
Abstract
Inflammation plays a vital role in the development of diabetic nephropathy, but the underlying regulatory mechanisms are only partially understood. Our previous studies demonstrated that, during acute inflammation, endothelial heparan sulfate (HS) contributes to the adhesion and transendothelial migration of leukocytes into perivascular tissues by direct interaction with l-selectin and the presentation of bound chemokines. In the current study, we aimed to assess the role of endothelial HS on chronic renal inflammation and fibrosis in a diabetic nephropathy mouse model. To reduce sulfation of HS specifically in the endothelium, we generated Ndst1f/fTie2Cre+ mice in which N-deacetylase/N-sulfotransferase-1 (Ndst1), the gene that initiates HS sulfation modifications in HS biosynthesis, was expressly ablated in endothelium. To induce diabetes, age-matched male Ndst1f/fTie2Cre- (wild type) and Ndst1f/fTie2Cre+ mice on a C57Bl/6J background were injected intraperitoneally with streptozotocin (STZ) (50 mg/kg) on five consecutive days (N = 10–11/group). Urine and plasma were collected. Four weeks after diabetes induction the animals were sacrificed and kidneys were analyzed by immunohistochemistry and qRT-PCR. Compared to healthy controls, diabetic Ndst1f/fTie2Cre- mice showed increased glomerular macrophage infiltration, mannose binding lectin complement deposition and glomerulosclerosis, whereas these pathological reactions were prevented significantly in the diabetic Ndst1f/fTie2Cre+ animals (all three p < 0.01). In addition, the expression of the podocyte damage marker desmin was significantly higher in the Ndst1f/fTie2Cre− group compared to the Ndst1f/fTie2Cre+ animals (p < 0.001), although both groups had comparable numbers of podocytes. In the cortical tubulo-interstitium, similar analyses show decreased interstitial macrophage accumulation in the diabetic Ndst1f/fTie2Cre+ animals compared to the diabetic Ndst1f/fTie2Cre− mice (p < 0.05). Diabetic Ndst1f/fTie2Cre+ animals also showed reduced interstitial fibrosis as evidenced by reduced density of αSMA-positive myofibroblasts (p < 0.01), diminished collagen III deposition (p < 0.001) and reduced mRNA expression of collagen I (p < 0.001) and fibronectin (p < 0.001). Our studies indicate a pivotal role of endothelial HS in the development of renal inflammation and fibrosis in diabetic nephropathy in mice. These results suggest that HS is a possible target for therapy in diabetic nephropathy.
Similar content being viewed by others


Introduction
Despite improvements in blood glucose control, progression towards diabetic nephropathy remains a major burden for diabetic patients and society [1, 2]. Inflammation is increasingly recognized as a key initiator of the histological changes seen in diabetic nephropathy leading to the hallmark histological changes, i.e., glomerulosclerosis, basement membrane thickening, and interstitial fibrosis [3]. Pro-inflammatory cytokines, chemokines, and growth factors such as interleukin (IL)-1, IL-6, IL-18, tumor necrosis factor, monocyte chemotactic protein (MCP)-1, RANTES, and transforming growth factor (TGF)-β are expressed in diabetic nephropathy and are thought to be involved in macrophage influx and play a role in fibrosis induction [3]. Targeting some of these chemokines and cytokines has already been shown to be effective in reducing clinical symptoms and histological changes in (experimental) diabetic nephropathy. MCP-1 deficiency in diabetic db/db mice resulted in less albuminuria, reduced interstitial macrophage influx, and reduced renal fibrosis [4]. Early clinical trials targeting inflammation in diabetic kidney disease reveal promising results [5]. As has been shown in general and for diabetic nephropathy specifically, the development of fibrosis is largely dependent on initiation by an inflammatory response, as has been reviewed by Kanasaki et al. [6]. It is thought that sustained inflammatory cell activation primes local fibroblasts, resulting in deposition of extracellular matrix (ECM) components [7]. This entails that inflammation is a major trigger and activator for the fibrotic response and indeed, MCP-1 deficiency, among other factors, results in reduced renal fibrosis in an animal model for type 2 diabetes [4]. Another important cytokine signaling network between inflammatory cells and fibrotic cells is TGF-β; targeting TGF-β has been demonstrated to reduce fibrosis and kidney hypertrophy in animal models for diabetic nephropathy [8,9,10]. Both MCP-1 and TGF-β bind heparan sulfate (HS) with high affinity [11, 12], which might indicate an important role for HS in the development of fibrosis in diabetic nephropathy and suggesting HS as a potential therapeutic target in diabetic nephropathy [11, 12].
Inflammation is characterized by the influx of leukocytes from the circulation to areas of tissue injury. During hyperglycemic conditions, like in diabetes, tissue damage results from alternative glucose metabolism, leading to the activation of endothelial cells in the glomerulus and the interstitium [13]. Activated endothelial cells produce ligands for l-selectin, which is expressed on leukocytes, causing leukocyte rolling and adhesion to the endothelium [14]. Our previous study identified HS expressed on the endothelium as a ligand for l-selectin and reduction of sulfation of endothelial HS diminishes the binding of l-selectin to these cells, which, in consequence, results in a reduced adhesion of leukocytes to the endothelium and reduced neutrophil infiltration in inflammation [15]. We also reported that binding of l-selectin in inflamed renal tissue occurs predominantly on the sub-endothelial basement membrane [12]. The involvement of sub-endothelial HS in leukocyte transmigration was proven in renal ischemia reperfusion experiments in which macrophage influx was reduced in mice deficient for the basement membrane hybrid Heparan sulfate proteoglycans (HSPGs)/collagens XV and XVIII [16]. Another important factor in leukocyte transmigration is the stimulation of leukocytes by binding of chemokines presented on the endothelium to G-protein-coupled receptors on leukocytes. The role of endothelial HS on chemokine-mediated leukocyte activation appears to be multifactorial [17]. There is an evidence that chemokines immobilized on endothelial HS, but not soluble chemokines, induce lymphocyte function-associated antigen (LFA-1) transformation on leukocytes enabling binding to endothelial expressed ICAM-1 and thereby facilitate firm adhesion [18]. It has also been shown that a chemotactic gradient of chemokines bound to endothelial HS facilitates leukocyte migration across the endothelium [19]. Furthermore, it has been shown that chemokine binding to HS results in chemokine oligomerization, which might increase the leukocyte activating potential of chemokines [20]. Finally, HS has been demonstrated to be involved in the transcytosis of chemokines across the endothelial layer in our previous study [15]. These chemokines are largely produced by perivascular leukocytes and for presentation on the luminal side of the endothelial cells they need to be transported through the endothelial layer. Studies, including our previous finding [15], have shown that this process is HS dependent [19, 21]. Taken together, these findings show the role of endothelial HS on l-selectin and chemokine-mediated recruitment of inflammatory cells.
In HS biosynthesis, the enzyme N-deacetylase/N-sulfotransferase-1 (Ndst1) initiates modification reactions to generate binding sites for protein ligands. Lack of the Ndst1 enzyme has been shown to result in an undersulfated and therefore biologically less active HS [22]. Ndst1 appears to be essential for mammalian development. Conventional knockout of Ndst1 in mice results in early post-natal death with abnormal lung and forebrain development defects [23, 24]. In previous studies, we generated a conditional Ndst1 (Ndst1f/f) mouse and crossbred with a transgenic mouse expressing Cre recombinase under the Tie2 tyrosine-kinase promoter (Tie2Cre) to generate the Ndst1f/f Tie2Cre+ mice in which Ndst1 is specifically ablated in endothelial cells [15, 25, 26]. In the current study, we induced diabetes in the Ndst1f/fTie2Cre+ mice and their littermate wildtype control Ndst1f/fTie2Cre− mice to directly assess the role of endothelial HS in development of renal inflammation and fibrosis in the streptozotocin (STZ)-induced diabetic nephropathy mouse model.
Materials and methods
Experimental animals
The Ndst1f/fTie2Cre+ mice were generated by breeding Ndst1f/f mice with transgenic Tie2Cre mice as previously described [15, 26]. All experimental mice were fully backcrossed to C57BL/6 background and handled according to guidelines of the University of Georgia Institutional Animal Care and Use Committee.
Induction of diabetes
Baseline plasma and 24 h urine samples were collected approximately 1 week before induction of diabetes. To induce diabetes, 7–11-week-old male mice received either 50 mg/kg STZ for diabetic animals (DB) or citrate buffer for healthy controls (HC) intraperitoneally on five consecutive days [27]. Animals were monitored for 2 weeks, where after 24 h urine was collected and blood glucose was measured. Animals responsive to the administered STZ with a blood glucose level over 300 mg/dl were included in the study. Four groups of mice were included in the study: HC/Ndst1f/fTie2Cre- (N = 3), HC/Ndst1f/fTie2Cre+ (N = 3), DB/Ndst1f/fTie2Cre− (N = 10), and DB/Ndst1f/fTie2Cre+ (N = 11). The animals were monitored on a daily basis for body weight, activity, and fur condition. After 4 weeks of monitoring, the animals were sacrificed and the organs were harvested (6 weeks after diabetes induction). Before sacrifice plasma and 24 h urine were collected.
Immunohistochemistry
Four micrometer frozen and formalin-fixed paraffin embedded kidney sections were used for immunohistochemical stainings. Details on fixation, antigen retrieval, antibodies, and conjugates are given in Table 1. All controls (omitting primary and/or secondary antibodies in various combinations) proved to be negative (not shown). For interstitial quantification of macrophages, T-cells, myofibroblasts, neutrophils and MCP1, ten photomicrographs per animal were randomly taken at a 200× magnification. Quantification of macrophages, T-cells, myofibroblasts, and MCP1 was performed using the MacBiophotonics ImageJ software (Rasband, W.S., ImageJ, U.S. National Institute of Health, Bethesda, Maryland, USA). Data was expressed as % of area stained for F4/80 (macrophages), CD3 (T-cells) and MCP1, and fold increase compared to control for αSMA (myofibroblasts). NimpR14+ neutrophils were counted manually and data was expressed as number of neutrophils per mm2 of tissue. To quantify WT-1+ podocytes ten cortical photomicrographs were taken randomly of every animal. Podocytes (by positively stained nuclei) were counted manually and data was expressed as mean number of podocytes per glomerulus. Glomerular macrophages were quantified in ten photomicrographs per animal which were randomly taken from the cortical region of the kidney at 200× magnification. Intraglomerular macrophages were counted manually and data was expressed as mean number of macrophages per glomerulus. Mesangial localization of the intraglomerular macrophages was shown by a double staining of collagen III with macrophage marker F4-80.
Deposition of collagen III, complement MBL-C, and C3 was quantified by analyzing ten randomly taken photomicrographs of the cortical region of the kidney per animal, at a magnification of 200×, with MacBiophotonics ImageJ software. MBL-C was co-stained with basement membrane HSPG agrin to better localize the distribution of MBL-C in the glomerulus. Data was expressed as mean % of area stained for the respective marker. Collagen III deposition in the glomerulus was quantified using 15 randomly taken photomicrographs of glomeruli at 400× magnification and analyzed with the MacBiophotonics ImageJ software. Data was expressed as the mean % of glomerular area stained for collagen III.
ELISA
Albumin concentration in the urine was determined using the following ELISA procedure. 96-wells plates were coated overnight with a goat anti-mouse albumin antibody (BETHYL Laboratories, Inc. Montgomery, Texas, USA) diluted 1:100 in 0.05 M Carbonate–bicarbonate at pH 9.6. After blocking with 50 mM Tris, 0.14 M NaCl, 1% BSA, pH 8.0, urinary samples were incubated for 1 h in various dilutions. Bound albumin was detected using an HRP conjugated goat anti-mouse albumin antibody diluted 1:75,000 (BETHYL Laboratories, Inc. Montgomery, Texas, USA). Binding was visualized using 3,3′,5,5′- tetramethylbenzidine (Sigma-Aldrich, Zwijndrecht, The Netherlands) where after 1 M H2SO4 was added and absorbance was measured at 450 nm (Benchmark Plus microplate spectrophotometer, BIORAD). Urinary albumin concentration was determined using a standard curve with known albumin concentrations.
Urinary creatinine was measured using the creatinine colorimetric assay kit (Cayman Chemicals, Ann Arbor, Michigan, USA) according to manufacturer’s instructions. Albumin values were adjusted for creatinine values by dividing the urinary albumin concentration by the urinary creatinine concentration resulting in the albumin/creatinine ratio (ACR).
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
RNA was isolated from 10 mg pieces of frozen kidney using the FavorPrep RNA Purification kit (Favorgen Biotech Corp, Vienna, Austria) according to the manufacturer protocol. The RNA concentration and integrity were determined by spectrophotometry (Nanodrop Technologies, Wilmington, DE). For qRT-PCR analysis, total RNA was reverse transcribed using the Qiagen reverse transcription kit (Venlo, the Netherlands) in accordance to the manufacturer’s protocol. qRT-PCR was performed in 384 wells plates (Applied Biosystems, Foster City, CA) with a final reaction volume of 10 µl consisting of 3 µl of cDNA, 5 µl of 2× SYBR Green Master (Bio-Rad, Veenendaal, The Netherlands), 0.08 µl 50 µM primer mix (0.4 µM) and 1.92 µl RNA free H20. All reactions were performed in triplicate. The primers used: desmin forward CAGGATCAACCTTCCTATCC, reverse CTGTCTTTTTGGTATGGACTTC, fibronectin forward CCTATAGGATTGGAGACACG, reverse GTTGGTAAATAGCTGTTCGG and collagen I forward CGTATCACCAAACTCAGAAG, reverse GAAGCAAAGTTTCCTCCAAG were all purchased from Sigma (Sigma, Zwijndrecht, The Netherlands). qRT-PCR reaction was performed using a ViiA7 Real-Time PCR system (AB Applied Biosystems). Differences in the expression of a gene of interest was determined by normalizing the mean Ct-value against the mean Ct value of ribosomal 36B4 housekeeping gene (forward: AAGCGCGTCCTGGGATTGTC and reverse: GCAGCCGCAAATGAGATGG) using ΔCt-method: ΔCt = Ct gene of interest—Ct mean 36B4. Relative expression of gene of interest was calculated as 2−(ΔCt). Control non-diabetic samples of both strains did not show differences for any of the transcripts and were represented in one group.
Statistics
Data is expressed as mean ± SEM. Gaussian distribution of the data was verified using the D’Agostino & Pearson omnibus normality test. One-way ANOVA was used to compare groups. Statistical significance was tested between the HC/Ndst1f/fTie2Cre- and DB/Ndst1f/fTie2Cre− groups and between the DB/Ndst1f/fTie2Cre− and DB/Ndst1f/fTie2Cre+ groups using Bonferroni multiple comparison test. When the variances were not equal a Welch’s correction was applied. p < 0.05 was considered statistically significant.
Results
Validation of the animal model
In our previous studies, we have observed that endothelial Ndst1 deletion leads to reduced N-sulfation and 2-O and 6-O sulfation modifications of endothelial HS [15, 25, 26]. To verify the under sulfated status of the endothelial HS in the Ndst1f/fTie2Cre+ animals, an FGF-2 binding assay was performed. The growth factor FGF-2 binds HS with a high affinity and the binding requires HS to be modified with N-O-sulfation and 2-O-sulfation [28, 29]. Ndst1f/fTie2Cre- mice clearly showed strong FGF-2 binding, predominantly with the peritubular capillaries (Fig. 1a). In contrast, FGF-2 binding was markedly reduced in the Ndst1f/fTie2Cre+ mice (Fig. 1b). This observation demonstrated that sulfation modification of endothelial HS in the peritubular vascular area is reduced in the Ndst1f/fTie2Cre+ kidney. After induction of diabetes, plasma glucose gradually increased over time, but no difference in glucose levels between the Ndst1f/fTie2Cre− and Ndst1f/fTie2Cre+ animals was seen (Fig. 1c), indicating that deficiency of endothelial HS does not prevent diabetes development in the mice. The mice were characterized by increased 24 h urinary volume accompanied by increased water intake (not shown). Urinary albumin excretion developed over time as shown by an increased albumin/creatinine ratio (ACR) between 2 and 6 weeks follow-up (p < 0.01). However, these parameters were not different between the Ndst1f/fTie2Cre− and the Ndst1f/fTie2Cre+ animals (Fig. 1d), indicating endothelial Ndst1 deficiency could neither prevent nor significantly reduce albuminuria. No increase in ACR was seen over time in HC mice, including both the Ndst1f/fTie2Cre− and the Ndst1f/fTie2Cre+ animals.

Validation of the model: FGF-2 binding, hyperglycemia and rising albumin/creatinine ratio (ACR). Binding of exogenous FGF-2 (in red fluorescence) with endothelial HS was reduced in the Ndst1f/fTie2Cre+ animals compared to the Ndst1f/fTie2Cre− mice (a, b) indicating reduced N-sulfation and 2-O-sulfation of HS. After administrating streptozotocin for 5 consecutive days plasma glucose starts to rise from baseline, reaching approximately 500 mg/dl at 6 weeks after the diabetes induction (c). No differences in blood glucose levels were observed between Ndst1f/fTie2Cre− and Ndst1f/fTie2Cre+ diabetic animals. The urinary albumin/creatinine ratio (ACR) starts to increase after induction of diabetes reaching significant level at 6 weeks (WT:p < 0.05 and KO: p < 0.01), showing albuminuria is developing. No significant difference in ACR, both at 2 and 6 weeks post diabetes induction, was seen between the Ndst1f/fTie2Cre− and Ndst1f/fTie2Cre+ group. In healthy control Ndst1f/fTie2Cre+ animals, no albuminuria is developing (d).
Glomerular inflammation and fibrosis in experimental diabetes is dependent on endothelial HS
Macrophage staining (F4-80 antibody) revealed that compared to healthy controls, glomerular macrophage influx was markedly increased in diabetic Ndst1f/fTie2Cre− mice, while no increase was observed in the diabetic Ndst1f/fTie2Cre+ animals (Fig. 2a–d). To demonstrate mesangial localization of the infiltrated macrophages in the glomerulus, macrophages were co-stained for collagen III, a constituent of the mesangial matrix. As shown in Fig. 2b it is evident that intraglomerular macrophages are in the mesangial area (open arrow). Sometimes periglomerular macrophages were also observed (bold arrows), however these were not included in the quantification.

Endothelial Ndst1 deficiency protects from glomerular macrophage influx, collagen deposition, MBL deposition and podocyte damage in STZ-diabetic kidney disease. a–d Glomerular macrophage influx (red) (white open arrow) was markedly reduced in the Ndst1f/fTie2Cre+ animals compared to the Ndst1f/fTie2Cre− group (p < 0.001). Collagen III was used to identify mesangial expansion. White bold arrows are used to show peri-glomerular macrophages (not quantified). e–h MBL-C deposition (green) (white arrows) in the kidney was reduced in the Ndst1f/fTie2Cre+ group compared to the positive Ndst1f/fTie2Cre− group (p < 0.01). Basement membrane HSPG agrin (red) was used to identify glomeruli. However, C3b deposition was minor and not different among the groups (inset H). i–k Routine PAS staining revealed increased mesangial expansion in Ndst1f/fTie2Cre− group compared to Ndst1f/fTie2Cre+ group. l Podocyte damage was measured by the mRNA expression of desmin and was reduced in the Ndst1f/fTie2Cre+ group compared to the Ndst1f/fTie2Cre− group (p < 0.001). However podocyte number did not differ among the groups. m–p Staining for collagen III revealed a strong reduction in the deposition in the Ndst1f/fTie2Cre+ group compared to the fibrotic Ndst1f/fTie2Cre− group (p < 0.001). Representative photomicrographs were taken at 400× magnification
Several reports have shown a role for Mannan Binding Lectin (MBL) pathway of complement-mediated damage in diabetes in both experimental models and human diabetes [30,31,32]. In the healthy control animals, few MBL-C staining was seen in glomeruli and more specifically in the glomerular mesangium (Fig. 2e). Signals were seen at the base of the glomeruli, where the efferent and afferent arterioles invade the glomerulus. In diabetic animals the deposition of the MBL-C was more widely distributed in the glomerulus (Fig. 2f). Moreover, the Ndst1f/fTie2Cre+ diabetic group showed a reduction in the MBL-C deposition compared to Ndst1f/fTie2Cre− diabetic animals (p < 0.01) (Fig. 2e–h). As a measure of complement activation, C3 deposition was assessed. However the increase in MBL-C deposition in the diabetic Ndst1f/fTie2Cre− group was not accompanied by increased C3 deposition as no differences were observed between both genotype diabetic groups in C3 staining (Fig. 2h, inset). C3 depositions were seen in both the glomerulus and in peri-tubular area, most probably in peri-tubular capillaries.
Routine PAS staining of the kidneys showed increased mesangial expansion in the diabetic Ndst1f/fTie2Cre− mice compared to non-diabetic control mice. However, only minor mesangial enlargements were observed in the diabetic Ndst1f/fTie2Cre+ mice (Fig. 2i–k). To confirm this observation, mesangial collagen III deposition was determined as a measure for glomerulosclerosis. Indeed, increased glomerular collagen III depositions were completely absent in the Ndst1f/fTie2Cre+ group in contrast to the Ndst1f/fTie2Cre− animals (p < 0.001) (Fig. 2m–p). To evaluate podocyte damage, mRNA levels of the podocyte damage marker desmin was quantified by qRT-PCR and revealed an increase in expression in diabetic Ndst1f/fTie2Cre− animals, which was lower in the Ndst1f/fTie2Cre+ mice (p < 0.001) (Fig. 2l). No diabetes-induced loss of podocyte numbers was observed among the groups as assessed by WT1-positive nuclei in the glomeruli (inset Fig.2l).
Collectively, these data show that glomerular influx of macrophages, deposition of lectin complement component MBL, glomerulosclerosis and podocyte injury in STZ-induced diabetic kidney disease in the mouse were completely prevented by Ndst1 deficiency in endothelial cells.
Tubulo-interstitial macrophage influx and fibrosis are reduced by endothelial Ndst1 deficiency in experimental diabetes
To evaluate the effect of the endothelial Ndst1 deficiency on tubulo-interstitial leukocyte influx under experimental diabetic conditions, density of macrophages, T-cells, and neutrophils in the interstitium were quantified using immunofluorescent staining. Diabetic conditions resulted in a significant influx of macrophages in the Ndst1f/fTie2Cre− mice. Interestingly, interstitial macrophage staining was significantly reduced in the diabetic Ndst1f/fTie2Cre+ group compared to the diabetic Ndst1f/fTie2Cre− group (p < 0.05) (Fig. 3a–d). Most macrophages were found in the peritubular compartment, while only a few macrophages were intratubular. To exclude that the reduced renal macrophage infiltration in diabetic Ndst1f/fTie2Cre+ mice was the result of reduced expression of chemoattractant, we evaluated the localization and expression of MCP1, a major chemoattractant for macrophages in renal tissues of the various groups of mice. In control tissue (both Ndst1f/fTie2Cre− and Ndst1f/fTie2Cre+ mice), MCP1 was mainly expressed by endothelial cells of arterioles and to a minor extent by the peritubular capillaries. In diabetic Ndst1f/fTie2Cre− mice, MCP1 expression was slightly increased, and surprisingly in the diabetic Ndst1f/fTie2Cre+ mice, renal endothelial expression of MCP1 was further increased, especially in the peritubular capillaries (Fig. 3e–h). Thus, despite increased expression of pro-inflammatory cytokine MCP1 in the diabetic Ndst1f/fTie2Cre+ mice, the reduced influx of macrophages in the same mice underlines the importance of endothelial HS for leukocyte entry. Diabetic condition was not associated with accumulation of T-cells and neutrophils in Ndst1f/fTie2Cre- mice and no differences were seen between Ndst1f/fTie2Cre+ and Ndst1f/fTie2Cre− animals (Fig. 3i–j).

Endothelial Ndst1 deficiency reduces interstitial inflammation under diabetic conditions.Kidney sections were stained for macrophages (F4/80), the chemoattractant MCP1, T-cells (CD3) and neutrophils (NimpR14). a–d Macrophage (green) influx in diabetes is increased in diabetic animals compared to health controls. However the increase is reduced in the Ndst1f/fTie2Cre+ group compared to the Ndst1f/fTie2Cre− group (p < 0.05). Agrin staining (red) was used to clarify the renal histology. e–h Endothelial MCP-1 is more expressed in diabetic groups, especially in the Ndst1f/fTie2Cre+ mice, despite a lower glomerular and tubulo-interstitial macrophage influx in latter group. i–j T-cells (i) and neutrophil (j) influx were unchanged between the Ndst1f/fTie2Cre- and Ndst1f/fTie2Cre+ group. Moreover compared to healthy control animals T-cell influx was not increased in the diabetic groups. Representative photomicrographs were taken at 200× magnification
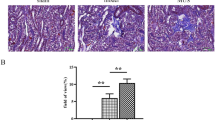
To assess whether or not endothelial HS is required for development of hyperglycemia-induced interstitial fibrosis, the kidney tissue sections from the diabetic mice were stained for expression of α-SMA, a key marker of myofibroblasts. Quantification showed that the accumulation of myofibroblasts was abundant in the interstitium of the Ndst1f/fTie2Cre− group, but was completely absent in the Ndst1f/fTie2Cre+ mice (Fig. 4a–d). Myofibroblasts are known producers of interstitial collagens. To find further support for the decrease of fibrosis in diabetic Ndst1f/fTie2Cre+ mice, collagen III deposition was determined. Anti-collagen III staining revealed a reduction in collagen III deposition in diabetic Ndst1f/fTie2Cre+ mice compared to the diabetic fibrotic Ndst1f/fTie2Cre− animals (p < 0.001) (Fig. 4e–h). In agreement with these findings, we also observed higher mRNA expression of collagen I and fibronectin, two additional fibrotic markers, in the diabetic Ndst1f/fTie2Cre− mice than in the diabetic Ndst1f/fTie2Cre+ mice (both p < 0.001) (Fig. 4i+j). Taken together, these data show that not only glomerular changes, but also tubulo-interstitial inflammation and fibrosis were prevented by endothelial Ndst1 deficiency.

Endothelial Ndst1 deficiency abolishes interstitial fibrosis.To assess the interstitial deposition of fibrotic components, sections were stained for myofibroblast marker αSMA and collagen III. a–d Although there was some variability, the increase in αSMA expression (red) was significantly reduced in the diabetic Ndst1f/fTie2Cre+ group compared to the diabetic Ndst1f/fTie2Cre− group (p < 0.05). Data was expressed as mean increase of αSMA expression compared to control. e–h Collagen III deposition was increased in the Ndst1f/fTie2Cre− group, but the increase was diminished in the Ndst1f/fTie2Cre+ group (p < 0.001). i, j At mRNA level both collagen I and fibronectin were expressed higher in the Ndst1f/fTie2Cre- group compared to the Ndst1f/fTie2Cre+ group (p < 0.01 and p < 0.001 respectively). The diabetic Ndst1f/fTie2Cre+ animals and healthy controls had comparable mRNA levels of collagen I and fibronectin. Representative photomicrographs were taken at 200× magnification
Discussion
In this study we demonstrate the crucial role of endothelial HS in diabetes-induced chronic renal inflammation and fibrosis. Endothelial Ndst1 deficiency resulted in a reduced macrophage accumulation both in the glomeruli and in the tubulo-interstitium of diabetic animals despite increased MCP1 expression. The reduction in cellular inflammation was accompanied by a reduced diabetes-induced glomerular MBL-C deposition in the Ndst1f/fTie2Cre+ mice. In addition, endothelial Ndst1 deficiency also associated with reduced renal fibrosis under diabetic conditions as shown by the decreased deposition of collagen III both in the tubulo-interstitium and in glomeruli and a reduced accumulation of myofibroblasts in the tubulo-interstitium.
It is generally accepted that endothelial HS proteoglycans play a pivotal role in leukocyte migration under inflammatory conditions where they act as a ligand for l-selectin and as docking structures for chemokines and cytokines, presenting them to high affinity receptors of leukocytes. As has been shown in other inflammatory disease models [15, 33, 34], endothelial Ndst1 deficiency leads to a reduced macrophage accumulation in our current study as well. Since, we showed reduced glomerular and tubulo-interstitial macrophage accumulation, this finding strongly supports a direct role of endothelial HSPGs on macrophage migration. Previously performed in vitro experiments have shown a strong reduction in macrophage, monocyte and neutrophil transmigration over Ndst1 deficient endothelium [15, 34, 35]. In vivo studies demonstrated that neutrophil transmigration is hampered in endothelial specific Ndst1 deficient mice in neutrophil-mediated acute inflammation and skin allergy models [15]. However, our chronic STZ-induced diabetes model is not characterized by neutrophil influx as can be seen in Fig. 3f. The difference is thought mostly due to different sets of chemokines that are involved in the various experimental conditions, such as MCP-1 is critical for macrophage transmigration [4], whereas IL-8, macrophage inflammatory protein (MIP)-2 and the CXCR2 ligand KC are more potent for neutrophil migration [15].
HSPGs produced in the endothelial cell are mainly deposited in the abluminal basement membrane but are also expressed on the apical side of endothelial cells [28]. We have shown before that predominantly basement membrane HSPGs are involved in l-selectin and chemokine binding under inflammatory conditions [12]. We also showed before that mice lacking two dominant endothelial basement membrane HSPGs, namely proteoglycan/collagen XV and XVIII hybrids, which are decorated mainly by HS side chains, showed strongly impaired neutrophil and macrophage transmigration in a renal ischemia/reperfusion model [16]. Therefore, we suggest that the protective effect of endothelial Ndst1 deficiency on diabetic inflammation is predominantly due to the expression of undersulfated HSPGs in the abluminal basement membrane of endothelial cells, although we cannot exclude a potential role of luminal expressed HSPGs.
The role of inflammation as a trigger and activator of renal fibrosis is rather well established [7, 36]. Interestingly, in our model, compared to the inhibitory effect on inflammation, a far stronger effect of endothelial Ndst1 deficiency was seen on ECM production, both in the tubulo-interstitium and in the glomerulus. And although endothelial Ndst1 deficiency has been shown before to reduce inflammation in a number of different models, such a clear anti-fibrotic effect of endothelial Ndst1 deficiency has not been shown before [15, 25, 33, 34, 37]. It is known that activated macrophages are a key regulator of fibroblast differentiation to collagen producing myofibroblasts in renal pathology [38] and this could be an explanation for the reduction in myofibroblasts seen in the endothelial Ndst1 deficient mice in our study. However, since the inhibitory effect of endothelial Ndst1 deficiency on fibrosis is far stronger compared to the effect on macrophage influx so we suggest other HS-dependent mechanisms play roles as well.
It has long been thought that renal myofibroblasts originated predominantly from resident fibroblasts, however recent work showed a significant contribution of circulation derived and CXCL16 dependent fibrocyte influx to ECM expansion in a renal fibrosis model [39]. Although the role of endothelial HSPGs on fibrocyte influx is unknown, CXCL16 has been demonstrated to be a heparin-binding chemokine, suggesting a role for endothelial HSPGs in CXCL16-mediated fibrocyte transmigration [40]. This could explain the dramatically decreased interstitial myofibroblast number and fibrosis in the endothelial Ndst1 deficient animals in our experiment. We also reasoned that a reduced macrophage infiltration in the kidneys of diabetic mice with endothelial HS deficiency might be the consequence of a reduced expresson of relevant chemoattractants. We thus evaluated MCP1, which is a dominant chemoattractant chemokine for monocytes/macrophages. Interestingly enough, renal MCP1 expression was increased in diabetic mice with endothelial HS deficiency compared to WT diabetic mice (Fig. 3e–h). Nevertheless, macrophage infiltration (both glomerular and tubulo-interstitial) was reduced in the diabetic endothelial Ndst1 deficient mice (Fig. 3a–d). This finding is identical to what we described earlier in a mouse renal ischemia/reperfusion model, where we showed that macrophage infiltration was significantly reduced in Coll XV/Coll XVIII (both basement membrane zone proteoglycans) deficient mice, despite increased MCP1 expression [16]. This finding again underlines the importance of functional endothelial HS to present chemokines towards rolling leukocytes, thereby facilitating the migration of leukocytes into the inflamed kidney. In addition, the endothelial cell itself is a source of ECM deposition. Under stimulus of heparan-binding factors, like TGF-β1, produced under hyperglycemic conditions, endothelial cells have been shown to undergo endothelial to mesenchymal transition (EndMT) [41]. Although the role of endothelial HSPG on EndMT is unknown, an inhibition in EndMT due to Ndst1 deficiency might be a possible explanation for the reduced ECM deposition observed in our study. Which of these mechanisms leads to the reduced ECM depositions in our study has not yet been fully determined. However, the severity of the reduction in ECM deposits might indicate involvement of endothelial HSPG in multiple pro-fibrotic processes under diabetic conditions.
We realized that the used Tie2Cre system also influences cells of the hematopoietic lineage and thus could influence data on inflammatory cell influx. However, in two individual studies we performed bone marrow transplantations from Ndst1 deficient donor Ndst1f/fTie2Cre+ mice into lethally radiated wildtype Ndst1f/fTie2Cre− recipients to generate hematopoietic Ndst1 deficient mice, and similarly from wildtype Ndst1f/fTie2Cre− donors into lethally radiated Ndst1f/fTie2Cre+ to generate endothelial Ndst1 deficient mice. We observed that deficiency of Ndst1 in hematopoietic cells does not alter peripheral blood cell count/differentiation and general hematopoiesis in bone marrow. Moreover, no effect of hematopoietic Ndst1 deficiency was observed in acute inflammation, skin allergy and in tumor angiogenesis, the later process involves inflammation too [15, 25]. These observations highlighted the dominant role of HS expressed by endothelial cells, not by hematopoietic cells in inflammation. In addition, in a very recent work from the Esko group [42], deletion of Ndst1 in macrophages leads to a proinflammatory phenotype by increasing expression of inflammatory chemokines and cytokines in diet-induced atherosclerosis, a chronic inflammatory disease. Whereas, in our diabetic model, we observed reduced macrophage infiltration in Ndst1f/fTie2Cre+ mice in which Ndst1 was deleted in both endothelial cells and macrophages, further highlighting endothelial, not macrophage HS, plays the major role in diabetic inflammation and fibrosis. Our result may be reasonably explained: in Ndst1f/fTie2Cre+ mice, although the chemokines/cytokines secretion from renal Ndst1-deficient macrophages might be increased, the chemokines/cytokines require endothelial HS to form an attractive gradient, to be transcytosed from abluminal-to-luminal side of endothelium and to present on endothelial luminal cell surface to fulfill macrophage infiltration into renal tissue, whereas all these endothelial HS-mediated processes were disrupted by the parallel endothelial Ndst1 deletion in the diabetic Ndst1f/fTie2Cre+ mice. This explanation is also strongly supported by our additional observation showing that even MCP-1 expression was increased, but macrophage infiltration, which mainly depends on MCP-1 in renal tissue, was reduced in our diabetic Ndst1f/fTie2Cre+ mice. In summary, observations in our previous and current studies plus current literature led us to believe that deficiency of endothelial, not hematopoietic HS, results in the inhibition of inflammation and fibrosis in our diabetic Ndst1f/fTie2Cre+ mice.
Despite strong effects of endothelial Ndst1 deficiency on inflammation and fibrosis, we could not show a significant improvement in urinary albumin excretion, which might be due to the relatively short follow-up. In recent years the paradigm that glomerular endothelial HSPG are directly involved in the glomerular filtration barrier has been challenged [43]. In agreement with this, our study did not find a significant protective effect of endothelial Ndst1 deficiency on proteinuria. However, we are aware of the evidence that Ndst1 deficient-endothelial cells still express undersulfated HS, and HS in basement membranes and ECM is also contributed by other type of cells, such as podocytes in the glomerulus and pericytes and/or vascular smooth muscle cells in non-glomerular vessels. Further studies, such as complete elimination of HS expression in endothelial cells and podocytes, will enable to clearly address the role of HS in glomerular filtration barrier. Nevertheless, our studies clearly demonstrate that endothelial HS plays an essential role in development of diabetes-induced renal inflammation and fibrosis, highlighting endothelial HS is a potential target for therapies to limit progression of diabetic nephropathy.
References
de Boer IH, Rue TC, Hall YN, et al. Temporal trends in the prevalence of diabetic kidney disease in the United States. JAMA. 2011;305:2532–9.
van den Brand JA. Diabetes mellitus as a cause of end-stage renal disease in europe: signs of improvement. Clin Kidney J. 2016;9:454–6.
Navarro-Gonzalez JF, Mora-Fernandez C, Muros de Fuentes M, et al. Inflammatory molecules and pathways in the pathogenesis of diabetic nephropathy. Nat Rev Nephrol. 2011;7:327–40.
Chow FY, Nikolic-Paterson DJ, Ma FY, et al. Monocyte chemoattractant protein-1-induced tissue inflammation is critical for the development of renal injury but not type 2 diabetes in obese db/db mice. Diabetologia. 2007;50:471–80.
Perez-Gomez MV, Sanchez-Nino MD, Sanz AB, et al. Targeting inflammation in diabetic kidney disease: early clinical trials. Expert Opin Investig Drugs. 2016;25:1045–58.
Kanasaki K, Taduri G, Koya D. Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol. 2013;4:7.
Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–96.
Sharma K, Deelman L, Madesh M, et al. Involvement of transforming growth factor-beta in regulation of calcium transients in diabetic vascular smooth muscle cells. Am J Physiol Ren Physiol. 2003;285:F1258–70.
Sharma K, Jin Y, Guo J, et al. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes. 1996;45:522–30.
Kim HW, Kim BC, Song CY, et al. Heterozygous mice for TGF-betaIIR gene are resistant to the progression of streptozotocin-induced diabetic nephropathy. Kidney Int. 2004;66:1859–65.
Rider CC. Heparin/heparan sulphate binding in the TGF-beta cytokine superfamily. Biochem Soc Trans. 2006;34:458–60.
Celie JW, Rutjes NW, Keuning ED, et al. Subendothelial heparan sulfate proteoglycans become major L-selectin and monocyte chemoattractant protein-1 ligands upon renal ischemia/reperfusion. Am J Pathol. 2007;170:1865–78.
Reidy K, Kang HM, Hostetter T, et al. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124:2333–40.
Lowe JB. Glycan-dependent leukocyte adhesion and recruitment in inflammation. Curr Opin Cell Biol. 2003;15:531–8.
Wang L, Fuster M, Sriramarao P, et al. Endothelial heparan sulfate deficiency impairs L-selectin- and chemokine-mediated neutrophil trafficking during inflammatory responses. Nat Immunol. 2005;6:902–10.
Zaferani A, Talsma DT, Yazdani S, et al. Basement membrane zone collagens XV and XVIII/proteoglycans mediate leukocyte influx in renal ischemia/reperfusion. PLoS One. 2014;9:e106732.
Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006;6:633–43.
Shamri R, Grabovsky V, Gauguet JM, et al. Lymphocyte arrest requires instantaneous induction of an extended LFA-1 conformation mediated by endothelium-bound chemokines. Nat Immunol. 2005;6:497–506.
Middleton J, Patterson AM, Gardner L, et al. Leukocyte extravasation: chemokine transport and presentation by the endothelium. Blood. 2002;100:3853–60.
Proudfoot AE, Handel TM, Johnson Z, et al. Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA. 2003;100:1885–90.
Middleton J, Neil S, Wintle J, et al. Transcytosis and surface presentation of IL-8 by venular endothelial cells. Cell. 1997;91:385–95.
Bame KJ, Esko JD. Undersulfated heparan sulfate in a chinese hamster ovary cell mutant defective in heparan sulfate N-sulfotransferase. J Biol Chem. 1989;264:8059–65.
Fan G, Xiao L, Cheng L, et al. Targeted disruption of NDST-1 gene leads to pulmonary hypoplasia and neonatal respiratory distress in mice. FEBS Lett. 2000;467:7–11.
Holmborn K, Ledin J, Smeds E, et al. Heparan sulfate synthesized by mouse embryonic stem cells deficient in NDST1 and NDST2 is 6-O-sulfated but contains no N-sulfate groups. J Biol Chem. 2004;279:42355–8.
Fuster MM, Wang L, Castagnola J, et al. Genetic alteration of endothelial heparan sulfate selectively inhibits tumor angiogenesis. J Cell Biol. 2007;177:539–49.
Zhang B, Xiao W, Qiu H, et al. Heparan sulfate deficiency disrupts developmental angiogenesis and causes congenital diaphragmatic hernia. J Clin Invest. 2014;124:209–21.
Like AA, Rossini AA. Streptozotocin-induced pancreatic insulitis: New model of diabetes mellitus. Science. 1976;193:415–7.
Ashikari-Hada S, Habuchi H, Kariya Y, et al. Characterization of growth factor-binding structures in heparin/heparan sulfate using an octasaccharide library. J Biol Chem. 2004;279:12346–54.
Jemth P, Kreuger J, Kusche-Gullberg M, et al. Biosynthetic oligosaccharide libraries for identification of protein-binding heparan sulfate motifs. exploring the structural diversity by screening for fibroblast growth factor (FGF)1 and FGF2 binding. J Biol Chem. 2002;277:30567–73.
Bouwman LH, Eerligh P, Terpstra OT, et al. Elevated levels of mannose-binding lectin at clinical manifestation of type 1 diabetes in juveniles. Diabetes. 2005;54:3002–6.
Hovind P, Hansen TK, Tarnow L, et al. Mannose-binding lectin as a predictor of microalbuminuria in type 1 diabetes: an inception cohort study. Diabetes. 2005;54:1523–7.
Ostergaard JA, Ruseva MM, Malik TH, et al. Increased autoreactivity of the complement-activating molecule mannan-binding lectin in a type 1 diabetes model. J Diabetes Res. 2016;2016:1825738.
Ge XN, Ha SG, Rao A, et al. Endothelial and leukocyte heparan sulfates regulate the development of allergen-induced airway remodeling in a mouse model. Glycobiology. 2014;24:715–27.
Rops AL, Loeven MA, van Gemst JJ, et al. Modulation of heparan sulfate in the glomerular endothelial glycocalyx decreases leukocyte influx during experimental glomerulonephritis. Kidney Int. 2014;86:932–42.
Floris S, van den Born J, van der Pol SM, et al. Heparan sulfate proteoglycans modulate monocyte migration across cerebral endothelium. J Neuropathol Exp Neurol. 2003;62:780–90.
Kanasaki K, Taduri G, Koya D. Diabetic nephropathy: the role of inflammation in fibroblast activation and kidney fibrosis. Front Endocrinol. 2013;4:7.
Zuberi RI, Ge XN, Jiang S, et al. Deficiency of endothelial heparan sulfates attenuates allergic airway inflammation. J Immunol. 2009;183:3971–9.
Nikolic-Paterson DJ, Wang S, Lan HY. Macrophages promote renal fibrosis through direct and indirect mechanisms. Kidney Int Suppl. 2014;4:34–8.
Chen G, Lin SC, Chen J, et al. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol. 2011;22:1876–86.
de Paz JL, Moseman EA, Noti C, et al. Profiling heparin-chemokine interactions using synthetic tools. ACS Chem Biol. 2007;2:735–44.
Cooley BC, Nevado J, Mellad J, et al. TGF-beta signaling mediates endothelial-to-mesenchymal transition (EndMT) during vein graft remodeling. Sci Transl Med. 2014;6:227ra34.
Gordts PL, Foley EM, Lawrence R, et al. Reducing macrophage proteoglycan sulfation increases atherosclerosis and obesity through enhanced type I interferon signaling. Cell Metab. 2014;20:813–26.
Goldberg S, Harvey SJ, Cunningham J, et al. Glomerular filtration is normal in the absence of both agrin and perlecan-heparan sulfate from the glomerular basement membrane. Nephrol Dial Transplant. 2009;24:2044–51.
Acknowledgements
The graduate school of Medical Sciences of the University Medical Center of Groningen provided financial support to DT in the form of PhD stipends. The research was supported by NIH grants HL-093339 and GM103390 to LW.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Talsma, D.T., Katta, K., Ettema, M.A.B. et al. Endothelial heparan sulfate deficiency reduces inflammation and fibrosis in murine diabetic nephropathy. Lab Invest 98, 427–438 (2018). https://doi.org/10.1038/s41374-017-0015-2
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41374-017-0015-2
This article is cited by
-
Heparanase is the possible link between monkeypox and Covid-19: robust candidature in the mystic and present perspective
AMB Express (2023)
-
Neutralization of excessive levels of active TGF-β1 reduces MSC recruitment and differentiation to mitigate peritendinous adhesion
Bone Research (2023)
-
Novel approach for quantification of multiple immunofluorescent signals using histograms and 2D plot profiling of whole-section panoramic images
Scientific Reports (2021)
