
Abstract
Rapid, high-throughput, timely, multiplex diagnosis of respiratory-tract infections still relies on laboratory infrastructure, sequential assays, and trained personnel, thereby delaying targeted therapy and outbreak containment. In this study, a Fully Automated rotary microfluidic platform (FA-RMP) for high-throughput multiplex respiratory tract pathogens detection was presented. FA-RMP enables a true “sample-in, result-out” workflow through the integration of swab lysis, reagent partitioning, lyophilized reverse transcription loop-mediated isothermal amplification (RT-LAMP), and moving-probe fluorescence read-out, all encapsulated with a disposable microfluidic cartridge and paired with a 9 kg, four-channel benchtop reader. The FA-RMP enables parallel processing of 16 independent reactions within 30 min, supporting simultaneous detection of up to 4 distinct clinical samples. Analytical validation using serially diluted Mycoplasma pneumoniae (MP) DNA established a limit of detection (LoD) of 50 copies µL−1 and a log-linear correlation between threshold time and template load (R2 = 0.9528). Testing with eight non-target respiratory pathogens yielded no amplification, confirming high analytical specificity. FA-RMP successfully detected the clinical samples with influenza A, influenza B, and MP, further demonstrating its robust multiplex detection capability. By integrating automated sample preparation, multiplex isothermal amplification and quantitative detection into a portable, high-throughput system, the platform delivers laboratory-grade performance at the point of care, serving as a scalable tool for routine respiratory pathogens screening and rapid epidemic response.

Similar content being viewed by others


Introduction
Respiratory tract infections (RTIs) remain a leading cause of global morbidity and mortality. In 2019, lower respiratory infections were the top infectious cause of death worldwide, accounting for roughly 2.6 million deaths and ranking among the top four causes of death overall1,2. Seasonal influenza, Mycoplasma pneumoniae (MP), SARS-CoV-2, and other respiratory pathogens frequently co-circulate, presenting overlapping and clinically indistinguishable symptoms3. Because these agents require different treatments and control measures, early and accurate diagnosis is essential to guide therapy and public health responses4. High-throughput, multi-pathogen testing is thus urgently needed to relieve pressure on hospitals and laboratories by quickly identifying the causative pathogens in large patient cohorts.
Reverse-transcription quantitative PCR (RT-qPCR) is considered as the gold standard for viral detection due to its high sensitivity and specificity5. However, RT-qPCR requires complex thermal cycling instrumentation and laboratory infrastructure, which may not be readily available in point-of-care settings. Moreover, RT-qPCR typically targets one pathogen per reaction (or a few using different fluorescence channels), and running numerous separate PCR tests can be time-consuming and resource-intensive6,7,8. Isothermal nucleic acid amplification techniques such as loop-mediated isothermal amplification (LAMP) have emerged as attractive alternatives to PCR for point-of-care diagnosis. LAMP eliminates thermal cycling constraints and retains high analytical sensitivity9. But its adoption is constrained by primer-design complexity and cross-primer interference that intensify in multiplex panels10. Moreover, most LAMP schemes still depend on manual nucleic-acid extraction, manual reagent loading, and fluorescence detection on external instruments—all of which undermine ease of use and convenience at the point of care.
Microfluidic technology has emerged as a powerful solution, integrating sample preparation, amplification, and detection into miniaturized, automated platforms. Recent advances in multiplex assays underscore the promise of microfluidic integration for respiratory diagnostics11,12,13,14. A finger-actuated disposable microfluidic system utilizing recombinase-based isothermal amplification for tuberculosis detection was introduced, which eliminates external power requirements but is inherently limited in quantitative precision and scalability due to manual actuation15. The other platform named Lab-Disk was presented in which centrifugal actuation, 39 °C isothermal incubation, and real-time fluorescence detection are tri-integrated within a single companion analyzer. However, the platform is limited to processing a single specimen per run, thus falling short of high-throughput, multi-sample requirements. And its dual-stage fluidic layout combined with active mixing, further elevates the complexity of chip design and fabrication16. An “ultrasimple” wedge-shaped microfluidic chip that encodes multiplex assays via the physical diameters of probe-functionalized microspheres, dispensing with both nucleic-acid amplification and optical components, was developed11. Nevertheless, its overall analytical sensitivity may lag behind amplification-based POCT platforms, and its scalability to high-throughput, multi-sample applications remains limited. Additionally, recent studies have demonstrated the potential of non-enzymatic signal amplification and digital microfluidic platform for multiplexed pathogen detection. However, practical implementations of these approaches are inherently constrained to single-sample detection per operational run17,18,19. Thus, state-of-the-art devices frequently compromise on at least one of three fronts: (i) throughput—many platforms handle a single specimen per run; (ii) actuation—manual finger pumps or complex centrifugal layouts limit point-of-care usability; and (iii) system integration—open-tube reagent handling or external optics jeopardize contamination control and portability.
In this study, a Fully Automated rotary microfluidic platform (FA-RMP) for high-throughput multiplex respiratory tract pathogens detection was developed to address these limitations. FA-RMP consists of microfluidic cartridges and 4 independent detection channels. The cartridges feature a rotary architecture that automatically performs sealed sample lysis, reagent mixing, and liquid partitioning. The platform with 4-channels enables liquid control, isothermal amplification, fluorescence detection and results analysis of up to four cartridges simultaneously. The developed platform can detect 50 copies/μL DNA of MP within 30 minutes and support the detection of 3 pathogens at the same time, which offer a promising tool for clinical, multiplex diagnosis of respiratory tract pathogens.
Materials and methods
Materials and reagents
Lyo-Ready™ Direct RNA/DNA LAMP Saliva 4 × reaction mixture was purchased from Meridian (Meridian Life Science Inc. USA.). 20× Eva Green was purchased from Biotium (Biotium, Inc., USA). RNase-free water was purchased and all LAMP primers were synthesized from Sangon Biotech Co. Ltd. (Shanghai, China). Nucleic acid release reagent was purchased from Tuoman (Tuoman Biotech, Shanghai). The standard cultures with nucleic acid of MP, influenza A (Flu A) and influenza B (Flu B) were obtained from Guangzhou Bondson Biotechnology Co., Ltd (Guangzhou, China). The process of LAMP lyophilization beads were obtained from Janyi (Shanghai Janyi Biotechnology Co., Ltd).
Design and fabrication of the microfluidic cartridge
The microfluidic cartridge used in this study was developed based on our previous research20. To enhance the integration capability for pathogens detection, a cylindrical module that can realize sample lysis by rotation was combined with the microfluidic cartridge in this study. The cartridge was designed using SolidWorks and fabricated by computer numerical control (CNC) machine. The cartridge consisted of a top cover, a base plate, and a sample lysis module. The sample lysis module was arranged along a circular track with a diameter of 36.8 mm, containing three cylindrical wells (diameter: 9.1 mm; depth: 15 mm). The LAMP mixture were pre-loaded as lyophilized beads and sealed within the reaction chambers of the cartridge.
Fabrication of the FA-RMP
The FA-RMP integrated several functional modules, including microfluidic cartridge, an optical detection module, and four individual modules incorporating temperature control stack. These components were enclosed in a compact shell measuring 340 mm × 255 mm × 230 mm (L × W × H) with a total mass of less than 9 kg, making it suitable for benchtop or portable clinical settings. The external shell was fabricated from reinforced acrylonitrile–butadiene–styrene (ABS) plastic to ensure both thermal and mechanical stability. The optical detection module comprised a mobile fluorescence-detection head mounted on a linear rail base. The fluorescence-detection head integrated an LED excitation source, two optical filters, a dichroic mirror, an objective lens, a focusing lens, and a multipixel photon counter. The temperature control stack consisted of heating plates, a cooling fan, and NTC thermistors supported precise ramp-up, isothermal holding, and active cooling. All subsystems were designed for seamless mechanical integration, and were controlled by embedded electronics and detection application software via a standard data interface. The FA-RMP supported up to four independent individual channels, allowing parallel detection of 4 samples.
Design of LAMP primers
The MP was selected as the target, and P1 gene of MP was chosen to be the amplification region. The sequences of P1 were retrieved from the NCBI database, and the LAMP primers were designed using PrimerExplorer V5 software. The primer sets were listed in Table 1.
Development and optimization of LAMP assay
All LAMP reactions were carried out in a total volume of 25 µL as follows: 6.25 µL of LAMP mixture, 1.25 µL EvaGreen fluorescent dye, 0.70 µL primers mixture, 1.00 µL MgSO₄ solution, 15.80 µL samples, and DEPC treated water. The LAMP mixture included the Bst DNA polymerase, buffer salts, and dNTPs. The primers mixture contained specific initial concentrations of three pairs of LAMP primers. To create a field-deployable cartridge, all LAMP amplification reagents were embedded in the reaction chambers of microfluidic cartridge as the lyophilized beads. The lyophilized beads were rehydrated by introducing the sample solution into the reaction chambers. The FA-RMP accommodated the microfluidic cartridge, provided precise temperature control and real-time fluorescence data acquisition. The standard LAMP protocol on the device was set as 65 °C for 30 min with optics recording fluorescence signals every 60 s. Prior to running experimental samples, a series of optimization experiments was conducted on the platform to determine the optimal reaction conditions for the LAMP assay.
Sensitivity and specificity of FA-RMP
To test the limit of detection (LoD) of FA-RMP, MP standard cultures with DNA were diluted from 5000 copies/μL to 20 copies/μL. Each dilution was tested in 3 independent replicate reactions on microfluidic cartridge. Genomic RNA/DNA panels of eight respiratory pathogens (Influenza A, Influenza B, Respiratory syncytial virus, Ureaplasma urealyticum, Chlamydia pneumoniae, Staphylococcus aureus, Legionella pneumophila, Streptococcus pneumoniae) were chosen to test the specificity of FA-RMP in this study.
Multi-target detection of respiratory pathogens
For multiplex detection, primers targeting MP, Flu A, and Flu B were prepared as lyophilized beads using and preloaded into reaction chambers of the microfluidic cartridge. Nasopharyngeal swab specimens confirmed positive for MP, influenza A, or influenza B were provided by Peking University Third Hospital. To comply with institutional biosafety protocols, the hospital heat-inactivated all specimens and processed them entirely within its certified biosafety level-2 laboratory. The specimens with pathogens were detected by the FA-RMP directly.
Data analysis
The real-time fluorescence intensity values were recorded, and the amplification rate was calculated. The endpoint fluorescence data were analyzed via one-way repeated-measures ANOVA (analysis of variance). The error bars of the fluorescence plots represent the standard error of independent replicates (n = 3). All experiments marked with n = 3 refer to technical replicates unless otherwise noted. Statistical significance tiers are denoted by symbols within the figures(“****” when p < 0.0001).
Results and discussion
Design and fabrication of the microfluidic cartridge
To ensure ease of use and full automation, the microfluidic cartridge was designed to perform on-chip nucleic acid lysis and LAMP amplification. The illustration of the microfluidic cartridge was displayed in Fig. 1a, and the LAMP reagent was preloaded into the reaction chambers of the cartridge as the lyophilized beads. The overall design and the description of the cartridge were displayed in Fig. 1b. The cartridge consists of a top cover, a reagent storage rotor, a base plate, a waterproof debubbler membrane, and two pistons. The base plate contains multiple microchannels and four independent reaction chambers, which enabled the detection for up to 4 targets within a single cartridge. The reagent storage rotor comprises three chambers: a lysis chamber (A), a sample loading chamber (B), and a mixing chamber (C). Rotation of the reagent rotor is driven by a motor, which in turn activates pistons to control liquid movement between chambers, enabling nucleic acid lysis, mixing, and eventual transfer to the amplification chambers.

a The digital image of the microfluidic cartridge and the lyophilized reagent beads were pre-loaded in the reaction chamber. b The overall illustration and the functional description of microfluidic cartridge. c The schematic diagram of the process for samples detection based on microfluidic cartridge
The overall cartridge functions and fluidic control process were displayed in Fig. 1c. After sample collection using a nasopharyngeal or oropharyngeal swab, the swab was broken off and inserted directly into the sample-loading chamber (Step 1). The chamber was then sealed by closing the cap integrated into the top cover. The flat-head piston pushed the lysis reagent from the lysis chamber into the sample-loading chamber via lysis buffer transfer microchannel, initiating nucleic acid extraction (Step 2 to Step 3). As the rotor continues to turn, the ball-head piston lifted under the guidance of a grooved cam structure, drawing the lysed sample into the mixing chamber (Step 4). Forward and reverse rotation of the rotor allowed the ball-head piston to perform repeated up-and-down strokes, ensuring thorough mixing (Step 5 to Step 6). Finally, continued rotation directed the mixed solution into an inlet channel, which leaded the liquid into the reaction chambers (Step 7). There, the lyophilized reagents were rehydrated, and the cartridge ready for amplification and fluorescence-based detection (Step 8).
Design and validation of the FA-RMP
The FA-RMP supports reverse-transcription LAMP (RT-LAMP) reactions within a consumable microfluidic chip, and integrates temperature control and fluorescence signal detection into a fully automated workflow. The system is capable of simultaneously processing up to four individual detection channels enabling high-throughput diagnosis.
As depicted in Fig. 2a, the platform integrates three subsystems: (i) microfluidic cartridge, (ii) a temperature-control stack, and (iii) a mobile fluorescence-detection head. After the user introduces the lysed sample into the chip inlet, a stepper motor drives the transmission component to rotate the upper groove of the microfluidic chip, thereby actuating a piston that sequentially allocates the sample to distinct chambers for nucleic-acid extraction and mixing and ultimately to the reaction chamber. The chip is clamped between upper and lower heater blocks to maintain isothermal amplification at 65 °C. The optical head is mounted on a linear rail and is moved beneath the chip by a second stepper motor to sequentially scan all wells, producing real-time fluorescence curves. Control software loaded on the system executes automated management of the entire process.

a The illustration of the FA-RMP consists of microfluidic cartridges, optical detection module, and 4-individual detection module. b The real-time temperature curve under the usual work condition for 30 min. c The linear relationship between the concentrations of standard fluorescent sodium solution and fluorescence intensity. d The consistency of the system by comparing the fluorescence intensity of different channels and chambers contained the 1 μM of fluorescent sodium solution. The error bars were obtained by calculating the results of three independent experiments (n = 3)
Accurate, uniform, and rapidly adjustable temperature is indispensable for isothermal LAMP assays. The system adopts a sandwich-type temperature-control module: during the heating phase, two polyimide-foil heaters clamp the chip’s amplification chamber between thermally conductive plates, while an NTC thermistor probe continuously acquires temperature data and feeds it to the control unit, providing feedback to a PID controller until the designated RT-LAMP temperature is reached. As the result shown in Fig. 2b, the system can maintain a constant temperature of 65 °C for 30 min at least. Real-time signal acquisition is accomplished by a compact optical head carried on a stepper-driven linear rail, which sequentially scans the four reaction regions. A dilution series of fluorescent sodium solution from 0.3125 to 5 μM was analyzed to evaluate the performance of intensity measurement of the optical module, and the result shown in Fig. 2c exhibited the linear correlation between the concentrations and fluorescence (R2 = 0.9820). The result displayed in Fig. 2d indicated that the fluorescence intensities of each chamber and channel was no significant difference, which contained the 1 μM of fluorescent sodium. The above results strongly proved the platform demonstrated in this study could support the needs of the pathogens detection.
Optimization of LAMP assay
Using the selected primers, the concentration of the primers in the LAMP reaction was optimized. The primers were evaluated by comparing the Tp value and the non-specific amplification signal of non-template control group. As the results shown in Fig. 3a, 60–80 μM of the inner primers (FIP/BIP) would lead to nonspecific signals due to the high-concentration primers form double-stranded structures and then bound by the fluorescence dye non-specifically21. The 20–40 μM of the primers with no significant differences in values of Tp and 20 μM was selected to minimize the risk of non-specific amplification while preserving efficient amplification performance. Likewise, outer primers (F3/B3) were optimal at 5 μM per test to prevent the risk of non-specific amplification under high concentration of primers as Fig. 3b, and loop primers at 10 μM per test as Fig. 3c.

a–c Optimization the concentration of inner primers, outer primers and loop primers. The concentrations in the figure represent the initial concentrations of primers added to the primer mixture, which are 10-80 μM for inner primers, 1–10 μM for outer primer, and 4–12 μM for loop primers, respectively. d Optimization the concentration of Mg2+ from 2 to 8 mM. The error bars represent the means ± s.d. from replicates (n = 3)
Mg2+ plays a critical role in regulating polymerase activity and optimizes the concentration of Mg2+ emerges as a viable strategy to enhance the performance of LAMP assays22. As shown in Fig. 3d, different concentrations of Mg2 (from 2 mM to 8 mM) were added in the reaction mixture, and the Tp values were obtained. The groups added with 8 mM and 6 mM Mg2+ exhibited the nonspecific signals, which may be attributed to the high concentration of Mg2+ causing incorrect binding between primer and template23. The optimal Mg2+ concentration was defined as the concentration that still achieved a rapid amplification of the positive sample without yielding any amplification in the negative control and 4 mM was finally chosen as the optimal concentration.
Optimization of LAMP assay on chip
To address the application requirements, the LAMP mixture was turned as lyophilized beads with advantages in terms of transportation, long-term storage stability, and compatibility. The efficiency of the LAMP lyophilized beads was validated by incubation at 65 °C for 30 min and the result was shown in Fig. 4. The standard cultures with MP DNA were pre-mixed with nucleic acid release solution in 10 min at room temperature and then detected as the sample. The amplification curves of Positive 1 (LAMP mixture) and Positive 2 (lyophilized beads) with MP DNA were standard S profiles, which indicated the LAMP lyophilized beads can realize the amplification and detection of MP accurately. The amplification curve of the group with positive 3 (lyophilized beads on chip) was same with the positive 2, indicating that the LAMP lyophilized beads could be utilized to detect the MP DNA released on-chip or off-chip.

Positive 1, LAMP reaction mixture with 1000 copies/μL of MP DNA. Positive 2, the lyophilized beads of LAMP with 1000 copies/μL of MP DNA. Positive 3, the lyophilized beads of LAMP with 1000 copies/μL of MP DNA on chip. NTC, non-template control. The error bars represent the means ± s.d. from replicates (n = 3)
Sensitivity and specificity of FA-RMP
To determine the limit of detection (LoD) of FA-RMP, each sample was serially diluted to different concentrations (5000–20 copies μL−1) and evaluated in 3 independent replicates per level on microfluidic cartridges under the optimized LAMP conditions. The platform reliably detected down to 50 copies μL−1 of MP culture samples (Fig. 5a). Linear regression of logarithm of MP concentration versus Tp yielded R2 = 0.9528 (Fig. 5b), indicating quantitative potential within the range 5000–50 copies μL−1.

a The sensitivity for detection of standard samples with different concentrations MP (from 5000 copies/μL to 20 copies/μL). b Linear relationship between the concentrations of standard samples and threshold times. c The specificity for detection of different types of pathogens. (****p < 0.0001 by ANOVA). Three replicates were run (n = 3) and the error bars represent the means ± s.d. from replicates
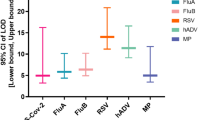
The detection specificity is crucial to evaluate the specificity of the FA-RMP. The screening primers of MP were preloaded and a variety of pathogens standard cultures including M. pneumoniae, Influenza A virus (Flu A), Influenza B virus (Flu B), Respiratory syncytial virus, Ureaplasma urealyticum, Chlamydia pneumoniae, Staphylococcus aureus, Legionella pneumophila, Streptococcus pneumoniae were chosen to test the specificity analysis of system and the intensity of the fluorescence in 30 min were obtained. The results shown in Fig. 5c indicate that the primer sets for M. pneumonia could only amplify the target pathogen and generate strong fluorescence signal intensity. There was no cross-reaction with other templates, which proved that the FA-RMP have great specificity.
Clinical testing for multiplex detection
The diagnostic procedure of the FA-RMP follows a streamlined sequence as shown in Fig. 6a. A nasopharyngeal or oropharyngeal swab is first collected from the individual. The swab tip is placed into the sample-loading chamber of a disposable microfluidic chip after breaking off the shaft, and the lid is closed to seal the chamber. The chip is then inserted into the benchtop instrument, where proper positioning is ensured by a mechanical interlock. The analysis is initiated by a single click within the dedicated software on the connected computer. The overall process of the FA-RMP for “samples-in, results-out” with microfluidic cartridge was shown in Fig. 6b, and the FA-RMP enabled automatically detection processes on-chip including nucleic acid release, isothermal amplification, and real-time fluorescence detection within ~30 min. The developed platform automatically operated by the software on the computer is shown in Fig. 6c.

a The workflow process of the FA-RMP for pathogen detection. b The schematic of the FA-RMP for the process of “Sample-in, Results-out”. c The FA-RMP automatically operated by the software on the computer
A total of 18 nasopharyngeal samples (healthy controls, n = 4; MP infected individuals, n = 12; Flu A infected individual, n = 1; Flu B infected individual, n = 1) were acquired from Peking University Third Hospital. The study protocol was reviewed and approved by the Peking University Third Hospital medical science research ethics committee (IRB approval number: 00006761-M2022102). To demonstrate the practicality and the consistency, the collected throat swabs of were detected by the developed platform (FA-RMP) and clinical standard device (7500 real time PCR, Thermo Fisher Scientific.). The result shown in Fig. 7 displayed the Tp value of the clinical samples under the two platforms. The reaction condition for qPCR was were configured following the the Mycoplasma pneumoniae detection kit (Sansure Biotech INC., China): initial denaturation at 94 °C for 5 min; 45 cycles of denaturation at 94 °C for 15 s, annealing at 57 °C for 30 s (with fluorescence acquisition). It also defined that the Tp value of sample ≤35 cycles were classified as positive and the results of all clinical samples based on qPCR were consistent with expectations. The FA-RMP is considered capable of identifying positive samples within 30 min, with Tp values greater than 30 uniformly classified as negative results. For the 12 positive samples detected by FA-RMP, all Tp values were below 20; negative samples exhibited no amplification trend. The testing results between FA-RMP and clinical standard device were 100%, which fully proved the clinical practicability of the FA-RMP.

The testing result of 16 clinical samples diagnosed by FA-RMP and the clinical standard device based on qPCR
Multiplex detection of Flu A, Flu B and MP
The clinical samples with Flu A, Flu B and MP were used to verify the ability of the developed platform for pathogens multiplexed detection. The microfluidic cartridges were prepared with sealing the microchambers and loading the nucleic acid release reagent and LAMP lyophilized beads. The LAMP primers for Flu A and Flu B were listed in the Table S1. The results displayed in Figs. S1, S2 proved the developed LAMP assays could be used to amplify M1 gene of Flu A and NA gene of Flu B with strong specificity.
After establishing the optimized conditions with the MP assay, we evaluated the ability of the system to perform simultaneous detection of multiple respiratory pathogens. The clinical samples were pretreated with the nucleic acid release reagent and then added into the microfluidic cartridge. As the results displayed in Fig. 8, We confirmed that known influenza A, influenza B, and MP samples were each detected specifically in the appropriate chamber with no cross-signal. This demonstrates the assay’s applicability to clinical specimens and its ability to multiplex targets on the chip.

a–f Show the real-time amplification curves of microfluidic cartridge chambers, in which lyophilized beads with lamp mixture were loaded. a–c Show the positive results of samples with Flu A, Flu B and MP respectively; d shows the positive result of samples with 3 pathogens. e, f Show the negative results of the samples
Conclusions
In this study, the FA-RMP, a fully automated rotary microfluidic platform that enables high-throughput multiplex respiratory pathogen detection was developed. By
Integrating on-chip samples lysis, precision fluid partitioning, and lyophilized RT-LAMP, the FA-RMP platform supports simultaneous detection of up to four distinct samples with three pathogen targets (including Mycoplasma pneumoniae, influenza A/B viruses) in under 30 min. The system demonstrated superior performance with a limit of detection down to 50 copies/μL, 100% specificity against 9 non-target pathogens. Notably, the all-in-one cartridge design eliminates the need for pre-processing or specialized equipment, enabling decentralized testing in resource-limited settings. This work provides a robust platform for rapid epidemiological surveillance and clinical decision-making, particularly in scenarios requiring simultaneous screening of co-circulating respiratory pathogens.
While the current validation focused on nasopharyngeal swabs, high-viscosity matrices such as sputum were not included. Subsequent development will include testing on such complex sample types to assess amplification delay or inhibition effects. Only a limited number of clinical specimens were analyzed in this study, reflecting the constrained availability during influenza season. Ongoing collaborations with multiple hospitals aim to increase sample size and broaden pathogen diversity. In parallel, the real-time fluorescence acquisition and modular chamber structure of FA-RMP provide a foundation for future integration of threshold-time, based quantification, and digital LAMP through reaction partitioning. In addition, the primers for other pathogens and multiplex isothermal amplification methods can be combined with FA-RMP to further enhance its detection performance and application value.
References
Kang, L. Y., Jing, W. Z., Liu, Q., Liu, J. & Liu, M. The trends of mortality, aetiologies and risk factors of lower respiratory infections in China from 1990 to 2019: findings from the Global Burden of Disease Study 2019. J. Infect. Public Health 15, 870–876 (2022).
Yu, X. R. et al. Estimating the global and regional burden of lower respiratory infections attributable to leading pathogens and the protective effectiveness of immunization programs. Int. J. Infect. Dis. 149, 107268 (2024).
Liu, Y. N. et al. Infection and co-infection patterns of community-acquired pneumonia in patients of different ages in China from 2009 to 2020: a national surveillance study. Lancet Microbe 4, E330–E339 (2023).
Leung, E. C. M. et al. Evaluation of the Xpert Xpress SARS-CoV-2/Flu/RSV assay for simultaneous detection of SARS-CoV-2, Influenza A and B viruses, and respiratory syncytial virus in nasopharyngeal specimens. J. Clin. Microbiol. 59, e02965–20 (2021).
Udugama, B. et al. Diagnosing COVID-19: the disease and tools for detection. Acs Nano 14, 3822–3835 (2020).
Schreckenberger, P. C. & McAdam, A. J. Point-counterpoint: large multiplex PCR panels should be first-line tests for detection of respiratory and intestinal pathogens. J. Clin. Microbiol. 53, 3110–3115 (2015).
Dutta, D. et al. COVID-19 diagnosis: a comprehensive review of the RT-qPCR method for detection of SARS-CoV-2. Diagnostics 12, 1503 (2022).
Juul, S. et al. Validation and advantages of using novel RT-qPCR melting curve analysis assays for the identification of SARS-CoV-2 variants. Sci. Rep. 12, 13069 (2022).
Dinh, P. H. & Seo, T. S. A comprehensive diagnostic platform leveraging voice-control feature for rapid SARS-CoV-2 detection using reverse transcription loop-mediated isothermal amplification. Sens. Actuators B Chem. 436, 137690 (2025).
Crego-Vicente, B., del Olmo, M. D., Muro, A. & Fernández-Soto, P. Multiplexing LAMP assays: a methodological review and diagnostic application. Int J. Mol. Sci. 25, 6374 (2024).
Tang, M. et al. Ultrasimple size encoded microfluidic chip for rapid simultaneous multiplex detection of DNA sequences. Biosens. Bioelectron. 253, 116172 (2024).
Fu, G. L., Li, X. J., Wang, W. H. & Hou, R. X. Multiplexed tri-mode visual outputs of immunoassay signals on a clip-magazine-assembled photothermal biosensing disk. Biosens. Bioelectron. 170, 112646 (2020).
Jin, J. L. et al. A real-time LAMP-based dual-sample microfluidic chip for rapid and simultaneous detection of multiple waterborne pathogenic bacteria from coastal waters. Anal. Methods 13, 2710–2721 (2021).
Xiao, Y. J. et al. Fully integrated and automated centrifugal microfluidic chip for point-of-care multiplexed molecular diagnostics. Biosens. Bioelectron. 255, 116240 (2024).
Wang, Z. Y. et al. A finger-driven disposable micro-platform based on isothermal amplification for the application of multiplexed and point-of-care diagnosis of tuberculosis. Biosens. Bioelectron. 195, 113663 (2022).
Dong, X. B. et al. A highly sensitive, real-time centrifugal microfluidic chip for multiplexed detection based on isothermal amplification. Talanta 268, 125319 (2024).
Lim, J. et al. TwinDemic detection: a non-enzymatic signal amplification system for on-site detection of multiple respiratory viruses. Sens Actuators B Chem. 424, 136933 (2025).
Bai, H. et al. A sample-to-answer digital microfluidic multiplexed PCR system for syndromic pathogen detection in respiratory tract infection. Lab Chip 25, 1552–1564 (2025).
Xie, R. B. et al. Rapid bacterial identification through multiplexed nucleic acid detection on a digital microfluidic platform for enhanced clinical intervention against infections. ACS Sens. 10, 2520–2530 (2025).
Zhang, D. G. Y. et al. Palm-shaped and one-step system (P-SOS) integrated with nucleic acid isothermal amplification to realize sample-to-results diagnosis of acute respiratory tract disease. Microchem. J. 212, 113480 (2025).
Kim, S. H., Lee, S. Y., Kim, U. & Oh, S. W. Diverse methods of reducing and confirming false-positive results of loop-mediated isothermal amplification assays: a review. Anal. Chim. Acta 1280, 341693 (2023).
Raddatz, B. W. et al. Development of an optimized colorimetric RT-LAMP for SARS-CoV-2 assay with enhanced procedure controls for remote diagnostics. Sci. Rep. 12, 21424 (2022).
Chandra, A., Keizerweerd, A. T., Que, Y. X. & Grisham, M. P. Loop-mediated isothermal amplification (LAMP) based detection of Colletotrichum falcatum causing red rot in sugarcane. Mol. Biol. Rep. 42, 1309–1316 (2015).
Author information
Authors and Affiliations
Contributions
Y. Deng helped analysis the importance of this study and revised the manuscript. W. Si helped design the clinical experiments. D. Zhang and A. Li designed the whole experiments, analyzed the data and wrote the manuscript. P. Ren and L. Zhang designed the device. B. Liu and S. Yu helped design the microfluidic cartridge. C. Li involved the data curation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, D., Li, A., Li, C. et al. A fully automated rotary microfluidic platform for high-throughput multiplex detection of respiratory tract pathogens. Microsyst Nanoeng 11, 186 (2025). https://doi.org/10.1038/s41378-025-01044-9
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41378-025-01044-9
