome translocation26,27. Formaldehyde is produced endogenously during one-carbon metabolism and demethylation reactions, or introduced through exposure to environmental toxins such as tobacco smoke28. In the liver, acetaldehyde is generated as a toxic intermediate during alcohol metabolism29. Alkylating agents, another reactive species, covalently modify nucleobases by adding alkyl groups, such as methyl groups, to oxygen or nitrogen atoms22. These agents can originate from both endogenous and exogenous sources, including metabolic by-products of lipid peroxidation and nitrosamines present in tobacco smoke30. Alkylated bases disrupt canonical Watson–Crick base-pairing interactions, thereby inducing ribosome stalling22,31. Finally, ultraviolet (UV) radiation is a physical source of nucleic acid damage, inducing dimerization of adjacent uracil nucleobases and the formation of RNA–RNA or RNA–protein crosslinks32. These RNA photolesions physically obstruct ribosome elongation during translation and have been shown to activate cellular pathways that respond to ribosome stalling33,34.
Stall-inducing motifs
Even in the absence of external insults, aberrant mRNAs that trigger ribosome stalling can arise from errors in mRNA processing. In addition, specific sequence motifs within intact transcripts can intrinsically slow elongation. Such pauses may result from inefficient A-site decoding, unfavorable peptide bond formation or nascent polypeptides that impede ribosomal function.
One prominent stall-inducing motif is the polyadenosine (poly(A)) tract. The polyadenylation signal (PAS) in pre-mRNAs triggers downstream cleavage and 3′ poly(A) extension35 (Fig. 1c). Poly(A) is critical for transcript stability and promotes translation efficiency36. Because translation terminates at stop codons, ribosomes do not reach the downstream poly(A) tail. However, if near-cognate PAS elements occur upstream of a stop codon, premature polyadenylation can generate mRNAs lacking a stop codon37 (Fig. 1c). Consequently, ribosomes translate into the poly(A) tail, producing polylysine tracts that stall elongation38.
Early in vitro translation studies using rabbit reticulocyte lysates showed that polybasic sequences such as polylysine and polyarginine slow translation through electrostatic interactions with the negatively charged ribosomal exit tunnel39. Subsequent cell-based studies found that stalling is stronger on polylysine tracts encoded by consecutive AAA codons than on equivalent tracts encoded by the alternative lysine codon AAG40,41, suggesting that the mRNA sequence also contributes to stalling. Later studies revealed that poly(A) mRNA adopts a helical conformation that hinders codon–anticodon pairing at the A-site, thereby inducing stalling42,43.
Other polyamino acid tracts can also slow elongation. Proline is a poor donor and acceptor in peptide bond formation due to its bulky cyclic side chain and imino character, which can cause ribosome stalling44,45. However, the universally conserved translation elongation factor eIF5A facilitates translation through consecutive proline residues46. Polytryptophan tracts have also been shown to induce ribosome stalling47. Although the precise molecular mechanism remains to be fully elucidated, the bulkiness of tryptophan side chains probably clogs the ribosomal exit tunnel47.
Codon usage is another key determinant of translation elongation rate. In general, optimal codons are decoded more rapidly than rare codons48. In Saccharomyces cerevisiae, repeats of the rare CGA codon have been widely used to induce ribosome stalling49. However, CGA-induced stalling is not solely attributable to codon rarity. Notably, the identity of the P-site tRNA can influence decoding efficiency at the A-site50; thus, effective translation inhibition requires two adjacent CGA codons occupying the P- and A-sites simultaneously. This neighboring codon effect is not limited to the CGA–CGA dicodon. For example, the CUC–CCG dicodon also reduces translation efficiency, suggesting that the cognate tRNA for the CUC codon in the P-site interferes with decoding of the CCG codon in the A-site50. By contrast, the reverse dicodon, CCG–CUC, does not reduce translation efficiency50 (Fig. 1d; inhibitory codon pairs), underscoring the importance of codon context (that is, sequence) in ribosome stalling rather than codon optimality alone. In yeast, 17 inhibitory codon pairs have been identified50. Notably, many of these include CGA and CUG codons, which are decoded via wobble base pairing: I·A for CGA and U·G for CUG.
Inosine, as in the I·A pair, is a common modification at the wobble position of tRNAs (nucleotide 34, the first base of the anticodon) arising from deamination of adenosine51. Although nucleotide modifications are most prevalent in tRNAs and rRNAs, mRNAs also carry several chemical marks, among which N6-methyladenosine (m6A) is the most abundant52 (Fig. 1d; m6A modification). m6A is deposited by a nuclear methyltransferase complex comprising METTL3 and METTL14 subunits, which methylates adenosine at the N6 position53. Although m6A-modified codons can still base pair with U in the anticodon, their decoding is slowed54, increasing ribosome collisions55.
Cellular responses to ribosome stalling
When ribosomes stall and collide, cells activate downstream pathways with two main functions: (1) resolving stalled ribosomes by splitting and recycling subunits, a process preceding the RQC pathway; and (2) preventing additional stalling events. If stalling persists, cells further engage broader stress responses, including the integrated stress response (ISR) and the ribotoxic stress response (RSR).
Resolution of stalled ribosomes
Canonical translation termination at stop codons is mediated by eukaryotic release factor 1 (eRF1), delivered to the ribosome by the translational GTPase eRF356. Upon eRF1 accommodation at the A-site, its conserved GGQ (Gly–Gly–Gln) motif catalyzes peptidyl-tRNA hydrolysis, releasing the polypeptide57. Termination is followed by ribosome recycling, during which the ATPase ABCE1 (Rli1 in yeast) dissociates the 80S ribosome into 60S and 40S subunits58,59. Because eRF1 specifically recognizes stop codons, it cannot resolve stalled ribosomes. Instead, a homologous protein, Pelota in metazoans and Dom34 in yeast, is delivered by the GTPase Hbs1 (an eRF3 homolog) to the empty A-site of ribosomes stalled at the 3′ end of truncated mRNAs60,61. As in canonical termination, the ATPase ABCE1/Rli1 mediates ribosome splitting62. However, unlike eRF1, Pelota/Dom34 lacks the GGQ motif required for peptidyl-tRNA hydrolysis, leaving the peptidyl-tRNA attached to the large ribosomal subunit.
If ribosomes stall within a coding sequence rather than at the 3′ end, the A-site may not be readily accessible to Pelota/Dom34. In such cases, ribosome collisions activate an alternative splitting mechanism. The unique 40S–40S interface formed between collided ribosomes is recognized by an E3 ubiquitin ligase3,4 (Fig. 2). In mammals, ZNF598 ubiquitylates eS10 (K138, K139) and uS10 (K4, K8)63,64, whereas in yeast, Hel2 targets uS10 at K6 and K865. While disomes (two collided ribosomes) represent the minimal substrate for ZNF598/Hel2 recognition, trisomes (three collided ribosomes) are targeted more efficiently for ubiquitylation66.
Ubiquitylated 40S ribosomal proteins in disomes or trisomes recruit the ribosome-splitting machinery ASC-1 complex (ASCC) in mammals or RQC-trigger (RQT) complex in yeast65,66,67. The ASCC/RQT complex comprises the RNA helicase ASCC3/Slh1, the ubiquitin-binding protein ASCC2/Cue3 and the zinc-finger protein TRIP4/Rqt4. Cue3 and Rqt4 bind Hel2-ubiquitylated ribosomal proteins68. Slh1 engages the 3′ mRNA overhang extending from the lead, stalled ribosome and exerts a pulling force on the transcript, powered by ATP hydrolysis. This action pulls the trailing ribosome forward, generating the force to split the stalled ribosome positioned between the RQT complex and the trailing ribosome69. Similar to Dom34-mediated splitting, the large ribosomal subunit split by the ASCC/RQT complex retains peptidyl-tRNA, which is subsequently degraded (Fig. 2; see also ‘RQC’ section). The ZNF598/Hel2-ubiquitylated small subunit is recycled by deubiquitinases: USP10 in mammals70 and Ubp2 and Ubp3 in yeast71,72.
Cellular mechanisms to minimize ribosome stalling
Even after stalled ribosomes are resolved, defective mRNAs may continue to be translated, causing recurrent stalling. To prevent this, cells activate additional collision-responsive pathways that suppress further stalling and limit the accumulation of aberrant translation products.
In human cells, EDF1 functions as a collision sensor that acts independently of ZNF598, recruiting the translational repressors GIGYF2 and 4EHP (eIF4E-homologous protein) to collided ribosomes73,74,75. The locally enriched 4EHP then outcompetes eIF4E for binding to the 5′ cap of the stall-inducing mRNA, thereby blocking translation initiation because 4EHP cannot interact with eIF4G76,77. This mechanism directly prevents further translation of the target mRNA, complementing ZNF598-mediated ribosome rescue. Because EDF1 is far more abundant than ZNF598, it may function as an upstream collision sensor74,75. However, ZNF598 activity does not depend on EDF174,75.
Stalled ribosomes also trigger mRNA decay pathways, including no-go decay and nonstop decay, which irreversibly eliminate faulty transcripts and thereby prevent further production of aberrant proteins. The mechanisms underlying these pathways have been reviewed elsewhere78. Notably, ribosome-induced mRNA decay not only safeguards against defective transcripts but may also regulate gene expression. For instance, m6A modification can destabilize mRNAs through a decay process mediated by YTH domain-containing family proteins (YTHDF)79,80. Notably, m6A marks within coding regions promote mRNA decay more strongly than those in untranslated regions81,82. As described earlier, m6A slows translation and induces ribosome collisions54,55. The collided ribosomes recruit YTHDF, enhancing their binding to m6A and thereby promoting mRNA decay under normal conditions55. Under stress conditions, however, global translation is reduced by the ISR (discussed in the following section), resulting in fewer ribosome collisions and, consequently, greater stability of m6A-modified mRNAs55. This mechanism may explain the increased abundance of certain m6A-modified transcripts during stress83,84. m6A-dependent mRNA decay can also alter transcript profiles in cancer cells. Translation of codons bearing m6A at the wobble position (the third nucleotide of the codon) is facilitated by a corresponding modification in the cognate tRNAs, in which U at the first anticodon position is modified to mcm5s2U (ref. 82). Thus, the balance between m6A and mcm5s2U modifications can determine mRNA stability. When mcm5s2U modification is insufficient, ribosomes stall and collide at m6A-modified codons, promoting mRNA decay82. Interestingly, cancer cells tend to reduce m6A levels while enhancing mcm5s2U modification, thereby stabilizing transcripts involved in oncogenic signaling pathways82.
When ribosome stalling arises from mRNA–protein crosslinks or from elongation or termination factors trapped within the ribosome, degradation of the stall-inducing proteins is promoted26,27,85,86. This pathway involves the additional collision sensor, GCN1, which associates with collided ribosomes and the E3 ubiquitin ligases RNF14 and RNF25. RNF25-mediated ubiquitylation of the small ribosomal subunit protein eS31 facilitates RNF14-driven polyubiquitylation of stall-inducing proteins, targeting them for proteasomal degradation. Notably, the RQC pathway is activated independently of the GCN1–RNF25–RNF14 axis26,27, underscoring how multiple quality control mechanisms cooperate to resolve ribosome stalling stress.
Stress response pathways
Cellular sensing of ribosome collisions can also activate stress response pathways. These include the ISR and the RSR (Fig. 2), which protect cells from persistent translational stress and influence cell-fate decisions.
The ISR is regulated by four upstream kinases, each activated by distinct stress signals87,88. One of these kinases, GCN2, was originally identified for its role in nutrient sensing89. During amino acid starvation, elevated levels of uncharged tRNAs activate yeast GCN2 through their direct binding to the histidyl-tRNA synthetase-like domain of GCN290,91. Activated GCN2 phosphorylates the α subunit of eIF2 (eIF2α)92. eIF2 forms a ternary complex with Met-tRNAiMet and GTP, which, together with other initiation factors, assembles onto the 40S subunit to form the 43S pre-initiation complex (PIC)93. As the PIC scans the 5′ untranslated region of the mRNA, the start codon is recognized through codon–anticodon pairing with Met-tRNAiMet, delivered by eIF293. This recognition triggers GTP hydrolysis by eIF2, leading to the dissociation of initiation factors and joining of the large ribosome subunit93. eIF2 bound to GDP is normally recycled to its active GTP-bound form by the guanine nucleotide exchange factor eIF2B, permitting subsequent rounds of translation initiation93. However, phosphorylated eIF2α inhibits eIF2B94,95, leading to a gradual depletion of functional eIF2-GTP and impairing PIC formation. As a result, the ISR suppresses global translation.
Notably, recent studies have identified collided ribosomes—rather than uncharged tRNAs—as critical activators of the ISR kinase GCN2 (Fig. 2). Early work using mice carrying mutations in a brain-specific tRNA (tRNAArg(UCU)) and in the putative ribosome recycling factor GTPBP2 revealed that ribosomal stalling can activate GCN2 independently of uncharged tRNA accumulation96. Subsequent studies showed that the ribosomal P-stalk is required in vivo and sufficient in vitro for GCN2 activation97,98, establishing a direct molecular link between the ribosome and ISR signaling. These findings support a model in which the P-stalk of stalled ribosomes activates GCN2. In actively translating ribosomes, however, the P-stalk, part of the GTPase-associated center, would be occupied by elongation factors (eEF1A and eEF2), preventing its interaction with GCN297,98. Importantly, titration of the ribosome inhibitor anisomycin revealed that GCN2 is activated by collided ribosomes rather than by stalled ribosomes themselves33. Intermediate concentrations of anisomycin, which induce ribosome collisions, activated GCN2, whereas higher concentrations that cause more severe stalling and thus fewer collisions failed to do so33. GCN1 has long been recognized as a critical co-activator of GCN299. Consistent with the notion that ribosome collisions act as an ISR trigger, cryo-electron microscopy analyses revealed that GCN1 associates with disomes100. Supporting this model, GCN1-selective ribosome profiling demonstrated enrichment of disome footprints101. More recently, Mbf1 (the yeast homolog of EDF1), which also binds collided ribosomes, was identified as an additional co-activator of Gcn2102. Interestingly, under normal conditions, yeast GCN2 associates with the nontranslating free 60S subunit as a homodimer103. Taken together, these findings suggest that GCN2, positioned in a stand-by state on the 60S subunit, can be redistributed to colliding ribosomes through its interaction with the critical co-activator GCN1. During activation, homodimeric GCN2 undergoes structural rearrangement and autophosphorylation104. Although uncharged tRNAs alone may not be sufficient to activate GCN2 without ribosome collisions, their accommodation at the A-site of stalled ribosomes can further enhance GCN2 activation105. The mechanistic details of GCN2 activation may differ depending on how ribosome collisions are induced106.
The RSR is another cellular pathway activated by ribosome stalling and collisions33,107 (Fig. 2). Its activation triggers a mitogen-activated protein kinase (MAPK) cascade that ultimately results in the activation of downstream MAPKs, including p38 and c-Jun N-terminal kinase (JNK). These kinases elicit cellular outcomes ranging from restoration of homeostasis to cell death, which may depend on the severity of stress33,34,108. ZAK, a MAPKKK, functions as the upstream activator of the RSR. Specifically, ZAKα, the longer of the two major ZAK isoforms, α and β, binds to ribosomes through its flexible C-terminal domain, where it is activated via autophosphorylation107,108. The kinase then dissociates from ribosomes and mediates downstream signaling107,108. At present, it remains unclear whether ZAKα specifically binds collided ribosome complexes. Mild ribosome stalling drives ZAKα-mediated p38 activation, which temporarily restricts cell cycle progression and regulates factors that resolve stalled ribosomes and prevent further stalling, thereby promoting recovery from translational stress34,108. By contrast, when cells experience severe stalling stress, for instance from extensive UV-induced mRNA damage, ZAKα-dependent activation of JNK can promote cell death33,108. Although UV radiation also activates the DNA damage response, the RSR—not the DNA damage response—primarily mediates early apoptosis in this context108.
Collided ribosomes may influence a broader spectrum of signaling pathways beyond the ISR and RSR, not necessarily through direct activation of protein kinases. One prominent example is the cGAS–STING pathway of the innate immune response. Upon viral infection, cytosolic foreign DNA is detected by cyclic GMP–AMP synthase (cGAS)109. Activated cGAS synthesizes the second messenger cyclic GMP–AMP110, a cyclic dinucleotide that binds to the cytosolic ligand-binding domain of the endoplasmic reticulum (ER) transmembrane protein STING (stimulator of interferon genes)111. Upon ligand binding, STING recruits TANK-binding kinase 1 (TBK1) and interferon regulatory factor 3 (IRF3), enabling TBK1-mediated phosphorylation and activation of IRF3112,113. The activated transcription factor IRF3 then drives the expression of type I interferons, which in turn suppress viral replication. Notably, collided ribosomes can interact with cGAS, enhancing its DNA-dependent synthesis of cyclic GMP–AMP114. During viral infection, the host translation system may experience increased ribosome collisions due to the heavy burden of viral protein production, which often relies on atypical translation mechanisms such as frameshifting and stop-codon readthrough115. Overall, ribosome collisions potentiate the cGAS–STING pathway by stimulating production of the second messenger.
Cross-talk between signaling pathways at stalled ribosomes
Ribosome collisions can activate three major pathways: the ISR, the RSR and the RQC (Fig. 2). The severity of collisions, including their frequency and duration, probably determines which pathway is engaged. Once ZNF598/Hel2 directs collided ribosomes into the RQC pathway, activation of the ISR and RSR is prevented because the signaling platform—the disome—is dismantled. For example, Hel2 has been shown to inhibit Gcn2-mediated phosphorylation of eIF2α116,117. Similarly, ZNF598 attenuates activation of the RSR pathway33. Consistent with this antagonistic relationship, activation of the ISR also suppresses both the RQC116 and RSR pathways33. By reducing global translation, the ISR lowers the frequency of ribosome collisions, thereby limiting the activation of collision-dependent pathways. Under mild translational stress, both the RQC and ISR pathways promote cellular protection and the restoration of proteostasis. However, under severe or prolonged stress, RSR-mediated activation of JNK can shift the balance toward apoptosis108.
RQC
RQC was first identified in S. cerevisiae5,6. It comprises several factors that act on the 60S ribosomal subunit, including Rqc2 (NEMF in mammals), the E3 RING ubiquitin ligase Ltn1 (Listerin/LTN1 in mammals), Rqc1 (TCF25 in mammals) and the AAA-ATPase Cdc48 (VCP in mammals). The 60S subunits that dissociate from stalled ribosomes retain a peptidyl-tRNA anchored at the P-site. The presence of this tRNA at the intersubunit interface is recognized by Rqc2/NEMF, which subsequently recruits Ltn1/LTN1 to the 60S subunit118,119 (Fig. 2). The C-terminal RING domain of Ltn1 is positioned near the ribosomal exit tunnel, where it ubiquitylates stalled polypeptides118,119. Rqc1/TCF25 promotes the formation of K48-linked polyubiquitin chains during this process120. The polyubiquitylated nascent polypeptides are subsequently released from the 60S. All tRNAs possess a conserved CCA sequence at their 3′ end, which is added during tRNA maturation. The CCA tail of the peptidyl-tRNA anchored on the 60S subunit is cleaved by the endonuclease Vms1/ANKZF1121. Cdc48/VCP then extracts polyubiquitylated stalled polypeptides from 60S for proteasomal degradation122. In human cells, tRNAs with truncated 3′ ends are restored by the CCA-adding enzyme tRNA nucleotidyl transferase 1 (TRNT1), enabling their subsequent aminoacylation123.
RQC and UFM1 at the ER
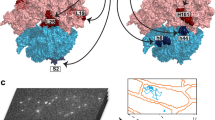
Secretory proteins are cotranslationally translocated into the ER. This process begins when the signal recognition particle binds to the signal sequence emerging from the ribosome and directs the translating ribosome to the ER membrane via interaction with the signal recognition particle receptor, an integral membrane protein complex124. The ribosome is then aligned with the SEC61 translocon, enabling nascent chains to be threaded directly into the ER lumen125. This tight coupling raises the question of whether the RQC can access and target stalled secretory polypeptides through the narrow gap between the ribosomal exit tunnel and the SEC61 translocon. Early studies demonstrated that stalled secretory polypeptides can be degraded by cytosolic RQC126,127. Subsequent work revealed that UFM1 conjugation (UFMylation) of the 60S ribosomal protein uL24 is essential for RQC at the ER128,129. UFM1 is a ubiquitin-like modifier whose conjugation requires an E3 ligase complex composed of three components: UFL1, DDRGK1 (also known as UFBP1) and CDK5RAP3130. DDRGK1, a transmembrane protein, anchors the UFM1 E3 ligase complex (E3UFM1) to the ER membrane, thereby restricting uL24 UFMylation to ribosomes at the ER rather than in the cytosol128,129,131. Because uL24 is positioned near the SEC61 translocon (Fig. 3a), its UFMylation has been proposed to weaken the interaction between 60S and SEC61 (Fig. 3b), thereby allowing the access of cytosolic RQC128. Subsequent studies revealed that a region of DDRGK1 within the E3UFM1 binds UFM1 conjugated to uL24 and occludes the contact site between the 60S subunit and SEC61, resulting in 60S detachment132,133 (Fig. 3c). Thus, the E3UFM1 functions not only as the writer but also as the reader of UFMylation. Cryo-electron microscopy structural analysis revealed that the E3UFM1 associates with 60S subunits lacking peptidyl-tRNA132,133 (Fig. 3b,c), suggesting that 60S subunits derived from canonical eRF1-dependent termination at stop codons are UFMylated to promote efficient 60S recycling after completion of secretory protein synthesis. UFMylation is prevented during elongation because the 40S subunit masks the intersubunit interface of the 60S, which is thought to be the key site for the initial binding of UFL1133, a subunit of the E3UFM1.

a A 60S ribosomal subunit remains associated with the ER translocon after completion of secretory-protein translation. Note that no peptidyl-tRNA remains attached. PDB ID: 8OJ0133. b The E3UFM1 complex associates with the 60S subunit bound to SEC61. UFM1 is conjugated to uL24, although the structure of this linkage has not been resolved. The UFL1 binding site overlaps with the 40S interface (see d for the position of the P-site), preventing UFMylation of 80S ribosomes. Note that the E3UFM1 remains bound to the 60S subunit after UFMylation. PDB ID: 8OJ0133. c UFMylation of uL24 induces a conformational change in DDRGK1 (which contains the long α-helix spanning the region between the ribosomal exit tunnel and SEC61), leading to detachment of the 60S subunit. PDB ID:8OJD133. d The UFL1 binding site at the intersubunit interface overlaps with that of NEMF (see c and d). The alternative conformation of UFL1 enables the E3UFM1 to co-assemble with NEMF on the same 60S subunit. PDB ID: 9GY4134. Note that the 60S subunit detached from SEC61 in c and d would remain tethered to the ER membrane, as DDRGK1 is a transmembrane protein. The structural details of DDRGK1 membrane insertion have not been resolved.
The elongated structure of the E3UFM1 wraps around the 60S subunit, spanning from the intersubunit interface to the exit tunnel132,133 (Fig. 3b). Notably, its binding site at the intersubunit interface overlaps with that of the RQC component NEMF, leaving it unclear how UFMylation and RQC are coordinated at the ER. A recent study showed that the UFL1 subunit in the E3UFM1 undergoes conformational adjustments in the presence of peptidyl-tRNA and NEMF134 (Fig. 3d). This structural flexibility allows the E3UFM1 and the RQC machinery to co-assemble on the same 60S subunit. Listerin can access stalled polypeptides once UFMylation displaces the 60S subunit from the SEC61 translocon. However, the detached 60S subunits—whether derived from canonical termination (Fig. 3c) or ribosome stalling (Fig. 3d)—remain associated with the E3UFM1. Subsequent deUFMylation of uL24 by UFSP2 ultimately recycles the 60S for reuse135. Failure of RQC at the SEC61 translocon not only disrupts ER homeostasis but may also impair global protein secretion by clogging the translocon136. Similarly, unresolved ribosome stalling during the translation of mitochondrial proteins can obstruct the translocase of the outer membrane (TOM) complex, thereby inhibiting mitochondrial protein import136.
CAT tail extension
Although the positioning of the Ltn1 RING domain near the ribosomal exit tunnel is well suited for ubiquitylating stalled polypeptides118,119, this activity is limited when lysine residues are not readily accessible near the tunnel. Remarkably, Rqc2/NEMF overcomes this limitation by extending stalled polypeptides that remain anchored to the 60S subunit (Fig. 4a). Rqc2/NEMF recruits aminoacyl-tRNAs to the A-site of the 60S subunit, enabling peptide bond formation at the peptidyl transferase center118,137,138. Consequently, stalled polypeptides can be elongated on the 60S subunit via a noncanonical form of translation that does not require the 40S subunit and mRNA templates. Because the ribosomal exit tunnel accommodates approximately 30–40 amino acids, lysine residues may, by chance, be buried within it. C-terminal elongation of stalled polypeptides can expose these hidden lysines beyond the tunnel, rendering them accessible for ubiquitylation by Ltn1139 (Fig. 4b). This C-terminal extension of stalled polypeptides was first identified in yeast, where Rqc2 selectively recruits alanyl- and threonyl-tRNAs118. Thus, the extension was termed the C-terminal Ala/Thr tail (CAT tail)118. Unlike canonical translation by 80S ribosomes, CAT tailing on the 60S subunit proceeds independently of GTP, suggesting that translational GTPases are not involved140. However, the process requires eIF5A, which binds to the E-site of the 60S subunit and facilitates peptidyl transfer141.

a Yeast Rqc2 and human NEMF extend the C-termini of stalled polypeptides on RQC-targeted 60S subunits without requiring an mRNA template. In yeast, Rqc2 specifically recruits tRNAAla(IGC) and tRNAThr(IGU), generating C-terminal Ala/Thr (CAT) tails. In humans, CAT tails consist predominantly of alanine. CAT tail elongation can expose previously buried lysine residues. b Ltn1 ubiquitylates lysine residues located near the ribosome exit tunnel. c If stalled polypeptides escape Ltn1-mediated ubiquitylation, their CAT tails can be recognized by alternative off-ribosome E3 ligases. d In yeast, undegraded CAT-tailed proteins form detergent-insoluble aggregates, driven by threonine residues. This aggregation may serve as a form of spatial quality control, benefiting cells under stress conditions, although chronic accumulation during aging can be toxic.
In yeast, tRNAAla(IGC) and tRNAThr(IGU) are selectively recruited by Rqc2 for CAT tailing118. In contrast to its selective recognition of incoming aminoacyl-tRNAs, Rqc2 does not discriminate among peptidyl-tRNAs, allowing CAT tailing to initiate on RQC-targeted polypeptides irrespective of their sequence context. During CAT tailing, competition between factors that promote tail elongation (Rqc2 and aminoacyl-tRNAs) and those that mediate peptidyl-tRNA cleavage (Vms1 and Arb1142) appears to determine when the process terminates, resulting in variation in CAT tail length. Notably, the amino acid composition of CAT tails differs among species. In Drosophila, CAT tails include amino acids beyond alanine and threonine143, whereas in humans, they consist predominantly of alanine residues137,138. Although CAT tails enhance Ltn1-mediated ubiquitylation (Fig. 4b), they also serve as degradation signals. If stalled polypeptides are released from 60S before Ltn1-dependent ubiquitylation, the CAT tails are recognized by alternative cytosolic E3 ligases, ensuring their clearance137,144 (Fig. 4c). In humans, off-ribosome degradation of CAT-tailed proteins may represent an extension of the C-end rule pathway, wherein the identity of the C-terminal amino acid determines protein stability145,146. Similarly, the yeast E3 ligase Hul5 has been shown to target a subset of CAT-tailed proteins144, although its recognition mechanism may differ from that of the human C-end rule. Notably, non-ubiquitylated CAT-tailed proteins can be released by peptidyl-tRNA hydrolase 1 (Ptrh1) rather than through CCA-end cleavage of the tRNA by ANKZF1147. This alternative release mechanism may enhance the efficiency of off-ribosome degradation by more effectively presenting alanine tails (that is, human CAT tails) without conjugated CCA nucleotides to cytosolic E3 ligases137.
In yeast, CAT-tailed proteins that escape degradation can accumulate as detergent-insoluble aggregates7,8 (Fig. 4d). However, the mechanism by which heterogeneous CAT tails of varying lengths and sequences co-aggregate remained unclear. A recent study demonstrated that threonine-rich polypeptides are particularly aggregation-prone148. Surprisingly, polythreonine exhibits a stronger aggregation propensity than polyglutamine148, which has been extensively studied for its involvement in human diseases, most notably Huntington’s disease. Among heterogeneous CAT tails, longer tails with higher threonine content can initiate aggregation. These initial aggregates exert a robust seeding effect on other threonine-enriched polypeptides, even when their length is short and threonine content is relatively low, leading to the co-aggregation of heterogeneous CAT tails148. In this context, the incorporation of aggregation-prone threonine residues into CAT tails appears counterintuitive if the primary function of CAT tails is to facilitate stalled polypeptide degradation by Ltn1 (Fig. 4b). Sequestering potentially toxic stalled polypeptides into aggregates could serve as a protective mechanism, particularly in rapidly dividing yeast cells149 (Fig. 4d). These aggregates may be cleared through autophagy or retained asymmetrically in mother cells to reset proteostasis in daughter cells150. However, if CAT-tailed aggregates accumulate beyond a certain threshold, due to aging or persistent stress, they can overwhelm cellular quality control systems and disrupt proteostasis10. Therefore, the composition of CAT tails warrants careful examination across organisms, as it may influence the fate and behavior of stalled polypeptides. Notably, mammalian CAT tails composed predominantly of alanine can also promote aggregation, even in the absence or at low abundance of threonine138,151.
Physiological impacts and therapeutic potential of RQC
An increased burden on protein quality control pathways, including RQC, in aged organisms can lead to proteostasis collapse and contribute to age-related protein aggregation disorders. Given its involvement in various diseases, RQC may also represent a potential therapeutic target.
Disorders associated with impaired RQC
Random mutagenesis using N-ethyl-N-nitrosourea (ENU) in mice first identified Ltn1 as a gene associated with a neurodegenerative disorder12. The corresponding protein, Listerin, was named after the ‘listing’ (leaning) phenotype observed in mutant mice. Mice carrying a homozygous hypomorphic allele, lister, which encodes a protein lacking 14 amino acids, were born without overt abnormalities. However, they progressively developed motor deficits associated with central nervous system (CNS) dysfunction and exhibited a substantially shortened lifespan. Similarly, ENU mutagenesis identified two missense mutations in mouse Nemf, R86S and R487G, whose homozygous expression also leads to progressive neuromuscular degeneration13. This phenotype is accompanied by atrophy of motor axons—and of sensory axons in the R86S variant—as well as compromised neuromuscular junction integrity. Similar mutations in yeast Rqc2 suggest that the NEMF R86S and R487G variants impair CAT tail extension while preserving core functions such as binding to large ribosomal subunits and recruiting LTN1. These hypomorphic Nemf mutations in mice highlight the importance of CAT tailing in maintaining neuromuscular integrity. Notably, NEMF-null mice display an earlier onset and more severe phenotypes than those carrying NEMF missense mutations[13]. Together, findings from Ltn1 and Nemf mutant models underscore the importance of RQC in resolving physiological translation challenges arising from ribosome stalling in vivo. Consistent with these observations, NEMF variants have also been associated with juvenile neuromuscular disorders in humans13,15. Beyond motor phenotypes, mutations in NEMF have been linked to intellectual disability, often accompanied by delayed speech development in early childhood13,16. Similarly, Ltn1-knockout mice exhibit cognitive impairments14. Although neuromuscular deficits were absent in this model—unlike in mice carrying the lister allele—these findings reinforce that the CNS is particularly susceptible to impaired RQC activity. Consistently, Ltn1 knockdown impairs neurite morphogenesis and reduces the viability of primary neurons138.
RQC involvement in C9orf72-linked ALS and FTD
In various neurodegenerative disease models, RQC has been identified as a disease modifier, as discussed below. However, it is also noteworthy that severe ribosome stalling can cause neurodegeneration even when RQC function remains intact. In mice, mutation of a CNS-specific tRNA combined with the loss of the putative ribosome rescue factors GTPBP1 or GTPBP2 induces ribosome stalling and, consequently, neurodegeneration152,153.
Chromosome 9 open reading frame 72 (C9orf72) is the most common genetic cause of amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD)154,155. The gene contains a hexanucleotide repeat sequence (GGGGCC, or G4C2) in its first intron154,155. In patients with ALS or FTD, pathological expansion of this repeat leads to bidirectional transcription, producing both sense (G4C2) and antisense (G2C4) transcripts156. Both transcripts can be translated in all reading frames through a noncanonical mechanism that does not require an AUG start codon157. Although the precise molecular details remain to be elucidated, the GC-rich repeat forms stable secondary structures that facilitate this atypical mode of translation initiation, known as repeat-associated non-AUG (RAN) translation158. RAN translation of G4C2 and G2C4 transcripts produces five distinct dipeptide repeat proteins: poly(GP), poly(PA), poly(GA), poly(PR) and poly(GR), which accumulate as intracellular aggregates in patients with C9orf72 expansions156,157. Among these, poly(GR) and poly(PR), both containing arginine residues, exhibit the highest cellular toxicity159,160. Notably, recent studies have implicated RQC in regulating RAN translation of G4C2 repeats161,162,163. During the synthesis of poly(GR) and poly(PR), the high density of positively charged arginine residues probably induces ribosome stalling through electrostatic interactions within the ribosomal exit tunnel164. In addition, the GC-rich G4C2 sequence can form stable RNA secondary structures165 that independently trigger ribosome stalling, regardless of the reading frame163. Consequently, activation of RQC by overexpressing ZNF598, NEMF, LTN1 or ANKZF1 reduces the levels of RAN translation products, whereas depletion of these RQC factors leads to further accumulation of dipeptide repeats163. Interestingly, ZNF598 protein expression, and consequently RQC activity, was found to be reduced in neurons derived from patients with ALS carrying C9orf72 G4C2 expansions161. These findings suggest that genetic or pharmacological enhancement of RQC could help mitigate the toxicity associated with C9orf72 RAN translation products.
Mitochondrial dysfunction
Mitochondrial dysfunction is a well-established feature of many neurodegenerative diseases. Notably, such dysfunction has been shown to reduce cytosolic levels of eRF1 and ABCE1, thereby impairing translation termination in human cell lines and Drosophila143. This cytosolic defect can, in turn, disrupt the mitochondrial proteome, as the vast majority of mitochondrial proteins are nuclear-encoded and translated in the cytosol166. Indeed, in Drosophila and human cell lines with mitochondrial dysfunction, C-I30, a nuclear-encoded core subunit of complex I in the electron transport chain, undergoes CAT tail extension, indicating ribosome stalling at stop codons caused by impaired cytosolic translation termination143. In addition, CAT-tailed C-I30 can be imported into mitochondria and incorporated into the complex I, thereby disrupting respiration and reducing ATP production143. Notably, modulation of RQC factor expression, such as repression of NEMF or overexpression of the endonuclease ANKZF1, decreases the production, and probably the proteotoxicity, of CAT-tailed C-I30143.
Cancer
5-Fluorouracil (5-FU) is a widely used chemotherapeutic agent for the treatment of solid tumors. Inside cells, 5-FU is converted into active metabolites that either inhibit thymidylate synthase, an enzyme in nucleotide biosynthesis, or become incorporated into DNA and RNA, thereby exerting cytotoxic effects on cancer cells167. Incorporation of 5-FU metabolites into mRNAs can induce ribosome stalling168. In addition, 5-FU treatment activates the mTOR signaling pathway, enhancing global mRNA translation and increasing the frequency of ribosome collisions, thereby contributing to its anticancer activity168. Notably, ZNF598-knockout cells are hypersensitive to 5-FU, potentially due to activation of the RSR168. Thus, therapeutic strategies that suppress the RQC pathway could potentiate the efficacy of 5-FU.
mRNA vaccines
RQC may also influence the efficacy and safety of mRNA vaccines. To increase stability and minimize innate immune response, modified ribonucleosides are commonly incorporated into mRNA vaccines169. A recent study showed that N1-methylpseudouridine, a modification used in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) mRNA vaccines, can induce ribosome stalling and subsequent frameshifting170. These findings suggest that RQC may help mitigate potential side effects arising from ribosome stalling during translation of mRNA vaccines. Translation of mRNA vaccines generates antigenic proteins, which are processed by the proteasome into peptides for presentation on major histocompatibility complex (MHC) class I molecules. In this context, it is noteworthy that RQC-mediated cotranslational degradation enhances MHC-I antigen presentation171 and may thereby improve vaccine efficacy.
Concluding remarks
Investigations into ribosome stalling-associated quality control mechanisms have led to several unexpected discoveries, including mRNA-independent translation (CAT tailing)118, threonine-based protein aggregation motifs148 and ribosomal ubiquitylation63,64,65. The emergence of the ribosome collision concept2 has further expanded our understanding of the molecular mechanisms underlying the ISR and the RSR33. Studies of ribosome stalling have also provided insights into a long-standing question—how SEC61-associated ribosomes are recycled—leading to the discovery of a UFM1-dependent 60S recycling mechanism132,133. Although the ribosome has been structurally characterized in great detail, investigations into ribosome stalling and collisions continue to uncover unexpected molecular events and regulatory principles. It is likely that many more unanticipated biological phenomena remain to be discovered in this context. Beyond its mechanistic complexity, ribosome stalling and collisions are increasingly recognized as contributors to neurological disorders, as well as other diseases and aging172. Thus, a deeper understanding of ribosome stalling biology holds great promise for advancing molecular medicine.
References
Hipp, M. S. & Hartl, F. U. Interplay of proteostasis capacity and protein aggregation: implications for cellular function and disease. J. Mol. Biol. 436, 168615 (2024).
Simms, C. L., Yan, L. L. & Zaher, H. S. Ribosome collision is critical for quality control during no-go decay. Mol. Cell 68, 361–373 (2017).
Juszkiewicz, S. et al. ZNF598 is a quality control sensor of collided ribosomes. Mol. Cell 72, 469–481 (2018).
Ikeuchi, K. et al. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 38, EMBJ2018100276 (2019).
Bengtson, M. H. & Joazeiro, C. A. Role of a ribosome-associated E3 ubiquitin ligase in protein quality control. Nature 467, 470–473 (2010).
Brandman, O. et al. A ribosome-bound quality control complex triggers degradation of nascent peptides and signals translation stress. Cell 151, 1042–1054 (2012).
Choe, Y. J. et al. Failure of RQC machinery causes protein aggregation and proteotoxic stress. Nature 531, 191–195 (2016).
Yonashiro, R. et al. The Rqc2/Tae2 subunit of the ribosome-associated quality control (RQC) complex marks ribosome-stalled nascent polypeptide chains for aggregation. eLife 5, e11794 (2016).
Izawa, T., Park, S. H., Zhao, L., Hartl, F. U. & Neupert, W. Cytosolic protein Vms1 links ribosome quality control to mitochondrial and cellular homeostasis. Cell 171, 890–903 (2017).
Stein, K. C., Morales-Polanco, F., van der Lienden, J., Rainbolt, T. K. & Frydman, J. Ageing exacerbates ribosome pausing to disrupt cotranslational proteostasis. Nature 601, 637–642 (2022).
Lee, J. et al. Pelota-mediated ribosome-associated quality control counteracts aging and age-associated pathologies across species. Proc. Natl Acad. Sci. USA 122, e2505217122 (2025).
Chu, J. et al. A mouse forward genetics screen identifies LISTERIN as an E3 ubiquitin ligase involved in neurodegeneration. Proc. Natl Acad. Sci. USA 106, 2097–2103 (2009).
Martin, P. B. et al. NEMF mutations that impair ribosome-associated quality control are associated with neuromuscular disease. Nat. Commun. 11, 4625 (2020).
Endo, R. et al. Dysregulation of ribosome-associated quality control elicits cognitive disorders via overaccumulation of TTC3. Proc. Natl Acad. Sci. USA 120, e2211522120 (2023).
Ahmed, A. et al. Biallelic loss-of-function variants in NEMF cause central nervous system impairment and axonal polyneuropathy. Hum. Genet. 140, 579–592 (2021).
Anazi, S. et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiatry 22, 615–624 (2017).
Shoemaker, C. J. & Green, R. Translation drives mRNA quality control. Nat. Struct. Mol. Biol. 19, 594–601 (2012).
Hofer, T. et al. Hydrogen peroxide causes greater oxidation in cellular RNA than in DNA. Biol. Chem. 386, 333–337 (2005).
Li, Z., Wu, J. & Deleo, C. J. RNA damage and surveillance under oxidative stress. IUBMB Life 58, 581–588 (2006).
Kauppila, T. E. S., Kauppila, J. H. K. & Larsson, N. G. Mammalian mitochondria and aging: an update. Cell Metab. 25, 57–71 (2017).
Sies, H. & Jones, D. P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 21, 363–383 (2020).
Simms, C. L. & Zaher, H. S. Quality control of chemically damaged RNA. Cell Mol. Life Sci. 73, 3639–3653 (2016).
Simms, C. L., Hudson, B. H., Mosior, J. W., Rangwala, A. S. & Zaher, H. S. An active role for the ribosome in determining the fate of oxidized mRNA. Cell Rep. 9, 1256–1264 (2014).
Tanaka, M. & Chock, P. B. Oxidative modifications of RNA and its potential roles in biosystem. Front. Mol. Biosci. 8, 685331 (2021).
Wheeler, H. B., Madrigal, A. A. & Chaim, I. A. Mapping the future of oxidative RNA damage in neurodegeneration: rethinking the status quo with new tools. Proc. Natl Acad. Sci. USA 121, e2317860121 (2024).
Zhao, S. et al. RNF14-dependent atypical ubiquitylation promotes translation-coupled resolution of RNA-protein crosslinks. Mol. Cell 83, 4290–4303 (2023).
Suryo Rahmanto, A. et al. K6-linked ubiquitylation marks formaldehyde-induced RNA-protein crosslinks for resolution. Mol. Cell 83, 4272–4289 (2023).
Tenney, L., Pham, V. N. & Chang, C. J. One carbon to rule them all: formaldehyde is a one-carbon signal connecting one-carbon metabolism and epigenetic methylation. ACS Chem. Biol. 19, 798–801 (2024).
Lieber, C. S. Alcohol and the liver: 1994 update. Gastroenterology 106, 1085–1105 (1994).
Klapacz, J. et al. Contributions of DNA repair and damage response pathways to the non-linear genotoxic responses of alkylating agents. Mutat. Res. Rev. Mutat. Res. 767, 77–91 (2016).
Yan, L. L., Simms, C. L., McLoughlin, F., Vierstra, R. D. & Zaher, H. S. Oxidation and alkylation stresses activate ribosome-quality control. Nat. Commun. 10, 5611 (2019).
Wurtmann, E. J. & Wolin, S. L. RNA under attack: cellular handling of RNA damage. Crit. Rev. Biochem. Mol. Biol. 44, 34–49 (2009).
Wu, C. C., Peterson, A., Zinshteyn, B., Regot, S. & Green, R. Ribosome collisions trigger general stress responses to regulate cell fate. Cell 182, 404–416 (2020).
Stoneley, M. et al. Unresolved stalled ribosome complexes restrict cell-cycle progression after genotoxic stress. Mol. Cell 82, 1557–1572(2022).
Gruber, A. J. & Zavolan, M. Alternative cleavage and polyadenylation in health and disease. Nat. Rev. Genet. 20, 599–614 (2019).
Passmore, L. A. & Coller, J. Roles of mRNA poly(A) tails in regulation of eukaryotic gene expression. Nat. Rev. Mol. Cell Biol. 23, 93–106 (2022).
Frischmeyer, P. A. et al. An mRNA surveillance mechanism that eliminates transcripts lacking termination codons. Science 295, 2258–2261 (2002).
Ito-Harashima, S., Kuroha, K., Tatematsu, T. & Inada, T. Translation of the poly(A) tail plays crucial roles in nonstop mRNA surveillance via translation repression and protein destabilization by proteasome in yeast. Genes Dev. 21, 519–524 (2007).
Lu, J. & Deutsch, C. Electrostatics in the ribosomal tunnel modulate chain elongation rates. J. Mol. Biol. 384, 73–86 (2008).
Arthur, L. et al. Translational control by lysine-encoding A-rich sequences. Sci. Adv. 1, e1500154 (2015).
Koutmou, K. S. et al. Ribosomes slide on lysine-encoding homopolymeric A stretches. eLife 4, e05534 (2015).
Chandrasekaran, V. et al. Mechanism of ribosome stalling during translation of a poly(A) tail. Nat. Struct. Mol. Biol. 26, 1132–1140 (2019).
Tesina, P. et al. Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 39, e103365 (2020).
Pavlov, M. Y. et al. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc. Natl Acad. Sci. USA 106, 50–54 (2009).
Melnikov, S. et al. Molecular insights into protein synthesis with proline residues. EMBO Rep. 17, 1776–1784 (2016).
Gutierrez, E. et al. eIF5A promotes translation of polyproline motifs. Mol. Cell 51, 35–45 (2013).
Mizuno, M. et al. The nascent polypeptide in the 60S subunit determines the Rqc2-dependency of ribosomal quality control. Nucleic Acids Res. 49, 2102–2113 (2021).
Liu, Y., Yang, Q. & Zhao, F. Synonymous but not silent: the codon usage code for gene expression and protein folding. Annu. Rev. Biochem. 90, 375–401 (2021).
Letzring, D. P., Dean, K. M. & Grayhack, E. J. Control of translation efficiency in yeast by codon-anticodon interactions. RNA 16, 2516–2528 (2010).
Gamble, C. E., Brule, C. E., Dean, K. M., Fields, S. & Grayhack, E. J. Adjacent codons act in concert to modulate translation efficiency in yeast. Cell 166, 679–690 (2016).
Suzuki, T. The expanding world of tRNA modifications and their disease relevance. Nat. Rev. Mol. Cell Biol. 22, 375–392 (2021).
Murakami, S. & Jaffrey, S. R. Hidden codes in mRNA: control of gene expression by m6A. Mol. Cell 82, 2236–2251 (2022).
Wang, X. et al. Structural basis of N6-adenosine methylation by the METTL3–METTL14 complex. Nature 534, 575–578 (2016).
Choi, J. et al. N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol. 23, 110–115 (2016).
Murakami, S. et al. m6A alters ribosome dynamics to initiate mRNA degradation. Cell 188, 3728–3743 (2025).
Alkalaeva, E. Z., Pisarev, A. V., Frolova, L. Y., Kisselev, L. L. & Pestova, T. V. In vitro reconstitution of eukaryotic translation reveals cooperativity between release factors eRF1 and eRF3. Cell 125, 1125–1136 (2006).
Aleksandrova, E. V. et al. Mechanism of release factor-mediated peptidyl-tRNA hydrolysis on the ribosome. Science 388, eads9030 (2025).
Pisarev, A. V. et al. The role of ABCE1 in eukaryotic posttermination ribosomal recycling. Mol. Cell 37, 196–210 (2010).
Becker, T. et al. Structural basis of highly conserved ribosome recycling in eukaryotes and archaea. Nature 482, 501–506 (2012).
Becker, T. et al. Structure of the no-go mRNA decay complex Dom34-Hbs1 bound to a stalled 80S ribosome. Nat. Struct. Mol. Biol. 18, 715–720 (2011).
Tsuboi, T. et al. Dom34:hbs1 plays a general role in quality-control systems by dissociation of a stalled ribosome at the 3′ end of aberrant mRNA. Mol. Cell 46, 518–529 (2012).
Pisareva, V. P., Skabkin, M. A., Hellen, C. U., Pestova, T. V. & Pisarev, A. V. Dissociation by Pelota, Hbs1 and ABCE1 of mammalian vacant 80S ribosomes and stalled elongation complexes. EMBO J. 30, 1804–1817 (2011).
Juszkiewicz, S. & Hegde, R. S. Initiation of quality control during poly(A) translation requires site-specific ribosome ubiquitination. Mol. Cell 65, 743–750 (2017).
Sundaramoorthy, E. et al. ZNF598 and RACK1 regulate mammalian ribosome-associated quality control function by mediating regulatory 40S ribosomal ubiquitylation. Mol. Cell 65, 751–760 (2017).
Matsuo, Y. et al. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat. Commun. 8, 159 (2017).
Matsuo, Y. et al. RQT complex dissociates ribosomes collided on endogenous RQC substrate SDD1. Nat. Struct. Mol. Biol. 27, 323–332 (2020).
Juszkiewicz, S., Speldewinde, S. H., Wan, L., Svejstrup, J. Q. & Hegde, R. S. The ASC-1 complex disassembles collided ribosomes. Mol. Cell 79, 603–614 e608 (2020).
Matsuo, Y., Uchihashi, T. & Inada, T. Decoding of the ubiquitin code for clearance of colliding ribosomes by the RQT complex. Nat. Commun. 14, 79 (2023).
Best, K. et al. Structural basis for clearing of ribosome collisions by the RQT complex. Nat. Commun. 14, 921 (2023).
Meyer, C., Garzia, A., Morozov, P., Molina, H. & Tuschl, T. The G3BP1-family-USP10 deubiquitinase complex rescues ubiquitinated 40S subunits of ribosomes stalled in translation from lysosomal degradation. Mol. Cell 77, 1193–1205 (2020).
Tomomatsu, S. et al. Polyubiquitin architecture editing on collided ribosomes maintains persistent RQC activity. EMBO J. 44, 6051–6077 (2025).
Scazzari, M., Zhang, Y., Moddemann, A. & Rospert, S. Stalled disomes marked by Hel2-dependent ubiquitin chains undergo Ubp2/Ubp3-mediated deubiquitination upon translational run-off. Commun. Biol. 8, 132 (2025).
Hickey, K. L. et al. GIGYF2 and 4EHP inhibit translation initiation of defective messenger RNAs to assist ribosome-associated quality control. Mol. Cell 79, 950–962 (2020).
Juszkiewicz, S. et al. Ribosome collisions trigger cis-acting feedback inhibition of translation initiation. eLife 9, e60038 (2020).
Sinha, N. K. et al. EDF1 coordinates cellular responses to ribosome collisions. eLife 9, e58828 (2020).
Rom, E. et al. Cloning and characterization of 4EHP, a novel mammalian eIF4E-related cap-binding protein. J. Biol. Chem. 273, 13104–13109 (1998).
Joshi, B., Cameron, A. & Jagus, R. Characterization of mammalian eIF4E-family members. Eur. J. Biochem. 271, 2189–2203 (2004).
Muller, M. B. D. et al. The ribosome as a platform to coordinate mRNA decay. Nucleic Acids Res. 53, gkaf049 (2025).
Wang, X. et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505, 117–120 (2014).
Park, O. H. et al. Endoribonucleolytic cleavage of m6A-containing RNAs by RNase P/MRP complex. Mol Cell 74, 494–507 (2019).
Zhou, Y. et al. m6A sites in the coding region trigger translation-dependent mRNA decay. Mol. Cell 84, 4576–4593 (2024).
Linder, B. et al. tRNA modifications tune m6A-dependent mRNA decay. Cell 188, 3715–3727 (2025).
Fry, N. J., Law, B. A., Ilkayeva, O. R., Holley, C. L. & Mansfield, K. D. N6-methyladenosine is required for the hypoxic stabilization of specific mRNAs. RNA 23, 1444–1455 (2017).
Rubio, R. M., Depledge, D. P., Bianco, C., Thompson, L. & Mohr, I. RNA m6A modification enzymes shape innate responses to DNA by regulating interferon beta. Genes Dev. 32, 1472–1484 (2018).
Gurzeler, L. A. et al. Drug-induced eRF1 degradation promotes readthrough and reveals a new branch of ribosome quality control. Cell Rep. 42, 113056 (2023).
Oltion, K. et al. An E3 ligase network engages GCN1 to promote the degradation of translation factors on stalled ribosomes. Cell 186, 346–362 e317 (2023).
Pakos-Zebrucka, K. et al. The integrated stress response. EMBO Rep. 17, 1374–1395 (2016).
Ryoo, H. D. The integrated stress response in metabolic adaptation. J. Biol. Chem. 300, 107151 (2024).
Hinnebusch, A. G. A hierarchy of trans-acting factors modulates translation of an activator of amino acid biosynthetic genes in Saccharomyces cerevisiae. Mol. Cell Biol. 5, 2349–2360 (1985).
Wek, R. C., Jackson, B. M. & Hinnebusch, A. G. Juxtaposition of domains homologous to protein kinases and histidyl-tRNA synthetases in GCN2 protein suggests a mechanism for coupling GCN4 expression to amino acid availability. Proc. Natl Acad. Sci. USA 86, 4579–4583 (1989).
Dong, J., Qiu, H., Garcia-Barrio, M., Anderson, J. & Hinnebusch, A. G. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol. Cell 6, 269–279 (2000).
Dever, T. E. et al. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell 68, 585–596 (1992).
Hinnebusch, A. G. The scanning mechanism of eukaryotic translation initiation. Annu. Rev. Biochem. 83, 779–812 (2014).
Kenner, L. R. et al. eIF2B-catalyzed nucleotide exchange and phosphoregulation by the integrated stress response. Science 364, 491–495 (2019).
Kashiwagi, K. et al. Structural basis for eIF2B inhibition in integrated stress response. Science 364, 495–499 (2019).
Ishimura, R., Nagy, G., Dotu, I., Chuang, J. H. & Ackerman, S. L. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. eLife 5, e14295 (2016).
Inglis, A. J. et al. Activation of GCN2 by the ribosomal P-stalk. Proc. Natl Acad. Sci. USA 116, 4946–4954 (2019).
Harding, H. P. et al. The ribosomal P-stalk couples amino acid starvation to GCN2 activation in mammalian cells. eLife 8, e50149 (2019).
Marton, M. J., Crouch, D. & Hinnebusch, A. G. GCN1, a translational activator of GCN4 in Saccharomyces cerevisiae, is required for phosphorylation of eukaryotic translation initiation factor 2 by protein kinase GCN2. Mol. Cell Biol. 13, 3541–3556 (1993).
Pochopien, A. A. et al. Structure of Gcn1 bound to stalled and colliding 80S ribosomes. Proc. Natl Acad. Sci. USA 118, e2022756118 (2021).
Muller, M. B. D., Kasturi, P., Jayaraj, G. G. & Hartl, F. U. Mechanisms of readthrough mitigation reveal principles of GCN1-mediated translational quality control. Cell 186, 3227–3244 (2023).
Kim, K. Q. et al. Multiprotein bridging factor 1 is required for robust activation of the integrated stress response on collided ribosomes. Mol. Cell 84, 4594–4611 (2024).
Paternoga, H. et al. Structure of a Gcn2 dimer in complex with the large 60S ribosomal subunit. Proc. Natl Acad. Sci. USA 122, e2415807122 (2025).
Masson, G. R. Towards a model of GCN2 activation. Biochem. Soc. Trans. 47, 1481–1488 (2019).
Zhou, C. et al. GCN1 couples GCN2 to ribosomal state to initiate amino acid response pathway signaling. Science 390, eads8728 (2025).
Gupta, R. & Hinnebusch, A. G. Differential requirements for P stalk components in activating yeast protein kinase Gcn2 by stalled ribosomes during stress. Proc. Natl Acad. Sci. USA 120, e2300521120 (2023).
Vind, A. C. et al. ZAKalpha recognizes stalled ribosomes through partially redundant sensor domains. Mol. Cell 78, 700–713 (2020).
Sinha, N. K. et al. The ribotoxic stress response drives UV-mediated cell death. Cell 187, 3652–3670 (2024).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP–AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP–AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Burdette, D. L. et al. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518 (2011).
Zhang, C. et al. Structural basis of STING binding with and phosphorylation by TBK1. Nature 567, 394–398 (2019).
Zhao, B. et al. A conserved PLPLRT/SD motif of STING mediates the recruitment and activation of TBK1. Nature 569, 718–722 (2019).
Wan, L. et al. Translation stress and collided ribosomes are co-activators of cGAS. Mol. Cell 81, 2808–2822 (2021).
Jaafar, Z. A. & Kieft, J. S. Viral RNA structure-based strategies to manipulate translation. Nat. Rev. Microbiol. 17, 110–123 (2019).
Yan, L. L. & Zaher, H. S. Ribosome quality control antagonizes the activation of the integrated stress response on colliding ribosomes. Mol. Cell 81, 614–628 e614 (2021).
Meydan, S. & Guydosh, N. R. Disome and trisome profiling reveal genome-wide targets of ribosome quality control. Mol. Cell 79, 588–602 e586 (2020).
Shen, P. S. et al. Protein synthesis. Rqc2p and 60S ribosomal subunits mediate mRNA-independent elongation of nascent chains. Science 347, 75–78 (2015).
Shao, S., Brown, A., Santhanam, B. & Hegde, R. S. Structure and assembly pathway of the ribosome quality control complex. Mol. Cell 57, 433–444 (2015).
Abaeva, I. S., Bulakhov, A. G., Hellen, C. U. T. & Pestova, T. V. The ribosome-associated quality control factor TCF25 imposes K48 specificity on Listerin-mediated ubiquitination of nascent chains by binding and specifically orienting the acceptor ubiquitin. Genes Dev. 39, 617–633 (2025).
Yip, M. C. J. et al. Mechanism for recycling tRNAs on stalled ribosomes. Nat. Struct. Mol. Biol. 26, 343–349 (2019).
Verma, R., Oania, R. S., Kolawa, N. J. & Deshaies, R. J. Cdc48/p97 promotes degradation of aberrant nascent polypeptides bound to the ribosome. eLife 2, e00308 (2013).
Yip, M. C. J., Savickas, S., Gygi, S. P. & Shao, S. ELAC1 repairs tRNAs cleaved during ribosome-associated quality control. Cell Rep. 30, 2106–2114 e2105 (2020).
Akopian, D., Shen, K., Zhang, X. & Shan, S. O. Signal recognition particle: an essential protein-targeting machine. Annu. Rev. Biochem. 82, 693–721 (2013).
Voorhees, R. M. & Hegde, R. S. Toward a structural understanding of co-translational protein translocation. Curr. Opin. Cell Biol. 41, 91–99 (2016).
von der Malsburg, K., Shao, S. & Hegde, R. S. The ribosome quality control pathway can access nascent polypeptides stalled at the Sec61 translocon. Mol. Biol. Cell 26, 2168–2180 (2015).
Crowder, J. J. et al. Rkr1/Ltn1 ubiquitin ligase-mediated degradation of translationally stalled endoplasmic reticulum proteins. J. Biol. Chem. 290, 18454–18466 (2015).
Scavone, F., Gumbin, S. C., Da Rosa, P. A. & Kopito, R. R. RPL26/uL24 UFMylation is essential for ribosome-associated quality control at the endoplasmic reticulum. Proc. Natl Acad. Sci. USA 120, e2220340120 (2023).
Ishimura, R. et al. Mechanistic insights into the roles of the UFM1 E3 ligase complex in ufmylation and ribosome-associated protein quality control. Sci. Adv. 9, eadh3635 (2023).
Peter, J. J. et al. A non-canonical scaffold-type E3 ligase complex mediates protein UFMylation. EMBO J. 41, e111015 (2022).
Wang, L. et al. UFMylation of RPL26 links translocation-associated quality control to endoplasmic reticulum protein homeostasis. Cell Res. 30, 5–20 (2020).
Makhlouf, L. et al. The UFM1 E3 ligase recognizes and releases 60S ribosomes from ER translocons. Nature 627, 437–444 (2024).
DaRosa, P. A. et al. UFM1 E3 ligase promotes recycling of 60S ribosomal subunits from the ER. Nature 627, 445–452 (2024).
Penchev, I. et al. UFMylation orchestrates spatiotemporal coordination of RQC at the ER. Sci. Adv. 11, eadv0435 (2025).
Millrine, D. et al. Human UFSP1 is an active protease that regulates UFM1 maturation and UFMylation. Cell Rep. 40, 111168 (2022).
Izawa, T. et al. Roles of dom34:hbs1 in nonstop protein clearance from translocators for normal organelle protein influx. Cell Rep. 2, 447–453 (2012).
Thrun, A. et al. Convergence of mammalian RQC and C-end rule proteolytic pathways via alanine tailing. Mol. Cell 81, 2112–2122 (2021).
Udagawa, T. et al. Failure to degrade CAT-tailed proteins disrupts neuronal morphogenesis and cell survival. Cell Rep. 34, 108599 (2021).
Kostova, K. K. et al. CAT-tailing as a fail-safe mechanism for efficient degradation of stalled nascent polypeptides. Science 357, 414–417 (2017).
Osuna, B. A., Howard, C. J., Kc, S., Frost, A. & Weinberg, D. E. In vitro analysis of RQC activities provides insights into the mechanism and function of CAT tailing. eLife 6, e27949 (2017).
Tesina, P. et al. Molecular basis of eIF5A-dependent CAT tailing in eukaryotic ribosome-associated quality control. Mol. Cell 83, 607–621 (2023).
Su, T. et al. Structure and function of Vms1 and Arb1 in RQC and mitochondrial proteome homeostasis. Nature 570, 538–542 (2019).
Wu, Z. et al. MISTERMINATE mechanistically links mitochondrial dysfunction with proteostasis failure. Mol. Cell 75, 835–848 (2019).
Sitron, C. S. & Brandman, O. CAT tails drive degradation of stalled polypeptides on and off the ribosome. Nat. Struct. Mol. Biol. 26, 450–459 (2019).
Lin, H. C. et al. C-terminal end-directed protein elimination by CRL2 ubiquitin ligases. Mol. Cell 70, 602–613 (2018).
Koren, I. et al. The eukaryotic proteome is shaped by E3 ubiquitin ligases targeting C-terminal degrons. Cell 173, 1622–1635 (2018).
Kuroha, K., Zinoviev, A., Hellen, C. U. T. & Pestova, T. V. Release of ubiquitinated and non-ubiquitinated nascent chains from stalled mammalian ribosomal complexes by ANKZF1 and Ptrh1. Mol. Cell 72, 286–302 (2018).
Chang, W. D., Yoon, M. J., Yeo, K. H. & Choe, Y. J. Threonine-rich carboxyl-terminal extension drives aggregation of stalled polypeptides. Mol. Cell 84, 4334–4349 (2024).
Sontag, E. M., Samant, R. S. & Frydman, J. Mechanisms and functions of spatial protein quality control. Annu. Rev. Biochem. 86, 97–122 (2017).
Aguilaniu, H., Gustafsson, L., Rigoulet, M. & Nystrom, T. Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299, 1751–1753 (2003).
Hung, H. C. et al. Poly-alanine-tailing is a modifier of neurodegeneration caused by Listerin mutation. Preprint at bioRxiv https://doi.org/10.1101/2024.08.24.608776 (2024).
Ishimura, R. et al. RNA function. Ribosome stalling induced by mutation of a CNS-specific tRNA causes neurodegeneration. Science 345, 455–459 (2014).
Terrey, M. et al. GTPBP1 resolves paused ribosomes to maintain neuronal homeostasis. eLife 9, e62731 (2020).
DeJesus-Hernandez, M. et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 72, 245–256 (2011).
Renton, A. E. et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 72, 257–268 (2011).
Zu, T. et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl Acad. Sci. USA 110, E4968–E4977 (2013).
Mori, K. et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science 339, 1335–1338 (2013).
Cleary, J. D., Pattamatta, A. & Ranum, L. P. W. Repeat-associated non-ATG (RAN) translation. J. Biol. Chem. 293, 16127–16141 (2018).
Kwon, I. et al. Poly-dipeptides encoded by the C9orf72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345, 1139–1145 (2014).
Mizielinska, S. et al. C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science 345, 1192–1194 (2014).
Park, J. et al. ZNF598 co-translationally titrates poly(GR) protein implicated in the pathogenesis of C9ORF72-associated ALS/FTD. Nucleic Acids Res. 49, 11294–11311 (2021).
Latallo, M. J. et al. Single-molecule imaging reveals distinct elongation and frameshifting dynamics between frames of expanded RNA repeats in C9ORF72-ALS/FTD. Nat. Commun. 14, 5581 (2023).
Tseng, Y. J. et al. Ribosomal quality control factors inhibit repeat-associated non-AUG translation from GC-rich repeats. Nucleic Acids Res. 52, 5928–5949 (2024).
Loveland, A. B. et al. Ribosome inhibition by C9ORF72-ALS/FTD-associated poly-PR and poly-GR proteins revealed by cryo-EM. Nat. Commun. 13, 2776 (2022).
Haeusler, A. R. et al. C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507, 195–200 (2014).
Pfanner, N., Warscheid, B. & Wiedemann, N. Mitochondrial proteins: from biogenesis to functional networks. Nat. Rev. Mol. Cell Biol. 20, 267–284 (2019).
Longley, D. B., Harkin, D. P. & Johnston, P. G. 5-fluorouracil: mechanisms of action and clinical strategies. Nat. Rev. Cancer 3, 330–338 (2003).
Chatterjee, S. et al. Ribosome quality control mitigates the cytotoxicity of ribosome collisions induced by 5-fluorouracil. Nucleic Acids Res. 52, 12534–12548 (2024).
Chaudhary, N., Weissman, D. & Whitehead, K. A. mRNA vaccines for infectious diseases: principles, delivery and clinical translation. Nat. Rev. Drug Discov. 20, 817–838 (2021).
Mulroney, T. E. et al. N1-methylpseudouridylation of mRNA causes +1 ribosomal frameshifting. Nature 625, 189–194 (2024).
Trentini, D. B. et al. Role for ribosome-associated quality control in sampling proteins for MHC class I-mediated antigen presentation. Proc. Natl Acad. Sci. USA 117, 4099–4108 (2020).
Di Fraia, D. et al. Altered translation elongation contributes to key hallmarks of aging in the killifish brain. Science 389, eadk3079 (2025).
Acknowledgements
We thank T. Chan Yew Kit for assistance with figure preparation. This work was supported by the Ministry of Education, Singapore, under its Academic Research Fund Tier 1 (RG114/23 and RT20/23; Y.-J.C.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chang, W.D., Choe, YJ. Quality control and signaling pathways at stalled ribosomes. Exp Mol Med 58, 82–93 (2026). https://doi.org/10.1038/s12276-025-01623-w
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s12276-025-01623-w
