
Abstract
FOXG1 (Forkhead Box G1) is a critical transcription factor for brain development, regulating progenitor cell proliferation, neuronal migration, and cortical circuit assembly. Pathogenic FOXG1 variants lead to FOXG1 syndrome, a neurodevelopmental disorder characterized by severe brain anomalies and cognitive impairments. Despite efforts to correlate genetic variants with clinical outcomes, the precise relationship remains elusive. Here, we analyzed clinical severity and brain anomalies in 14 individuals with FOXG1 variants, investigating how these variants impact FOXG1’s properties and functions. We uncovered a strong correlation between the severity of brain anomalies in affected individuals and functional alterations of these variants. Variants with very low protein expression were associated with moderate-to-severe brain anomalies. A luciferase reporter assay was used to assess the ability of FOXG1 variants to repress COUP-TFI (NR2F1) expression-a function of FOXG1 validated through single-cell RNA-sequencing (scRNA-seq). Variants losing COUP-TFI repression ability by binding to COUP-TFI’s enhancer region consistently caused moderate-to-severe brain anomalies. Furthermore, in utero electroporation (IUE) in embryonic mouse brains was employed to study their impact on neuronal migration and differentiation. Electroporation of wild-type Foxg1 delayed neuronal migration and altered their cell fate. Remarkably, variants associated with moderate-to-severe brain anomalies impaired these functions, while those with mild brain anomalies caused partial impairment. Thus, by combining protein expression, COUP-TFI repression, and neuronal migration assays, we developed a patient stratification paradigm for predicting the severity of FOXG1 syndrome. This workflow successfully differentiated 92.3% of cases, facilitating early diagnosis and guiding future therapeutic interventions.
Similar content being viewed by others


Introduction
FOXG1, a transcription factor in the Forkhead (FOX) family, plays a crucial role in brain development [1, 2]. FOXG1 is predominantly expressed in the cerebral cortex, governing multiple developmental processes, such as neural progenitor cell proliferation, neuronal migration, and cell fate determination [3,4,5,6,7,8]. In early cortical progenitors, FOXG1 induces cell cycle re-entry, upregulates progenitor genes such as Pax6, and suppresses Cajal-Retzius cell production [1, 4, 9]. FOXG1’s specific spatial expression pattern and interaction with FGF8 are crucial for cerebral patterning [10, 11]. Additionally, FOXG1 regulates the cell fate of projection neurons by inhibiting the expression of layer VI gene Tbr1, leading to de-repression of the expression of layer V gene Ctip2, thus facilitating the transition from producing layer VI neurons to layer V neurons [12,13,14]. Later, FOXG1 downregulation in young neurons causes the de-repression of COUP-TFI (also known as NR2F1), leading to the production of layer IV sensory recipient neurons from SATB2-expressing upper-layer projection neurons [7]. These findings underscore FOXG1’s diverse roles in cerebral cortex development and the potential impacts of FOXG1 mutations.
Heterozygous variants of FOXG1 can lead to FOXG1 syndrome, a rare neurodevelopmental disorder previously described as a congenital form of Rett syndrome [15], with an estimated incidence of 1 in 30,000 [16]. Individuals with FOXG1 syndrome may exhibit microcephaly, simplified gyral patterns, underdeveloped frontal cortex, and hypogenesis or agenesis of the corpus callosum [17, 18]. They also suffer from severe intellectual disability, epilepsy, hyperkinetic movement disorders, sleep disorders, and deficits in language and motor skills [19, 20]. Genetically, FOXG1 syndrome involves de novo mutations, primarily loss-of-function (LOF) variants resulting in FOXG1 haploinsufficiency [21], while a few cases with duplication of FOXG1 may lead to elevated FOXG1 levels [22]. Pathogenic variants impact various FOXG1 protein domains, leading to diverse clinical manifestations [17,18,19,20, 23]. Specific genotypes, such as truncating variants in the N-terminus, tend to be associated with more severe clinical features and brain anomalies, while milder phenotypes are associated with missense variants in the Forkhead DNA binding domain (FBD) [18,19,20]. However, this relationship is not absolute, as certain genotypes may exhibit variable clinical and imaging characteristics [17, 19]. The genetic variability among patients poses a challenge in dissecting genotype-phenotype associations [24].
Despite advances in next-generation sequencing (NGS), determining the impact of identified variants remains challenging [25], as a healthy individual’s exome contains numerous variations compared to the standard genome. Various databases and tools, including gnomAD [26] (https://gnomad.broadinstitute.org/), SIFT [27] (https://sift.bii.a-star.edu.sg/), PolyPhen [28] (http://genetics.bwh.harvard.edu/pph2/), and VEP [29] (https://www.ensembl.org/vep), assist in identifying potential disease-causing variations, though the methodology is subject to ongoing refinement. Importantly, functional assays capable of distinguishing between benign and pathogenic variants in many genetic diseases, including FOXG1 syndrome, are still lacking.
This study investigates how pathogenic FOXG1 variants with different severities impact the properties and functions of the protein, particularly in cortical development. We uncovered a strong correlation between the severity of brain anomalies and functional alterations. Consequently, we have established a workflow of function assays for predicting brain anomalies and clinical severity resulting from FOXG1 variants.
Materials and methods
Recruitment of FOXG1 syndrome cases
We enrolled individuals with genetically confirmed FOXG1 syndrome in our hospital and integrated data from previously reported cases from the literature to investigate the impact of FOXG1 variants in different domains. Inclusion criteria were: (1) a pathogenic or likely pathogenic intragenic FOXG1 variant defined by American College of Medical Genetics and Genomics (ACMG) guidelines, and (2) comprehensive clinical descriptions with cranial MRI scans (Supplementary Fig. 1).
Clinical characterization
We used the FOXG1 syndrome clinical severity scoring system [19] to assess the impact on overall functioning and daily life systematically. The scoring system included 17 phenotypic items across four categories: somatic growth (4 items), motor and speech development (4 items, if applicable), behavior (3 items), and neurological features (6 items). Each item was rated on a 0 to 2-point scale, and the clinical severity score (CSS) was the average of these items, ranging from 0–2 [19]. Severity was classified as mild (0–0.67), moderate (0.68–1.33), and severe (1.34–2).
Neuroimaging characterization
Brain MRI images were analyzed by two independent pediatric neurologists, W.-T. Lee and L.-C. Wong, and scored using the FOXG1 brain MRI severity score (MRI SS), adapted from M. Pringsheim et al. [18] (Supplementary Table 1). This scoring system assessed critical factors like simplified gyral patterns, basal ganglia hypoplasia, enlargement of inner CSF spaces, anomalies in the corpus callosum, and frontal lobe hypoplasia. Each of these items was assigned a rating scale, with scores ranging from 0–1 or 0–3, depending on the specific item under consideration. Total scores ranged from 0–6, with severity levels categorized as normal (0), mild (1–2), moderate (3–4), and severe (5–6).
Constructs
The human FOXG1 (NM_005249.5) and mouse Foxg1 (NM_001160112.1) are highly conserved, sharing 93.74% similarity. To ensure compatibility with mouse experiments, we constructed a plasmid containing mouse Foxg1 cDNA tagged with Myc-tag at its N-terminus, based on pCMV and pCAG vectors. To create the FOXG1 syndrome-associated variants, mutagenesis was performed following the manufacturer’s instructions using QuikChange II Site-Directed Mutagenesis Kit. Sequence alignment was performed using the protein BLAST algorithm (National Institutes of Health, USA).
Western blotting
Cells were lysed using RIPA buffer containing 10% protease and sonicated. Following sonication, the lysate rotated at 4 °C for 1 h on a rotor. The supernatant was collected after centrifugation at 15,000 g for 15 min at 4 °C. Protein concentrations were determined using the BCA assay (Thermo Fisher Scientific, USA). Proteins were separated by electrophoresis on a 0.1% SDS and 10% acrylamide gel, transferred to a PVDF membrane, and probed with primary antibodies: anti-BF-1 (Takara; diluted 1:5000), anti-Myc (Thermo Fisher Scientific; diluted 1:5000), and mouse anti-GAPDH (GeneTex; diluted 1:5000). The primary antibodies were detected with horseradish peroxidase (HRP)-conjugated secondary antibodies: anti-mouse (GeneTex; diluted 1:10000) and anti-rabbit (Sigma Aldrich; diluted 1:10000). Signals were developed with ECL reagent (Millipore) and detected by Luminescence/Fluorescence Imaging system Amersham Imager 680 (GE). Protein band intensities were analyzed using ImageJ software (NIH, USA). Three independent experiments from three batches of transfected cells were performed.
Immunofluorescence staining of cultured cells
HEK293T cells (60,019), purchased from BCRC, were fixed in 4% paraformaldehyde, washed with phosphate-buffered saline (PBS), and permeabilized in 0.1% in PBST at 4 °C for 15 min, followed by blocking at room temperature for 20 min in PBS with 2% bovine serum albumin and 4% normal goat serum. Subsequently, cells were incubated for up to 2 h or overnight with primary antibodies: anti-BF-1 (Takara; diluted 1:500) for detecting FOXG1 and anti-Myc (Thermo Fisher Scientific; diluted 1:500) for detecting Myc-tag. After PBS washes twice, cells were incubated in secondary antibodies: anti-mouse (GeneTex; diluted 1:10000) and anti-rabbit (Sigma Aldrich; diluted 1:10000) for 1 h, followed by a 15-min DAPI staining. Images were acquired using a confocal microscope (Zeiss LSM700, Germany).
scRNA-seq analysis
For mouse gene profiling, we utilized our previously published scRNA-seq dataset from C57BL/6 embryonic mouse brains at embryonic day (E) 13.5 and E15.5 of mixed sex [30]. Additionally, we analyzed scRNA-seq data from CD-1 mouse embryos, also of mixed sex, spanning developmental stages E10, E12, E14, E15, E16, and E18 [31]. For human gene profiling, we employed scRNA-seq data from the future somatosensory area of the human embryonic forebrain at gestational week (GW) 18 [32], including samples of mixed sex. Analysis of single cell transcriptomics was performed using Seurat, an R-based toolkit [33]. Cells from the dorsal pallium were selected based on the expression of neocortical markers, such as EMX1 and EMX2. Cell identity, including progenitor, precursor, and neuron populations, was determined through cell cycle scoring using Seurat’s built-in functions, alongside the expression of key markers, including progenitor genes such as Pax6/PAX6, precursor genes such as Tbr2/TBR2, and neuronal genes such as Nex/NEX.
Luciferase reporter assay
The luciferase reporter assay followed the methods in previous reports [7, 34]. Briefly, the COUP-TFI gene was cloned into pGL4.1 with or without FOXG1 binding site PBS1. pRL-TK, containing the Renilla luciferase, served as an internal standard for transfection efficiency. U87-MG cells (Food Industry Research and Development Institute, Taiwan) were co-transfected with reporter plasmid (0.5 µg) and pRL-TK (0.05 µg) using Lipofetamine 3000 (Invitrogen). After 48 h, cells were harvested for luciferase activity measurement by the Dual-Luciferase Assay System (Promega). Signals were measured using TECAN 200/200Pro. Relative luciferase units (RLUs) were calculated by normalizing luciferase activity to Renilla activity, which was further normalized to cells with the reporter plasmid containing COUP-TFI promoter. All values of luciferase assay were normalized by pGL4.1, and FOXG1 variants were further normalized by WT FOXG1. Three independent experiments from three batches of transfected cells were performed.
In utero electroporation (IUE)
IUE was performed according to previous studies [34,35,36,37,38]. Briefly, Foxg1 (1 μg/μl) and GFP constructs (0.5 μg/μl) were co-injected into the lateral ventricle of embryonic brains of ICR mice (BioLASCO) at E13.75 and electroporated with a diameter 5 mm forceps electrode, which transmitted five electric pulses at 40 V for 50 ms at 1-s interval through the uterine wall by an electroporation generator (Harvard Apparatus). Brains of the electroporated embryos of mixed sex were harvested after the electroporation at E16.75 or P7 and subsequently perfused with PBS and 4% paraformaldehyde (PFA) for tissue fixation. Three independent experiments from three animals for each group were performed. The order of injection was randomized and blinded in each IUE experiment.
Immunofluorescence staining of brain slices
Brain slices were permeabilized in PBST (PBS with 0.2% Triton X-100) for 30 min. After blocking with 10% normal goat serum/5% BSA/0.2% PBST solution, slices were incubated with the primary antibody at 4 °C for 36 h. Slices were then incubated with the secondary antibody at room temperature for 2 h, and stained with DAPI for 1 h. Stained brain slices were mounted in Prolonged Gold Antifade reagent (Life Technologies, USA) and stored at 4 °C until imaging, all handled in darkness. Images were acquired using a confocal microscope (Zeiss LSM700, Germany) with 5X and 20X objectives and analyzed with ImageJ FIJI software (NIH, USA). Primary antibodies used include rabbit anti-TBR1 (ab31940, Abcam), rabbit anti-BF1 (M227, Takara), mouse anti-RORβ (N7927, R&D), and mouse anti-BRN2 (sc-393324, Santa Cruz Biotechnology).
Statistical analysis
For statistical analysis, Prism (GraphPad) was used to conduct One- and Two-way ANOVA. Dunnett’s multiple comparisons test and uncorrected Fisher’s LSD are used as post-hoc tests. Data were shown as the mean ± S.E.M. All data were obtained from 3 independent experiments. A significant difference was defined as p < 0.05.
Results
Clinical, genetic, and image characteristics in individuals with FOXG1 syndrome
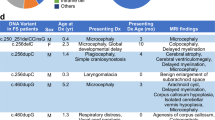
We collected clinical, brain MRI, and genetic data from 14 FOXG1 syndrome cases, with 5 from our hospital and 9 from the literature [17, 18, 39, 40]. All cases carried intragenic heterozygous FOXG1 variants, including p.Gln86AspfsTer34 (Q86Dfs*34), p.Gln86ArgfsTer106 (Q86Rfs*106), p.Glu136Ter (E136X), p.Pro182Gln (P182Q), p.Asn187Lys (N187K), p.Ile194Ser (I194S), p.Phe215Leu (F215L), p.Arg230His (R230H), p.Leu235His (L235H), p.Gly252Val (G252V), p.Gly252Asp (G252D), p.Trp255LeufsTer156 (W255Lfs*156), p.Tyr307Ter (Y307X), and p.Tyr416Ter (Y416X) (Table 1). These variants covered different FOXG1 domains (Fig. 1A), including 3 N-terminal early terminations (2 frameshift and 1 nonsense), 9 variants within or near the FBD and CS domains (8 missense and 1 frameshift), and 2 C-terminal nonsense variants (Table 1, Fig. 1A).

A Localization of the 14 FOXG1 variants analyzed in this study. Three variants lead to early termination at the N terminus, and three variants result in truncation at the C terminus, while the remaining 8 variants are missense variants within or in proximity to the CS and FBD domains. Variants causing different severities of brain anomalies are color-coded: Red for severe, green for moderate, blue for mild, and black for normal. B Protein expression of FOXG1 and its variants in transfected cells, determined by western blotting with Myc antibody. GAPDH served as the loading control. C Bar graph showing the relative expression of FOXG1 variants compared to WT FOXG1 in ascending order of expression (n = 3 independent transfections). D Bar graph presenting the relative expression of FOXG1 variants on a log2 scale (n = 3 independent transfections). Error bars: S.E.M. *p < 0.05, **p < 0.01, ****p < 0.0001. One-way ANOVA; post-hoc: Uncorrected Fisher’s LSD.
The predominant clinical manifestation was microcephaly, affecting 13 out of 14 cases. A comprehensive analysis of brain MRIs (Table 1, Supplementary Fig. 1) revealed distinct neuroimaging features, with anomalies in the corpus callosum (11/14) and a simplified gyral pattern (11/14) being the most prevalent. This was followed by hypoplasia of the frontal lobes (9/14), enlargement of inner CSF spaces (8/14), and hypoplasia of the basal ganglia relative to the thalami (7/14). Anomalies within the corpus callosum were further categorized as partial agenesis in 7 individuals and thinning in 4 individuals.
Clinical severity ranged from mild to severe, with N-terminal variants presenting moderate to severe clinical severity, while those with pathogenic variants within the FBD domain exhibited heterogeneous severity, ranging from mild to severe. Individuals with variants within the C-terminal region displayed moderate clinical severity. The brain MRI severity score, reflecting the summation of brain anomalies, also ranged from mild to severe (Table 1). Interestingly, N-terminal variants correlated with severe brain MRI anomalies, whereas FBD domain variants exhibited heterogeneous severity. Variants in the C-terminal region were linked to moderate-to-severe brain anomalies. Although there was some correlation between variant location and clinical features, they did not provide a perfect prediction of severity.
Low-expressing Foxg1 variants are linked to moderate-to-severe brain anomalies
To investigate the effects of different variants on FOXG1 expression, we cloned and transfected Myc-tagged Foxg1 (pCMV-Myc-Foxg1) into U87-MG human glioblastoma cells. The FOXG1 protein localized correctly within the nucleus (Supplementary Fig. 2). Subsequently, disease-associated variants were generated and transfected into U87-MG cells, and protein expression was analyzed after two days in culture (DIV) by western blotting (Fig. 1B). Among the nonsense variants, E136X exhibited very low protein expression, whereas Y307X and Y416X showed higher expression compared to wild-type (WT) FOXG1. For missense variants, I194S, F215L, R230H, and G252V displayed lower expression, while others (P182Q, N187K, L235H, and G252D) exhibited levels similar to or higher than WT FOXG1. All frameshift variants (Q86Dfs*34, Q86Rfs*106, W255Lfs*156) consistently displayed very low expression (Fig. 1B–D).
Protein expression levels were categorized as very low (Q86Dfs*34, Q86Rfs*106, E136X, R203H, G252V, W255Lfs*156), low (I194S, F215L), normal (P182Q, N187K, L235H), and high (G252D, Y416X, Y307X). Interestingly, variants with very low expression correlated with moderate-to-severe MRI findings, suggesting a link between reduced expression and severe phenotypes. This assay alone identified 54.5% (6/11) of cases with moderate-to-severe brain anomalies. However, due to its low correlations with the brain MRI and clinical severity scores (R2 = 0.0072 and 0.046, respectively; Supplementary Fig. 3A), additional assays are necessary for a more accurate prediction of FOXG1 syndrome severity.
Failure to repress the COUP-TFI promoter is associated with moderate-to-severe brain anomalies
Given FOXG1’s established role in repressing COUP-TFI (NR2F1) expression [7], we analyzed Foxg1 and Nr2f1 expression using our scRNA-seq data from mouse cortices at E13.5 and E15.5 [30]. Additionally, scRNA-seq data from mouse embryos spanning E10–E18 were examined [31] (Fig. 2, Supplementary Fig. 4). Cells were categorized into three main types: progenitor (i.e., radial glial cells), precursor (i.e., intermediate progenitors), and neuron, based on various cell markers, including Pax6, Emx1, Eomes, Neurod6, Tbr1, Bcl11b, Satb2, and Fabp7 (Fig. 2A) [30]. We found that, at both stages, Foxg1 was predominantly expressed in neurons, with minor expression in progenitors and precursors, while Nr2f1 showed neuron-specific expression (Fig. 2B). Interestingly, Foxg1 and Nr2f1 expression appeared mutually exclusive (Fig. 2C), consistent with Foxg1’s role in repressing Nr2f1 expression during cortical development [7]. We extended this analysis using scRNA-seq data from the human embryonic forebrain at GW18 [32], where we observed a similar mutual exclusion between FOXG1 and NR2F1, confirming the conserved nature of this regulatory mechanism between mouse and human cortical development.

A UMAP plots depicting scRNA-seq results of the cortices from mouse embryos at E13.5 and E15.5 (top); mouse embryos at E10, 12, 14, 15, 16 and 18 (middle); and human fetus at GW18 (bottom). Three developmental states (progenitors, precursors, and neurons) are identified based on their marker genes. B Heat maps illustrating the expression of Foxg1/FOXG1 (red) and COUP-TF1 (Nr2f1/NR2F1, green), revealing distinct expression patterns. Foxg1/FOXG1 and Nr2f1/NR2F1 expression exhibit mutually exclusive expression patterns. C Scatter plots of Foxg1/FOXG1 and Nr2f1/NR2F1 expression in these cells shows relatively few cells co-expressing Foxg1/FOXG1 and Nr2f1/NR2F1. D Diagram outlining the luciferase reporter assay. Luciferase expression is driven by the COUP-TFI promoter (COUPTF1pro) and its enhancer-like region PBS1. Co-transfection of Foxg1 and the reporter construct allows FOXG1 to bind to the PBS1 region, repressing the expression of downstream genes. E Verification of WT FOXG1 repression of COUP-TFI, requiring PBS1. Cells transfected with Foxg1 along with the Luciferase reporter driven by COUPTF1pro without PBS1 exhibited high luciferase activity. In contrast, the inclusion of PBS1 reduced the luciferase activity, confirming a repression function of FOXG1 (n = 3 independent transfections). F Luciferase activity of cells transfected with Foxg1 variants along with the luciferase reporter driven by the COUP-TFI promoter and PBS1. While the expression of Y416X, L235H, Y307X, P182Q, G252D, and F215L resulted in similarly low luciferase activity as WT FOXG1, the expression of other variants showed significantly higher luciferase activity, indicating impaired COUP-TFI repression function (n = 3 independent transfections). Error bars: S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001. ****p < 0.0001. One-way ANOVA; post-hoc: Uncorrected Fisher’s LSD.
To assess the impact of FOXG1 variants on the ability to repress COUP-TFI expression, we used a luciferase reporter construct with the COUP-TFI promoter and COUP-TFI enhancer PBS1 (Fig. 2D). Previous studies showed FOXG1 binding to PBS1 represses the promoter activity, downregulating COUP-TFI [7]. In this “COUP-TFI repression assay,” the reporter construct and Foxg1 cDNA were co-transfected into U87-MG cells. WT FOXG1 led to low luciferase activity, consistent with its repressor role in COUP-TFI expression (Fig. 2E) [7].
Comparing luciferase luminescence among cells expressing FOXG1 variants, 4 variants (P182Q, L235H, Y307X, and Y416X) showed low activities similar to WT FOXG1 (Fig. 2F). In contrast, G252D and F215L, associated with mild MRI severity, exhibited slightly increased activity, suggesting partial LOF. The remaining 8 FOXG1 variants, associated with moderate (E136X, N187K, I194S, R230H, G252V) or severe (Q86Dfs*34, Q86Rfs*106, W255Lfs*156) MRI severity scores, displayed elevated activity, indicating LOF in COUP-TFI repression. Notably, all 6 variants with very low protein expression exhibited significantly higher luciferase activity compared to WT FOXG1 (p < 0.05, Fig. 2F), consistent with expected LOF effects. Among other variants, only N187K and I194S showed LOF in the repression of luciferase activity, both associated with moderate MRI severity, regardless of their expression levels (N187K: normal; I194S: low). While correlations between COUP-TFI repression and the brain MRI (R2 = 0.085) and clinical severity (R2 = 0.090) scores remained low (Supplementary Fig. 3B), this additional assay increased sensitivity in detecting variants causing moderate-to-severe brain anomaly from 54.5% (6/11) to 72.7% (8/11).
Impact on neuronal migration correlates with brain MRI severity
Previously, FOXG1 overexpression in the developing mouse cortex at E13.75 delayed neuronal migration and attenuated layer IV neuronal fate [7]. To investigate how FOXG1 variants impact these functions, we used IUE to introduce constructs overexpressing WT FOXG1 or its variants, along with GFP plasmid, into neural progenitors at E13.75 (Fig. 3). Three days post-IUE, brain sections stained with TBR1 (T-Box Brain Transcription Factor 1) as a marker for the deep cortical plate (CP) showed that, in control brains electroporated with the empty vector, about half of the GFP+ cells had migrated from the ventricular zone (VZ) to the CP, with some cells in transit within the intermediate zone (IZ) and VZ. Notably, upon electroporation of WT Foxg1, most electroporated GFP+ cells were distributed in the IZ and VZ, with very few cells (<10%) reaching the CP, consistent with previous studies [7].

A Neuronal distribution of brains electroporated with Foxg1 and the pathogenic variants. Mouse brains were electroporated with GFP along with Foxg1 WT or its variants at E13.75. Neuronal cell distribution was examined 3 days after IUE in brain slices stained with the layer marker TBR1 (red) and DAPI (blue). While more than half of the cells electroporated with the empty vector migrated to the CP, FOXG1 overexpression altered neuronal migration, causing most cells to accumulate in the VZ and IZ. Cells electroporated with pathogenic Foxg1 variants displayed varying degrees of migration alteration. Bar = 100 μm. B The bar graph shows cell distributions in the VZ, IZ, and CP after IUE. C The bar graph shows cell distributions in the CP after IUE. Dashed horizontal lines indicate 10 and 25% of cells distributed in the CP. Error bars: S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001. ****p < 0.0001. Two-way ANOVA, post-hoc: Dunnett’s multiple comparisons test (n = 3 independent IUE experiments).
IUE of disease-associated FOXG1 variants showed varied effects (Fig. 3). Brains electroporated with 6 variants with very low expression (Q86Dfs*34, Q86Rfs*106, E136X, R203H, G252V, W255Lfs*156) showed many GFP+ cells in the CP (>25%), similar to brains electroporated with the vector only, indicating functional impairment. Among the two other variants showing LOF in COUP-TFI repression, I194S overexpression also resulted in > 25% of cells reaching the CP. Curiously, N187K overexpression led to very few cells reaching the CP (<10%), suggesting retained functionality in affecting neuronal migration. Of the remaining 6 variants without LOF in COUP-TFI repression, P182Q and Y307X exhibited similar percentages of cells in the CP compared to WT Foxg1 (<10% in all 3 groups, p > 0.05), suggesting relatively normal function. F215L and G252D resulted in 10–25% of cells migrating to the CP, suggesting some LOF. L235H and Y416X led to > 25% of GFP+ cells in the CP, suggesting a greater LOF. Collectively, our findings reveal a good correlation between functional impairment in these variants and the severity of brain anomalies (R2 = 0.405, Supplementary Fig. 3C). Combining these three functional assays increased the prediction rate for variants causing moderate-to-severe brain anomaly to 90.9% (10/11), except for the Y307X variant, which, despite minimal LOF in all three assays, was associated with severe brain anomalies.
Correlation between brain MRI severity and altered cell fate
To investigate the impact of FOXG1 variants on neuronal cell fate, brains after IUE at E13.75 were stained with layer-specific markers at P7, including BRN2 (Brain-specific homeobox/POU domain protein 2) for layer II/III and RORβ (Retinoic acid-related orphan receptor beta) for layer IV (Fig. 4). In control brains, most GFP+ cells reached layer IV and expressed RORβ but not BRN2. In contrast, FOXG1 overexpression shifted cells to layers II/III, V, and VI, predominantly RORβ- and BRN2+, consistent with a mutually repressive interaction between RORβ and BRN2 [41].

Mouse brains were electroporated with GFP (green) along with Foxg1 variants or the empty vector at E13.75. Brain sections at P7 were immunostained with the layer II/III marker BRN2 or layer IV marker RORβ, and counterstained with DAPI (blue). A Neurons electroporated with the empty vector were mostly BRN2- and distributed in layer IV, while FOXG1-expressing neurons were redistributed to layer II/III and expressed BRN2 (red). Neurons expressing pathogenic Foxg1 variants displayed variable effects on this fate change. The lower panels show high-magnification images with separated channels. Arrows indicate BRN2+ neurons. Bars = 100 μm. The bar graph shows the percentage of BRN2+ neurons among GFP+ cells after overexpressing Foxg1 WT and its variants. The percentage of BRN2+ cells inversely correlates with the severity of brain malformations. B Conversely, most neurons electroporated with the empty vector were RORβ+ (red), while neurons expressing FOXG1 were redistributed to layer II/III and became RORβ-. Neurons expressing pathogenic variants displayed varying effects on this fate change. Lower panels show the high-magnification images with separated channels. Arrows indicate RORβ+ neurons. Bars = 100 μm. The bar graph shows the percentage of RORβ+ among GFP+ cells after overexpressing FOXG1 WT and its variants. The percentage of RORβ+ cells correlates well with the severity of brain malformations. Error bars: S.E.M. *p < 0.05, **p < 0.01, ***p < 0.001. ****p < 0.0001. One-way ANOVA, post-hoc: Uncorrected Fisher’s LSD (n = 3 independent IUE).
Brains electroporated with Foxg1 variants causing normal (P182Q), mild (F215L), and moderate (N187K) brain anomalies showed a slight but significant reduction in BRN2+ cells compared to Foxg1 WT-electroporated brains (Fig. 4), indicating some LOF in altering neuronal differentiation. Notably, brains electroporated with variants leading to severe brain anomalies (L235H and W255Lfs*156) exhibited even greater reductions in BRN2+ cells, suggesting disrupted FOXG1 function. Conversely, brains electroporated with normal-to-moderate variants (P182Q, F215L, and N187K) showed partial LOF in directing neurons from RORβ+ to RORβ-, whereas severe variants (L235H and W255Lfs*156) lost most of this function (Fig. 4). Although we assessed only a subset of the 14 variants spanning different severity levels due to the complexity of this assay, these results suggest a correlation between the ability to alter cell fate and the severity of brain anomalies in FOXG1 syndrome.
Clinical severity prediction workflow for FOXG1 syndrome
Based on these functional assays, we developed a flowchart to predict the severity of brain anomalies resulting from FOXG1 variants (Fig. 5A). FOXG1 variants initially classified as very low expressers (<50% of WT expression) typically lead to severe (Q86Dsf*34, Q86Rfs*106, and W255Lfs*156) or moderate (E136X, R230H, and G252V) brain anomalies. This assay identified 54.5% (6/11) of moderate-to-severe cases and no (0/2) mild cases. Further refinement is achieved through a COUP-TFI repression assay. For variants with normal or high expression levels, those exhibiting impaired COUP-TFI repression ability (N187K and I194S) resulted in moderate brain anomalies, increasing the sensitivity of detecting moderate-to-severe cases from 54.5% (6/11) to 72.7 (8/11). FOXG1 variants demonstrating relatively normal COUP-TFI repression ability should undergo a further assessment of their impact on neuronal migration through IUE. In cases of WT and variants leading to normal MRI (P182Q), fewer than 10% of cells typically reach the CP. Variants leading to severe (L235H) and moderate (Y416X) brain anomalies usually allow a higher percentage of cells (>25%) to reach the CP, while mild variants (e.g., F215L and G252D) result in 10–25% of cells reaching the CP. Among these variants, only Y307X, associated with severe brain anomalies, does not align with our prediction model. Combining all three assays resulted in a sensitivity of 90.9% (10/11) for moderate-to-severe cases, 100% (2/2) for mild cases, and an overall sensitivity of 92.3% (12/13).

A The expression levels of FOXG1 variants are first examined, and variants with very low expression (<50%) tend to cause severe to moderate brain anomalies. This categorization can be further refined through the COUP-TFI repression assay. While all very low expression variants exhibit impaired repression, among variants with normal to high expression levels, those showing impaired COUP-TFI repression tend to lead to moderate brain anomalies. For FOXG1 variants demonstrating relatively normal COUP-TFI repression ability, their effects on neuronal migration and differentiation are further assessed through IUE. In cases of WT FOXG1 and variants that are associated with normal MRI, fewer than 10% of cells typically reach the CP. Variants leading to severe and moderate brain anomalies usually result in a higher percentage of cells (>25%) reaching the CP, while mild variants lead to 10–25% of cells reaching the CP. It’s worth noting that among these variants, only Y307X, which is associated with severe brain anomalies, does not align with our prediction model. #: variants that did not entirely fit the model. B PCA of FOXG1 variant severity. Data from protein expression, COUP-TFI repression, and neuronal migration assays were plotted in a three-dimensional scatter plot, with severity groups color-coded. Clustering analysis effectively differentiates normal, mild, moderate, and severe brain anomalies, with the exception of N187K and Y307X, which remain outliers.
Interestingly, there are only moderate correlations between these 3 assays (Supplementary Fig. 5), suggesting each functional assay provides valuable insights into the properties and functions of different variants. To further validate our workflow, we applied principal component analysis (PCA) to visualize the clustering of variants based on FOXG1 protein expression, COUP-TFI repression in the luciferase assay, and the percentage of cells reaching the CP in the migration assay. This analysis revealed a clear separation of normal, mild, moderate, and severe MRI phenotypes, with the exception of N187K and Y307X (Fig. 5B), reinforcing the robustness of our predictive framework.
Discussion
In this study, we investigated the impact of FOXG1 variants on brain development to predict the severity of FOXG1 syndrome. We identified a correlation between the molecular and cellular effects of FOXG1 mutations and brain malformations rather than the clinical severity score, as clinical severity may be influenced by age, broader neurological functions, and other factors beyond cortical development. By assessing clinical symptoms, brain MRI findings (Table 1), protein expression levels (Fig. 1), COUP-TFI repression (Fig. 2), neuronal migration (Fig. 3), and differentiation (Fig. 4) associated with different Foxg1 variants, we developed a workflow to predict the severity of brain anomalies (Fig. 5). This approach accurately identified 90.9% (10/11) of cases with moderate-to-severe brain anomalies and correctly distinguished all mild and normal cases (3/3). This diagnostic workflow could prove valuable for predicting the pathogenicity of newly identified FOXG1 variants through NGS during prenatal or neonatal genetic screening.
The FOXG1 variants observed in FOXG1 syndrome display diverse prognoses (Table 1) [18,19,20]. Protein expression analysis provided insights into the functional impacts of these variants (Fig. 1). FOXG1 is initially expressed in cortical progenitor cells (E8.5–E10.5) and later in neurons (E13) [4]. Once expressed, FOXG1 promotes cortical progenitor proliferation and guides their differentiation into projection neurons while orchestrating the sequential generation of deep-layer (DL) and upper-layer (UL) neurons [13]. FOXG1 functions in a spatiotemporal manner, as demonstrated by its suppression by EGR2 in early postmitotic stages, which leads to COUP-TFI upregulation and facilitates L4 neuron specification [7]. In our scRNA-seq analysis at E13.5 and E15.5, Foxg1 expression was predominantly observed in neurons, with lower levels in progenitors and precursors (Fig. 2B). This pattern, along with previous studies, suggests potential differences in the temporal dynamics of FOXG1 mRNA and protein expression across developmental timepoints. Interestingly, pathogenic variants with very low expression were generally associated with moderate to severe brain anomalies, most of which also impaired COUP-TFI repression (Fig. 2), indicating disruptions in FOXG1’s DNA binding function and its regulation of target genes. Notably, some variants with normal protein levels still impaired COUP-TFI repression, emphasizing that protein expression alone does not determine functional outcomes.
FOXG1 overexpression dramatically altered neuronal distribution (Fig. 3) and the expression of layer markers (Fig. 4), consistent with its role in regulating neuronal migration and cell fate [3]. Disease-associated Foxg1 variants with very low protein expression all resulted in cell distributions similar to those in brains electroporated with the empty vector. Among the variants with normal expression but LOF in COUP-TFI repression, I194S exhibited LOF in altering neuronal migration, whereas N187K delayed neuronal migration similar to WT FOXG1. This inconsistency may be caused by the compensatory effect of the N187K variant acting on other downstream targets critical for neuronal migration in mice. Nevertheless, N187K caused moderate brain anomalies in humans, correctly correlated with LOF in COUP-TFI repression, emphasizing the importance of the COUP-TFI repression assay. Among the variants that showed neither very low expression nor LOF in COUP-TFI repression, their ability to alter neuronal migration correlated well with the severity of brain anomalies (Figs. 3, 5). Intriguingly, the nonsense variant Y307X, associated with severe brain anomalies, showed no defects in any of these functional assays, suggesting that additional assays are needed to more accurately predict clinical severity based on functional loss.
With the increase use of NGS, whole exome sequencing (WES) is frequently employed to identify genetic causes of neurological disorders, especially when a genetic origin is suspected. Some parents opt for prenatal or neonatal NGS screening to assess genetic disorder risks, but distinguishing between benign and pathogenic variants remains a challenge. While ongoing efforts aim to refine methodologies using databases and software tools, our study proposes a cost-effective approach to predict possible pathogenic variants of FOXG1. The three assays can be performed simultaneously within a two-week timeframe, offering valuable tools for early diagnosis and treatment stratification. This workflow could also estimate the probability of brain anomalies for individuals carrying variants that do not show abnormality in all three assays (currently with a false-negative rate of 7.69%). However, the IUE assay, in particular, requires specialized expertise and equipment that may not be readily available in diagnostic laboratories. Future studies should focus on developing alternative, more clinically accessible approaches that retain the core functional insights provided by these assays while facilitating broader implementation in clinical practice.
A promising application of these assays is to stratify individuals for early treatment. A recent study demonstrated the effectiveness of a CRISPR/Cas9 system coupled with adeno-associated virus (AAV) in repairing FOXG1 variants in human cell lines, including hiPSCs [42]. This approach normalized PAX6 expression levels in developing neurons, laying the foundation for novel personalized therapies for FOXG1 syndrome. Despite challenges in applying treatments due to FOXG1’s critical role during early brain development, early prediction of potential variant effects may assist in risk-benefit assessments for early gene therapy in FOXG1 syndrome.
Study limitations
This study has several limitations that should be acknowledged. First, the sample size is small, comprising only 14 individuals with FOXG1 syndrome, which is a consequence of the disorder’s rarity. The wide age range of the participants (14–263 months) introduces variability that may influence clinical severity scores. Additionally, due to the small sample size, multiple testing corrections, such as Bonferroni or FDR, were not applied, as they would further reduce statistical power. Consequently, the correlation coefficients (R² values) between functional assays and clinical severity were relatively low. Another limitation is that only a single MRI series was available for most individuals collected from the literature, which may affect the consistency of imaging-based severity assessments. Furthermore, although we identified a strong correlation between FOXG1 variants and brain malformations, the relationship with broader clinical phenotypes-including somatic growth, motor and speech development, behavior, and neurological features-remained less defined, suggesting that FOXG1 may exert additional functions at different developmental stages. Lastly, due to cost constraints, all experiments were performed in triplicate, and while ANOVA was used for statistical analysis, larger replication studies are needed to strengthen the robustness of these findings. Despite these limitations, our proposed workflow provides a valuable framework for predicting the severity of brain anomalies in FOXG1 syndrome and may contribute to future diagnostic and therapeutic strategies.
Data availability
All data contained in this study are available from the corresponding author upon reasonable request.
References
Xuan S, Baptista CA, Balas G, Tao W, Soares VC, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–52.
Tao W, Lai E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron. 1992;8:957–66.
Miyoshi G, Fishell G. Dynamic FoxG1 expression coordinates the integration of multipolar pyramidal neuron precursors into the cortical plate. Neuron. 2012;74:1045–58.
Hanashima C, Li SC, Shen L, Lai E, Fishell G. Foxg1 suppresses early cortical cell fate. Science. 2004;303:56–59.
Kumamoto T, Toma K, Gunadi, McKenna WL, Kasukawa T, Katzman S, et al. Foxg1 coordinates the switch from nonradially to radially migrating glutamatergic subtypes in the neocortex through spatiotemporal repression. Cell Rep. 2013;3:931–45.
Cargnin F, Kwon JS, Katzman S, Chen B, Lee JW, Lee SK. FOXG1 orchestrates neocortical organization and cortico-cortical connections. Neuron. 2018;100:1083–96.e1085
Hou PS, Miyoshi G, Hanashima C. Sensory cortex wiring requires preselection of short- and long-range projection neurons through an Egr-Foxg1-COUP-TFI network. Nat Commun. 2019;10:3581.
Hanashima C, Shen L, Li SC, Lai E. Brain factor-1 controls the proliferation and differentiation of neocortical progenitor cells through independent mechanisms. J Neurosci. 2002;22:6526–36.
Manuel MN, Martynoga B, Molinek MD, Quinn JC, Kroemmer C, Mason JO, et al. The transcription factor Foxg1 regulates telencephalic progenitor proliferation cell autonomously, in part by controlling Pax6 expression levels. Neural Develop. 2011;6:1–12.
Storm EE, Garel S, Borello U, Hebert JM, Martinez S, McConnell SK, et al. Dose-dependent functions of Fgf8 in regulating telencephalic patterning centers. Development. 2006;133:1831–44.
Shimamura K, Rubenstein JL. Inductive interactions direct early regionalization of the mouse forebrain. Development. 1997;124:2709–18.
Toma K, Hanashima C. Switching modes in corticogenesis: mechanisms of neuronal subtype transitions and integration in the cerebral cortex. Front Neurosci. 2015;9:274.
Toma K, Kumamoto T, Hanashima C. The timing of upper-layer neurogenesis is conferred by sequential derepression and negative feedback from deep-layer neurons. J Neurosci. 2014;34:13259–76.
Kumamoto T, Hanashima C. Neuronal subtype specification in establishing mammalian neocortical circuits. Neurosci Res. 2014;86:37–49.
Wong LC, Singh S, Wang HP, Hsu CJ, Hu SC, Lee WT. FOXG1-related syndrome: from clinical to molecular genetics and pathogenic mechanisms. Int J Mol Sci. 2019;20:4176.
Lopez-Rivera JA, Perez-Palma E, Symonds J, Lindy AS, McKnight DA, Leu C, et al. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain. 2020;143:1099–105.
Vegas N, Cavallin M, Maillard C, Boddaert N, Toulouse J, Schaefer E, et al. Delineating <em>FOXG1</em> syndrome: from congenital microcephaly to hyperkinetic encephalopathy. Neurol Genet. 2018;4:e281.
Pringsheim M, Mitter D, Schröder S, Warthemann R, Plümacher K, Kluger G, et al. Structural brain anomalies in patients with FOXG1 syndrome and in Foxg1+/− mice. Ann Clin Transl Neurol. 2019;6:655–68.
Mitter D, Pringsheim M, Kaulisch M, Plümacher KS, Schröder S, Warthemann R, et al. FOXG1 syndrome: genotype-phenotype association in 83 patients with FOXG1 variants. Genet Med. 2018;20:98–108.
Wong LC, Huang CH, Chou WY, Hsu CJ, Tsai WC, Lee WT. The clinical and sleep manifestations in children with FOXG1 syndrome. Autism Res. 2023;16:953–66.
Shoichet SA, Kunde S-A, Viertel P, Schell-Apacik C, von Voss H, Tommerup N, et al. Haploinsufficiency of novel FOXG1B variants in a patient with severe mental retardation, brain malformations and microcephaly. Hum Genet. 2005;117:536–44.
Brunetti-Pierri N, Paciorkowski AR, Ciccone R, Mina ED, Bonaglia MC, Borgatti R, et al. Duplications of FOXG1 in 14q12 are associated with developmental epilepsy, mental retardation, and severe speech impairment. Eur J Hum Genet. 2011;19:102–7.
Brimble E, Reyes KG, Kuhathaas K, Devinsky O, Ruzhnikov MRZ, Ortiz-Gonzalez XR, et al. Expanding genotype-phenotype correlations in FOXG1 syndrome: results from a patient registry. Orphanet J Rare Dis. 2023;18:149.
Akol I, Gather F, Vogel T. Paving therapeutic avenues for FOXG1 syndrome: untangling genotypes and phenotypes from a molecular perspective. Int J Mol Sci. 2022;23:954.
Ehrhart F, Sangani NB, Curfs LMG. Current developments in the genetics of Rett and Rett-like syndrome. Curr Opin Psychiatry. 2018;31:103–8.
Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434–43.
Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–74.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, et al. The ensembl variant effect predictor. Genome Biol. 2016;17:122.
Tsai MH, Ke HC, Lin WC, Nian FS, Huang CW, Cheng HY, et al. Novel lissencephaly-associated NDEL1 variant reveals distinct roles of NDE1 and NDEL1 in nucleokinesis and human cortical malformations. Acta Neuropathol. 2024;147:13.
Ruan X, Kang B, Qi C, Lin W, Wang J, Zhang X. Progenitor cell diversity in the developing mouse neocortex. Proc Natl Acad Sci USA. 2021;118:e2018866118.
Bhaduri A, Sandoval-Espinosa C, Otero-Garcia M, Oh I, Yin R, Eze UC, et al. An atlas of cortical arealization identifies dynamic molecular signatures. Nature. 2021;598:200–4.
Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. 2018;36:411–20.
Chang CH, Zanini M, Shirvani H, Cheng JS, Yu H, Feng CH, et al. Atoh1 controls primary cilia formation to allow for SHH-triggered granule neuron progenitor proliferation. Dev Cell. 2019;48:184–99.e185
Tsai MH, Muir AM, Wang WJ, Kang YN, Yang KC, Chao NH, et al. Pathogenic variants in CEP85L cause sporadic and familial posterior predominant lissencephaly. Neuron. 2020;106:237–45.e238
Tsai MH, Cheng HY, Nian FS, Liu C, Chao NH, Chiang KL, et al. Impairment in dynein-mediated nuclear translocation by BICD2 C-terminal truncation leads to neuronal migration defect and human brain malformation. Acta Neuropathol Commun. 2020;8:106.
Huang CW, Lee KY, Lin PT, Nian FS, Cheng HY, Chang CH, et al. Muscleblind-like 2 knockout shifts adducin 1 isoform expression and alters dendritic spine dynamics of cortical neurons during brain development. Neuropathol Appl Neurobiol. 2023;49:e12890.
Chen JL, Chang CH, Tsai JW. Gli2 rescues delays in brain development induced by Kif3a dysfunction. Cereb Cortex. 2019;29:751–64.
Bahi-Buisson N, Nectoux J, Girard B, Van Esch H, De Ravel T, Boddaert N, et al. Revisiting the phenotype associated with FOXG1 mutations: two novel cases of congenital Rett variant. Neurogenetics. 2010;11:241–9.
Takahashi S, Matsumoto N, Okayama A, Suzuki N, Araki A, Okajima K, et al. FOXG1 mutations in Japanese patients with the congenital variant of Rett syndrome. Clin Genet. 2012;82:569–73.
Oishi K, Aramaki M, Nakajima K. Mutually repressive interaction between Brn1/2 and Rorb contributes to the establishment of neocortical layer 2/3 and layer 4. Proc Natl Acad Sci USA. 2016;113:3371–6.
Croci S, Carriero ML, Capitani K, Daga S, Donati F, Papa FT, et al. AAV-mediated FOXG1 gene editing in human Rett primary cells. Eur J Hum Genet. 2020;28:1446–58.
Acknowledgements
The authors would like to express their gratitude to the affected individuals and their families for participating in this study. We also appreciate Dr. Fang-Shin Nian (University of Liège) for valuable comments and insights.
Funding
This study was funded by grants from National Science and Technology Council (110-2314-B-002 -293 -MY3) to L.-C.W.; and (110-2628-B-A49A-506, 111-2628-B-A49-016, 112-2628-B-A49-005, and 112-2314-B-A49-075), National Health Research Institutes (NHRI-EX109-10904NI), and the Brain Research Center, National Yang Ming Chiao Tung University (NYCU) through the Featured Areas Research Center Program within the framework of the Higher Education Sprout Project by the Ministry of Education (MOE) to J.-W.T. The scRNA-seq was supported and performed at the Genomics Center for Clinical and Biotechnological Applications of the National Core Facility for Biopharmaceuticals (NCFB), Taiwan. Open Access funding enabled and organized by National Yang Ming Chiao Tung University.
Author information
Authors and Affiliations
Contributions
This manuscript was designed and conceptualized by T-YL, L-CW, P-SH, M-HL, W-TL, and J-WT Clinical data analysis was performed by L-CW and W-TL. Experiments were conducted and analyzed by T-YL, P-SH, C-KW, H-YC, H-JZ, C-YT, M-HL, and J-WT. Manuscript writing and editing was performed by T-YL, L-CW, P-SH, W-TL and J-WT. Manuscript was reviewed by all authors.
Corresponding authors
Ethics declarations
Ethics approval
All experiments and animal maintenance were conducted in compliance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at National Yang-Ming University. The human study received approval from the Ethical Committee of the National Taiwan University Hospital (202012180RINB), and the committee waived the requirement for informed consent.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, TY., Wong, LC., Hou, PS. et al. Functional defects in FOXG1 variants predict the severity of brain anomalies in FOXG1 syndrome. Mol Psychiatry 30, 4824–4835 (2025). https://doi.org/10.1038/s41380-025-03077-y
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41380-025-03077-y
