
Abstract
Allogeneic hematopoietic stem cell transplantation (HSCT) has significantly improved the outcome of children with high-risk (HR) acute myeloid leukemia (AML). Implementing allogeneic HSCT depends on numerous factors, including adverse cytogenetics, molecular abnormalities, poor response to first-line treatment, or relapsed or primary refractory disease. In HR AML, allogeneic HSCT is considered to be the consolidation strategy of choice in first complete remission (CR1) and offers the best chance of cure for patients with relapsed disease. Advances in donor/recipient typing, conditioning regimens, graft-versus-host-disease (GvHD) management, and supportive care have contributed to this improvement in overall—and transplant—outcome. This review will comprehensively discuss indications for HSCT and its modalities in pediatric AML by examining past, current, and future strategies for disease- and response-related stratification. We will examine the key importance of low/negative measurable residual disease (MRD) before transplantation and discuss conditioning regimens and graft variables, as well as novel approaches to harness the graft-versus-leukemia (GvL) effect, including targeted immunotherapy. The review will also address toxicities associated with HSCT, GvHD prophylaxis, and the management of treatment failure. Ultimately, this review seeks to inform clinical practice and highlights how improved outcomes have been achieved through the collective efforts of international study groups.
Similar content being viewed by others
HSCT in pediatric AML: Where do we come from?
Historically, allogeneic hematopoietic stem cell transplantation (HSCT) has been employed as a consolidation strategy in pediatric acute myeloid leukemia (AML) in first complete remission (CR1) when a human leukocyte antigen (HLA)-identical sibling donor was available [1,2,3]. Although a reduction in the relapse rate (RR) was observed, this benefit was offset by the risk of transplant-related mortality (TRM) [4]. Over time, optimizing disease risk stratification, pre-transplant induction or reinduction chemotherapy strategies, and supportive care resulted in significantly improved survival rates [5,6,7]. Studies have shown that HSCT improves overall survival (OS) and disease-free survival (DFS) compared to chemotherapy alone, largely due to a lower RR, in patients with high-risk (HR) features. As a result, both survival outcomes and disease control—at least in certain patient subsets—can be improved by HSCT [8]. This has resulted in a shift in approach, where the decision to proceed with HSCT in CR1 is now primarily driven by a risk assessment that considers disease characteristics and treatment response [4, 6, 9,10,11,12].
Despite these improvements achieved through the joint efforts of international study groups (AIEOP, BFM-AML SG, COG, JPLSG, MRC/NCRI, EORTC-CLG, NOPHO, PPLLSG, and SJCRH), the role of HSCT as a consolidation strategy for children and adolescents with newly diagnosed AML in CR1, and in particular the question as to which cytogenetics/molecular aberration constitutes HR disease [13, 14], remain controversial due to a paucity of data and the absence of randomized clinical trials comparing HSCT with other post-remission therapies [15,16,17].
In this article, we present current recommendations for patient eligibility for HSCT in CR1, donor selection, stem cell graft choices, conditioning regimens, and the management of both acute and late toxicities (Fig. 1). We also discuss the importance of achieving low-level or negative measurable residual disease (MRD) prior to HSCT, as well as maintenance strategies following HSCT. Additionally, we explore potential future directions for HSCT in pediatric AML and identify key areas for further clinical research.

Schematic representation of the variable influencing eligibility criteria, donor and graft selection, conditioning regimens, and graft-versus-host-disease prophylaxis choices in children with AML.
To transplant or not to transplant in CR1?
The criteria for considering HSCT instead of multimodal chemotherapy alone as consolidation treatment, and for identifying which patients will benefit more from HSCT, are still debated questions. Historically, the decision was based on a disease-specific risk assessment and individual patient characteristics, and guided by a general benchmark for improved DFS. For adults, HSCT was typically considered if the expected DFS was predicted to improve by at least 10% compared to chemotherapy alone. For children, a more stringent, though not evidence-based, threshold of a 30% improvement in DFS was often used [16, 18]. Currently, the decision is generally guided by molecular and cytogenetic risk factors and treatment response. This section focuses on the current criteria indicating the need for HSCT in CR1.
The geneticists’ point of view
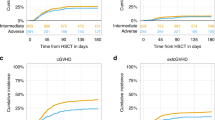
Approximately 30–35% of children with AML have a cytogenetic profile that could be considered as HR and an indication for HSCT in CR1. While cytogenetics/molecular aberrations that constitute HR disease vary between cooperative study groups, there is growing consensus, and as an example, we will detail the HR cytogenetic criteria of the (Berlin—Frankfurt—Münster) AML-BFM study group in this section (Fig. 2) [15, 19,20,21,22]. Genetic abnormalities considered HR have evolved over time and will continue to evolve, and the clinician is advised to keep abreast of changes. Furthermore, the question as to whether genetic factors or treatment response should primarily guide transplant decisions in CR1 is a subject of debate.

High-risk features in pediatric AML are defined by cytogenetic and molecular abnormalities, as well as poor response to treatment as indicated by measurable residual disease: 1Karyotypic Abnormalities: Complex karyotype (≥3 aberrations including at least one structural abnormality), excluding cases with recurrent translocations; monosomal karyotype, such as monosomy 7 or deletion 5q (-7, -5/del(5q)). 2Chromosomal Translocations: t(16;21)(p11;q22)→FUS::ERG; t(9;22)(q34;q11.2) → BCR::ABL1; t(6;9)(p22;q34) → DEK::NUP214; t(7;12)(q36;p13) → MNX1::ETV6; inv(3)(q21q26)/t(3;3)(q21;q26) → RPN1::MECOM; inv(16)(p13q24) → CBFA2T3::GLIS2; t(5;11)(q35;p15.5) → NUP98::NSD1; t(11;12)(p15;p13) → NUP98::KDM5A; 12p abnormalities; 11q23/KMT2A rearrangements, including: t(4;11)(q21;q23) → KMT2A::AFF1; t(6;11)(q27;q23) → KMT2A::AFDN; t(10;11)(p12;q23)→KMT2A::MLLT10. 3Monogenic HSCT Classifiers: FLT3-ITD with an allelic ratio (AR) ≥ 0.5, either alone or in combination with other recurrent abnormalities or NPM1 mutations. 4Measurable Residual Disease (MRD): Multiparametric flow cytometry (MFD)-MRD ≥0.1% after first or second induction or (if MFC-MRD result is not available/ informative) blast count ≥5% at second induction. HR high-risk, KMT2A Histone-Lysin-N-Methyltransferase 2A, AFD Afadin, Adherents Junction Formation Factor, MLLT10 Histone Lysine Methyltransferase DOT1L Cofactor, FUS RNA Binding Protein, ERG ETS transcription factor, BCR Breakpoint cluster region, ABL1 Abelson Murine Leukemia Viral Oncogene Homolog 1, DEK protoonko-gene, NUP nucleoporin, MNX1 Motor Neuron And Pancreas Homeobox 1, ETV6 ETS Variant Transcription Factor 6, RPN1 Ribophorin I, MECOM MDS1 And EVI1 Complex Locus, FLT3 FMS-like tyrosine kinase 3, ITD internal tandem duplication, NPM1 nucleophosmin 1, WT1 Wilms Tumor 1, CBFAT3 CBFA2/RUNX1 Partner Transcriptional Co-Repressor 3, GLIS2 GLIS Family Zinc Finger 2, NSD1 Nuclear Receptor Binding SET Domain Protein 1, KDM5A Lysine Demethylase 5A, MFC multiparametric flow cytometry, MRD measurable residual disease, MRD.
HR cytogenetic features
A subset of cytogenetic abnormalities correlates with an increased risk of disease recurrence and induction failure, thereby negatively impacting survival [23]. The Medical Research Council)-AML group as well as the AML-BFM study group defined a monosomal karyotype, including monosomy -5, -7, del(5q), abnormal 3q, and 12p abnormalities as indicators of poor prognosis [19, 20, 24,25,26]. Monosomy 7 or del(7q) is associated with poor outcomes, but 5-year OS is highly variable based on other cytogenetic events, which is not addressed by most studies. 5-year OS for -7 can range from 5% with co-occurring -5/del(5q), inv(3), or +21, to 35% in patients without additional unfavorable cytogenetics [24, 25]. In contrast, deletion of 7q [del(7q)] shows a somewhat better prognosis, with a 5-year OS of 51%, and is considered more favorable than monosomy 7 [25], but variability dependent on co-occurring abnormalities is retained.
Similarly, monosomy 5 or del(5q) are associated with poor outcome with 5-year OS of 27% and 23%, respectively, where it has to be noted that the majority of cases harbor additional abnormalities and isolated -5/del5q is too rare to assign an individual prognostic impact. Of note, improved outcomes have been reported in patients with monosomy 7 or monosomy 5 when undergoing HSCT, but patients remain a poor risk group [23].
Abnormalities of chromosome 3q alone have not been reported to impact prognosis [19]. However, there is a strong association of -7/del(7q) and 3q abnormalities, and these patients belong to the group of highest risk, since the 5-year OS is as low as 5% for inv(3)/-7. In addition, AML with inv(3)(q21q26.2)/t(3;3)(q21;q26.2), which occurs in ~1% of newly diagnosed pediatric AML, has a very poor prognosis [27,28,29,30]. Survival of patients with abnormalities of 12p ranges from 22% to 35% [19, 20]. Overall, the sparsity of patients with these rare abnormalities and the heterogeneity of confounding factors included in the various studies make it difficult to precisely quantify their prognosis. Nevertheless, most patients with abnormalities of chromosome 5, 7, or 12p have to be regarded as HR and are likely to benefit from HSCT.
Some, but not all, cooperative study groups consider a complex karyotype (three or more unrelated chromosomal abnormalities in the absence of other class-defining recurring genetic abnormalities) to also confer an HR of relapse, with an event-free survival probability (EFS) of only 30% to 40% [19, 20].
11q23/KMT2A (formerly MLL) rearrangements (KMT2A-R), which are highly prevalent in pediatric cohorts, accounting for 20-24% of AML cases, are associated with a heterogeneous outcome, depending on the fusion partner involved [19, 24, 31, 32]. In retrospective analyses, which include data from 756 patients with KMT2A-R AML, t(4;11)(q21;q23.3)/KMT2A::MLLT2, t(6;11)(q27;q23)/KMT2A::MLLT4, t(10;11)(p12;q23)/KMT2A::MLLT10, and t(10;11)(p11.2;q23)/KMT2A::ABI1 were associated with a dismal prognosis when treated with chemotherapy alone [31,32,33,34,35].
Yuen et al. reported a 5-year EFS of 49% and OS of 67% in pediatric AML patients with 11q23/KMT2A-R, which were significantly worse compared to those of patients without such rearrangements. Among the subtypes, patients with t(6;11)(q27;q23)/KMT2A::MLLT4 had particularly poor outcomes, with a 5-year EFS of 13% and OS of 24%. Those with t(10;11)(p12;q23)/KMT2A:: MLLT10 showed a 5-year EFS of 23% and OS of 57% [36]. A retrospective study of >750 children with KMT2A-R treated by European and US cooperative groups found that t(4;11)(q21;q23), t(6;11)(q27;q23), t(10;11)(q12;q23), and t(10;11)(p11.2;q23) were associated with a 5-year EFS of only 10% to 40% [31]. To note, introducing Gemtuzumab Ozogamicin (GO) into treatment regimens and incorporating MRD improved prognosis for both HR and non-HR KMT2A-R patients [32, 34].
A number of rare chromosomal translocations are associated with a poor prognosis and are considered an indication for transplantation in CR1. These include the FUS::ERG translocation resulting from t(16;21)(p11;q22), which is associated with an extremely poor prognosis [37] and the t(9;22)/BCR::ABL1 translocation [38,39,40]. Patients with FUS::ERG-positive AML are often primary refractory, or relapse quickly, and in Children’s Oncology Group (COG) clinical trials, 100% of transplanted patients succumbed to their disease [41]. The median survival time of patients with Philadelphia chromosome-positive AML was reported to be 7.5 months [42]. t(6;9)(p22;q34)/DEK::NUP214, often associated with FLT3 internal tandem duplication (FLT3/ITD) (~40% of cases), has a high RR with chemotherapy alone and is also considered an indication for an allograft in CR1 [43,44,45]. Data in both pediatric and adult populations, although obtained in small cohorts, show an OS higher than 50% for this subtype following HSCT in CR1 [43, 44].
Furthermore, cryptic gene fusions, including NUP98-rearrangements—t(5;11)(q35;p15)/NUP98::NSD1, t(11;12)(p15;p13)/NUP98::KDM5A—, inv16/CBFA2T3::GLIS2 and t(7;12)(q36;p13)/MNX1::ETV6 predict a poor outcome [46,47,48,49,50]. The 5-year OS for NUP98 fusions was 35% versus 64% in a reference group [51]. Specifically, NUP98-NSD1 patients had an OS of 36% and EFS of 17%, while NUP98-KDM5A patients had an OS of 30% and EFS of 25%. RR was also significantly higher: 64% for NUP98-NSD1 and 68% for NUP98-KDM5A, as compared to the reference group. Treatment response, measured by 5-year DFS post-induction, was lower in all NUP98 subtypes (27% vs. 52% in a reference group): NSD1 and KDM5A (28%) [51]. CBFA2T3-GLIS2 AML, an extremely aggressive AML subtype occurring in very young children, has a poor prognosis, with 5-year OS ranging from 14% to 42% and 5-year EFS ranging from 8% to 33% [52]. The patients with t(7;12) had a 3-year EFS of 24-43% [53]. These high failure rates highlight the need for new treatment modalities. An example is CBFA2T3-GLIS2 AML, where luveltamab tazevibulin, an antibody-drug conjugate (ADC), is currently being employed as a bridge to transplant and/or maintenance therapy (MT) post-transplantation (Supplementary Table 1). Finally, the t(8;16)(p11;p13)/KAT6A::CREBBP rearrangement has an important age-dependent impact on prognosis: in very young infants/neonates this translocation can be associated with spontaneous remission supporting a watch-and-wait strategy, whilst in older children the prognosis is poor and represents a criterion for HSCT in CR1 [54].
There is a very limited number of monogenic HSCT classifiers: only the internal tandem duplications in FLT3 (FLT3-ITD), which occur in ~10–20% of pediatric AML patients, either alone or with WT1 co-mutations, have proven to be of prognostic significance in de novo disease and at relapse [55,56,57]. In this context, patients with both NUP98 gene fusion co-expression and FLT3/ITD mutation have an EFS of only 13%, compared to 31% in patients with the FLT3/ITD mutation alone [58]. FLT3-ITD may not be HR in the presence of a low allelic ratio (AR < 0.5) or concomitant NPM1 mutations, but it can have an unfavorable prognostic value when associated with a high allelic ratio (HAR > 0.5) and without an NPM1 mutation [57, 59,60,61,62,63]. A COG study demonstrated that HAR FLT3/ITD mutation is associated with a 16% 4-year progression-free survival (PFS) rate and an 83% RR, which are significantly worse compared to those with FLT3 wild-type (FLT3/WT) [61]. However, it will be crucial to assess the prognostic value of FLT3-ITD AR when accounting for co-occurring mutations [57, 64, 65] and the significance of HAR is not as strong as initially found when considering the broader mutational landscape, but more comprehensive studies are needed. In a recent study, high FLT3-ITD AR retained its prognostic significance when accounting for NPM1, CEBPA, or WT1 status and patients with HAR did benefit from HSCT [57].
AML with myelodysplasia-related changes (MRC) is a subtype characterized by both blast count and genetic features. According to the 5th edition of the World Health Organization (WHO) Classification of Myeloid Neoplasms, this condition is now classified as “AML, myelodysplasia-related (AML-MR),” retaining the criterion of over 20% blasts, which distinguishes it from myelodysplastic syndrome (MDS). In the ITCC (International Therapeutic Classification of Cancer) system, the category of AML-MRC has been replaced by two distinct entities: AML with myelodysplasia-related cytogenetic abnormalities and AML with myelodysplasia-related gene mutations, neither requiring dysplastic features. Both the ITCC and WHO classifications include a complex karyotype as a defining feature of AML-MR or AML with myelodysplasia-related cytogenetic abnormalities [66].
Genetic features (chromosome abnormalities and genetic mutations) include unbalanced abnormalities such as del(5)/t(5q), -7, del(11q), del(12p)/t(12p), 13/del(13q), i(17q), del17p/t(17p), -idic(X)(q13). Additionally, mutations in genes such as TP53, ASXL1, BCOR, EZH2, SF3B1, SRSF2, STAG2, U2AF1, and ZRSR2 are commonly observed [14, 66]. Patients with AML-MRC must undergo HSCT that can be preceded by a pre-transplant treatment, aimed at reducing the blast percentage prior to transplantation [67].
Therapy-related (t)-AML (t-AML) and secondary AML (s-AML) are indications for HSCT, irrespective if they arise from a germline mutation and/or are chemotherapy- or radiotherapy-related [68]. Gene mutations seen in t-AML and s-AML include activating mutations in tyrosine kinase RAS/BRAF pathways leading to an increase in cell proliferation, inactivating mutations in genes encoding hematopoietic transcription factors resulting in disrupted cell differentiation, and inactivating mutations in the tumor suppressor gene TP53. Currently, 10–20% of diagnosed AML and MDS cases are therapy-related, making t-AML the most common secondary malignancy in adults [69] and accounting for 1–3% of cases in children [68]. Retrospective studies have shown the challenges of determining the optimal preparative regimen [70]. Induction response rates and OS are poor, and children with t-AML have an HR of relapse and toxicity as a result of previous chemotherapy exposure for their initial disease. The choice of induction therapy must consider the cumulative drug dose—especially of anthracyclines (see below, section “Chasing CR: how to treat relapsed or secondary refractory pediatric AML?”)—and the often-impaired regenerative capacity of the bone marrow (BM). Due to the HR of additional toxicity, patients should proceed to HSCT after minimal induction chemotherapy, generally following 1 or 2 cycles of chemotherapy to induce remission [70,71,72]. Children achieving CR with full hematological recovery, CR with partial regeneration (CRp), or with no evidence of leukemia (NEL) have a considerable chance of being cured by HSCT. However, patients who fail to achieve morphological CR (<5% blasts) are unlikely to benefit from HSCT [70, 73].
At the time of diagnosis of t-MDS/AML, patients may present with a concomitant active neoplastic disorder [74], however, thorough results on HSCT for secondary hematologic malignancies in the context of an active primary solid tumor have not been reported in pediatric patients and will therefore not be further discussed in this review.
Measurable residual disease monitoring as the “go to solution”
Some study groups place greater emphasis on MRD rather than cytogenetics or molecular biology when determining treatment options in first-line therapy [75].
The European Leukemia Net-MRD Working Party has provided guidelines for standardizing MRD assessment by either multiparametric flow cytometry (MFC-MRD) or molecular biology methods, including recommendations on the methodologies, time points for assessment, MRD thresholds, and definition of response [76, 77]. Commonly, the leukemia-associated aberrant immunophenotype (LAIP) and/or different-from-normal phenotype (DfN) are used for MRD assessment in children with AML (Fig. 2). However, molecular MRD monitoring by reverse-transcription quantitative PCR is often used in patients with FLT3-ITD, NPM1c, and fusion genes like RUNX1::RUNX1T1 and CBFB::MYH11 [76]. Molecular markers for MRD should be chosen based on their genetic stability in the leukemic clone [78].
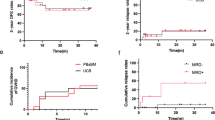
Early assessment of MRD—during and after each course of induction, at the end-of-induction (EOI), and prior to consolidation therapy—helps in identifying patients most likely to benefit from therapy intensification, including HSCT [79, 80]. HR patients with EOI MRD > 1% showed a trend toward improved OS with consolidative HSCT compared to those without HSCT: 44% versus 23% [79]. In this study, MFC-MRD was applied as risk-stratification criteria together with genetic features to a cohort of 232 children consecutively enrolled in the AML02 multicenter trial [79]. MRD-positivity, defined as ≥0.1% of the mononuclear BM cells after induction 1, was associated with an unfavorable outcome in HR AML. Moreover, any MRD-positivity after induction 2 was predictive of an adverse outcome. This combined approach (MFC-MRD and genetic features) showed a 3-year EFS and OS of 63% and 71%, respectively. 80% (155 of 193) of patients achieved MRD of <0.1% after induction 2, and the cumulative incidence of relapse (CIR) for this group was 17%. MRD of ≥1% after induction 1 was the only significant independent adverse prognostic factor for both EFS and OS.
Moreover, the persistence of MFC-detectable MRD after EOI has been associated with inferior EFS and OS, as demonstrated in the Nordic Society of Pediatric Hematology and Oncology (NOPHO)-AML 2004 trial [81], and the COG AAML03P1 protocol [82]. In 2016, Tierens et al. retrospectively analyzed MFC-MRD prognostic impact (≥0.1% leukemic events were considered MRD-positive) at two different time points (day 15 of induction therapy and before consolidation therapy) in a cohort of 201 children enrolled in the NOPHO-AML 2004 trial [81]. In a multivariate analysis, only MFC-MRD-positivity before consolidation therapy was associated with an unfavorable outcome, with a strong impact both on EFS and OS. Recent findings further suggest a predictive role for combined blood (day 8) and BM (day 22) MRD [83].
European cooperative groups have also identified MFC-MRD 0.1% after EOI as an independent prognostic feature for relapse-free survival (RFS) and OS [84, 85]. In a retrospective study on the prognostic role of MFC-MRD in a cohort of 142 pediatric AML patients treated according to the Associazione Italiana di Ematologia e Oncologia Pediatrica (AIEOP)-AML 2002/01 trial [85], respective MRD levels (<0.1% vs. ≥0.1%) after induction 1 correlated with 8-year DFS (73% vs. 35%) and OS (82% vs. 51%). Similar results were observed for MRD levels after induction 2 (8-year DFS: 68% for MRD < 0.1% vs. 21% for MRD ≥ 0.1%; 8-year OS: 77% for MRD < 0.1% vs. 55% for MRD ≥ 0.1%). In a multivariate analysis, MRD ≥ 0.1% after induction 1 was associated with an adverse outcome.
These findings suggest that intensification of therapy through HSCT may improve outcomes in patients with a suboptimal treatment response at the EOI.
In recent years, several studies have explored real-time quantitative polymerase chain reaction (qPCR)-based molecular MRD as a predictor of relapse. However, the lack of standardized protocols, cut-offs, and time points—especially in pediatric settings—has limited its routine use before HSCT. An international retrospective (I)-BFM-AML study supports the clinical utility of qPCR-MRD in transplant management as a potential alternative to MFC [86]. Benetton et al. identified 2.1 × 10−4 as the most informative cut-off, distinguishing patients with low relapse risk (10.4%) and excellent OS (82.8%). A higher cut-off of 1 × 10−2 identified a subgroup with poor OS (<40%). These findings highlight molecular advances in improving sensitivity and specificity, making qPCR-MRD a potentially valuable complement to MFC assessment in guiding clinical decisions for HSCT in CR1, while its prognostic significance post-transplant remains unclear.
Altogether, recent meta-analyses have demonstrated that HSCT in CR1 improved OS and DFS with reduced RR compared to chemotherapy alone in 1448 HR pediatric AML patients treated between 1998 and 2017 [8]. Recommendations to proceed to HSCT in CR1 in these patients were based on multiple parameters, including unfavorable genetic alterations (see section “AML with myelodysplasia-related changes”), failure to achieve CR, or poor response to first-line treatment as measured by morphology and EOI MRD, which were based on each trial risk assessment [8]. Hence, most groups use a combination of genetic abnormalities and treatment response as detected by MFC-MRD to define patients as HR and offer them to HSCT. In particular, EOI MRD-positivity identifies additional patients at HR [84, 85, 87, 88], thereby guiding the selection of candidates for HSCT in CR1 [79].
Emerging methods for MRD monitoring include droplet digital PCR and next-generation sequencing (NGS), integrating genetic and transcriptomic analyses with circulating tumor DNA mirroring the genomic information from BM [89,90,91]. These technologies may provide the foundation for more precise HR definitions, and MRD-based risk stratification, potentially enabling treatment intensification of poor induction responders with HSCT in the future.
How to deal with primary induction failure and primary refractory disease?
Treatment response, irrespective of genetic risk factors, is a strong predictor of outcome.
Achieving a deep MRD response is optimal prior to allograft [86]. However, patients with persistent blasts may still be curable by HSCT: patients with >30% AML blasts in the BM prior to HSCT have a leukemia-free survival (LFS) of 10%. An LFS of 10% in patients with a disease that is invariably fatal without HSCT may be acceptable to most families [92].
For patients with primary induction failure (PIF) or refractory disease, the prognosis is poor without HSCT [93,94,95]. Patients with PIF may benefit from intensifying therapy and/or from novel therapeutic approaches, if available, to achieve morphological remission prior to HSCT [96, 97]. Achieving blast reduction is crucial for optimizing outcomes, as discussed in the next section and shown in Fig. 3.

Retrospective analyses have demonstrated that HSCT in CR1 improves OS with reduced RR in high-risk and r/r pediatric AML patients. The prognostic significance of NR or MRD-positivity before HSCT, as well as the association of subsequent HSCT with poorer survival outcomes, has been confirmed by various study groups. Numeric details are provided below: 1AML-BFM (2010-2012): 5-year pOS: 76% [183]; AIEOP-2002/01: 8-year pOS: 74% [205]. 2AML-BFM (2011-2012): CIR: 25.1% (SE 3.9), NR: 12.3% (SE 2.8) [183]; AIEOP-2002/01: CIR: 17% [205]. 3AML-BFM 2004/2012 and AML-BFM registry 2012: 5-year pOS 54.5%, SE = 4.4; COG (AAML0531 and AAML1031) (2013–2017): 5-year pOS 40%; 5-year pOS 24%, MRD+, COG: 5-year pOS 41%, MRD− [106]. 4BFM 2004/2012 and AML-BFM registry 2012: NR patients: 5-year pOS 26.7%, SE = 9.0 [106]. 5AML-SCT-BFM: CR1/CR2: 4-year pOS and pEFS 61 and 70%, CIR 22%, NRM 15% [107]. 62-year pLFS: CR 33%, NR 19%, 8-year pLFS: CR 24%, NR, 10% [206]. 4-year pOS w HSCT: 31% w/o 3%, CIR and NRM at 4 years: 45% and 22% [177]. *Consider maintenance therapy. AML acute myeloid leukemia, AML-MRC AML-myelodysplasia-related changes, PIF primary induction failure, s/t-AML secondary/therapy-related AML, r/r AML relapsed/refractory AML, MRD measurable residual disease, CR complete remission, NR no response, BFM Berlin–Frankfurt–Münster Study Group, COG Children’s Oncology Group, HSCT hematopoietic stem cell transplant, OS overall survival, RR relapse rate, CIR cumulative incidence of relapse, SE standard error, LFS leukemia-free survival, NRM non-relapse mortality.
Chasing CR: How to treat relapsed or secondary refractory pediatric AML?
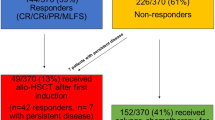
Approximately 30–40% of patients with de novo childhood AML experience leukemia relapse. These patients should be considered for HSCT in the second CR (CR2) [98,99,100,101]. Achieving CR2 in relapsed or secondary refractory patients is essential for optimizing post-HSCT outcomes. Several key factors must be carefully considered for increasing the likelihood of achieving CR2. These include the site of relapse, the time from the initial diagnosis to relapse, the patient’s response to reinduction therapy, and the cumulative anthracycline dose received (Table 1). Additionally, the availability of novel chemotherapeutic agents and the immunophenotype or presence of specific mutations/cytogenetics, which may identify appropriate targeted therapies, are important considerations in choosing effective strategies for these patients (Table 1 and Supplementary Table 1) [98, 102,103,104,105].
Outcomes for HSCT in children with relapsed AML have improved over time: In the COG cohorts AAML0531 and AAML1031, the 5-year pOS was 33% and 37%, respectively. For patients relapsing between 2013 and 2017, the 5-year pOS was 40% [106]. Similarly, the BFM study group reported an improvement in 5-year OS from 39% in 2009–2013 to 49% in 2013–2017 [106]. Patients in CR2 treated on the AML-SCT-BFM 2007 trial, which focused on conditioning regimens, had a 4-year EFS of 46% and a RR of 27% [107]. Next, a multicenter retrospective analysis of 343 children with acute leukemia (AL) treated between 2010 and 2015 demonstrated that HLA-haploidentical related α/β T-cell- and B-cell-depleted transplantation (α/β-haploidentical-HSCT) was equally effective as matched unrelated donor (MUD) HSCT [108]. This finding supports the use of α/β-haploidentical-HSCT for children with AL who either lack a matched donor or require urgent transplantation without sufficient time to identify a suitable MUD. The 5-year probability of LFS for either MUD, mismatched unrelated donor (MMUD), or α/β-haploidentical-HSCT transplantations was 67%, 55%, and 62%, respectively, with all patients being in morphological remission and receiving myeloablative conditioning (MAC) regimens. Notably, compared to MMUD-HSCT, children treated with α/β-haploidentical-HSCT had a lower incidence of grades II–IV acute and chronic graft-versus-host-disease (aGvHD/cGvHD), as well as a lower cumulative incidence (CI) of non-relapse mortality (NRM). The probability of cGvHD-free/relapse-free (GRFS) was 34% after MMUD-HSCT versus 58% after α/β-haploidentical-HSCT, while NRM rates were 28% and 9%, respectively [108]. More recently, the European Society for Blood and Marrow Transplantation reported a matched-pairs analysis of children with AML in CR1 or CR2 who underwent either MUD HSCT with anti-thymocyte globulin (ATG) (N = 253) or unmanipulated haploidentical-HSCT with post-transplant cyclophosphamide (PT-CY) (N = 95) following MAC conditioning between 2011 and 2021 [109]. Despite a higher incidence of grade III-IV aGVHD in the haploidentical group, no significant differences were observed in 2-year OS (78% vs. 71%), LFS (72 vs. 69%), CI of relapse (19% vs. 19%) NRM (8% vs. 11%) and GRFS (60% vs. 54%) between the MUD and haploidentical-HSCT groups, respectively. This study indicates that haploidentical-HSCT with PT-CY is a suitable alternative for children with AML who lack a matched donor [109].
Obtaining deep CR in relapsed or primary refractory patients is crucial to improve outcomes, as reported in a prospective study in 123 children who proceeded to HSCT in CR with a significantly better DFS in MRD-negative compared to MRD-positive patients [110]. In independent cooperative group studies conducted by COG, 765 of 852 (90%) patients were monitored by flow cytometry for residual disease (RD) [106]. The 5-year OS probability following relapse for patients who were RD-positive at the EOI was 24% (n = 222) and 41% for those who were RD-negative (n = 543, p < 0.001).
According to Wang et al., the OS rates of 37 pediatric AML patients treated with matched family donor (MFD) HSCT in CR1 after induction therapy with the Chinese Children’s Leukemia Group–AML2015 regimen were 89%, 75%, and 75%, while the RRs were 11%, 24%, and 33% at 1, 3, and 5 years post-HSCT, respectively [111]. 14 patients (37%) received donor lymphocyte infusions (DLI) due to positive MRD post-transplantation, and relapse was recorded in 9 patients (3–46 months post-HSCT), with a second HSCT performed in 5 patients. Notably, Homoharringtonine-based induction therapy was superior to Etoposide-based induction therapy, which highlights the need for more effective therapeutic strategies for patients who relapse after HSCT. In line with this, Tierens et al. reported on a phase III study using an intensified, response-guided induction regimen containing liposomal Daunorubicin (DNX) or Mitoxantrone, along with MRD-based risk stratification on day 22 after induction 1 and again after induction 2 [112]. Patients with poor induction response (Mitoxantrone had a superior anti-leukemic effect) were treated with HSCT. 5-year EFS and OS rates were 77% and 83%, respectively, for HR patients with HSCT (85%) [112]. Intensification of induction, risk stratification on the basis of treatment response, and treatment intensification with HSCT in HR patients led to improved HSCT outcomes.
Despite the major benefit of achieving CR, there is evidence supporting the use of a consolidative HSCT in the absence of CR in subsets of patients: the BFM study group reported an OS rate of 27% at 5 years in children with ≥5% residual leukemic blasts after two courses of reinduction with DNX + Fludarabin + Cytarabin (DNX-FLA ± FLA), suggesting that HSCT has a role post-relapse even in the absence of a morphological CR [106]. However, advanced disease at HSCT continues to be associated with poorer survival outcomes in children [113, 114], and MRD remains a strong prognostic factor prior to HSCT, significantly influencing outcomes [115].
State-of-the-art multimodal chemotherapy and targeted therapies offer the potential to achieve CR prior to HSCT in relapsed AML patients. Ongoing efforts for improvement are highlighted by the multitude of studies in this pediatric cohort (Supplementary Table 1). Importantly, new compounds currently under development may offer more personalized therapies to these patients; however, more trials focusing on children are still urgently required (Fig. 4).

Immunotherapy initiatives include ADC antibody (Abs)-drug conjugates, bispecific Abs, mAbs monoclonal Abs, CI checkpoint inhibitors, and cellular therapies (DLI donor lymphocyte infusion, CTL cytotoxic T lymphocytes, CAR chimeric antigen receptor approaches, NK natural killer, CIK cytokine-induced killer). Targeted therapies are summarized as RTK receptor tyrosine kinase inhibitors, BH3 mimetics selective small-molecule B-cell lymphoma 2 (Bcl-2) Homology 3, and inhibitors involved in protein degradation. WT1 Wilms tumor protein, PRAME preferentially expressed antigen in melanoma, ADGRE2 adhesion G protein-coupled receptor E2, FLT3 FMS‐like tyrosine kinase 3, TIM-3 T cell immunoglobulin and mucin-domain containing-3, GO gemtuzumab ozogamicin, PVEK pivekimab sunirine, CTLA-4 cytotoxic T-lymphocyte-associated protein 4, PD-1 programmed cell death protein 1, XPO1 exportin 1, HDACs histone deacetylases, HMA DNA hypomethylating agents, BTK Bruton’s tyrosine kinase, JAK Janus kinase, CDK cyclin-dependent kinase, mTOR mammalian target of rapamycin, IDH isocitrate dehydrogenase, FOLR1 folate receptor alpha, MDM2 mouse double minute 2 homolog, PROTAC proteolysis targeting chimera.
Bridging to HSCT in the short-term
Whilst achieving CR is important prior to HSCT, delaying HSCT is a case of concern in the relapsed/refractory (r/r) disease setting. Reasons for postponing HSCT may include an unexpected delay in donor availability, e.g., due to temporary inability to donate or inability to undergo anesthesia or mobilization of peripheral stem cells, withdrawal of consent to donate, limited number of collection centers, etc. In these circumstances, it is crucial to maintain CR, and short-term bridging therapy is advised. Currently, the choice of pre-transplant bridging therapies includes consolidation with high-dose (HD)-Ara-C monotherapy, GO (although the risk of liver veno-occlusive disease cannot be scotomized), Venetoclax/Azacytidine (VEN/AZA) or low-intensity consolidation with LD-Ara-C and Thioguanine, depending on disease dynamics and the time span to be bridged. In the absence of relevant studies, individualized patient management should be considered, particularly for heavily chemotherapy pretreated patients with persistent disease prior to HSCT (Fig. 4). This includes targeted therapies and, where applicable and/or available in the pre- and/or post-transplant setting (in case of a prior HSCT) [116], cellular immunotherapies such as DLI, cytotoxic T lymphocytes (CTL), natural killer (NK) cells, cytokine-induced killer (CIK) cells or, in the future, their chimeric antigen receptor (CAR) modified counterparts, including CAR-T, CAR-NK, and CAR-CIK cells.
What do we need to consider when choosing the donor stem cells? A debate on the best stem cell donor and stem cell source
The choice of donor is complicated by limited clinical data which are often outdated and involve small AML patient cohorts [108]. When discussing graft selection, two interconnected aspects must be addressed simultaneously: HLA matching and the choice of stem cell source.
HLA-identical sibling donors have historically been the preferred donor, balancing relapse occurrence and the risk of developing GvHD. Currently, the outcome of patients transplanted from an HLA-identical sibling, or a fully matched unrelated volunteer (URD) is comparable [117].
In the absence of an HLA-MFD, an immediate search for a MUD should be initiated at diagnosis for patients with AML-MRC, t-AML, s-AML, r/r AML, or HR AML (Figs. 3 and 5). Several studies have found that the killer-cell immunoglobulin-like receptor (KIR) alloreactivity in the donor/recipient pair is associated with AML outcome [118,119,120,121]. However, recent data from children with acute lymphoblastic leukemia (ALL; n = 372) or AML (n = 344) who received well-matched, T cell–replete, or in vivo T cell–depleted, URD transplantation, and who were reported to the Center for International Blood and Marrow Transplant Research (CIBMTR) between 2005 and 2016, showed that KIR ligand mismatch, KIR gene content (Cen B), KIR2DS1 mismatching, and Cen B/telomeric A were not significantly associated with relapse or DFS in the AML subgroup undergoing well-matched URD transplantations [122].

The choice of donor and conditioning regimes for patients with an indication for allogeneic HSCT for pAML. Conditioning regimes are considered “reduced intensity/toxicity (RIC)” if the dose of Busulfan is less than 8 mg/kg PO or IV, Melphalan is less than 150 mg/m², total body irradiation (TBI) dose is ≤500 as a single dose, or 800 cGy administered as fractionated doses. Other regimens are considered myeloablative (MAC). pAML pediatric acute myeloid leukemia, HR high-risk, AML-MRC AML-myelodysplasia-related changes, s/t-AML secondary/therapy-related AML, r/r AML relapsed/refractory AML, MRD measurable residual disease, RD residual disease, CR complete remission, HSCT hematopoietic stem cell transplant, HLA human leukocyte antigen, MSD matched sibling donor (second choice in case of a minor child), MFD matched family donor, MUD matched unrelated donor, MMFD mismatched family donor, MMUD mismatched unrelated donor, BM bone marrow, PBSC peripheral blood stem cells, UCB umbilical cord blood, GvHD graft-versus-host disease, GvL graft-versus leukemia, CNI calcineurin inhibitor, MTX methotrexate, MMF mycophenolate mofetil, MAC myeloablative conditioning, RIC reduced intensity conditioning, Bu Busulfan, Cy Cyclophosphamide, Mel Melphalan, Treo Treosulfan, Flu Fludarabine, TT Thiotepa, (LD)-TBI (low dose) total body irradiation, Clo Clofarabine.
Additionally, in the absence of a matched donor for HR AML, the use of mismatched/haploidentical family donors (MMFD), MMUD, and unrelated umbilical cord blood (UCB), HSCT should be considered. In this context, Fierro-Pineda et al. reported similar feasibility, safety and efficacy rates for matched donor and haploidentical-HSCT allografts in a small cohort of 15 patients with AML or MDS [123]. Wang et al. and Ciurea et al. both reported an equivalent DFS of 75% for haploidentical versus matched sibling (MSD) HSCT in CR1 in larger cohorts of pediatric and adult patients with intermediate- and HR AML [124, 125]. Moreover, a retrospective review of children with AML in CR1 reported a 75% OS for haploidentical recipients [126]. However, more confirmatory results are urgently needed in the pediatric AML setting. Clear recommendations on which HLA-haploidentical-HSCT approach should be preferred cannot yet be made, although promising results have recently been published with the use of α/β T cell- and B-cell-depleted allografts [108, 109, 127].
The second consideration of graft selection is the stem cell source. Both unmanipulated BM and peripheral blood stem cells (PBSC) are generally acceptable options (Fig. 5). However, caution on the use of PBSC is advised in adolescents and in transplants from female donors to male recipients due to an increased risk of GvHD. In cases of mismatched HSCT, the use of unmanipulated PBSC should be approached with even greater caution, and ex vivo or in vivo T cell depletion (TCD) may be necessary to mitigate the GvHD risk [128].
While UCB is less frequently used in the last years, it remains a potential stem cell source for HSCT [129]. Recipients of UCB-HSCT (UCBT) may be at higher risk for early and increased TRM, delayed platelet and neutrophil engraftment, and slower immune reconstitution compared with recipients of BM and PBSC grafts [130]. However, UCB recipients have a reduced risk of cGvHD and the limitation of delayed neutrophil engraftment has improved in more recent experience [131,132,133,134]. Recent OS and EFS outcomes of single (s)UCBT in children with HR and refractory AML were 71% (95%CI 62%-77%) and 72% (95%CI 64%-78%), respectively [134]. The guidelines from the National Marrow Donor Program and the CIBMTR recommend the following criteria for sUCBT: a minimum of 8/8 high-resolution HLA typing (HLA-A, HLA-B, HLA-C, and HLA-DRB1), ≥4/6 typing for HLA-A and HLA-B antigens, high-resolution HLA-DRB1 typing, ≥4/8 high-resolution typing, a total nucleated cell (TNC) count ≥2.5 × 107/kg, and a CD34+ cell count ≥1.5 × 105/kg [135]. HLA-C and KIR combinations have been reported to significantly impact RFS in UCBTs for AML [136]. Patients receiving a 7/8 ABCDR-matched graft with a single HLA-C mismatch experienced significantly poorer RFS than 8/8 matched UCBTs (P = 0.04). The 5-year cumulative recurrence rates of CR1, CR2, and non-responder (NR) groups were 5%, 19%, and 30%. This suggests that, for pediatric patients with AML, UCBT may be a suitable alternative option.
In this respect, outcome comparisons of MFD, MUD, and unrelated UCBT in AML showed no difference in RR or LFS but improved cGvHD-free survival [137] and, in particular, EFS [138] for unrelated UCBT. Horgan et al. suggested that UCBT without serotherapy could be the optimal transplant option for children with MRD-positive myeloid malignancy [138]. Furthermore, current evidence from a large multicenter retrospective study involving 316 recipients of MSD, MUD, UCB, and double UCB transplants demonstrated no significant differences in RR, LFS, or NRM based on the stem cell source.
The choice between BM or PBSC is influenced by collection logistics, and extreme weight differences between donor and recipient, which could limit the ability to obtain adequate numbers of hematopoietic stem cells from BM. PBSC grafts compared with BM are associated with higher rates of cGvHD. If the use of unmanipulated PBSC is necessary, PT-Cy can be employed as an effective in vivo T cell-depletion strategy to mitigate the risk of cGvHD. PBSC grafts are generally not recommended as the preferred stem cell source, particularly from unrelated donor transplants [139,140,141].
Despite significant challenges, more prospective studies to advance our understanding of the best suitable stem cell sources, the optimal number of cells to infuse, and individualized donor-recipient matching [142] would be desirable and may help refine GvHD prophylaxis and tailor allograft approaches to improve outcomes in allogeneic HSCT.
How is optimal HSCT performed in terms of conditioning?
After donor selection, determining the appropriate conditioning regimen is another critical step. Conditioning regimes are separated into “reduced intensity/toxicity (RIC)” with Busulfan (Bu) dosing of <8 mg/kg PO or IV equivalent, Melphalan (Mel) dosing <150 mg/m², total body irradiation (TBI) dose of ≤500 cGy as a single dose, or 800 cGy administered as fractionated doses [143]. In contrast, regimens are considered MAC for any higher dosing. MAC is generally recommended for children with AML. The choice of conditioning regimen should be carefully balanced taking into consideration potential toxicities, the individual patient’s prior treatments and current performance status, and any existing infectious complications at the time of transplantation.
A large retrospective CIBMTR study found no difference in OS or LFS between TBI-based and non-TBI-based MAC regimens [144]. In view of the risk of increased late effects associated with TBI, MAC conditioning with Bu, Cyclophosphamide (Cy) and Mel may be used for patients transplanted in CR1 and CR2 [139]. It is recommended to use weight- and a pharmacokinetically-adjusted dose of Bu to minimize toxicity and maximize efficacy. In the AML-SCT-BFM 2007 trial, the 4-year EFS, OS, CIR, and TRM rates were 61%, 70%, 22%, and 15%, respectively [107]. Notably, TRM varied significantly by age: it was 9% (SE 3%) in children under 12 years, but increased to 31% (SE 9%) in children aged 12 years and older [107]. However, contemporary HSCT regimens and improved HLA typing have largely mitigated these age-related differences, making HSCT with either Bu or Treosulfan (Treo) conditioning safer for older pediatric and young adults (AYA) aged 16-20 [145].
In response to these outcomes, the current trial being conducted by AIEOP/BFM is randomizing patients to receive either Bu/Cy/Mel or Treo, Fludarabine (Flu), and Thiotepa (Thio). This trial could hopefully help determine the optimal MAC regimen, not limited to patients with AML older than 12 years. Additionally, the ongoing prospective, randomized SCRIPT-AML study by the NOPHO-DBH (Dutch-Belgian-Hong Kong) consortium is testing the hypothesis that increasing Bu exposure to achieve a cumulative area under the curve of 90 mg*h/L and replacing alkylating agents (Cy, Mel) with antimetabolites (Clofarabine(Clo), Flu) could improve outcomes.
Whilst MAC is the regimen of choice for most patients, a subset of patients—particularly those with underlying genetic disorders—who are at risk of significantly higher TRM rates should be considered for RIC regimens. This special category includes children with Down syndrome, MDS/leukemia predisposition syndromes, children with an inherited bone marrow failure syndrome such as Fanconi anemia, Schwachman-Diamond syndrome, congenital amegakaryocytic thrombocytopenia, Diamond-Blackfan anemia, dyskeratosis congenita, and severe congenital neutropenia.
Similarly, children with significant comorbidities or those undergoing a second HSCT may also benefit from RIC instead of MAC conditioning. RIC regimens include combinations such as Bu/Flu, Flu/Mel, or regimens incorporating Clo or Treo [146]. In CD33+ pediatric AML patients in CR1/CR2, the pOS and EFS at 5 years following RIC HSCT (Bu/Flu) and GO consolidation were 61% and 78%, respectively [147]. Children with poorly responding primary disease or relapse who underwent early HSCT after a cytoreductive regimen with Flu, Amsacrine, and Cy, followed by RIC conditioning and prophylactic DLI, had 4-year EFS and OS rates of 49% and 53%, respectively. CIR was 38%, and TRM was 11% [107]. These findings support the benefit of consolidative HSCT in subsets of patients, even in the absence of CR, as previously reported in the section “Chasing CR: how to treat relapsed or secondary refractory pediatric AML?”.
Two retrospective studies of 141 and 34 children with AML [144, 148] found no significant difference between RIC and MAC in terms of OS, RR, and TRM. However, ongoing improvements in HSCT procedures, leading to declining TRM rates, may shift this balance. When managed carefully, MAC conditioning may improve long-term survival, even in heavily pretreated patients and those at HR for relapse, such as patients with refractory AML and t-AML.
Janus’ face: graft-versus-leukemia (GvL) and GvHD
While the donor immune cell-mediated anti-leukemia effect is essential to a favorable HSCT outcome, acute and chronic GvHD are major contributors to mortality and morbidity, and the benefit of GvL must be balanced against the risks of GvHD (Fig. 5). This is particularly important in the presence of HLA disparity between donor and recipient and the use of PBSCs. Therefore, ex vivo TCD of the graft, such as the α/β TCD, is commonly used in pediatric patients undergoing transplantation from an HLA-haploidentical donor. As noted in paragraph 4, α/β-haploidentical-HSCT can be handled successfully and is a promising option for patients with no other graft available [108, 109].
In vivo, T-cell depleting serotherapy is administered pre-transplant to patients receiving grafts from MUD, MMFD, or 5–6/8 matched UCB, but is not typically used for those transplanted from an MFD or receiving a 6–8/8 matched UCB [138].
All patients given an unmanipulated graft usually receive immunosuppression (IS) with calcineurin inhibitors (CNI) for GvHD prevention starting prior to stem cell infusion. Patients receiving grafts from an MMD or PBSC, or unrelated UCB, should receive additional prophylaxis with either MMF (mycophenolate mofetil) or short-course Methotrexate (MTX). Short-course MTX is used for in vivo depletion of proliferating alloreactive T cells following HSCT in this setting. Patients who receive unmanipulated BM from a haploidentical parent can also be successfully managed with an in vivo TCD/modulation approach using PT-Cy.
In the absence of GvHD, MMF can be stopped at day 28 post-transplant, and CNI tapered over 4–6 weeks from day 60 (MFD), day 100 (MUD), or earlier if mixed chimerism or MRD is detected. This enhances the benefits of GvL (see section “Losing a battle is not losing the war: maintenance therapy post-transplant and second HSCT”). Optimization of anti-infective and supportive care treatment that does not disturb the gastrointestinal microbiome (GM)—thereby decreasing toxicity, relapse, and GvHD rates—should be considered in future patient management [149, 150].
As GM alterations are linked to leukemogenesis and treatment-related complications, especially during HSCT, various approaches can be used to modulate GM in children, including nutritional interventions, fecal microbiota transplantation, and prebiotics [151, 152]. While there is strong scientific rationale and emerging clinical interest, caution is needed when using probiotics or microbial agents mentioned above. One of the most effective ways to influence GM is by modulating antibiotic use with strategies such as narrow-spectrum antibiotics and optimizing treatment timing and duration. These approaches should be explored in future pediatric clinical studies.
Losing a battle is not losing the war: maintenance therapy post-transplant and second HSCT
From 10% to 60% of patients relapse within the first year of HSCT depending on risk criteria as reported by Shahn et al. [153, 154]. A number of options have the potential to maintain remission post-HSCT (Figs. 3 and 4). These include withdrawal of immunosuppression, DLI, and—if available and applicable—CTLs, NK cells, CIK cells, or, in the future, CAR-T, CAR-NK, and CAR-CIK immune cell therapy. These later immunotherapies are specific/targeted therapies compared to bulk population DLI but are not yet available to most patients [155,156,157,158,159,160,161,162,163,164]. Hypomethylating agents (HMAs) (Decitabine and Azacytidine), alone or combined with DLI or rhG-CSF [165, 166], tyrosine kinase inhibitors (TKIs) (for FLT3-mutated AMLs) [167] and Venetoclax are frequently used for managing leukemia relapse after HSCT. TKI maintenance after HSCT is a promising treatment option for HR AML, leading to long-term remission with minimal side effects. For patients with FLT3-positive AML who initially received Midostaurin, a switch to Sorafenib should be considered after transplantation. Sorafenib is, to date, the only inhibitor that demonstrated efficacy in improving both PFS and OS as post-HSCT MT [168,169,170,171]. However, due to their improved safety profile and higher efficacy, second-generation FLT3 inhibitors such as Quizartinib or Gilteritinib can be used post-transplant, if available. Levis et al. showed that Gilteritinib was beneficial as post-HSCT maintenance in adults with FLT3-ITD AML who had detectable MRD, resulting in a significant RFS benefit, particularly in this MRD-positive subgroup [172]. However, the timing and duration of treatment with TKIs after HSCT have not yet been determined for children. In a retrospective study, 15 pediatric patients were treated with Sorafenib given for a median of 100 days post-HSCT and extended for a period of 18 months in some patients [173]. The addition of the FLT3 inhibitor Quizartinib to pre-transplant chemotherapy, in combination with post-transplant maintenance, will be studied in FLT3+ pediatric patients within the CHIP-AML protocol as part of a linked “Quizartinib” trial (recently approved and currently recruiting). Several targeted agents are being investigated in clinical trials (see Supplementary Table 1), with more in development for patients who relapse after a second HSCT and face a poor prognosis. However, there are insufficient data to clearly recommend which post-HSCT targeted-/immune-therapy should be used, and the decision is based on physician choice and drug availability when participation in a clinical trial is not possible.
A second HSCT may be considered in patients who have relapsed post-HSCT, and have achieved a significant reduction in blast count with reinduction chemotherapy (as defined above in the section “Chasing CR: how to treat relapsed or secondary refractory pediatric AML?”. and shown in Fig. 3), and have an acceptable performance status.
Retrospective studies on children who underwent a second HSCT have demonstrated a long-term DFS ranging between 10% and 50% [174,175,176]. However, the 5-year OS was only 15% [55], when patients with a poor response to reinduction therapy and an early relapse (<1 year) are included. Patients who relapsed >1 year after the first HSCT and who had a good response to reinduction chemotherapy had a better survival rate of 24–35% [176]. These findings were provided by the following studies: in a large cohort of 122 patients, 4-year OS, NRM, and CIR of 31%, 22%, and 45% were reported after the second HSCT [177]. In a smaller cohort of 46 children, a 5-year OS of 41.7% was reported, which increased with an inter-HSCT interval of >2 years (63% vs. 27%; p = 0.01) [175]. Furthermore, in a multicenter national analysis of mismatched T-repleted UCB, an impressive 2-year EFS of 69% was noted in a cohort of r/r AML patients with a previous history of HSCT (n = 24) [138].
Consequently, achieving CR following reinduction therapy is critical. Additionally, post-transplantation MT could be considered to maintain remission for more than 12 months after HSCT and to avoid inter-HSCT intervals of less than 6 months to reduce the risk of relapse and toxicity [174,175,176]. Conditioning for a second HSCT must be selected carefully based on the patient’s prior history and comorbidities. If a third complete remission can be achieved after a second HSCT, a 5-year OS of 40% has been reported [56]. No child or AYA with multiple relapsed AML has ever been reported to survive after a third HSCT (Fig. 3).
Concluding remarks
This review highlights the current approach to HSCT in AML achieved through consecutive improvements of several study groups [6, 35, 106, 107, 178,179,180,181,182,183,184,185,186,187,188,189,190,191]. The internationally agreed principles for diagnosis, risk stratification, treatment response, and pre- and post-transplant management provide guidelines and recommendations for the treatment of AML patients with r/r or HR disease in CR1, both currently and in the future [5, 6, 192, 193].
Although survival rates have gradually improved in recent decades, there is still an urgent need for more effective and less toxic induction therapies and conditioning regimens for patients undergoing HSCT, in order to minimize both acute and late toxicity, and thereby further extend the use of HSCT as a curative approach in pediatric AML. Incorporating molecular disease characterization (epigenomic, genomic, transcriptomic) and MRD monitoring—including NGS—will continue to refine risk stratification and therapeutic decisions in CR1 and relapsed disease, and highlight opportunities for studies aiming to define timing of HSCT, optimal remission status before HSCT, clinical parameter-orientated donor selection and conditioning, and GvHD regimens, towards a more individualized patient management including the use of targeted therapies and cellular immunotherapy.
References
Egan G, Chopra Y, Mourad S, Chiang KY, Hitzler J. Treatment of acute myeloid leukemia in children: a practical perspective. Pediatr Blood Cancer. 2021;68:e28979.
Bleakley M, Shaw PJ, Nielsen JM. Allogeneic bone marrow transplantation for childhood relapsed acute lymphoblastic leukemia: comparison of outcome in patients with and without a matched family donor. Bone Marrow Transplant. 2002;30:1–7.
Bleakley M, Lau L, Shaw PJ, Kaufman A. Bone marrow transplantation for paediatric AML in first remission: a systematic review and meta-analysis. Bone Marrow Transplant. 2002;29:843–52.
Niewerth D, Creutzig U, Bierings MB, Kaspers GJ. A review on allogeneic stem cell transplantation for newly diagnosed pediatric acute myeloid leukemia. Blood. 2010;116:2205–14.
Zwaan CM, Kolb EA, Reinhardt D, Abrahamsson J, Adachi S, Aplenc R, et al. Collaborative efforts driving progress in pediatric acute myeloid leukemia. J Clin Oncol. 2015;33:2949–62.
Creutzig U, van den Heuvel-Eibrink MM, Gibson B, Dworzak MN, Adachi S, de Bont E, et al. Diagnosis and management of acute myeloid leukemia in children and adolescents: recommendations from an international expert panel. Blood. 2012;120:3187–205.
Lie SO, Abrahamsson J, Clausen N, Forestier E, Hasle H, Hovi L, et al. Treatment stratification based on initial in vivo response in acute myeloid leukaemia in children without Down’s syndrome: results of NOPHO-AML trials. Br J Haematol. 2003;122:217–25.
Masetti R, Muratore E, Gori D, Prete A, Locatelli F. Allogeneic hematopoietic stem cell transplantation for pediatric acute myeloid leukemia in first complete remission: a meta-analysis. Ann Hematol. 2022;101:2497–506.
Merli P, Algeri M, Del Bufalo F, Locatelli F. Hematopoietic stem cell transplantation in pediatric acute lymphoblastic leukemia. Curr Hematol Malig Rep. 2019;14:94–105.
Lee DH, Kwon YJ, Lim J, Kim Y, Han K, Chung NG, et al. Comparable outcomes of HLA-matched unrelated and HLA-identical sibling donor bone marrow transplantation for childhood acute myeloid leukemia in first remission. Pediatr Transplant. 2009;13:210–6.
Moore J, Nivison-Smith I, Goh K, Ma D, Bradstock K, Szer J, et al. Equivalent survival for sibling and unrelated donor allogeneic stem cell transplantation for acute myelogenous leukemia. Biol Blood Marrow Transplant. 2007;13:601–7.
Horan JT, Alonzo TA, Lyman GH, Gerbing RB, Lange BJ, Ravindranath Y, et al. Impact of disease risk on efficacy of matched related bone marrow transplantation for pediatric acute myeloid leukemia: the Children’s Oncology Group. J Clin Oncol. 2008;26:5797–801.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19.
Algeri M, Merli P, Locatelli F, Pagliara D. The role of allogeneic hematopoietic stem cell transplantation in pediatric leukemia. J Clin Med. 2021;10:3790.
Hasle H. A critical review of which children with acute myeloid leukaemia need stem cell procedures. Br J Haematol. 2014;166:23–33.
Kelly MJ, Horan JT, Alonzo TA, Eapen M, Gerbing RB, He W, et al. Comparable survival for pediatric acute myeloid leukemia with poor-risk cytogenetics following chemotherapy, matched related donor, or unrelated donor transplantation. Pediatr Blood Cancer. 2014;61:269–75.
Cornelissen JJ, Gratwohl A, Schlenk RF, Sierra J, Bornhauser M, Juliusson G, et al. The European LeukemiaNet AML Working Party consensus statement on allogeneic HSCT for patients with AML in remission: an integrated-risk adapted approach. Nat Rev Clin Oncol. 2012;9:579–90.
Harrison CJ, Hills RK, Moorman AV, Grimwade DJ, Hann I, Webb DK, et al. Cytogenetics of childhood acute myeloid leukemia: United Kingdom Medical Research Council Treatment trials AML 10 and 12. J Clin Oncol. 2010;28:2674–81.
von Neuhoff C, Reinhardt D, Sander A, Zimmermann M, Bradtke J, Betts DR, et al. Prognostic impact of specific chromosomal aberrations in a large group of pediatric patients with acute myeloid leukemia treated uniformly according to trial AML-BFM 98. J Clin Oncol. 2010;28:2682–9.
Gamis AS, Alonzo TA, Meshinchi S, Sung L, Gerbing RB, Raimondi SC, et al. Gemtuzumab ozogamicin in children and adolescents with de novo acute myeloid leukemia improves event-free survival by reducing relapse risk: results from the randomized phase III Children’s Oncology Group trial AAML0531. J Clin Oncol. 2014;32:3021–32.
Burnett AK, Wheatley K, Goldstone AH, Stevens RF, Hann IM, Rees JH, et al. The value of allogeneic bone marrow transplant in patients with acute myeloid leukaemia at differing risk of relapse: results of the UK MRC AML 10 trial. Br J Haematol. 2002;118:385–400.
Sharma A, Galimard JE, Pryce A, Bhoopalan SV, Dalissier A, Dalle JH, et al. Cytogenetic abnormalities predict survival after allogeneic hematopoietic stem cell transplantation for pediatric acute myeloid leukemia: a PDWP/EBMT study. Bone Marrow Transplant. 2024;59:451–8.
Grimwade D, Walker H, Oliver F, Wheatley K, Harrison C, Harrison G. et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92:2322–33.
Hasle H, Alonzo TA, Auvrignon A, Behar C, Chang M, Creutzig U, et al. Monosomy 7 and deletion 7q in children and adolescents with acute myeloid leukemia: an international retrospective study. Blood. 2007;109:4641–7.
Quessada J, Cuccuini W, Saultier P, Loosveld M, Harrison CJ, Lafage-Pochitaloff M. Cytogenetics of pediatric acute myeloid leukemia: a review of the current knowledge. Genes. 2021;12:924.
Richard-Carpentier G, Rausch CR, Sasaki K, Hammond D, Morita K, Takahashi K, et al. Characteristics and clinical outcomes of patients with acute myeloid leukemia with inv(3)(q21q26.2) or t(3;3)(q21;q26.2). Haematologica. 2023;108:2331–42.
Groschel S, Sanders MA, Hoogenboezem R, de Wit E, Bouwman BAM, Erpelinck C, et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell. 2014;157:369–81.
Yamazaki H, Suzuki M, Otsuki A, Shimizu R, Bresnick EH, Engel JD, et al. A remote GATA2 hematopoietic enhancer drives leukemogenesis in inv(3)(q21;q26) by activating EVI1 expression. Cancer Cell. 2014;25:415–27.
Blakemore C, Damodharan S, Puccetti D. Inv(3) acute myeloid leukemia in a young adult and review of the literature. Case Rep Oncol Med. 2023;2023:6628492.
Balgobind BV, Raimondi SC, Harbott J, Zimmermann M, Alonzo TA, Auvrignon A, et al. Novel prognostic subgroups in childhood 11q23/MLL-rearranged acute myeloid leukemia: results of an international retrospective study. Blood. 2009;114:2489–96.
Pollard JA, Guest E, Alonzo TA, Gerbing RB, Loken MR, Brodersen LE, et al. Gemtuzumab ozogamicin improves event-free survival and reduces relapse in pediatric KMT2A-rearranged AML: results from the Phase III Children’s Oncology Group Trial AAML0531. J Clin Oncol. 2021;39:3149–60.
Conneely SE, Rau RE. The genomics of acute myeloid leukemia in children. Cancer Metastasis Rev. 2020;39:189–209.
van Weelderen RE, Klein K, Harrison CJ, Jiang Y, Abrahamsson J, Arad-Cohen N, et al. Measurable residual disease and fusion partner independently predict survival and relapse risk in childhood KMT2A-rearranged acute myeloid leukemia: a study by the International Berlin-Frankfurt-Munster Study Group. J Clin Oncol. 2023;41:2963–74.
Klusmann JH, Reinhardt D, Zimmermann M, Kremens B, Vormoor J, Dworzak M, et al. The role of matched sibling donor allogeneic stem cell transplantation in pediatric high-risk acute myeloid leukemia: results from the AML-BFM 98 study. Haematologica. 2012;97:21–29.
Yuen KY, Liu Y, Zhou YZ, Wang Y, Zhou DH, Fang JP, et al. Mutational landscape and clinical outcome of pediatric acute myeloid leukemia with 11q23/KMT2A rearrangements. Cancer Med. 2023;12:1418–30.
Byrne M, Danielson N, Sengsayadeth S, Rasche A, Culos K, Gatwood K, et al. The use of venetoclax-based salvage therapy for post-hematopoietic cell transplantation relapse of acute myeloid leukemia. Am J Hematol. 2020;95:1006–14.
Piedimonte M, Ottone T, Alfonso V, Ferrari A, Conte E, Divona M, et al. A rare BCR-ABL1 transcript in Philadelphia-positive acute myeloid leukemia: case report and literature review. BMC Cancer. 2019;19:50.
Bacher U, Haferlach T, Alpermann T, Zenger M, Hochhaus A, Beelen DW, et al. Subclones with the t(9;22)/BCR-ABL1 rearrangement occur in AML and seem to cooperate with distinct genetic alterations. Br J Haematol. 2011;152:713–20.
Neuendorff NR, Burmeister T, Dorken B, Westermann J. BCR-ABL-positive acute myeloid leukemia: a new entity? Analysis of clinical and molecular features. Ann Hematol. 2016;95:1211–21.
Buteyn NJ, Burke CG, Sartori VJ, Deering-Gardner E, DeBruine ZJ, Kamarudin D, et al. EZH2-driven immune evasion defines high-risk pediatric AML with t(16;21) FUS::ERG gene fusion. bioRxiv [Preprint]. 2024. https://doi.org/10.1101/2024.05.14.594150.
Zhang Z, Sun C, Cai L, Chen Y, Zhou X, Chen S, et al. Prognostic factors in Philadelphia chromosome-positive acute myeloid leukemia using fluorescence in situ hybridization. Clin Lab. 2024;70. https://doi.org/10.7754/Clin.Lab.2023.230645
Tarlock K, Alonzo TA, Moraleda PP, Gerbing RB, Raimondi SC, Hirsch BA, et al. Acute myeloid leukaemia (AML) with t(6;9)(p23;q34) is associated with poor outcome in childhood AML regardless of FLT3-ITD status: a report from the Children’s Oncology Group. Br J Haematol. 2014;166:254–9.
Ishiyama K, Takami A, Kanda Y, Nakao S, Hidaka M, Maeda T, et al. Allogeneic hematopoietic stem cell transplantation for acute myeloid leukemia with t(6;9)(p23;q34) dramatically improves the patient prognosis: a matched-pair analysis. Leukemia. 2012;26:461–4.
Sandahl JD, Coenen EA, Forestier E, Harbott J, Johansson B, Kerndrup G, et al. t(6;9)(p22;q34)/DEK-NUP214-rearranged pediatric myeloid leukemia: an international study of 62 patients. Haematologica. 2014;99:865–72.
Hollink IH, van den Heuvel-Eibrink MM, Arentsen-Peters ST, Pratcorona M, Abbas S, Kuipers JE, et al. NUP98/NSD1 characterizes a novel poor prognostic group in acute myeloid leukemia with a distinct HOX gene expression pattern. Blood. 2011;118:3645–56.
Ostronoff F, Othus M, Gerbing RB, Loken MR, Raimondi SC, Hirsch BA, et al. NUP98/NSD1 and FLT3/ITD coexpression is more prevalent in younger AML patients and leads to induction failure: a COG and SWOG report. Blood. 2014;124:2400–7.
Shiba N, Ichikawa H, Taki T, Park MJ, Jo A, Mitani S, et al. NUP98-NSD1 gene fusion and its related gene expression signature are strongly associated with a poor prognosis in pediatric acute myeloid leukemia. Genes Chromosomes Cancer. 2013;52:683–93.
Hara Y, Shiba N, Ohki K, Tabuchi K, Yamato G, Park MJ, et al. Prognostic impact of specific molecular profiles in pediatric acute megakaryoblastic leukemia in non-Down syndrome. Genes Chromosomes Cancer. 2017;56:394–404.
de Rooij JD, Zwaan CM, van den Heuvel-Eibrink M. Pediatric AML: from biology to clinical management. J Clin Med. 2015;4:127–49.
Bertrums EJM, Smith JL, Harmon L, Ries RE, Wang YJ, Alonzo TA, et al. Comprehensive molecular and clinical characterization of NUP98 fusions in pediatric acute myeloid leukemia. Haematologica. 2023;108:2044–58.
de Rooij JD, Masetti R, van den Heuvel-Eibrink MM, Cayuela JM, Trka J, Reinhardt D, et al. Recurrent abnormalities can be used for risk group stratification in pediatric AMKL: a retrospective intergroup study. Blood. 2016;127:3424–30.
Espersen ADL, Noren-Nystrom U, Abrahamsson J, Ha SY, Pronk CJ, Jahnukainen K, et al. Acute myeloid leukemia (AML) with t(7;12)(q36;p13) is associated with infancy and trisomy 19: data from Nordic Society for Pediatric Hematology and Oncology (NOPHO-AML) and review of the literature. Genes Chromosomes Cancer. 2018;57:359–65.
Coenen EA, Zwaan CM, Reinhardt D, Harrison CJ, Haas OA, de Haas V, et al. Pediatric acute myeloid leukemia with t(8;16)(p11;p13), a distinct clinical and biological entity: a collaborative study by the International-Berlin-Frankfurt-Munster AML-study group. Blood. 2013;122:2704–13.
Creutzig U, Zimmermann M, Dworzak MN, Gibson B, Tamminga R, Abrahamsson J, et al. The prognostic significance of early treatment response in pediatric relapsed acute myeloid leukemia: results of the international study Relapsed AML 2001/01. Haematologica. 2014;99:1472–8.
Ciurea SO, Zhang MJ, Bacigalupo AA, Bashey A, Appelbaum FR, Aljitawi OS, et al. Haploidentical transplant with posttransplant cyclophosphamide vs matched unrelated donor transplant for acute myeloid leukemia. Blood. 2015;126:1033–40.
Qiu KY, Liao XY, Liu Y, Huang K, Li Y, Fang JP, et al. Poor outcome of pediatric patients with acute myeloid leukemia harboring high FLT3/ITD allelic ratios. Nat Commun. 2022;13:3679.
Wang JW, Yu L, Yang XG, Xu LH. NUP98::NSD1 and FLT3/ITD co-expression is an independent predictor of poor prognosis in pediatric AML patients. BMC Pediatr. 2024;24:547.
Bazarbachi A, Bug G, Baron F, Brissot E, Ciceri F, Dalle IA, et al. Clinical practice recommendation on hematopoietic stem cell transplantation for acute myeloid leukemia patients with FLT3-internal tandem duplication: a position statement from the Acute Leukemia Working Party of the European Society for Blood and Marrow Transplantation. Haematologica. 2020;105:1507–16.
Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, Balgobind BV, Arentsen-Peters ST, Alders M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009;113:5951–60.
Meshinchi S, Alonzo TA, Stirewalt DL, Zwaan M, Zimmerman M, Reinhardt D, et al. Clinical implications of FLT3 mutations in pediatric AML. Blood. 2006;108:3654–61.
Manara E, Basso G, Zampini M, Buldini B, Tregnago C, Rondelli R, et al. Characterization of children with FLT3-ITD acute myeloid leukemia: a report from the AIEOP AML-2002 Study Group. Leukemia. 2017;31:18–25.
Schechter T, Gassas A, Chen H, Pollard J, Meshinchi S, Zaidman I, et al. The outcome of allogeneic hematopoietic cell transplantation for children with FMS-like tyrosine kinase 3 internal tandem duplication-positive acute myelogenous leukemia. Biol Blood Marrow Transplant. 2015;21:172–5.
Schlenk RF, Kayser S, Bullinger L, Kobbe G, Casper J, Ringhoffer M, et al. Differential impact of allelic ratio and insertion site in FLT3-ITD-positive AML with respect to allogeneic transplantation. Blood. 2014;124:3441–9.
Pratcorona M, Brunet S, Nomdedeu J, Ribera JM, Tormo M, Duarte R, et al. Favorable outcome of patients with acute myeloid leukemia harboring a low-allelic burden FLT3-ITD mutation and concomitant NPM1 mutation: relevance to post-remission therapy. Blood. 2013;121:2734–8.
Zhou Q, Zhao D, Zarif M, Davidson MB, Minden MD, Tierens A, et al. A real-world analysis of clinical outcomes in AML with myelodysplasia-related changes: a comparison of ICC and WHO-HAEM5 criteria. Blood Adv. 2024;8:1760–71.
Locatelli F, Strahm B. How I treat myelodysplastic syndromes of childhood. Blood. 2018;131:1406–14.
Hossain MJ, Xie L, Caywood EH. Prognostic factors of childhood and adolescent acute myeloid leukemia (AML) survival: evidence from four decades of US population data. Cancer Epidemiol. 2015;39:720–6.
Pedersen-Bjergaard J, Andersen MT, Andersen MK. Genetic pathways in the pathogenesis of therapy-related myelodysplasia and acute myeloid leukemia. Hematol Am Soc Hematol Educ Program. 2007;1:392–7.
Sendker S, Schneider M, Antoniou E, Neumann D, Niktoreh N, Dirksen U, et al. Improved outcome and therapeutic directions in pediatric therapy-related AML: recommendations by the AML-BFM Study Group. Blood Adv. 2025;9:2831–41.
Kobos R, Steinherz PG, Kernan NA, Prockop SE, Scaradavou A, Small TN, et al. Allogeneic hematopoietic stem cell transplantation for pediatric patients with treatment-related myelodysplastic syndrome or acute myelogenous leukemia. Biol Blood Marrow Transplant. 2012;18:473–80.
Gassas A, Sivaprakasam P, Cummins M, Breslin P, Patrick K, Slatter M, et al. High transplant-related mortality associated with haematopoietic stem cell transplantation for paediatric therapy-related acute myeloid leukaemia (t-AML). A study on behalf of the United Kingdom Paediatric Blood and Bone Marrow Transplant Group. Bone Marrow Transplant. 2018;53:1165–9.
Madanat YF, Gerds AT. Can allogeneic hematopoietic cell transplant cure therapy-related acute leukemia? Best Pract Res Clin Haematol. 2019;32:104–13.
Annereau M, Willekens C, El Halabi L, Chahine C, Saada V, Auger N, et al. Use of 5-azacitidine for therapy-related myeloid neoplasms in patients with concomitant active neoplastic disease. Leuk Res. 2017;55:58–64.
Schuurhuis GJ, Heuser M, Freeman S, Bene MC, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131:1275–91.
Heuser M, Freeman SD, Ossenkoppele GJ, Buccisano F, Hourigan CS, Ngai LL, et al. 2021 Update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138:2753–67.
Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424–47.
Tuval A, Shlush LI. Evolutionary trajectory of leukemic clones and its clinical implications. Haematologica. 2019;104:872–80.
Rubnitz JE, Inaba H, Dahl G, Ribeiro RC, Bowman WP, Taub J, et al. Minimal residual disease-directed therapy for childhood acute myeloid leukaemia: results of the AML02 multicentre trial. Lancet Oncol. 2010;11:543–52.
Buldini B, Maurer-Granofszky M, Varotto E, Dworzak MN. Flow-cytometric monitoring of minimal residual disease in pediatric patients with acute myeloid leukemia: recent advances and future strategies. Front Pediatr. 2019;7:412.
Tierens A, Bjorklund E, Siitonen S, Marquart HV, Wulff-Juergensen G, Pelliniemi TT, et al. Residual disease detected by flow cytometry is an independent predictor of survival in childhood acute myeloid leukaemia; results of the NOPHO-AML 2004 study. Br J Haematol. 2016;174:600–9.
Loken MR, Alonzo TA, Pardo L, Gerbing RB, Raimondi SC, Hirsch BA, et al. Residual disease detected by multidimensional flow cytometry signifies high relapse risk in patients with de novo acute myeloid leukemia: a report from Children’s Oncology Group. Blood. 2012;120:1581–8.
Karol SE, Coustan-Smith E, Pounds S, Wang L, Inaba H, Ribeiro RC, et al. Clinical impact of minimal residual disease in blood and bone marrow of children with acute myeloid leukemia. Blood Adv. 2023;7:3651–7.
van der Velden VH, van der Sluijs-Geling A, Gibson BE, te Marvelde JG, Hoogeveen PG, Hop WC, et al. Clinical significance of flowcytometric minimal residual disease detection in pediatric acute myeloid leukemia patients treated according to the DCOG ANLL97/MRC AML12 protocol. Leukemia. 2010;24:1599–606.
Buldini B, Rizzati F, Masetti R, Fagioli F, Menna G, Micalizzi C, et al. Prognostic significance of flow-cytometry evaluation of minimal residual disease in children with acute myeloid leukaemia treated according to the AIEOP-AML 2002/01 study protocol. Br J Haematol. 2017;177:116–26.
Benetton M, Merli P, Walter C, Hansen M, Da Ros A, Polato K, et al. Molecular measurable residual disease assessment before hematopoietic stem cell transplantation in pediatric acute myeloid leukemia patients: a retrospective study by the I-BFM Study Group. Biomedicines. 2022;10:1530.
MRD-AML-BFM Study Group, Langebrake C, Creutzig U, Dworzak M, Hrusak O, Mejstrikova E, et al. Residual disease monitoring in childhood acute myeloid leukemia by multiparameter flow cytometry: the MRD-AML-BFM Study Group. J Clin Oncol. 2006;24:3686–92.
Sievers EL, Lange BJ, Alonzo TA, Gerbing RB, Bernstein ID, Smith FO, et al. Immunophenotypic evidence of leukemia after induction therapy predicts relapse: results from a prospective Children’s Cancer Group study of 252 patients with acute myeloid leukemia. Blood. 2003;101:3398–406.
Meena JP, Pathak N, Gupta AK, Bakhshi S, Gupta R, Makkar H, et al. Molecular evaluation of gene mutation profiles and copy number variations in pediatric acute myeloid leukemia. Leuk Res. 2022;122:106954.
Ruan M, Liu L, Qi B, Chen X, Chang L, Zhang A, et al. Targeted next-generation sequencing of circulating tumor DNA, bone marrow, and peripheral blood mononuclear cells in pediatric AML. Front Oncol. 2021;11:666470.
Chen X, Liu L, Zhang A, Yi M, Lan Y, Zheng Z, et al. Droplet digital PCR for genetic mutations monitoring predicts relapse risk in pediatric acute myeloid leukemia. Int J Hematol. 2022;116:669–77.
O’Hare P, Lucchini G, Cummins M, Veys P, Potter M, Lawson S, et al. Allogeneic stem cell transplantation for refractory acute myeloid leukemia in pediatric patients: the UK experience. Bone Marrow Transplant. 2017;52:825–31.
Quarello P, Fagioli F, Basso G, Putti MC, Berger M, Luciani M, et al. Outcome of children with acute myeloid leukaemia (AML) experiencing primary induction failure in the AIEOP AML 2002/01 clinical trial. Br J Haematol. 2015;171:566–73.
Nemecek ER, Gooley TA, Woolfrey AE, Carpenter PA, Matthews DC, Sanders JE. Outcome of allogeneic bone marrow transplantation for children with advanced acute myeloid leukemia. Bone Marrow Transplant. 2004;34:799–806.
Duval M, Klein JP, He W, Cahn JY, Cairo M, Camitta BM, et al. Hematopoietic stem-cell transplantation for acute leukemia in relapse or primary induction failure. J Clin Oncol. 2010;28:3730–8.
Okamoto Y, Kudo K, Tabuchi K, Tomizawa D, Taga T, Goto H, et al. Hematopoietic stem-cell transplantation in children with refractory acute myeloid leukemia. Bone Marrow Transplant. 2019;54:1489–98.
Gorman MF, Ji L, Ko RH, Barnette P, Bostrom B, Hutchinson R, et al. Outcome for children treated for relapsed or refractory acute myelogenous leukemia (rAML): a Therapeutic Advances in Childhood Leukemia (TACL) Consortium study. Pediatr Blood Cancer. 2010;55:421–9.
Egan G, Tasian SK. Relapsed pediatric acute myeloid leukaemia: state-of-the-art in 2023. Haematologica. 2023;108:2275–88.
Moritake H. [Pediatric acute myeloid leukemia]. Rinsho Ketsueki. 2024;65:928–36.
Strachan DC, Gu CJ, Kita R, Anderson EK, Richardson MA, Yam G, et al. Ex vivo drug sensitivity correlates with clinical response and supports personalized therapy in pediatric AML. Cancers. 2022;14:6240.
Wijnen NE, Koedijk JB, Klein K, Luesink M, Goemans BF, Zwaan CM, et al. Treating CD33-positive de novo acute myeloid leukemia in pediatric patients: focus on the clinical value of gemtuzumab ozogamicin. Onco Targets Ther. 2023;16:297–308.
Takahashi T, Lake AJ, Wachter F, Calderon FA, Dandoy C, Keating AK. Effects of total body irradiation on hematopoietic cell transplantation outcomes in pediatric acute myeloid leukemia with prior central nervous system involvement. Transplant Cell Ther. 2024;30:812 e811–2.
Wang Y, Yang X, Liu Y, Li Y. A review of common immunotherapy and nano immunotherapy for acute myeloid leukemia. Front Immunol. 2025;16:1505247.
Bubb QR, Balood M, Seir GE, Swartzrock L, Haslett E, Ho K, et al. Development of multivalent CAR T cells as dual immunotherapy and conditioning agents. Mol Ther Oncol. 2025;33:200944.
Syed YY. Revumenib: first approval. Drugs. 2025;85:577–83.
Rasche M, Zimmermann M, Steidel E, Alonzo T, Aplenc R, Bourquin JP, et al. Survival following relapse in children with acute myeloid leukemia: a report from AML-BFM and COG. Cancers. 2021;13:2336.
Sauer MG, Lang PJ, Albert MH, Bader P, Creutzig U, Eyrich M, et al. Hematopoietic stem cell transplantation for children with acute myeloid leukemia-results of the AML SCT-BFM 2007 trial. Leukemia. 2020;34:613–24.
Bertaina A, Zecca M, Buldini B, Sacchi N, Algeri M, Saglio F, et al. Unrelated donor vs HLA-haploidentical alpha/beta T-cell- and B-cell-depleted HSCT in children with acute leukemia. Blood. 2018;132:2594–607.
Ruggeri A, Santoro N, Galimard JE, Kalwak K, Algeri M, Zubarovskaya L, et al. Matched unrelated donor transplantation versus haploidentical transplantation with post-transplant cyclophosphamide in children with acute myeloid leukemia: a PDWP-EBMT study. Haematologica. 2024;109:2122–30.
Jacobsohn DA, Loken MR, Fei M, Adams A, Brodersen LE, Logan BR, et al. Outcomes of measurable residual disease in pediatric acute myeloid leukemia before and after hematopoietic stem cell transplant: validation of difference from normal flow cytometry with chimerism studies and Wilms tumor 1 gene expression. Biol Blood Marrow Transplant. 2018;24:2040–6.
Wang B, Wen X, Zhang R, Zhu G, Wu Y, Zhang Y, et al. Homoharringtonine-based induction therapy reduces the recurrence rate of pediatric acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. Cell Transplant. 2023;32:9636897231183559.
Tierens A, Arad-Cohen N, Cheuk D, De Moerloose B, Fernandez Navarro JM, Hasle H, et al. Mitoxantrone versus liposomal daunorubicin in induction of pediatric AML with risk stratification based on flow cytometry measurement of residual disease. J Clin Oncol. 2024;42:2174–85.
Bitan M, Ahn KW, Millard HR, Pulsipher MA, Abdel-Azim H, Auletta JJ, et al. Personalized prognostic risk score for long-term survival for children with acute leukemia after allogeneic transplantation. Biol Blood Marrow Transplant. 2017;23:1523–30.
Pochon C, Detrait M, Dalle JH, Michel G, Dhedin N, Chalandon Y, et al. Improved outcome in children compared to adolescents and young adults after allogeneic hematopoietic stem cell transplant for acute myeloid leukemia: a retrospective study from the Francophone Society of Bone Marrow Transplantation and Cell Therapy (SFGM-TC). J Cancer Res Clin Oncol. 2022;148:2083–97.
Gilleece MH, Shimoni A, Labopin M, Robinson S, Beelen D, Socie G, et al. Measurable residual disease status and outcome of transplant in acute myeloid leukemia in second complete remission: a study by the acute leukemia working party of the EBMT. Blood Cancer J. 2021;11:88.
Renard C, Corbel A, Paillard C, Pochon C, Schneider P, Simon N, et al. Preventive and therapeutic strategies for relapse after hematopoietic stem cell transplant for pediatric AML (SFGM-TC)]. Bull Cancer. 2025;112:S135–S145.
Gibson BES, Sauer MG, Amrolia P. Acute myeloid leukemia in children. In: Carreras E, Dufour C, Mohty M, Kroger N, editors. The EBMT handbook: hematopoietic stem cell transplantation and cellular therapies. 7th ed. Springer, Cham; 2019. p. 523–30.
Venstrom JM, Pittari G, Gooley TA, Chewning JH, Spellman S, Haagenson M, et al. HLA-C-dependent prevention of leukemia relapse by donor activating KIR2DS1. N Engl J Med. 2012;367:805–16.
Locatelli F, Bauquet A, Palumbo G, Moretta F, Bertaina A. Negative depletion of alpha/beta+ T cells and of CD19+ B lymphocytes: a novel frontier to optimize the effect of innate immunity in HLA-mismatched hematopoietic stem cell transplantation. Immunol Lett. 2013;155:21–23.
Bernson E, Hallner A, Sander FE, Wilsson O, Werlenius O, Rydstrom A, et al. Impact of killer-immunoglobulin-like receptor and human leukocyte antigen genotypes on the efficacy of immunotherapy in acute myeloid leukemia. Leukemia. 2017;31:2552–9.
Bakhtiari T, Ahmadvand M, Yaghmaie M, Sadeghi A, Mousavi SA, Rostami T, et al. Investigation of KIR/HLA relationship and other clinical variables after T-cell-replete haploidentical bone marrow transplantation in patients with acute myeloid leukemia (AML). BMC Immunol. 2023;24:10.
Verneris MR, Miller JS, Hsu KC, Wang T, Sees JA, Paczesny S, et al. Investigation of donor KIR content and matching in children undergoing hematopoietic cell transplantation for acute leukemia. Blood Adv. 2020;4:1350–6.
Fierro-Pineda JC, Tsai HL, Blackford A, Cluster A, Caywood E, Dalal J, et al. Prospective PTCTC trial of myeloablative haplo-BMT with posttransplant cyclophosphamide for pediatric acute leukemias. Blood Adv. 2023;7:5639–48.
Wang Y, Liu QF, Xu LP, Liu KY, Zhang XH, Ma X, et al. Haploidentical vs identical-sibling transplant for AML in remission: a multicenter, prospective study. Blood. 2015;125:3956–62.
Ciurea SO, Bayraktar UD. No donor”? Consider a haploidentical transplant. Blood Rev. 2015;29:63–70.
Mo XD, Zhang XH, Xu LP, Wang Y, Yan CH, Chen H, et al. Unmanipulated haploidentical hematopoietic stem cell transplantation in first complete remission can abrogate the poor outcomes of children with acute myeloid leukemia resistant to the first course of induction chemotherapy. Biol Blood Marrow Transplant. 2016;22:2235–42.
Merli P, Algeri M, Galaverna F, Bertaina V, Lucarelli B, Boccieri E, et al. TCRalphabeta/CD19 cell-depleted HLA-haploidentical transplantation to treat pediatric acute leukemia: updated final analysis. Blood. 2024;143:279–89.
Kim BK, Hong KT, Choi JY, Kim H, Park HJ, Kang HJ. Comparable outcomes of allogeneic peripheral blood versus bone marrow hematopoietic stem cell transplantation from a sibling donor for pediatric patients. Ann Hematol. 2024;103:2051–8.
Kurtzberg J, Troy JD, Page KM, El Ayoubi HR, Volt F, Maria Scigliuolo G, et al. Unrelated donor cord blood transplantation in children: lessons learned over 3 decades. Stem Cells Transl Med. 2023;12:26–38.
Shi Y, Wu J, Wang Z, Zhang L, Wang Z, Zhang M, et al. Efficacy and safety of geptanolimab (GB226) for relapsed or refractory peripheral T cell lymphoma: an open-label phase 2 study (Gxplore-002). J Hematol Oncol. 2021;14:12.
Tang X, Chen J, Fang J, Sun X, Qin MQ, Li J, et al. Similar outcomes of allogeneic hematopoietic cell transplantation from unrelated donor and umbilical cord blood vs. sibling donor for pediatric acute myeloid leukemia: Multicenter experience in China. Pediatr Transplant. 2015;19:413–21.
Zheng C, Zhu X, Tang B, Yao W, Song K, Tong J, et al. Comparative analysis of unrelated cord blood transplantation and HLA-matched sibling hematopoietic stem cell transplantation in children with high-risk or advanced acute leukemia. Ann Hematol. 2015;94:473–80.
Visentin S, Auquier P, Bertrand Y, Baruchel A, Tabone MD, Pochon C, et al. The impact of donor type on long-term health status and quality of life after allogeneic hematopoietic stem cell transplantation for childhood acute leukemia: a Leucemie de l’Enfant et de L’Adolescent Study. Biol Blood Marrow Transplant. 2016;22:2003–10.
Chen EL, Liu HL, Geng LQ, Tang BL, Zhu XY, Yao W, et al. [Unrelated cord blood stem cell transplantation for high-risk/refractory childhood acute myeloid leukemia: a clinical analysis of 160 cases]. Zhonghua Xue Ye Xue Za Zhi. 2021;42:549–54.
Dehn J, Spellman S, Hurley CK, Shaw BE, Barker JN, Burns LJ, et al. Selection of unrelated donors and cord blood units for hematopoietic cell transplantation: guidelines from the NMDP/CIBMTR. Blood. 2019;134:924–34.
Rivera-Franco MM, Wynn L, Volt F, Hernandez D, Cappelli B, Scigliuolo GM, et al. Unsupervised clustering analysis of regimen and HLA characteristics in pediatric umbilical cord blood transplantation. Transplant Cell Ther. 2024;30:910.e1–15.
Keating AK, Langenhorst J, Wagner JE, Page KM, Veys P, Wynn RF, et al. The influence of stem cell source on transplant outcomes for pediatric patients with acute myeloid leukemia. Blood Adv. 2019;3:1118–28.
Horgan C, Mullanfiroze K, Rauthan A, Patrick K, Butt NA, Mirci-Danicar O, et al. T-cell replete cord transplants give superior outcomes in high-risk and relapsed/refractory pediatric myeloid malignancy. Blood Adv. 2023;7:2155–65.
Lucchini G, Labopin M, Beohou E, Dalissier A, Dalle JH, Cornish J, et al. Impact of conditioning regimen on outcomes for children with acute myeloid leukemia undergoing transplantation in first complete remission. An analysis on behalf of the Pediatric Disease Working Party of the European Group for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2017;23:467–74.
Shinzato A, Tabuchi K, Atsuta Y, Inoue M, Inagaki J, Yabe H, et al. PBSCT is associated with poorer survival and increased chronic GvHD than BMT in Japanese paediatric patients with acute leukaemia and an HLA-matched sibling donor. Pediatr Blood Cancer. 2013;60:1513–9.
Anasetti C, Logan BR, Lee SJ, Waller EK, Weisdorf DJ, Wingard JR, et al. Peripheral-blood stem cells versus bone marrow from unrelated donors. N Engl J Med. 2012;367:1487–96.
Cieri N, Hookeri N, Stromhaug K, Li L, Keating J, Diaz-Fernandez P, et al. Systematic identification of minor histocompatibility antigens predicts outcomes of allogeneic hematopoietic cell transplantation. Nat Biotechnol. 2024;43:1–12.
Bacigalupo A, Ballen K, Rizzo D, Giralt S, Lazarus H, Ho V, et al. Defining the intensity of conditioning regimens: working definitions. Biol Blood Marrow Transplant. 2009;15:1628–33.
Ishida H, Adachi S, Hasegawa D, Okamoto Y, Goto H, Inagaki J, et al. Comparison of a fludarabine and melphalan combination-based reduced toxicity conditioning with myeloablative conditioning by radiation and/or busulfan in acute myeloid leukemia in Japanese children and adolescents. Pediatr Blood Cancer. 2015;62:883–9.
Tomizawa D, Tanaka S, Kondo T, Hashii Y, Arai Y, Kudo K, et al. Allogeneic hematopoietic stem cell transplantation for adolescents and young adults with acute myeloid leukemia. Biol Blood Marrow Transplant. 2017;23:1515–22.
Burroughs LM, Shimamura A, Talano JA, Domm JA, Baker KK, Delaney C, et al. Allogeneic hematopoietic cell transplantation using Treosulfan-based conditioning for treatment of marrow failure disorders. Biol Blood Marrow Transplant. 2017;23:1669–77.
Zahler S, Bhatia M, Ricci A, Roy S, Morris E, Harrison L, et al. A phase I study of reduced-intensity conditioning and allogeneic stem cell transplantation followed by dose escalation of targeted consolidation immunotherapy with gemtuzumab ozogamicin in children and adolescents with CD33+ acute myeloid leukemia. Biol Blood Marrow Transplant. 2016;22:698–704.
Bitan M, He W, Zhang MJ, Abdel-Azim H, Ayas MF, Bielorai B, et al. Transplantation for children with acute myeloid leukemia: a comparison of outcomes with reduced intensity and myeloablative regimens. Blood. 2014;123:1615–20.
McMahon S, Sahasrabhojane P, Kim J, Franklin S, Chang CC, Jenq RR, et al. Contribution of the oral and gastrointestinal microbiomes to bloodstream infections in leukemia patients. Microbiol Spectr. 2023;11:e0041523.
Masetti R, Muratore E, Leardini D, Zama D, Turroni S, Brigidi P, et al. Gut microbiome in pediatric acute leukemia: from predisposition to cure. Blood Adv. 2021;5:4619–29.
Masetti R, Leardini D, Muratore E, Fabbrini M, D’Amico F, Zama D, et al. Gut microbiota diversity before allogeneic hematopoietic stem cell transplantation as a predictor of mortality in children. Blood. 2023;142:1387–98.
Jozefczuk P, Bilinski J, Minkowska A, Laguna P. Gut microbiome in children undergoing hematopoietic stem cell transplantation. Best Pract Res Clin Gastroenterol. 2024;72:101955.
Dahlberg A, Leisenring W, Bleakley M, Meshinchi S, Baker KS, Summers C, et al. Prognosis of relapse after hematopoietic cell transplant (HCT) for treatment of leukemia or myelodysplastic syndrome (MDS) in children. Bone Marrow Transplant. 2019;54:1337–45.
Shah NN, Borowitz MJ, Steinberg SM, Robey NC, Gamper CJ, Symons HJ, et al. Factors predictive of relapse of acute leukemia in children after allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2014;20:1033–9.
Gorczynska E, Turkiewicz D, Toporski J, Kalwak K, Rybka B, Ryczan R, et al. Prompt initiation of immunotherapy in children with an increasing number of autologous cells after allogeneic HCT can induce complete donor-type chimerism: a report of 14 children. Bone Marrow Transplant. 2004;33:211–7.
Rettinger E, Willasch AM, Kreyenberg H, Borkhardt A, Holter W, Kremens B, et al. Preemptive immunotherapy in childhood acute myeloid leukemia for patients showing evidence of mixed chimerism after allogeneic stem cell transplantation. Blood. 2011;118:5681–8.
Bader P, Kreyenberg H, Hoelle W, Dueckers G, Kremens B, Dilloo D, et al. Increasing mixed chimerism defines a high-risk group of childhood acute myelogenous leukemia patients after allogeneic stem cell transplantation where pre-emptive immunotherapy may be effective. Bone Marrow Transplant. 2004;33:815–21.
Wang QS, Wang Y, Lv HY, Han QW, Fan H, Guo B, et al. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol Ther. 2015;23:184–91.
Kenderian SS, Ruella M, Shestova O, Klichinsky M, Aikawa V, Morrissette JJ, et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29:1637–47.
Pizzitola I, Anjos-Afonso F, Rouault-Pierre K, Lassailly F, Tettamanti S, Spinelli O, et al. Chimeric antigen receptors against CD33/CD123 antigens efficiently target primary acute myeloid leukemia cells in vivo. Leukemia. 2014;28:1596–605.
Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. 2019;25:1064–72.
Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. 2018;131:108–20.
Nguyen R, Wu H, Pounds S, Inaba H, Ribeiro RC, Cullins D, et al. A phase II clinical trial of adoptive transfer of haploidentical natural killer cells for consolidation therapy of pediatric acute myeloid leukemia. J Immunother Cancer. 2019;7:81.
Bremm M, Pfeffermann LM, Cappel C, Katzki V, Erben S, Betz S, et al. Improving clinical manufacturing of IL-15 activated cytokine-induced killer (CIK) cells. Front Immunol. 2019;10:1218.
Gao L, Zhang Y, Wang S, Kong P, Su Y, Hu J, et al. Effect of rhG-CSF combined with decitabine prophylaxis on relapse of patients with high-risk MRD-negative AML after HSCT: an open-label, multicenter, randomized controlled trial. J Clin Oncol. 2020;38:4249–59.
Tamura A, Ishida T, Saito A, Yamamoto N, Yokoi T, Uemura S, et al. Low-dose azacitidine maintenance therapy after allogeneic stem cell transplantation for high-risk pediatric acute myeloid leukemia. Pediatr Blood Cancer. 2018;65:e27284.
Pollard JA, Alonzo TA, Gerbing R, Brown P, Fox E, Choi J, et al. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ acute myeloid leukemia: a report from the Children’s Oncology Group Protocol AAML1031. J Clin Oncol. 2022;40:2023–35.
Alati C, Pitea M, Mico MC, Marafioti V, Greve B, Pratico G, et al. Optimizing maintenance therapy in acute myeloid leukemia: where do we stand in the year 2024? Expert Rev Hematol. 2024;17:515–25.
Xuan L, Wang Y, Yang K, Shao R, Huang F, Fan Z, et al. Sorafenib maintenance after allogeneic haemopoietic stem-cell transplantation in patients with FLT3-ITD acute myeloid leukaemia: long-term follow-up of an open-label, multicentre, randomised, phase 3 trial. Lancet Haematol. 2023;10:e600–e611.
Biavasco F, Zeiser R. FLT3-inhibitor therapy for prevention and treatment of relapse after allogeneic hematopoietic cell transplantation. Int J Hematol. 2022;116:341–50.
Senapati J, Kadia TM. Which FLT3 inhibitor for treatment of AML? Curr Treat Options Oncol. 2022;23:359–80.
Levis MJ, Hamadani M, Logan B, Jones RJ, Singh AK, Litzow M, et al. Gilteritinib as post-transplant maintenance for AML with internal tandem duplication mutation of FLT3. J Clin Oncol. 2024;42:1766–75.
Tarlock K, Chang B, Cooper T, Gross T, Gupta S, Neudorf S, et al. Sorafenib treatment following hematopoietic stem cell transplant in pediatric FLT3/ITD acute myeloid leukemia. Pediatr Blood Cancer. 2015;62:1048–54.
Meshinchi S, Leisenring WM, Carpenter PA, Woolfrey AE, Sievers EL, Radich JP, et al. Survival after second hematopoietic stem cell transplantation for recurrent pediatric acute myeloid leukemia. Biol Blood Marrow Transplant. 2003;9:706–13.
Taga T, Murakami Y, Tabuchi K, Adachi S, Tomizawa D, Kojima Y, et al. Role of second transplantation for children with acute myeloid leukemia following posttransplantation relapse. Pediatr Blood Cancer. 2016;63:701–5.
Yaniv I, Krauss AC, Beohou E, Dalissier A, Corbacioglu S, Zecca M, et al. Second hematopoietic stem cell transplantation for post-transplantation relapsed acute leukemia in children: a retrospective EBMT-PDWP Study. Biol Blood Marrow Transplant. 2018;24:1629–42.
Uden T, Bertaina A, Abrahamsson J, Ansari M, Balduzzi A, Bourquin JP, et al. Outcome of children relapsing after first allogeneic haematopoietic stem cell transplantation for acute myeloid leukaemia: a retrospective I-BFM analysis of 333 children. Br J Haematol. 2020;189:745–50.
Creutzig U, Ritter J, Schellong G. Identification of two risk groups in childhood acute myelogenous leukemia after therapy intensification in study AML-BFM-83 as compared with study AML-BFM-78. AML-BFM Study Group. Blood. 1990;75:1932–40.
Creutzig U, Zimmermann M, Ritter J, Reinhardt D, Hermann J, Henze G, et al. Treatment strategies and long-term results in paediatric patients treated in four consecutive AML-BFM trials. Leukemia. 2005;19:2030–42.
Creutzig U, Dworzak M, Zimmermann M, Bourquin JP, Gruhn B, Fleischhack G, et al. Randomised introduction of 2-CDA as intensification during consolidation for children with high-risk AML-results from Study AML-BFM 2004. Klin Padiatr. 2015;227:116–22.
Rasche M, von Neuhoff C, Dworzak M, Bourquin JP, Bradtke J, Gohring G, et al. Genotype-outcome correlations in pediatric AML: the impact of a monosomal karyotype in trial AML-BFM 2004. Leukemia. 2017;31:2807–14.
Creutzig U, Zimmermann M, Bourquin JP, Dworzak MN, Fleischhack G, Graf N, et al. Randomized trial comparing liposomal daunorubicin with idarubicin as induction for pediatric acute myeloid leukemia: results from Study AML-BFM 2004. Blood. 2013;122:37–43.
Rasche M, Zimmermann M, Borschel L, Bourquin JP, Dworzak M, Klingebiel T, et al. Successes and challenges in the treatment of pediatric acute myeloid leukemia: a retrospective analysis of the AML-BFM trials from 1987 to 2012. Leukemia. 2018;32:2167–77.
Uffmann M, Rasche M, Zimmermann M, von Neuhoff C, Creutzig U, Dworzak M, et al. Therapy reduction in patients with Down syndrome and myeloid leukemia: the international ML-DS 2006 trial. Blood. 2017;129:3314–21.
Zwaan CM, Reinhardt D, Corbacioglu S, van Wering ER, Bokkerink JP, Tissing WJ, et al. Gemtuzumab ozogamicin: first clinical experiences in children with relapsed/refractory acute myeloid leukemia treated on compassionate-use basis. Blood. 2003;101:3868–71.
Creutzig U, Semmler J, Kaspers GL, Reinhardt D, Zimmermann M. Re-induction with L-DNR/FLAG improves response after AML relapse, but not long-term survival. Klin Padiatr. 2014;226:323–31.
Niktoreh N, Lerius B, Zimmermann M, Gruhn B, Escherich G, Bourquin JP, et al. Gemtuzumab ozogamicin in children with relapsed or refractory acute myeloid leukemia: a report by Berlin-Frankfurt-Munster study group. Haematologica. 2019;104:120–7.
Creutzig U, Dworzak M, von Neuhoff N, Rasche M, Reinhardt D. [Acute promyelocytic leukemia: new treatment strategies with ATRA and ATO—AML-BFM-recommendations]. Klin Padiatr. 2018;230:299–304.
Niktoreh N, Walter C, Zimmermann M, von Neuhoff C, von Neuhoff N, Rasche M, et al. Mutated WT1, FLT3-ITD, and NUP98-NSD1 fusion in various combinations define a poor prognostic group in pediatric acute myeloid leukemia. J Oncol. 2019;2019:1609128.
Rasche M, Steidel E, Zimmermann M, Bourquin JP, Boztug H, Janotova I, et al. Second relapse of pediatric patients with acute myeloid leukemia: a report on current treatment strategies and outcome of the AML-BFM Study Group. Cancers. 2021;13:789.
Mann G, Reinhardt D, Ritter J, Hermann J, Schmitt K, Gadner H, et al. Treatment with all-trans retinoic acid in acute promyelocytic leukemia reduces early deaths in children. Ann Hematol. 2001;80:417–22.
Creutzig U, Zimmermann M, Bourquin JP, Dworzak MN, Kremens B, Lehrnbecher T, et al. Favorable outcome in infants with AML after intensive first- and second-line treatment: an AML-BFM study group report. Leukemia. 2012;26:654–61.
Creutzig U, Kutny MA, Barr R, Schlenk RF, Ribeiro RC. Acute myelogenous leukemia in adolescents and young adults. Pediatr Blood Cancer. 2018;65:e27089.
Goyal SD, Zhang MJ, Wang HL, Akpek G, Copelan EA, Freytes C, et al. Allogeneic hematopoietic cell transplant for AML: no impact of pre-transplant extramedullary disease on outcome. Bone Marrow Transplant. 2015;50:1057–62.
Stove HK, Sandahl JD, Abrahamsson J, Asdahl PH, Forestier E, Ha SY, et al. Extramedullary leukemia in children with acute myeloid leukemia: a population-based cohort study from the Nordic Society of Pediatric Hematology and Oncology (NOPHO). Pediatr Blood Cancer. 2017;64. https://doi.org/10.1002/pbc.26520.
Dusenbery KE, Howells WB, Arthur DC, Alonzo T, Lee JW, Kobrinsky N, et al. Extramedullary leukemia in children with newly diagnosed acute myeloid leukemia: a report from the Children’s Cancer Group. J Pediatr Hematol Oncol. 2003;25:760–8.
Nakayama H, Tabuchi K, Tawa A, Tsukimoto I, Tsuchida M, Morimoto A, et al. Outcome of children with relapsed acute myeloid leukemia following initial therapy under the AML99 protocol. Int J Hematol. 2014;100:171–9.
Karlsson L, Forestier E, Hasle H, Jahnukainen K, Jonsson OG, Lausen B, et al. Outcome after intensive reinduction therapy and allogeneic stem cell transplant in paediatric relapsed acute myeloid leukaemia. Br J Haematol. 2017;178:592–602.
Fagioli F, Zecca M, Locatelli F, Lanino E, Uderzo C, Di Bartolomeo P, et al. Allogeneic stem cell transplantation for children with acute myeloid leukemia in second complete remission. J Pediatr Hematol Oncol. 2008;30:575–83.
Webb DK, Wheatley K, Harrison G, Stevens RF, Hann IM. Outcome for children with relapsed acute myeloid leukaemia following initial therapy in the Medical Research Council (MRC) AML 10 trial. MRC Childhood Leukaemia Working Party. Leukemia. 1999;13:25–31.
Moritake H, Tanaka S, Miyamura T, Nakayama H, Shiba N, Shimada A, et al. The outcomes of relapsed acute myeloid leukemia in children: results from the Japanese Pediatric Leukemia/Lymphoma Study Group AML-05R study. Pediatr Blood Cancer. 2021;68:e28736.
Aladjidi N, Auvrignon A, Leblanc T, Perel Y, Benard A, Bordigoni P, et al. Outcome in children with relapsed acute myeloid leukemia after initial treatment with the French Leucemie Aique Myeloide Enfant (LAME) 89/91 protocol of the French Society of Pediatric Hematology and Immunology. J Clin Oncol. 2003;21:4377–85.
Wells RJ, Adams MT, Alonzo TA, Arceci RJ, Buckley J, Buxton AB, et al. Mitoxantrone and cytarabine induction, high-dose cytarabine, and etoposide intensification for pediatric patients with relapsed or refractory acute myeloid leukemia: Children’s Cancer Group Study 2951. J Clin Oncol. 2003;21:2940–7.
McCarthy AJ, Pitcher LA, Hann IM, Oakhill A. FLAG (fludarabine, high-dose cytarabine, and G-CSF) for refractory and high-risk relapsed acute leukemia in children. Med Pediatr Oncol. 1999;32:411–5.
Locatelli F, Masetti R, Rondelli R, Zecca M, Fagioli F, Rovelli A, et al. Outcome of children with high-risk acute myeloid leukemia given autologous or allogeneic hematopoietic cell transplantation in the aieop AML-2002/01 study. Bone Marrow Transplant. 2015;50:181–8.
Lund TC, Ahn KW, Tecca HR, Hilgers MV, Abdel-Azim H, Abraham A, et al. Outcomes after second hematopoietic cell transplantation in children and young adults with relapsed acute leukemia. Biol Blood Marrow Transplant. 2019;25:301–6.
Acknowledgements
This work was supported by the by the Frankfurt Cancer Institute (ER, DH, JHK), by the Mildred-Scheel-Nachwuchszentrum (MSNZ) (70113301, ER), by grants from the parents Association “Hilfe für Krebskranke Kinder e.V.” as part of the C3OMBAT-AML consortium (ER, DH, JHK). DH is supported by the Frankfurt Foundation for Children with Cancer. Figures 1, 2, 4 and 5 were created in BioRender: https://BioRender.com/saa9nj7, https://BioRender.com/jga7qly, https://BioRender.com/ioa2s8u, https://BioRender.com/v7ks5pw.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Consortia
Contributions
ER, DH, BG, and FL wrote the manuscript. MS, DT, KK, KK, DR, and J-HK contributed to the interpretation of data, critically reviewed the manuscript, and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
KK acknowledges research support from Jazz Pharmaceuticals, travel grants from Sobi and Medac, and receives honoraria from Vertex. BG declares honoraria from Vertex. JHK acknowledges advisory roles with Bluebird Bio, Novartis, Roche, and Jazz Pharmaceuticals. FL participated in advisory boards for Amgen, Sanofi, and Vertex; speaker’s bureau for Amgen, Gilead, Miltenyi, Novartis, Sanofi, and SOBI. All other authors declare that the manuscript was written in the absence of any commercial or financial relationships that could be perceived as a potential conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rettinger, E., Heckl, D., Gibson, B. et al. The hallmarks of hematopoietic stem cell transplantation for pediatric acute myeloid leukemia. Leukemia 39, 2313–2328 (2025). https://doi.org/10.1038/s41375-025-02685-5
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41375-025-02685-5
