
Abstract
During cortical development, newly born neurons migrate radially or tangentially from their origin to expand the cortex. Simultaneously, neuron-derived factors support angiogenesis, and an elaborate network of blood cerebral vessels develops in the cortex. Traditionally, blood cerebral vessels were considered to support the growing cortex or migrating neurons by providing nutrients and oxygen. However, recent studies have shed light on Endothelial cells’ influence on cortical development; they guide neuronal migration by providing molecular cues and structural support. Here, we review the current understanding of how CNS cerebral vessels support neurogenesis, neuronal migration, and the formation of the six-layer cortical structure during development. Additionally, we explore current knowledge regarding the vascular role in neurodevelopmental disorders, including autism spectrum disorder, attention deficit hyperactivity disorder, and schizophrenia.
Similar content being viewed by others

Introduction
The human brain is complex, and while it is not the largest, it has a larger cerebral cortex than other mammals relative to its size. This difference is thought to be a critical factor in human cognitive abilities [1,2,3]. There are different theories about why some animals have a larger cerebral cortex than others. One theory is that a larger cerebral cortex allows more neurons and connections, leading to more extraordinary cognitive ability [3, 4]. However, this theory does not always hold true, as some animals with smaller brains may have more neurons than animals with larger brains [5, 6]. Another theory is that the way neurons are connected in the brain is more important than the number of neurons themselves [7,8,9]. This theory suggests that the human brain may have a more efficient way of connecting neurons, which allows us to think and learn more effectively. Researchers are still trying to understand exactly how the size and structure of the cerebral cortex relate to cognitive ability. However, it is clear that the cerebral cortex plays an important role in human intelligence.
The cerebral cortex can be divided based on laminar organization into two types: the neocortex, which is larger and has six cell layers, and the allocortex, which is much smaller and has three or four layers. In the human brain, the allocortex accounts for just 10% of the cortex, with the neocortex making up 90% [10]. The neocortex is known for its six layers, with layers I, III, and V having lower cell density than layers II, IV, and VI [11]. The neocortex is further divided into four lobes: the frontal lobe, parietal lobe, temporal lobe, and occipital lobe [12, 13]. The frontal lobe, the most prominent lobe of the cerebral cortex, is responsible for a wide range of critical functions. It governs our ability to plan and make decisions, including complex strategies in new situations [14, 15]. Additionally, it controls speech production through an area called Broca’s area [16]. Furthermore, the frontal lobe is essential for personality and emotional regulation. Damage to this area can lead to significant changes in behavior and social interactions. Finally, the frontal lobe plays a role in movement control through its motor cortex. In contrast, the parietal lobe focuses on processing sensory information regarding touch, position, and pain [17]. It also integrates this information with other senses like vision and hearing, allowing for complex functions like spatial recognition and object manipulation [18,19,20]. The temporal lobe deals primarily with hearing and auditory processing [21, 22]. It also includes Wernicke’s area, which is crucial for understanding language [23, 24]. Beyond that, the temporal lobe is involved in memory, particularly long-term memories related to facts and events [declarative memory]. Finally, the occipital lobe, the smallest of the four, is responsible for vision [25]. It receives and interprets raw visual data, allowing us to recognize and distinguish objects in the world. The allocortex has three or four layers and includes structures such as the hippocampus and olfactory cortex. It is involved in more primitive functions, including memory formation and olfaction [26].
The intricate development and function of the brain’s complex cortical structure have long fascinated scientists. Despite constituting only 2% of total body mass, the human brain consumes approximately 20% of the body’s resting energy [27]. This substantial energy demand is sustained by an extensive vascular network, which continuously supplies oxygen and nutrients to neurons while facilitating the removal of metabolic waste products. By 4–8 weeks of gestation, blood vessels from the perineural vascular plexus [PNVP) begin to penetrate the human neural tube [28], a process that parallels vascular invasion in mice around embryonic day (E) 9.5–11.5 [29]. This temporal alignment underscores the intricate interdependence between vascular and neural development, as the expanding central nervous system (CNS) relies on an emerging vascular network for oxygen and nutrient supply [30, 31].
The relationship between the vascular and CNS systems in the contexts of neurogenesis, and neurodevelopmental disorders has been reviewed separately [32,33,34,35,36,37,38,39,40]. However, comprehensive reviews that specifically address vascular contributions to CNS development, particularly in relation to neurogenesis, neuronal migration [both radial and tangential), the establishment of cortical architecture, and their roles in neurodevelopmental disorders, are not consolidated in a single source. This review aims to fill this gap. By collecting current knowledge on vascular influences in neurogenesis and cortical organization, this work highlights key mechanistic insights and potential research directions, ultimately seeking to encourage further research into neurodevelopmental disorders through the lens of vascular–neural interactions during development.
Cortical development
An in-depth discussion of this topic is beyond the scope of the present review, as this topic has been thoroughly reviewed previously [39, 41,42,43,44,45,46,47,48,49]; however, for continuity and readability, we will briefly discuss the process of cortical development.
The process of cortical development, including neural tube formation, neurogenesis, and neuronal migration, is evolutionarily conserved across mammals, with humans exhibiting similar fundamental mechanisms that govern the formation and organization of the cerebral cortex. During early embryonic development, a flat sheet of cells called the neural plate folds inwards to form a hollow tube, the neural tube. This closure process is crucial for brain and spinal cord development [50,51,52,53]. Even before complete closure, the front end of the neural tube bulges into three distinct regions. The forebrain will eventually give rise to the cerebral cortex, thalamus, hypothalamus, and other important structures [54,55,56]. The midbrain region will develop into parts involved in vision, hearing, and motor control [57, 58]. The hindbrain will become the pons, medulla oblongata, and cerebellum, responsible for functions like breathing, heart rate, and balance [57, 59,60,61]. The forebrain further subdivides into secondary vesicles, including the telencephalon. The telencephalon itself has two main parts: the dorsal telencephalon, which is the key region for cortical development [62] and will eventually become the convoluted outer layer of the brain we know as the cerebral cortex, and the ventral telencephalon, which gives rise to other important structures like the basal ganglia and the amygdala.
Before the onset of neurogenesis, the neuroepithelium is composed of neuroepithelial stem cells that undergo symmetric divisions that expand the neuroepithelial surface [63,64,65]. The development of neocortex circuitry relies on evolutionarily conserved and stereotypical histogenic processes [66]. These events include neural stem cell proliferation, the production of neurons and glia, neuronal migration, neuronal differentiation (both molecularly and structurally), axon pathfinding and target innervation, synaptogenesis, and maturation [66].
Neuronal migration during cortical development is a complex process essential for organizing the layers of the cerebral cortex. Here, special cells called radial glial cells (RGCs) take center stage [41, 64]. They first divide rapidly, then switch to an asymmetric division, creating one new RGC and one new projection neuron [67,68,69,70]. The first neurons born in the germinal ventricular zone (VZ) embark on a journey towards the surface of the developing brain, forming a primordial layer called the preplate at about E11.5 [71,72,73]. This initial layer is subsequently split by the arrival of a wave of new neurons (E13), creating the superficial marginal zone (future layer I) and the deeper subplate [74,75,76]. In the generation of layers II–VI, interestingly, cortical neuron generation follows an “inside-out” sequence. Early-born neurons move to reside in the deeper layers, while later-born ones climb upwards past their younger siblings to settle in more superficial layers [77]. The subplate, separated from the VZ by the intermediate zone (future white matter), witnesses another key development - the emergence of the subventricular zone (SVZ) between itself and the VZ [45]. This zone acts as a secondary proliferative region, primarily generating glial cells to support the developing cortex. The SVZ expands significantly in late embryonic and early postnatal stages as the VZ diminishes.
The asymmetric division of radial glial stem cells enhances the density of neuronal progenitors in the VZ in humans, and later, these cells migrate rapidly to form the SVZ [78]. These cells have the potential to divide and migrate into the cortex. Based on the localizsed density of cell layers, the SVZ is distributed into two zones: the inner SVZ (iSVZ) and the outer SVZ (oSVZ) in humans and primates [79, 80]. In humans, the SVZ develops between gestational weeks (GW) 12 and 16 and is organized into two distinct regions: the iSVZ and the oSVZs, with the oSVZ playing a critical role in cortical expansion and size determination during brain development [81]. While mice primarily possess a single type of radial glia [82], humans exhibit three primary subtypes: apical radial glia (aRG), located in the VZ, which extend processes from the ventricular surface to the pial surface and undergo asymmetric divisions to generate neurons and intermediate progenitor cells [83]; truncated radial glia (tRG), derived from aRG, characterized by shorter processes that do not span the entire cortical wall and contribute to gliogenesis during later stages of cortical development [84]; and outer radial glia (oRG), also known as basal radial glia, situated in the oSVZ, which possess a long basal fiber that often extends to the pial surface but lacks an apical attachment to the ventricular surface [82, 85]. Within the oSVZ, oRG cells exhibit random cleavage patterns and divide asymmetrically to produce a variety of neuronal cell types, significantly contributing to cortical expansion and size determination [81]. Gyrogenesis, the process of cortical folding, occurs in humans during the third trimester, driven by differential growth rates between the cortex and subcortical structures, increased neuronal production, and cytoskeletal dynamics [86], whereas this folding is absent in mice due to their smaller brain size and simpler developmental program [87].
Generally, two types of migration have been identified in developing brains: radial glial migration, in which the neural progenitors (NPs) migrate out of the origin to their final location using the glial cell in the developing brain that serves as both a scaffold and a guide for migrating neurons. They extend long processes from the VZ to the pial surface, providing a pathway along which neurons migrate radially from the VZ to the outer layers of the cerebral cortex [83, 88, 89]. Another type of migration is tangential migration, where interneurons born in the ganglionic eminence migrate tangentially to the dorsal cortex [41].
Vascular development in the cortex
In the developing mouse brain, the vascular network emerges through a tightly orchestrated interplay of cellular precursors and molecular cues, beginning with the hemangioblast a pluripotent mesodermal progenitor arising in the yolk sac around E 6.5–7.5 [90]. These cells, derived from embryonic stem cells, differentiate peripherally into angioblasts endothelial precursors while centrally giving rise to hematopoietic cells [91, 92]. Vasculogenesis, the de novo formation of blood vessels, commences as angioblasts, induced by endoderm-derived signals such as fibroblast growth factor (FGF) and bone morphogenetic protein 4 (BMP4), migrate and aggregate into blood islands within the mesoderm [91, 93]. By E8.5, angiogenic factors like vascular endothelial growth factor (VEGF), secreted by the dorsal ectoderm of the newly formed neural tube, recruit fetal liver kinase 1 (FLK1)-positive angioblasts from the pre-somitic mesoderm to establish the PNVP encircling the neural tube [94], forming the primary vascular scaffold before cardiac function begins. This extracerebral plexus, established via vasculogenesis, precedes the brain-specific vascularization [95].
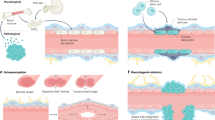
A day after the PNVP forms, cerebral vessels sprout into the neural tube at E9.5, forming the intraneural vascular plexus (INVP) [94]. From E10.5 onward, vascular sprouts from the pial surface invade the neuroepithelium, extending toward the ventricles to form a complex network The prevailing notion for CNS angiogenesis posited that pial cerebral vessels, derived from the perineural plexus encircling the neural tube, undergo passive sprouting into the brain parenchyma [96]. These cerebral vessels were thought to extend radial branches towards the ventricles, where the neurogenic zones, the VZ, and SVZ, are situated. Surface pial cerebral vessels surround the entire brain by E9.5. During E10-E11, these cerebral vessels progressively perforate into the developing brain as the periventricular vascular plexus advances into the dorsal telencephalon [97] Fig. 1.

A Pial cerebral cerebral vessels or perineural vascular plexus (PNVP) formation begins at embryonic day (E)10.5, followed by its sprouting in the developing cortex by E12.5. Moreover, by this time, a periventricular vascular plexus originates in the ventral telencephalon. At E15.5, the angiogenic sprouting completes and spreads throughout the CNS; at 17.5, these angiogenic cerebral cerebral vessels elongate and stabilize. B Simultaneously, by E10.5, the radial glial cells (neural stem cells raised from the neuroepithelial cells) divide symmetrically to increase the pool of these radial glial cells in the avascular, hypoxic zone. Later, these radial glial cells divide asymmetrically to form 1 basal progenitor and 1 neuron or 1 intermediate progenitor (E12.5). Gradually, radial cells form layers of neurons by E15.5. By E17.5, the remaining layers of neurons are formed and possess astrocytes in the developing cortex. The Cajal-al-Retzius cells could be seen at the early embryonic stage (E10.5) and gradually expand over the later developmental stages. These cells support the radial glial structure and assist neuronal migration during cortical development. Interneurons migrate along two tangential routes, located in the marginal zone (MZ) and above the subventricular zone (SVZ). As development progresses, interneurons undergo a transition to a radial-oriented migration pattern to reach their final laminar positions and integrate into neuronal circuits alongside excitatory neurons.
This notion was challenged by studies suggesting that pial and periventricular cerebral vessels originate differently and follow separate developmental timelines. Periventricular cerebral vessels in the ventral telencephalon are believed to stem from a basal cerebral vessel, likely originating from pharyngeal arch arteries, located on the telencephalic vesicle’s floor within the basal ganglia primordium [98]. By E9, pial cerebral vessels begin to encircle the telencephalon, while a distinct group of periventricular cerebral vessels is confined to the ventral telencephalon. Between E9-E10, the basal cerebral vessel matures, giving rise to periventricular branches that extend ventrally to dorsally and laterally to medially, eventually forming a vascular lattice in the dorsal telencephalon [96, 97].
Vascular quantification during embryonic brain development revealed that cerebral vessel density in the cortical plate first rises sharply from E13.5 to E15.5 but stays relatively stable after E15.5 [99]. The same study reports that during the early stages of embryonic development, there is a significant increase in cerebral vessel branch point frequency from E13.5 to E15.5 and E16.5, indicating that sprouting is a predominant growth mode. However, by E17.5, the branch point frequency decreases significantly. Thus, it is suggested that the later stages of vascular development in the cortex primarily involve elongating and stabilizing existing cerebral vessels [99].
During embryonic development, human cranial vasculature initiates around 21 days post-fertilization, prior to the primordial heart tube beating (approximately 22–23 days) and the initiation of neural tube closure [95]. Light and electron microscopy studies of human embryos suggest that microvasculature is present within the neural tube as early as the fifth GW [100]. Angiogenesis begins first in the caudal portions of the midbrain and brainstem, progressing rostrally to include cortical vascular invasion by the sixth to seventh GWs (Gestation Week) [95]. By approximately 52 days, all components of the Circle of Willis are readily distinguishable [95]. At the early stage, PNVP forms around the neural tube, serving as the initial vascular network from which angiogenesis proceeds. By the fourth week of development, the brain is supplied by the internal carotid and basilar arteries, marking the establishment of major arterial trunks [101]. By the seventh week, the cerebral hemispheres begin to be vascularized [102], ensuring that the expanding brain tissue receives adequate oxygen and nutrients. A more detailed summary of human embryonic vascular development has recently been reviewed by Boutom et al.; Bertulli and Robert. [95, 103].
Vascular Roles in the CNS
The cerebral vessels occupy around 20–30% of the total brain volume [104]. This complex, dense network of vessels’ primary function is to supply nutrients and oxygen to neurons, glial cells, astrocytes, and other non-neural cells during development and maintenance [105, 106]. They are vital for transporting oxygenated blood and essential nutrients to the brain and other tissues [104, 105]. Additionally, they eliminate carbon dioxide (CO2) and other metabolic byproducts or wastes from these tissues and transport the hormones and signaling molecules among the organs and tissues. The microvasculature of the parenchymal drains the deoxygenated blood centrifugally from the surface of the ventricular ependymal layer towards the pial at the marginal cortex through venules and veins [104]. To gain a deeper understanding of the vascular role in oxygen and nutrient support, we recommend consulting review articles [38, 94, 107,108,109].
Blood-Brain Barrier (BBB)
CNS vessels are distinct from peripheral vessels due to the presence of a semi-permeable barrier, the BBB, which restricts the entry of toxins, pathogens, large biomolecules, and many drugs into the brain [104, 105]. The BBB is primarily formed by endothelial cells (ECs) lining the cerebral microvasculature, which are supported by astrocytic end-feet and pericytes [110]. These endothelial cells, which form the luminal lining of blood vessels, are connected by a network of adherens junctions, gap junctions, and tight junctions [111]. Adherens junctions, mediated by vascular endothelial (VE) cadherins and associated scaffolding proteins such as α-, β-, and p120-catenin, connect the cells to one another and anchor them to the actin cytoskeleton, thereby reinforcing cell-to-cell adhesion and maintaining the integrity of tight junctions. Gap junctions, composed of connexins like Cx40 and Cx43, facilitate intercellular communication among endothelial cells, pericytes, and astrocytes, which is essential for coordinated vascular function [111].
Tight junctions are critical for controlling paracellular permeability across the BBB. These junctions are formed by transmembrane proteins such as claudins, occludin, and zonula occludens (ZO) proteins, which regulate the diffusion of substances between cells [112]. Claudin-5 is a key component of the tight junctions in the BBB, with its mRNA levels reported to be significantly higher than those of other claudin isoforms found in brain microvessels [113]. The PDZ domain at the C-terminus of claudin-5 interacts with ZO-1 and ZO-2, which assists in anchoring claudin-5 at the tight junctions [114]. Occludin, which is highly expressed in CNS ECs compared to non-neuronal tissues, plays a regulatory role in modulating paracellular transport and is closely associated with claudin-based strands [115]. The ZO proteins (ZO-1, ZO-2, and ZO-3) belong to the membrane-associated guanylate kinase (MAGUK) family and are essential for the assembly and stabilization of the tight junction complex [116]. Specifically, ZO-1, with a molecular weight of 225 kDa, interacts with claudins, occludin, and junction adhesion molecules (JAMs), and its dimerization with ZO-2 provides the structural framework necessary for the polymerization of claudin strands [111].
Matrix metalloproteinases (MMPs) have a key role in the regulation of BBB integrity, function, and remodeling under physiological and pathological conditions [111]. Specifically, the zinc-dependent endopeptidases MMP-2 and MMP-9 are accountable for the proteolytic degradation of tight junction proteins occludin, claudins, and ZO-1, which are essential for BBB integrity [117]. Pathologically, particularly under oxidative stress conditions, reactive oxygen species (ROS) function to activate MMPs, and accessory factors such as hypoxia-inducible factor-1α (HIF1α) and amyloid-β enhance their activity in the central nervous system [111]. This proteolytic breakdown of BBB proteins by MMP-2 and MMP-9 is a critical mechanism that culminates in BBB disruption and ensuing cerebral dysfunction [111].
Even though tight junctions restrict paracellular movements, the necessary molecular transfer is facilitated by specialized transporters. Key transporters include Glucose Transporter 1 (GLUT1) for glucose, L-type Amino Acid Transporter 1 (LAT1) for essential amino acids, P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP) for drug efflux, Organic Anion Transporting Polypeptides (OATPs) for organic anions, and Major Facilitator Superfamily Domain-containing Protein 2a (Mfsd2a) for Docosahexaenoic Acid (DHA) uptake [118,119,120,121]. These transporters ensure essential nutrients enter the brain while keeping harmful substances out. Alternatively, substances can traverse the barrier by transcytosis, this mechanism involves the encapsulation of molecules into endocytic vesicles that move across the endothelial cell and release their contents on the opposite side [122, 123]. However, compared to peripheral ECs, CNS ECs possess relatively low transcytosis [105, 124].
In mice, BBB maturation occurs around mouse E15.5, as indicated by the restricted permeability of a 10 kDa tracer in the developing brain [125]. BBB formation is tightly regulated by neural developmental programs, with Wnt ligands (Wnt7a, Wnt7b) secreted by NPs activating β-catenin signaling in ECs to drive barrier establishment [126]. Additionally, subventricular neuroectodermal cells produce VEGF, directing angiogenesis through a VEGF concentration gradient [127]. Impaired neural-derived VEGF disrupts vessel density and leads to structural malformations, particularly in the cortex and retina [127]. Astrocytes secrete Sonic Hedgehog (Shh), while BBB ECsexpress Hedgehog (Hh) receptors, collectively promoting BBB formation and maintaining its integrity throughout embryonic development and adulthood [128].
In human embryos, research utilizing electron microscopy, horseradish peroxidase tracing, and advanced freeze-fracture techniques indicates that the BBB effectively restricts proteins and large molecules from the earliest developmental stages [129]. In the human telencephalon, BBB formation starts early in prenatal life, with critical structural and molecular characteristics appearing between 12 and 18 weeks of gestation [130]. Analysis of the cortical plate microvasculature through immunohistochemistry and electron microscopy shows that the BBB-specific glucose transporter GLUT1 is present at both time points, mainly along the abluminal and lateral surfaces of endothelial cells. At 12 weeks, ECs are linked by short junctions featuring discrete membrane fusion sites, whereas by 18 weeks, these evolve into elaborate tight junctions [130]. Tight junction proteins, such as occludin and claudin-5, are observed in ECs at 12 weeks, initially distributed in the cytoplasm before relocating to cell boundaries, where they form incomplete, patchy junctions [131]. By 18 weeks, these tight junctions develop into sophisticated, nearly continuous networks, closely resembling those of the adult BBB [131]. Nevertheless, some research suggests that the overall levels of key transmembrane tight junction proteins (occludin, claudin-5, and JAM-1) remain relatively stable between 16 and 40 weeks of gestation, showing no marked increase in expression over this period [132].
Neurovascular coupling
Another important function of the blood vessels is involved in neurovascular coupling that is regulated in terms of neuronal activity. The mechanism by which neuronal activity regulates the cerebral blood flow (CBF) for using oxygen is known as neurovascular coupling [133]. CBF, blood oxygen level dependents (BOLD), glucose, and regulatory molecules are crucial for assessing defects in neurovascular coupling. Several studies use functional Magnetic resonance imaging (MRI) to understand the effect of neuronal function by CBF in various CNS diseases via measuring the BOLD signals [109, 134,135,136,137,138,139,140]. The components of the neurovascular unit, including neurons, astrocytes, BBB, ECs, and pericytes control the cerebral blood for maintaining neuronal activity [141]. Molecular signaling such as neuronal Nitric Oxide Synthase (nNOS), glutamate and N-methyl-D-aspartate (NMDA) interaction in neurons, and glutamate and prostaglandin crosstalk are known to regulate neurovascular coupling [142,143,144]. This is required to fulfill the growing demands of oxygen which is always higher than the demands for proper neuronal function in the brain, known as hyperemia. Moreover, it is associated with the waste-removal process of the brain.
Vascular contribution to neurogenesis
The transition of proliferative and multipotent neural stem cells (NSCs) into fully differentiated neurons is called neurogenesis. In mice, cortical neurogenesis begins around E 9–10, where neuroepithelial cells begin to acquire features associated with RGCs and continue until birth, primarily within the VZ and SVZ of the developing brain [145]. In humans, cortical neurogenesis follows a similar but more prolonged timeline, starting around GW 7 and continuing until mid-gestation [146]. Even though neurogenesis and angiogenesis share the same developmental timeline, little is known about how cerebral vessels influence embryonic neurogenesis.
The early developed VZ is avascular, where neural epithelial cells transform into apical radial glial cells (aRGCs). These aRGCs divide symmetrically and proliferate, increasing their density in this hypoxic, avascular niche [147]. This hypoxic zone induces the expression of hypoxia-inducible factor-1α (HIF-1α) in NSCs or aRGCs and elevates the expression of VEGF, which attracts cerebral vessel growth toward them [147, 148]. In both mice and humans, nascent blood vessels in the embryonic brain extend filopodia toward the ventricular surface to establish contact with apical progenitors. Experimental enhancement of filopodia density through endothelial cell-specific KO of sphingosine-1-phosphate receptor (S1P1) or Recombination Signal Binding Protein for Immunoglobulin Kappa J Region (RBPJ) (a Notch transcription factor) prolongs NP mitosis and promotes neuronal differentiation. Additionally, VEGF-A further induces filopodia growth [149]. However, VEGF is also reported to promote neurogenesis. Angiogenesis alleviates hypoxia in the neurogenic zone and promotes neural stem cell differentiation [150]. Furthermore, when vascular development is disrupted during CNS development, cortical NSC expansion increases at the expense of NSC differentiation. This switch is controlled by HIF-1α in NSCs [38].
Through co-culture experiments with endothelial and perivascular cells and both embryonic and adult NSCs, it has been reported that NSCs respond to soluble factors released by ECs [151]. These factors promote the self-renewal of NSCs, enabling extensive production of projection neurons and interneurons in vitro. However, this interaction inhibits differentiation, as newly produced stem cells differentiate only in the absence of ECs [151]; this result contradicted Lange et al. [150], and Chou et al. [152], where angiogenesis or EC co-culture promotes NSC differentiation [150, 152]. The contradiction in the studies likely stems from context-dependent roles. Shen et al. use the mouse embryonic stem cells and adult endothelial cell lines, which are not in the developmental stage [153]. In contrast, Lange et al. [150] use in vivo studies to show that the release of hypoxia by angiogenesis triggers the differentiation of NSCs [150]. Chou et al. [152] were using adult human brain ECs with human NSCs [152]. This discrepancy may also be due to differences in experimental conditions, models, or signaling environments. Additionally, vascular smooth muscle cells do not support this self-renewal and differentiation process [153].
Many studies indicate that vascular ECs enhance neuronal proliferation, neurite outgrowth, synaptic activity, and increased action potential and neuronal functions [154,155,156,157]. These functions are largely attributed to the signaling molecules secreted by the vascular ECs, such as the VEGF, Brain-Derived Neurotrophic Factor (BDNF), Nitric oxide, artemin, and endothelin [156, 158, 159]. The co-culture experiments of the ECs and the neurons demonstrated increased neuron generation by the secretory molecules Notch-1 and Hes1 in embryonic brain ECs [153]. Ex-vivo results prove enhanced neurogenesis and neuronal migration while studying the co-cultured subependymal zone explants [156, 160]. Additionally, the interaction of brain ECs and the neurons increased the action potential of the neurons and the neuronal pre-Vesicular Glutamate Transporter 1 and postsynaptic activity (Postsynaptic Density Protein 95) in the co-culture [156].
Vascularization of brain organoids
The derivation of region-specific neural organoids from human pluripotent stem cells has greatly enhanced in vitro modeling of the CNS, closely recapitulating the unique neuronal and glial cell types that are hallmarks of different brain regions including the forebrain, hippocampus, thalamus, retina, midbrain, cerebellum, choroid plexus, and spinal cord [95]. Yet one of the biggest limitations of these 3D structures is the lack of a functional vascular system, limiting their growth because of hypoxia-induced cell death with HIF-1α-positive centers but also diminishing their physiological relevance [161].
To overcome these challenges, various bioengineering approaches have been established to incorporate ECs together with other members of the neurovascular unit (NVU) into organoids. Strategies such as physical co-culture with ECs, co-derivation of ECs from organoids [162], and angiogenesis induction from pre-patterned vascular networks have shown encouraging results. For example, co-culturing human pluripotent stem cells (hPSCs) with human umbilical vein endothelial cells (HUVECs) produces cortical organoids that contain vascular networks with a viability of over 200 days and are able to anastomose with host vasculature upon transplantation into the mouse cortex [157]. In line with this, culturing cerebral organoids in Matrigel with hPSC-derived ECs was found to vascularize into a cluster of differentiation (CD)-31 positive vessels surrounding VZ/SVZ-like structures upon transplantation(157]. Despite these advances, the vascular plexuses developed in vitro do not generally reproduce the in vivo pattern of CNS vascularization, where angiogenesis is initiated from the PNVP and develops inward.
State-of-the-art methods have attempted to enhance vascular imitation by introducing (NVU) stromal components—pericytes and microglia—through assembloid models or genetic reprogramming techniques. Assembloids created through the fusion of neural spheroids with mesodermal aggregates or hPSC-derived endothelial and stromal cell spheroids not only improve vascularization and microglial infiltration but also trigger expression of BBB-specific genes GLUT-1, claudin-5, and ZO-1 [163]. At the same time, co-derivation methods with VEGF-directed EC differentiation [164] or with ETV2 reprogramming generate organoids containing perfusable vascular networks with BBB-like features, including the development of tight junctions and the presence of active efflux transporter activity [165].
Transplantation of vascularized organoids into adult mice has demonstrated the organization of intact NVU elements, including a developed basement membrane and astrocytic localization of water channels like aquaporin-4 and Kir4.1 [166], showing the potential role of astrocytes in vascular maturation. Furthermore, vascularized organoids demonstrate activation of Wnt signaling, a critical pathway for angiogenesis in the CNS, and expression of a number of BBB-forming genes such as VE-cadherin, occludin, and claudin-5 [166]. The vascularized constructs are also found to exhibit enhanced neuronal proliferation with greater expression of Paired Box 6 (Pax6) and phosphorylated Vimentin (p-VIM), and migratory neuron markers Doublecortin (DCX) and early cortical neuron markers T-Box Brain Transcription Factor 1 (TBR1) compared to non-vascularized organoids [167]. Current methodologies enable the engineering of vascular-like tissue in brain organoids that replicates important features of the BBB and NVU, they nonetheless fail to fully reproduce the anatomical and functional complexity of CNS vasculature in vivo.
Vascular contribution to radial glial migration
Even though several studies support the role of radial glia in angiogenesis and vascular patterning in the cortex [99, 168, 169], research on the direct influence of blood cerebral vessels on radial glia-guided neuronal migration has yet to be explored. Two distinct progenitor cell types, radial glia and intermediate progenitors, participate in cortical neurogenesis. Radial glia, bipolar cells with radial fibers that extend to the pial surface, divide asymmetrically at the ventricular surface with vertical cleavage planes to produce a radial glial cell and either a neuron (direct neurogenesis) or an intermediate progenitor cell [170]. Intermediate progenitor cells are multipolar cells, express the T-box transcription factor 2 (TBR2), and migrate from the ventricular surface to undergo symmetrical divisions [170]. Through a series of confocal imaging in the embryonic brain at E13 and E14, the study showed that intermediate progenitor cells are spatially and temporally associated with blood cerebral vessels in the embryonic cortex during neurogenesis. The appearance of TBR2 cells correlates with the appearance of vascularization, and TBR2 cells are aligned with the honeycomb pattern of the SVZ vascular plexus when they are at the ventricular surface [170].
Reelin-Disabled-1 (Dab1) signaling in NPs is important for cortical lamination [171]. A recent study investigated this signaling pathway’s role in ECs. Compromising endothelial Dab1 during early embryonic development results in poorly defined cortical layers and misplaced cells in the marginal zone. TBR1 and CUX1-positive cells migrated past their respective layers toward the marginal zone in the E17.5 brain [172]. However, endothelial Dab1 deletion didn’t affect neurogenesis or the intermediate progenitor cell positioning, suggesting a functional interaction with postmitotic neurons. Postnatal deletion of Dab1 causes post-mitotic neurons to migrate to layer I. Furthermore, more CUX1-positive cells were observed in layer IV of Postnatal P8 brains [172]. Together, these studies suggest that projection neurons stay close to the angiogenic cerebral vessels in a developing cortex, and the cerebral vessels can provide instructive and permissive cues for radial glial-supported neuronal migration and cortical lamination.
Vascular contribution to tangential migration
The cortical interneurons originate from multiple progenitor pools within the ventral telencephalon subpallium [173]. Distinct proliferative regions in this area include the medial ganglionic eminence (MGE), which gives rise to parvalbumin (PV)-expressing cortical interneurons, somatostatin (SST)-expressing cortical interneurons, and the projection neurons of the globus pallidus (GP); the caudal ganglionic eminence (CGE), which gives rise to vasoactive intestinal peptide (VIP)-expressing cortical interneurons and Reelin-expressing cortical interneurons; and the lateral ganglionic eminence (LGE), which gives rise to olfactory bulb interneurons and the medium spiny projection neurons of the striatum [173].
Unlike projection neurons, cortical interneurons must travel a long tangential route to integrate into their final laminar allocation in the cortex [174]. During this journey, interneurons follow distinct migratory routes that converge in the dorsal cerebral cortex. Using Glutamic Acid Decarboxylase 65 (GAD65) -GFP reporter mice, it has been demonstrated that the dual gamma-aminobutyric acid (GABA) stream formation occurs deep within the ventral telencephalon. Interestingly, the vascular network in the cortex forms a day ahead of interneuron migration, and they share a common path [29]. The interneuron stream splits into two streams in the ventral telencephalon, with each stream entering one of two corridors created by the pial and periventricular vascular networks. Additionally, periventricular ECs express the GABAA receptor Gabrb3, while Pial ECs express Gabra2 and use secreted levels of GABA as a molecular cue to guide the two streams of interneuron migration [175]. EC-specific deletion of Gabrb3 and vesicular GABA transporter (Vgat) results in defective angiogenesis, indicating defective migration of interneurons to the cortex [175]. Another recent study in neonate mice reports that glutamate controls the cerebral vessel-associated migration of interneurons. Stimulation of glutamate results in the tissue plasminogen activator (t-pa) and mmp-9 activity in the pial migratory route, which is shown to be mediated via the N-methyl-D-aspartate receptor (NMDAR) mechanism [176]. Further deletion of the glutamate ionotropic receptor NMDA type subunit 1 (Grin1) results in a reduction in the population of interneurons in the superficial cortical layer [176].
VEGFs and their receptors (VEGFRs) play crucial roles in regulating vasculogenesis and angiogenesis [177]. Neuronal progenitors in the VZ are considered a source of VEGF(147). Deletion of Vegf from neuronal progenitors leads to impaired angiogenesis. This highlights the importance of endothelial Vegf in interneuron migration [159]. Embryonic deletion of endothelial Vegfa results in reduced migration of GABA neurons to the cortex [154].
In another study, the ubiquitous expression of the Vegfa120 isoform was used to disrupt its signaling gradient. Despite an aberrant vascular network, interneurons still managed to reach the dorsal cortex during mid-phases of corticogenesis [178]. These changes were not attributed to altered medial ganglionic eminence (MGE) progenitor proliferation or changes in their viability. Further confirming the findings of Li et al. [154], endothelial deletion of Vegfa alters cortical interneuron numbers, their intracortical distribution, and their spatial proximity to cerebral blood vessels [154]. This suggests that interneurons can respond to chemotropic sources of Vegfa.
Emphasizing the close association of cerebral vessels and interneuron migration, an increase in vascularization of the MGE and the underlying mantle region was observed during the period when mouse interneuron migration was initiated. In the embryonic mouse brain, vascular density was modulated by manipulating APC Down-Regulated 1 (Apcdd1), a negative regulator of Wnt/β-catenin signaling. Loss of function (LOF) of Apcdd1 resulted in reduced vascular density in the medial ganglionic eminence (MGE), while gain of function (GOF) increased vascular density. Reduced vascular density was linked to diminished migration of calbindin-positive interneurons to the cortex, whereas increased vascular density promoted their migration. Moreover, the study demonstrated that two EC-derived factors—Secreted Protein Acidic and Rich in Cysteine (SPARC) and Serine Protease Inhibitor Clade E Member 1 (SerpinE1)—play a role in promoting interneuron migration in vitro, as their removal impaired the endothelial cells’ ability to enhance migration. In addition, human stem cell-derived interneurons xenografted into the mouse cortex exhibited increased migratory behavior and morphological changes following pretreatment with SPARC and SerpinE1 [179].
Interestingly, vascular association with radial glial progenitors (RGPs) in the ventral telencephalon influences the proliferation and migration of interneurons. Distinctly organized RGPs are found in the ventral telencephalon (MGE/Preoptic Area (PoA)), where most neocortical interneurons are born [180]. During early embryonic stages (E11.5-12.5), RGPs are anchored to the pial basement membrane. However, these RGPs become progressively anchored to periventricular cerebral vessels as development progresses. Deletion of Integrin β1 from RGPs disrupts this association and causes a decrease in progenitor division in the MGE/PoA [180]. This deletion also leads to a substantial loss of PV-expressing interneurons and, to a lesser degree, Somatostatin-expressing interneurons in the neocortex.
Vascular contribution to neurodevelopmental disorders
Due to the critical role of vascular structure in neurogenesis, neuronal migration, and cortical development, any alterations in vascular functions may significantly impact normal brain development. Our objective is to gather literature linking vascular abnormalities with major neurodevelopmental disorders, autism, ADHD, and schizophrenia.
Autism
Autism spectrum disorder (ASD) is a neurodevelopmental condition characterized by difficulties in social interaction and communication, alongside repetitive behaviors and distinctive patterns of learning and attention. The neuropathological changes associated with ASD begin early in development and persist into childhood. ASD is associated with organizational defects in the cortex, hippocampus, brainstem, and cerebellum.
Recent research has underscored the critical role of cerebrovascular dysfunction in ASD (Fig. 2). A study focusing on the 16p11.2 deletion, a genetic variant associated with ASD, found significant vascular abnormalities in a mouse model [181]. These mice exhibited altered vascular growth, which was linked to the neurological and behavioral symptoms characteristic of ASD. Specifically, the study revealed reduced vascular density and abnormal endothelial cell morphology in the brains of these male mice [181]. These findings suggest that vascular dysfunction contributes to the pathophysiology of ASD, emphasizing the importance of cerebrovascular health in understanding and potentially mitigating ASD symptoms.
Further genetic studies have identified mutations in the Slc7a5 gene, which encodes a protein transporter found in the BBB that regulates branched-chain amino acid (BCAA) levels in the brain [182]. Deleting this transporter in mice led to abnormal brain development and behavior, while directly administering BCAAs in the brain improved behavior in these mice [182]. This indicates that Slc7a5 mutations may contribute to ASD by affecting BCAA levels in the brain. Moreover, studies in rats with autism induced by valproic acid showed that memantine treatment improved BBB permeability, social behavior, communication, intestinal motility, serotonin levels, and decreased anxiety and inflammation [183].
The neuropathological changes in ASD show widespread brain disorganization, affecting regions such as the cortex, hypothalamus, brainstem, and cerebellum [124,125,126,127,128,129,130,131]. There is evidence of disrupted neurogenesis, abnormal brain development, and irregular neuronal migration in young brains affected by ASD [184, 185]. It is reported that in children with ASD, cortical neurons tend to be more numerous, densely packed [184, 186], and smaller [186,187,188,189].
Despite its significance in cortical development, the importance of cerebrovascular contributions to ASD has been consistently underestimated. A postmortem study in the autism brain showed (this is the first cellular study of blood cerebral vessels in ASD brains) that angiogenesis persists in children and young adults with ASD past the time it ceases in typically developing individuals [190]. Non-angiogenic differentiation markers (Ulex Europaeus Agglutinin-1 (UEA-1), CD146, vimentin, and α-SMA) were similar in ASD and control samples, whereas angiogenic markers (nestin and CD34) were found to be highly increased in ASD and almost nonexistent in controls [190]. Therefore, nestin-positive pericytes and CD34-positive ECs detect angiogenesis and were both found to be increased in ASD.
The link between the neural activity and the subsequent changes in CBF is termed neurovascular coupling. When neural activity is increased, multiple mechanisms interplay to increase blood flow. These include vasoactive agents (ie, nitric oxide, potassium ions, hydrogen ions, adenosine, and carbon dioxide). Functional MRI (fMRI) is a powerful tool used to measure neuronal activity by detecting changes in blood flow, allowing researchers to visualize and understand brain function in real-time. The deficits of monitoring errors were determined using fMRI by comparing the Go and No-go tasks performed in 8–12-year-old autistic children to the typical developing children (TD) [191]. Unlike TD, autistic children showed high BOLD signals in the anterior medial prefrontal cortex and the left superior temporal gyrus, suggesting high neuronal activity in autistic children [191]. Moreover, the resting cerebral blood flow (rCBF) measured by pulsed arterial spin labeling MRI was higher in the frontal white and grey matter in the subcortical region of the ASD participants than in the TD [192]. Another study with diffuse correlation spectroscopy (DCS) indicates a change in the rCBF in the right and left hemispheres of ASD children versus TD children [193]. These findings support the alterations in the cerebrovascular components in ASD patients, comparing the defects in the BBB, angiogenesis, cerebral blood flow, and neurovascular uncoupling. As the neuronal activity regulates the CBF, its mediator astrocytes play a crucial role in communication between neuronal connectivity and blood flow regulation. Interestingly, a study pointed out an increase in the expression of astrocytic markers connexin 43 in Brodmann’s Area 9 (BA9)-superior frontal cortex and decreased aquaporin-4 in the cerebellum of the patients with autism, indicating abnormal astrocyte connections in the NVU which is critical for the neurovascular coupling [194].
ADHD
Attention Deficit/Hyperactivity Disorder (ADHD) is a neurodevelopmental disorder characterized by inattention, hyperactivity, and impulsiveness. The way in which CNS vasculature impacts the development and progression of ADHD and its influence on the cortex is not clear in disease pathology (Fig. 2).
In an animal model of ADHD, Spontaneously Hypertensive Rats (SHR), elevated levels of MMP2 and MMP9 and decreased levels of tight junction proteins (ZO-1 and occludin) were observed [160], indicating BBB structural damage due to neuroinflammation and excessive autophagy. In the same animal model, higher levels of monocarboxylate transporter 1 (MCT1) were found in the hippocampus, particularly at the BBB ECs [195]. This study linked this upregulation to a compensatory mechanism where hyperactivity in ADHD may increase skeletal muscle lactate production and transport it into the brain to mitigate energy deficits.
Neuropathological defects in the ADHD brain include poor connectivity between the supplementary motor area and the left amygdala, as well as reduced activity in the superior and left medial frontal gyri, which was also related to ADHD [196]. Data from the Healthy Brain Network showed that ADHD children had diminished network efficiency and structural connectivity in default mode networks, with advanced structure-functional coupling but reduced cognitive flexibility. Medication improved neural flexibility in ADHD participants [197].
Aiming to investigate the serum levels of BBB tight junction proteins claudin-5, occludin, zonulin, and tricellulin in children with ADHD compared to controls, the study by Ferahkaya et al. revealed significantly lower levels of claudin-5 and tricellulin in the ADHD group, indicating compromised BBB integrity [198]. No significant differences were observed for zonulin and occludin. These results suggest that alterations in specific tight junction proteins may contribute to BBB dysfunction in ADHD.
For understanding the cerebrovascular defects in ADHD, there are several reports that describe the change in the cerebral blood flow and defective neurovascular coupling. Compared to TD children, those with ADHD exhibit lower whole-brain CBF(CBF) amplitude of low-frequency fluctuation (ALFF) [196]. Regionally, ADHD children show reduced CBF-ALFF coupling in the bilateral thalamus, right parahippocampal gyrus, and the default mode network, including the left anterior cingulate. Decreased coupling is also observed in the executive control network, while increased coupling is seen in the attention and somatosensory networks [196]. Another study examined brain dysfunction in adults with ADHD by monitoring changes in blood flow and functional connectivity. Adults with ADHD showed reduced resting-state CBF in large-scale networks like the somatomotor, ventral attentional, and limbic networks compared to controls [199]. Hypoperfusion in the left putamen and left amygdala was linked to ADHD symptoms.
Other studies underscore the importance of Prefrontal blood flow dysregulation in the disorder’s pathogenesis and treatment. The first study by Spalletta et al. evaluated young drug-naive ADHD outpatients without structural brain abnormalities using single photon emission computerized tomography (SPECT) [200]. The findings revealed decreased rCBF in the left dorsolateral prefrontal cortex (DLPFC) compared to the right DLPFC. There were significant correlations between age and rCBF in both the dorsolateral and orbital prefrontal cortex regions. Higher severity of clinical symptoms and neuropsychological attention impairment were associated with higher right relative rCBF and lower left relative rCBF [200]. The second study by Lee et al. investigated the effects of long-term methylphenidate treatment on rCBF in ADHD children. Drug-naive ADHD children showed decreased rCBF in the right orbitofrontal cortex and middle temporal gyrus, with increased rCBF in the dorsomedial prefrontal and somatosensory areas bilaterally. Post-treatment, methylphenidate normalized rCBF in the orbitofrontal cortex and somatosensory areas and induced a significant reduction in rCBF in the right striatum [201]. These changes were associated with improved ADHD symptoms, suggesting that methylphenidate enhances attentional control and motor response by modulating aberrant vascular activity in healthy adults.
Methylphenidate is an FDA-approved stimulant primarily used to treat ADHD in children and narcolepsy in adults [202]. It blocks catecholamine transporters and acts as a dopamine agonist in the basal ganglia [203]. While its efficacy and safety in treating ADHD in both children and adults are well documented, it is currently under investigation for potential applications in children with ASD [204, 205]. Methylphenidate has notable effects on CBF. Studies have shown that methylphenidate increases CBF in specific brain regions, which is associated with its therapeutic effects. Methylphenidate enhances CBF in the prefrontal cortex, a region critical for attention and executive functions, and this increase is linked to improved cognitive performance and symptom relief in individuals with ADHD [206, 207]. The drug’s action on norepinephrine and dopamine transporters leads to increased availability of these neurotransmitters, which in turn affects neurovascular coupling, ensuring that active brain regions receive more blood flow, supporting enhanced neuronal activity [208]. The impact of methylphenidate on CBF can vary with age, with younger individuals experiencing different patterns of blood flow changes compared to adults, reflecting developmental differences in brain function and structure [206, 209, 210]. Chronic use of methylphenidate has been associated with sustained changes in CBF, which may contribute to its long-term efficacy and safety profile. However, more research is needed to fully understand these effects [206].

An aberrant angiogenesis pattern is observed in ASD. Abnormal CBF is reported in both ASD and ADHD. BBB is reported to be compromised in both conditions, as identified by the lower expression of Claudin-3, -5, and -12 in ASD. In contrast, ADHD demonstrates changes with Claudin-5, occludin, ZO1, and tricellulin. Elevated levels of the inflammatory marker MMP9 are involved in both ASD and ADHD, while MMP2 is unique to ADHD. Additionally, genetic changes in 16p11.2 and SLC7A5 contribute to vascular pathology in ASD.
Schizophrenia (SZ)
SZ affects approximately 1% of the global population and is characterized by hallucinations, delusions, alogia (poverty of speech), anhedonia (loss of pleasure), negative thoughts, and cognitive impairments in memory and learning [211]. Typically emerging in adolescence or early adulthood, SZ is linked to developmental abnormalities in early brain growth, positioning it as a neurodevelopmental disorder [212].
There is a genetic association between vascular malformation and SZ development. The 22q11.2 deletion syndrome (22qDS) is a genetic disorder characterized by a deletion on chromosome 22q11.2, leading to variable phenotypic expressions, including a significantly increased risk of developing SZ in mice [212]. This genetic syndrome can increase the likelihood of SZ by more than 20-fold. 22q11 deletion syndrome (22q11DS) is associated with a 75% reduction in claudin-5 expression in cerebral ECs, implicating BBB integrity and vascular health in SZ pathology [212]. In animal models, AAV-mediated reduction of claudin-5 expression induces anxiety-like behaviors and cognitive deficits, underscoring the critical role of BBB dysfunction in SZ [212].
Microarray hybridization analysis in mouse E-13.5 CNS ECs revealed periventricular ECs gene profile classification enriched in disease categories like SZ, nervous system diseases, epilepsy, autism, mood, and depressive disorders [29]. ECs are crucial in vascular development in the CNS.
Researchers are utilizing patient-derived induced pluripotent stem cells to gain deeper insights into the pathology of SZ. Research has increasingly focused on the connection between angiogenesis, SZ, and vascular endothelial growth factor A (VEGFA). Elevated serum VEGFA levels have been observed in patients with SZ and are associated with structural abnormalities in the prefrontal cortex [213]. Studies using SZ patient-derived induced pluripotent stem cells (iPSCs) have demonstrated impaired angiogenesis; conditioned media from SZ-iPSCs was notably less effective at inducing angiogenesis in vitro and in vivo in chicken embryos, compared to control media [214]. Recent organoid studies derived from SZ patients have shown a higher proportion of ECs, along with an increased expression of VEGFA and alterations in angiogenic pathways compared to controls [215]. These humanized models also revealed BBB disruption, evidenced by increased permeability and altered distribution of tight junction proteins such as claudin-5 and ZO-1 due to the higher expression of VEGFA [216]. Also, astrocytes generated from SZ patient’s IPSCs induced an inflammatory response, which in turn altered vascular development in the chick embryo [217].
Due to the abnormal vascular structure or neuronal activity, the blood flow changes. There is a reduction of CBF in the frontal, occipital, and parietal lobes of SZ individuals, which was investigated by a noninvasive technique, arterial spin labeling [211]. Katsel P., in 2017, described developmental CNS abnormalities and altered CBF in SZ [218]. Moreover, Patients with SZ demonstrate reduced cerebral blood volume and cerebral vessel abnormality in the CNS, which is observed specifically in the visual and prefrontal cortex of both hemispheres [219].
The evidence linking vascular abnormalities to SZ underscores the critical role of vascular development in the CNS for understanding SZ pathology. Impaired angiogenesis, BBB dysfunction, and altered CBF observed in SZ highlight the need for further exploration of endothelial cell function and vascular-related pathways. Investigating these mechanisms through animal- and patient-derived models may uncover therapeutic targets to address neurovascular defects that contribute to SZ.
Conclusion and future perspective
Current evidence supports the significance and influence of CNS vascular structure in all aspects of cortical development, including neurogenesis, neuronal differentiation, and migration. However, we are beginning to understand the underlying mechanisms mediating vascular-regulated cortical development. As discussed in the review, we have substantial knowledge of how cerebral vessels develop in the embryonic brain. Additionally, bulk mRNA sequencing and single-cell RNA sequencing techniques have provided information on the genetic profile of CNS ECs during development and adulthood compared to peripheral ECs [220,221,222,223,224]. We now have information on the unique set of genes expressed by CNS ECs of arteries, veins, and capillaries, which partially explains the heterogeneity among brain ECs [221, 225]. However, the molecular mechanisms driving this heterogeneity have not been identified. The role of neurovascular interaction during development and adulthood in driving spatiotemporal transcriptomic heterogeneity is also not yet understood.
In CNS, pericytes appear around E 9.5 in mice. Pericytes are essential for the stabilization and maturation of cerebral blood vessels, as well as for maintaining the BBB. Some recent studies demonstrate that they regulate neuronal survival, neurogenesis, and axonal development and affect neurovascular coupling [226,227,228]. A glycoprotein, vitronectin, expressed by the pericytes, regulates neurogenesis. The treatment with vitronectin antibody or in a knockout in mice ablated the neurogenesis and reduced the expression of pro-neurogenic CNTF [226]. Another molecule of pericytes, pleiotrophin knockout, impaired the pericytes and induced neuronal loss in mice [227]. Interestingly, pericyte degeneration has been associated with reduced oxygen supply and hampered neuronal excitability in mice [228]. To our knowledge, no study has reported the role of pericytes in neuronal migration and their organization in the developing cortex.
The contribution of CNS ECs to neurogenesis has been primarily elucidated through in vitro studies. Vascular models that inhibit cerebral vessel growth are not ideal for studying neurogenesis, as a hypoxic environment is known to induce neurogenesis. CNS EC-specific Cre animal models and viral models are now available and can be utilized to study neurovascular interactions and validate the in vitro neurogenesis findings.
Moreover, the interaction between blood cerebral vessels and radially migrating neurons, or projection neurons, is not well studied. During cortical development, the number of projection neurons is significantly higher, and they are in close contact with blood cerebral vessels. According to Segarra et al., cerebral vessels participate in the migration of projection neurons through the Dab1-Reelin pathway [172]. However, an alternative argument suggests that Dab1 EC knockout affects normal vascular development, which may indirectly influence neurogenesis and neuron migration. Additionally, this study did not examine interneurons, leaving it unclear whether the observed effects are general or specific to projection neurons.
Finally, several brain disorders are accompanied by defects in cortical development (e.g., Autism, ADHD, epilepsy) [229,230,231,232,233,234]. Understanding the vascular role and the precise mechanisms of neurovascular communication that are disturbed in these conditions is crucial. Elucidating these mechanisms can provide insights into how vascular abnormalities contribute to cortical malformations and inform the development of targeted therapies to correct or mitigate these defects. Additionally, many neurological diseases injure the normal cortical layering (e.g., stroke, TBI Alzheimer’s disease, and the damage is permanent [235,236,237]. NP transplantation can be a promising approach; however, it has not successfully integrated neurons into the desired cortical layers. Several studies have claimed that transplanted NPs stay close to cerebral vessels [238,239,240]. However, the adult cerebral vessels likely lack the developmental mechanisms to orchestrate the migration of these NPs in the cortex. To overcome this challenge, it is crucial to understand the neurovascular interactions that orchestrate the migration of NPs in the cortex and to utilize these developmental mechanisms to achieve cortical regeneration.
Despite the crucial role of CNS cerebral vessels in neurogenesis, neuronal migration, and cortical development, our understanding of their involvement in neurodevelopmental disorders such as autism and ADHD is still in its infancy. Investigating neuronal or cortical defects in embryonic vascular models is essential for gaining deeper insights into these diseases. Additionally, testing new vascular deletion models is important for elucidating their significance in the pathology of neurodevelopmental disorders.
References
Hofman MA. Size and shape of the cerebral cortex in mammals. II. The cortical volume. Brain Behav Evol. 1988;32:17–26.
Rilling JK, Insel TR. The primate neocortex in comparative perspective using magnetic resonance imaging. J Hum Evol. 1999;37:191–223.
S H-H. The remarkable, yet not extraordinary, human brain as a scaled-up primate brain and its associated cost. In: Georg F Striedter JCA, and Francisco JA, editor. In the light of evolution: volume VI: brain and behavior VI. Washington (DC): National Academies Press (US); 2013.
Herculano-Houzel S. The human brain in numbers: a linearly scaled-up primate brain. Front Hum Neurosci. 2009;3:31.
Marino L. A comparison of encephalization between odontocete cetaceans and anthropoid primates. Brain Behav Evol. 1998;51:230–8.
Tower DB. Structural and functional organization of mammalian cerebral cortex; the correlation of neurone density with brain size; cortical neurone density in the fin whale (Balaenoptera physalus L.) with a note on the cortical neurone density in the Indian elephant. J Comp Neurol. 1954;101:19–51.
Dicke U, Roth G. Neuronal factors determining high intelligence. Philos Trans R Soc Lond B Biol Sci. 2016;371:20150180.
Miller EN, Sherwood CC. What single neurons can tell us. Elife. 2019;8:e44560.
Genç E, Fraenz C, Schlüter C, Friedrich P, Hossiep R, Voelkle MC, et al. Diffusion markers of dendritic density and arborization in gray matter predict differences in intelligence. Nat Commun. 2018;9:1905.
Strominger NL, Demarest RJ, Laemle LB. Cerebral cortex. Noback’s human nervous system, seventh edition: structure and function. Totowa, NJ: Humana Press; 2012. p. 429–51.
Thomson AM. Neocortical layer 6, a review. Front Neuroanat. 2010;4:13.
El-Baba RM, Schury MP. Neuroanatomy, frontal cortex. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Vachha BA, Massoud TF, Huang SY. Anatomy of the cerebral cortex, lobes, and cerebellum. Neuroimaging Clin N Am. 2022;32:463–73.
Chayer C, Freedman M. Frontal lobe functions. Curr Neurol Neurosci Rep. 2001;1:547–52.
Catani M. The anatomy of the human frontal lobe. Handb Clin Neurol. 2019;163:95–122.
Flinker A, Korzeniewska A, Shestyuk AY, Franaszczuk PJ, Dronkers NF, Knight RT, et al. Redefining the role of Broca’s area in speech. Proc Natl Acad Sci USA. 2015;112:2871–5.
Berlucchi G, Vallar G. The history of the neurophysiology and neurology of the parietal lobe. Handb Clin Neurol. 2018;151:3–30.
Battelli L, Alvarez GA, Carlson T, Pascual-Leone A. The role of the parietal lobe in visual extinction studied with transcranial magnetic stimulation. J Cogn Neurosci. 2009;21:1946–55.
Berryhill ME, Olson IR. The right parietal lobe is critical for visual working memory. Neuropsychologia. 2008;46:1767–74.
Cohen YE, Russ BE, Gifford GW 3rd. Auditory processing in the posterior parietal cortex. Behav Cogn Neurosci Rev. 2005;4:218–31.
Patel A, Biso G, Fowler JB. Neuroanatomy, temporal lobe. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Kiernan JA. Anatomy of the temporal lobe. Epilepsy Res Treat. 2012;2012:176157.
Javed K, Reddy V, Das JM, Wroten M. Neuroanatomy, wernicke area. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Binder JR. The wernicke area: modern evidence and a reinterpretation. Neurology. 2015;85:2170–5.
Rehman A, Al Khalili Y. Neuroanatomy, occipital lobe. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Insausti R, Muñoz-López M, Insausti AM, Artacho-Pérula E. The human periallocortex: layer pattern in presubiculum, parasubiculum and entorhinal cortex. A review. Front Neuroanat. 2017;11:84.
Balasubramanian V. Brain power. Proc Natl Acad Sci USA. 2021;118:e2107022118.
Marín-Padilla M. The human brain intracerebral microvascular system: development and structure. Front Neuroanat. 2012;6:38.
Won C, Lin Z, Kumar TP, Li S, Ding L, Elkhal A, et al. Autonomous vascular networks synchronize GABA neuron migration in the embryonic forebrain. Nat Commun. 2013;4:2149.
Eichmann A, Thomas JL. Molecular parallels between neural and vascular development. Cold Spring Harb Perspect Med. 2013;3:a006551.
Ribatti D, Guidolin D. Morphogenesis of vascular and neuronal networks and the relationships between their remodeling processes. Brain Res Bull. 2022;186:62–9.
Crouch EE, Joseph T, Marsan E, Huang EJ. Disentangling brain vasculature in neurogenesis and neurodegeneration using single-cell transcriptomics. Trends Neurosci. 2023;46:551–65.
Ishihara K, Takata K, Mizutani KI. Involvement of an aberrant vascular system in neurodevelopmental, neuropsychiatric, and neuro-degenerative diseases. Life. 2023;13:221.
Karakatsani A, Shah B, Ruiz de Almodovar C. Blood vessels as regulators of neural stem cell properties. Front Mol Neurosci. 2019;12:85.
Manzo J, Hernández-Aguilar ME, Toledo-Cárdenas MR, Herrera-Covarrubias D, Coria-Avila GA. Dysregulation of neural tube vascular development as an aetiological factor in autism spectrum disorder: insights from valproic acid exposure. J Physiol. 2025. https://doi.org/10.1113/JP286899
Ouellette J, Crouch EE, Morel JL, Coelho-Santos V, Lacoste B. A vascular-centric approach to autism spectrum disorders. Neurosci Insights. 2024;19:26331055241235921.
Ouellette J, Lacoste B. From neurodevelopmental to neurodegenerative disorders: the vascular continuum. Front Aging Neurosci. 2021;13:749026.
Paredes I, Himmels P, Ruiz de Almodóvar C. Neurovascular communication during CNS development. Dev Cell. 2018;45:10–32.
Vogenstahl J, Parrilla M, Acker-Palmer A, Segarra M. Vascular Regulation of developmental neurogenesis. Front Cell Dev Biol. 2022;10:890852.
Wang Y, Yu S, Li M. Neurovascular crosstalk and cerebrovascular alterations: an underestimated therapeutic target in autism spectrum disorders. Front Cell Neurosci. 2023;17:1226580.
Ayala R, Shu T, Tsai LH. Trekking across the brain: the journey of neuronal migration. Cell. 2007;128:29–43.
Hatanaka Y, Hirata T. How do cortical excitatory neurons terminate their migration at the right place? critical roles of environmental elements. Front Cell Dev Biol. 2020;8:596708.
Luhmann HJ, Fukuda A, Kilb W. Control of cortical neuronal migration by glutamate and GABA. Front Cell Neurosci. 2015;9:4.
Meyerink BL, Tiwari NK, Pilaz LJ. Ariadne’s thread in the developing cerebral cortex: mechanisms enabling the guiding role of the radial glia basal process during neuron migration. Cells. 2020;10:3.
Nadarajah B, Parnavelas JG. Modes of neuronal migration in the developing cerebral cortex. Nat Rev Neurosci. 2002;3:423–32.
Nishimura YV, Nabeshima YI, Kawauchi T. Morphological and molecular basis of cytoplasmic dilation and swelling in cortical migrating neurons. Brain Sci. 2017;7:87.
Silva CG, Peyre E, Nguyen L. Cell migration promotes dynamic cellular interactions to control cerebral cortex morphogenesis. Nat Rev Neurosci. 2019;20:318–29.
Subramanian L, Calcagnotto ME, Paredes MF. Cortical malformations: lessons in human brain development. Front Cell Neurosci. 2019;13:576.
Fujioka T, Kaneko N, Sawamoto K. Blood vessels as a scaffold for neuronal migration. Neurochem Int. 2019;126:69–73.
Agirman G, Broix L, Nguyen L. Cerebral cortex development: an outside-in perspective. FEBS Lett. 2017;591:3978–92.
Kuwar Chhetri P, Das JM. Neuroanatomy, neural tube development and stages. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Singh R, Munakomi S. Embryology, neural tube. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Engelhardt DM, Martyr CA, Niswander L. Pathogenesis of neural tube defects: the regulation and disruption of cellular processes underlying neural tube closure. WIREs Mech Dis. 2022;14:e1559.
Blackshaw S, Scholpp S, Placzek M, Ingraham H, Simerly R, Shimogori T. Molecular pathways controlling development of thalamus and hypothalamus: from neural specification to circuit formation. J Neurosci. 2010;30:14925–30.
Dennis D, Picketts D, Slack RS, Schuurmans C. Forebrain neurogenesis: from embryo to adult. Trends Dev Biol. 2016;9:77–90.
Monuki ES Induction and patterning in the telencephalon. In: Pfaff DW, Volkow ND, Rubenstein JL, editors. Neuroscience in the 21st century: from basic to clinical. Cham: Springer International Publishing; 2022. pp. 193-215.
Caminero F, Cascella M. Neuroanatomy, mesencephalon midbrain. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Ruchalski K, Hathout GM. A medley of midbrain maladies: a brief review of midbrain anatomy and syndromology for radiologists. Radiol Res Pract. 2012;2012:258524.
Pujades C. The multiple functions of hindbrain boundary cells: tinkering boundaries? Semin Cell Dev Biol. 2020;107:179–89.
Rinaman L. Hindbrain noradrenergic A2 neurons: diverse roles in autonomic, endocrine, cognitive, and behavioral functions. Am J Physiol Regul Integr Comp Physiol. 2011;300:R222–35.
Basinger H, Hogg JP. Neuroanatomy, brainstem. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Ekonomou A, Kazanis I, Malas S, Wood H, Alifragis P, Denaxa M, et al. Neuronal migration and ventral subtype identity in the telencephalon depend on SOX1. PLoS Biol. 2005;3:e186.
Arai Y, Taverna E. Neural progenitor cell polarity and cortical development. Front Cell Neurosci. 2017;11:384.
Casas Gimeno G, Paridaen J. The symmetry of neural stem cell and progenitor divisions in the vertebrate brain. Front Cell Dev Biol. 2022;10:885269.
Akula SK, Exposito-Alonso D, Walsh CA. Shaping the brain: the emergence of cortical structure and folding. Dev Cell. 2023;58:2836–49.
Kast RJ, Levitt P. Precision in the development of neocortical architecture: from progenitors to cortical networks. Prog Neurobiol. 2019;175:77–95.
Noctor SC, Flint AC, Weissman TA, Wong WS, Clinton BK, Kriegstein AR. Dividing precursor cells of the embryonic cortical ventricular zone have morphological and molecular characteristics of radial glia. J Neurosci. 2002;22:3161–73.
Taverna E, Götz M, Huttner WB. The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol. 2014;30:465–502.
Matsuzaki F, Shitamukai A. Cell division modes and cleavage planes of neural progenitors during mammalian cortical development. Cold Spring Harb Perspect Biol. 2015;7:a015719.
Coquand L, Brunet Avalos C, Macé AS, Farcy S, Di Cicco A, Lampic M, et al. A cell fate decision map reveals abundant direct neurogenesis bypassing intermediate progenitors in the human developing neocortex. Nat Cell Biol. 2024;26:698–709.
Marin-Padilla M. Dual origin of the mammalian neocortex and evolution of the cortical plate. Anat Embryol. 1978;152:109–26.
Yoshinaga S, Shin M, Kitazawa A, Ishii K, Tanuma M, Kasai A, et al. Comprehensive characterization of migration profiles of murine cerebral cortical neurons during development using FlashTag labeling. iScience. 2021;24:102277.
Kwan KY, Sestan N, Anton ES. Transcriptional co-regulation of neuronal migration and laminar identity in the neocortex. Development. 2012;139:1535–46.
Molliver ME, Kostović I, van der Loos H. The development of synapses in cerebral cortex of the human fetus. Brain Res. 1973;50:403–7.
Gao Z, Godbout R. Reelin-disabled-1 signaling in neuronal migration: splicing takes the stage. Cell Mol Life Sci. 2013;70:2319–29.
Govek EE, Hatten ME, Van Aelst L. The role of Rho GTPase proteins in CNS neuronal migration. Dev Neurobiol. 2011;71:528–53.
Mukhtar T, Taylor V. Untangling cortical complexity during development. J Exp Neurosci. 2018;12:1179069518759332.
Zarzor MS, Blumcke I, Budday S. Exploring the role of the outer subventricular zone during cortical folding through a physics-based model. Elife. 2023;12:e82925.
Dehay C, Kennedy H, Kosik KS. The outer subventricular zone and primate-specific cortical complexification. Neuron. 2015;85:683–94.
Ortega JA, Memi F, Radonjic N, Filipovic R, Bagasrawala I, Zecevic N, et al. The subventricular zone: a key player in human neocortical development. Neuroscientist. 2018;24:156–70.
Fietz SA, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish JL, et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010;13:690–9.
Yang Z. The principle of cortical development and evolution. Neurosci Bull. 2025;41:461–85.
Ferent J, Zaidi D, Francis F. Extracellular control of radial glia proliferation and scaffolding during cortical development and pathology. Front Cell Dev Biol. 2020;8:578341.
Bilgic M, Wu Q, Suetsugu T, Shitamukai A, Tsunekawa Y, Shimogori T, et al. Truncated radial glia as a common precursor in the late corticogenesis of gyrencephalic mammals. Elife. 2023;12:RP91406.
Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, et al. Molecular identity of human outer radial glia during cortical development. Cell. 2015;163:55–67.
Wilson S, Christiaens D, Yun H, Uus A, Cordero-Grande L, Karolis V, et al. Dynamic changes in subplate and cortical plate microstructure at the onset of cortical folding in vivo. bioRxiv. [Preprint]. 2024. Available from: https://www.biorxiv.org/content/10.1101/2023.10.16.562524v1.
de Vareilles H, Rivière D, Mangin JF, Dubois J. Development of cortical folds in the human brain: an attempt to review biological hypotheses, early neuroimaging investigations and functional correlates. Dev Cogn Neurosci. 2023;61:101249.
Jinnou H, Sawada M, Kawase K, Kaneko N, Herranz-Pérez V, Miyamoto T, et al. Radial glial fibers promote neuronal migration and functional recovery after neonatal brain injury. Cell Stem Cell. 2018;22:128–37.e9.
Miranda-Negrón Y, García-Arrarás JE. Radial glia and radial glia-like cells: their role in neurogenesis and regeneration. Front Neurosci. 2022;16:1006037.
Furuta C, Ema H, Takayanagi S, Ogaeri T, Okamura D, Matsui Y, et al. Discordant developmental waves of angioblasts and hemangioblasts in the early gastrulating mouse embryo. Development. 2006;133:2771–9.
Koduru SV, Leberfinger AN, Pasic D, Forghani A, Lince S, Hayes DJ, et al. Cellular based strategies for microvascular engineering. Stem Cell Rev Rep. 2019;15:218–40.
Cao N, Yao ZX. The hemangioblast: from concept to authentication. Anat Rec. 2011;294:580–8.
Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–95.
Peguera B, Segarra M, Acker-Palmer A. Neurovascular crosstalk coordinates the central nervous system development. Curr Opin Neurobiol. 2021;69:202–13.
Boutom SM, Silva TP, Palecek SP, Shusta EV, Fernandes TG, Ashton RS. Central nervous system vascularization in human embryos and neural organoids. Cell Rep. 2024;43:115068.
Joshua SG, Karen KH A vascular perspective on neurogenesis. In: Luca B, editor. Neural stem cells. Rijeka: IntechOpen; 2013. Ch. 8.
Puelles L, Martínez-Marin R, Melgarejo-Otalora P, Ayad A, Valavanis A, Ferran JL. Patterned vascularization of embryonic mouse forebrain, and neuromeric topology of major human subarachnoidal arterial branches: a prosomeric mapping. Front Neuroanat. 2019;13:59.
Vasudevan A, Long JE, Crandall JE, Rubenstein JL, Bhide PG. Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nat Neurosci. 2008;11:429–39.
Ma S, Kwon HJ, Johng H, Zang K, Huang Z. Radial glial neural progenitors regulate nascent brain vascular network stabilization via inhibition of Wnt signaling. PLoS Biol. 2013;11:e1001469.
Allsopp G, Gamble HJ. Light and electron microscopic observations on the development of the blood vascular system of the human brain. J Anat. 1979;128:461–77.
Javed K, Reddy V, Das JM. Neuroanatomy, posterior cerebral arteries. StatPearls. Treasure Island (FL): StatPearls Publishing; 2025.
Ritsuko KP, Kurjak A. Fetal brain vascularity visualized by conventional 2D and 3D power doppler technology. Donald Sch J Ultrasound Obstet Gynecol. 2010;4:249–58.
Bertulli L, Robert T. Embryological development of the human cranio-facial arterial system: a pictorial review. Surg Radiol Anat. 2021;43:961–73.
Agarwal N, Carare RO. Cerebral vessels: an overview of anatomy, physiology, and role in the drainage of fluids and solutes. Front Neurol. 2020;11:611485.
Daneman R, Prat A. The blood-brain barrier. Cold Spring Harb Perspect Biol. 2015;7:a020412.
Konan LM, Reddy V, Mesfin FB. Neuroanatomy, cerebral blood supply. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Bonkowsky JL, Son JH. Hypoxia and connectivity in the developing vertebrate nervous system. Dis Model Mech. 2018;11:dmm037127.
Rademakers T, Horvath JM, van, Blitterswijk CA, LaPointe VLS. Oxygen and nutrient delivery in tissue engineering: approaches to graft vascularization. J Tissue Eng Regen Med. 2019;13:1815–29.
Cui C, Jiang X, Wang Y, Li C, Lin Z, Wei Y, et al. Cerebral hypoxia-induced molecular alterations and their impact on the physiology of neurons and dendritic spines: a comprehensive review. Cell Mol Neurobiol. 2024;44:58.
Dotiwala AK, McCausland C, Samra NS. Anatomy, head and neck: blood brain barrier. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Stamatovic SM, Johnson AM, Keep RF, Andjelkovic AV. Junctional proteins of the blood-brain barrier: new insights into function and dysfunction. Tissue Barriers. 2016;4:e1154641.
Kadry H, Noorani B, Cucullo L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS. 2020;17:69.
Ohtsuki S, Yamaguchi H, Katsukura Y, Asashima T, Terasaki T. mRNA expression levels of tight junction protein genes in mouse brain capillary endothelial cells highly purified by magnetic cell sorting. J Neurochem. 2008;104:147–54.
Dithmer S, Blasig IE, Fraser PA, Qin Z, Haseloff RF. The basic requirement of tight junction proteins in blood-brain barrier function and their role in pathologies. Int J Mol Sci. 2024;25:5601.
Cording J, Berg J, Käding N, Bellmann C, Tscheik C, Westphal JK, et al. In tight junctions, claudins regulate the interactions between occludin, tricellulin and marvelD3, which, inversely, modulate claudin oligomerization. J Cell Sci. 2013;126:554–64.
Fanning AS, Anderson JM. Zonula occludens-1 and -2 are cytosolic scaffolds that regulate the assembly of cellular junctions. Ann N Y Acad Sci. 2009;1165:113–20.
Zheng X, Ren B, Gao Y. Tight junction proteins related to blood-brain barrier and their regulatory signaling pathways in ischemic stroke. Biomed Pharmacother. 2023;165:115272.
Chaves JCS, Dando SJ, White AR, Oikari LE. Blood-brain barrier transporters: an overview of function, dysfunction in Alzheimer’s disease and strategies for treatment. Biochim Biophys Acta Mol Basis Dis. 2024;1870:166967.
Chua GL, Tan BC, Loke RYJ, He M, Chin CF, Wong BH, et al. Mfsd2a utilizes a flippase mechanism to mediate omega-3 fatty acid lysolipid transport. Proc Natl Acad Sci USA. 2023;120:e2215290120.
Veszelka S, Tóth A, Walter FR, Tóth AE, Gróf I, Mészáros M, et al. Comparison of a rat primary cell-based blood-brain barrier model with epithelial and brain endothelial cell lines: gene expression and drug transport. Front Mol Neurosci. 2018;11:166.
Nguyen C, Lei HT, Lai LTF, Gallenito MJ, Mu X, Matthies D, et al. Lipid flipping in the omega-3 fatty-acid transporter. Nat Commun. 2023;14:2571.
Ayloo S, Gu C. Transcytosis at the blood-brain barrier. Curr Opin Neurobiol. 2019;57:32–8.
Pulgar VM. Transcytosis to cross the blood brain barrier, new advancements and challenges. Front Neurosci. 2018;12:1019.
Villaseñor R, Lampe J, Schwaninger M, Collin L. Intracellular transport and regulation of transcytosis across the blood-brain barrier. Cell Mol Life Sci. 2019;76:1081–92.
Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, et al. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature. 2014;509:507–11.
Daneman R, Agalliu D, Zhou L, Kuhnert F, Kuo CJ, Barres BA. Wnt/beta-catenin signaling is required for CNS, but not non-CNS, angiogenesis. Proc Natl Acad Sci USA. 2009;106:641–6.
Gerhardt H. VEGF and endothelial guidance in angiogenic sprouting. Organogenesis. 2008;4:241–6.
Blanchette M, Daneman R. Formation and maintenance of the BBB. Mech Dev. 2015;138:8–16.
Mollgøard K, Saunders NR. Complex tight junctions of epithelial and of endothelial cells in early foetal brain. J Neurocytol. 1975;4:453–68.
Bertossi M, Virgintino D, Errede M, Roncali L. Immunohistochemical and ultrastructural characterization of cortical plate microvasculature in the human fetus telencephalon. Microvasc Res. 1999;58:49–61.
Virgintino D, Robertson D, Benagiano V, Errede M, Bertossi M, Ambrosi G, et al. Immunogold cytochemistry of the blood-brain barrier glucose transporter GLUT1 and endogenous albumin in the developing human brain. Brain Res Dev Brain Res. 2000;123:95–101.
Ballabh P, Hu F, Kumarasiri M, Braun A, Nedergaard M. Development of tight junction molecules in blood vessels of germinal matrix, cerebral cortex, and white matter. Pediatr Res. 2005;58:791–8.
Cauli B, Hamel E. Revisiting the role of neurons in neurovascular coupling. Front Neuroenergetics. 2010;2:9.
Cantin S, Villien M, Moreaud O, Tropres I, Keignart S, Chipon E, et al. Impaired cerebral vasoreactivity to CO2 in Alzheimer’s disease using BOLD fMRI. Neuroimage. 2011;58:579–87.
Cao Y, Welch KM, Aurora S, Vikingstad EM. Functional MRI-BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56:548–54.
Dichter GS. Functional magnetic resonance imaging of autism spectrum disorders. Dialogues Clin Neurosci. 2012;14:319–51.
Ekstrom A. How and when the fMRI BOLD signal relates to underlying neural activity: the danger in dissociation. Brain Res Rev. 2010;62:233–44.
Logothetis NK. The underpinnings of the BOLD functional magnetic resonance imaging signal. J Neurosci. 2003;23:3963–71.
Röder CH, Hoogendam JM, van der Veen FM. FMRI, antipsychotics and schizophrenia. Influence of different antipsychotics on BOLD-signal. Curr Pharm Des. 2010;16:2012–25.
Nordahl CW, Mello M, Shen AM, Shen MD, Vismara LA, Li D, et al. Methods for acquiring MRI data in children with autism spectrum disorder and intellectual impairment without the use of sedation. J Neurodev Disord. 2016;8:20.
McConnell HL, Mishra A. Cells of the blood-brain barrier: an overview of the neurovascular unit in health and disease. Methods Mol Biol. 2022;2492:3–24.
Guerra G, Lucariello A, Perna A, Botta L, De Luca A, Moccia F. The role of endothelial Ca(2+) signaling in Neurovascular coupling: a view from the lumen. Int J Mol Sci. 2018;19:938.
Petzold GC, Murthy VN. Role of astrocytes in neurovascular coupling. Neuron. 2011;71:782–97.
Courtney MJ, Li LL, Lai YY. Mechanisms of NOS1AP action on NMDA receptor-nNOS signaling. Front Cell Neurosci. 2014;8:252.
Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–84.
Budday S, Steinmann P, Kuhl E. Physical biology of human brain development. Front Cell Neurosci. 2015;9:257.
Stepien BK, Wielockx B. From vessels to neurons-the role of Hypoxia pathway proteins in embryonic neurogenesis. Cells. 2024;13:621.
Komabayashi-Suzuki M, Yamanishi E, Watanabe C, Okamura M, Tabata H, Iwai R, et al. Spatiotemporally dependent vascularization is differently utilized among neural progenitor subtypes during neocortical development. Cell Rep. 2019;29:1113–29.e5.
Di Marco B, Crouch EE, Shah B, Duman C, Paredes MF, Ruiz de Almodovar C, et al. Reciprocal interaction between vascular filopodia and neural stem cells shapes neurogenesis in the ventral telencephalon. Cell Rep. 2020;33:108256.
Lange C, Turrero Garcia M, Decimo I, Bifari F, Eelen G, Quaegebeur A, et al. Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. Embo J. 2016;35:924–41.
Winkelman MA, Koppes AN, Koppes RA, Dai G. Bioengineering the neurovascular niche to study the interaction of neural stem cells and endothelial cells. APL Bioeng. 2021;5:011507.
Chou CH, Sinden JD, Couraud PO, Modo M. In vitro modeling of the neurovascular environment by coculturing adult human brain endothelial cells with human neural stem cells. PLoS One. 2014;9:e106346.
Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, et al. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science. 2004;304:1338–40.
Li S, Haigh K, Haigh JJ, Vasudevan A. Endothelial VEGF sculpts cortical cytoarchitecture. J Neurosci. 2013;33:14809–15.
Tsai HH, Niu J, Munji R, Davalos D, Chang J, Zhang H, et al. Oligodendrocyte precursors migrate along vasculature in the developing nervous system. Science. 2016;351:379–84.
Wu KW, Mo JL, Kou ZW, Liu Q, Lv LL, Lei Y, et al. Neurovascular interaction promotes the morphological and functional maturation of cortical neurons. Front Cell Neurosci. 2017;11:290.
Shi Y, Sun L, Wang M, Liu J, Zhong S, Li R, et al. Vascularized human cortical organoids (vOrganoids) model cortical development in vivo. PLoS Biol. 2020;18:e3000705.
Colucci-D’Amato L, Speranza L, Volpicelli F. Neurotrophic factor BDNF, physiological functions and therapeutic potential in depression, neurodegeneration and brain cancer. Int J Mol Sci. 2020;21:7777.
Rosenstein JM, Krum JM, Ruhrberg C. VEGF in the nervous system. Organogenesis. 2010;6:107–14.
Leventhal C, Rafii S, Rafii D, Shahar A, Goldman SA. Endothelial trophic support of neuronal production and recruitment from the adult mammalian subependyma. Mol Cell Neurosci. 1999;13:450–64.
Rizzuti M, Melzi V, Brambilla L, Quetti L, Sali L, Ottoboni L, et al. Shaping the neurovascular unit exploiting human brain organoids. Mol Neurobiol. 2024;61:6642–57.
Kusuma S, Shen YI, Hanjaya-Putra D, Mali P, Cheng L, Gerecht S. Self-organized vascular networks from human pluripotent stem cells in a synthetic matrix. Proc Natl Acad Sci USA. 2013;110:12601–6.
Dao L, You Z, Lu L, Xu T, Sarkar AK, Zhu H, et al. Modeling blood-brain barrier formation and cerebral cavernous malformations in human PSC-derived organoids. Cell Stem Cell. 2024;31:818–33.e11.
Nwokoye PN, Abilez OJ. Bioengineering methods for vascularizing organoids. Cell Rep Methods. 2024;4:100779.
Cakir B, Xiang Y, Tanaka Y, Kural MH, Parent M, Kang YJ, et al. Engineering of human brain organoids with a functional vascular-like system. Nat Methods. 2019;16:1169–75.
Kistemaker L, van Bodegraven EJ, de Vries HE, Hol EM. Vascularized human brain organoids: current possibilities and prospects. Trends Biotechnol. 2025;43:1275–85.
Sun XY, Ju XC, Li Y, Zeng PM, Wu J, Zhou YY, et al. Generation of vascularized brain organoids to study neurovascular interactions. Elife. 2022;11:e76707.
Biswas S, Cottarelli A, Agalliu D. Neuronal and glial regulation of CNS angiogenesis and barriergenesis. Development. 2020;147:dev182279.
Morimoto K, Tabata H, Takahashi R, Nakajima K. Interactions between neural cells and blood vessels in central nervous system development. Bioessays. 2024;46:e2300091.
Javaherian A, Kriegstein A. A stem cell niche for intermediate progenitor cells of the embryonic cortex. Cereb Cortex. 2009;19(Suppl 1(Suppl 1)):i70–7.
Franco SJ, Martinez-Garay I, Gil-Sanz C, Harkins-Perry SR, Müller U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482–97.
Segarra M, Aburto MR, Cop F, Llaó-Cid C, Härtl R, Damm M, et al. Endothelial Dab1 signaling orchestrates neuro-glia-vessel communication in the central nervous system. Science. 2018;361:eaao2861.
Lim L, Mi D, Llorca A, Marín O. Development and functional diversification of cortical interneurons. Neuron. 2018;100:294–313.
Llorca A, Deogracias R. Origin, development, and synaptogenesis of cortical interneurons. Front Neurosci. 2022;16:929469.
Li S, Kumar TP, Joshee S, Kirschstein T, Subburaju S, Khalili JS, et al. Endothelial cell-derived GABA signaling modulates neuronal migration and postnatal behavior. Cell Res. 2018;28:221–48.
Léger C, Dupré N, Aligny C, Bénard M, Lebon A, Henry V, et al. Glutamate controls vessel-associated migration of GABA interneurons from the pial migratory route via NMDA receptors and endothelial protease activation. Cell Mol Life Sci. 2020;77:1959–86.
Shibuya M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer. 2011;2:1097–105.
Barber M, Andrews WD, Memi F, Gardener P, Ciantar D, Tata M, et al. Vascular-derived vegfa promotes cortical interneuron migration and proximity to the vasculature in the developing forebrain. Cereb Cortex. 2018;28:2577–93.
Genestine M, Ambriz D, Crabtree GW, Dummer P, Molotkova A, Quintero M, et al. Vascular-derived SPARC and SerpinE1 regulate interneuron tangential migration and accelerate functional maturation of human stem cell-derived interneurons. Elife. 2021;10:e56063.
Tan X, Liu WA, Zhang XJ, Shi W, Ren SQ, Li Z, et al. Vascular influence on ventral telencephalic progenitors and neocortical interneuron production. Dev Cell. 2016;36:624–38.
Ouellette J, Toussay X, Comin CH, Costa LDF, Ho M, Lacalle-Aurioles M, et al. Vascular contributions to 16p11.2 deletion autism syndrome modeled in mice. Nat Neurosci. 2020;23:1090–101.
Tărlungeanu DC, Deliu E, Dotter CP, Kara M, Janiesch PC, Scalise M, et al. Impaired amino acid transport at the blood brain barrier is a cause of autism spectrum disorder. Cell. 2016;167:1481–94.e18.
Kumar H, Sharma B. Memantine ameliorates autistic behavior, biochemistry & blood brain barrier impairments in rats. Brain Res Bull. 2016;124:27–39.
Courchesne E. Brainstem, cerebellar and limbic neuroanatomical abnormalities in autism. Curr Opin Neurobiol. 1997;7:269–78.
Wegiel J, Kuchna I, Nowicki K, Imaki H, Wegiel J, Marchi E, et al. The neuropathology of autism: defects of neurogenesis and neuronal migration, and dysplastic changes. Acta Neuropathol. 2010;119:755–70.
Casanova MF, El-Baz AS, Kamat SS, Dombroski BA, Khalifa F, Elnakib A, et al. Focal cortical dysplasias in autism spectrum disorders. Acta Neuropathol Commun. 2013;1:67.
Jacot-Descombes S, Uppal N, Wicinski B, Santos M, Schmeidler J, Giannakopoulos P, et al. Decreased pyramidal neuron size in Brodmann areas 44 and 45 in patients with autism. Acta Neuropathol. 2012;124:67–79.
van Kooten IA, Palmen SJ, von Cappeln P, Steinbusch HW, Korr H, Heinsen H, et al. Neurons in the fusiform gyrus are fewer and smaller in autism. Brain. 2008;131:987–99.
Fatemi SH, Halt AR, Realmuto G, Earle J, Kist DA, Thuras P, et al. Purkinje cell size is reduced in cerebellum of patients with autism. Cell Mol Neurobiol. 2002;22:171–5.
Azmitia EC, Saccomano ZT, Alzoobaee MF, Boldrini M, Whitaker-Azmitia PM. Persistent angiogenesis in the autism brain: an immunocytochemical study of postmortem cortex, brainstem and cerebellum. J Autism Dev Disord. 2016;46:1307–18.
Goldberg MC, Spinelli S, Joel S, Pekar JJ, Denckla MB, Mostofsky SH. Children with high functioning autism show increased prefrontal and temporal cortex activity during error monitoring. Dev Cogn Neurosci. 2011;1:47–56.
Peterson BS, Zargarian A, Peterson JB, Goh S, Sawardekar S, Williams SCR, et al. Hyperperfusion of frontal white and subcortical gray matter in autism spectrum disorder. Biol Psychiatry. 2019;85:584–95.
Lin F, Huang W, Lu S, Li J. Cerebral blood flow measured by diffuse correlation spectroscopy in children with autism spectrum disorder. J Biophotonics. 2023;16:e202300151.
Fatemi SH, Folsom TD, Reutiman TJ, Lee S. Expression of astrocytic markers aquaporin 4 and connexin 43 is altered in brains of subjects with autism. Synapse. 2008;62:501–7.
Pellerin L, Pellegri G, Martin JL, Magistretti PJ. Expression of monocarboxylate transporter mRNAs in mouse brain: support for a distinct role of lactate as an energy substrate for the neonatal vs. adult brain. Proc Natl Acad Sci USA. 1998;95:3990–5.
Su S, Zhao J, Dai Y, Lin L, Zhou Q, Yan Z, et al. Altered neurovascular coupling in the children with attention-deficit/hyperactivity disorder: a comprehensive fMRI analysis. Eur Child Adolesc Psychiatry. 2024;33:1081–91.
Lee D, Quattrocki Knight E, Song H, Lee S, Pae C, Yoo S, et al. Differential structure-function network coupling in the inattentive and combined types of attention deficit hyperactivity disorder. PLoS One. 2021;16:e0260295.
Ferahkaya H, Akca OF, Kılınç I, Baysal T. Claudin-5, occludin, zonulin and tricellulin levels of children with attention deficit/hyperactivity disorder. Eur Psychiatry. 2023;66:S96–7. Suppl 1
Tan YW, Liu L, Wang YF, Li HM, Pan MR, Zhao MJ, et al. Alterations of cerebral perfusion and functional brain connectivity in medication-naïve male adults with attention-deficit/hyperactivity disorder. CNS Neurosci Ther. 2020;26:197–206.
Spalletta G, Pasini A, Pau F, Guido G, Menghini L, Caltagirone C. Prefrontal blood flow dysregulation in drug naive ADHD children without structural abnormalities. J Neural Transm. 2001;108:1203–16.
Lee JS, Kim BN, Kang E, Lee DS, Kim YK, Chung JK, et al. Regional cerebral blood flow in children with attention deficit hyperactivity disorder: comparison before and after methylphenidate treatment. Hum Brain Mapp. 2005;24:157–64.
Verghese C, Patel P, Abdijadid S. Methylphenidate. StatPearls. Treasure Island (FL): StatPearls Publishing; 2024.
Costa A, Riedel M, Pogarell O, Menzel-Zelnitschek F, Schwarz M, Reiser M, et al. Methylphenidate effects on neural activity during response inhibition in healthy humans. Cereb Cortex. 2013;23:1179–89.
McCracken JT, Badashova KK, Posey DJ, Aman MG, Scahill L, Tierney E, et al. Positive effects of methylphenidate on hyperactivity are moderated by monoaminergic gene variants in children with autism spectrum disorders. Pharmacogenomics J. 2014;14:295–302.
Ventura P, de Giambattista C, Spagnoletta L, Trerotoli P, Cavone M, Di Gioia A, et al. Methylphenidate in autism spectrum disorder: a long-term follow up naturalistic study. J Clin Med. 2020;9:2566.
Schrantee A, Bouziane C, Bron EE, Klein S, Bottelier MA, Kooij JJS, et al. Long-term effects of stimulant exposure on cerebral blood flow response to methylphenidate and behavior in attention-deficit hyperactivity disorder. Brain Imaging Behav. 2018;12:402–10.
Monden Y, Dan H, Nagashima M, Dan I, Kyutoku Y, Okamoto M, et al. Clinically-oriented monitoring of acute effects of methylphenidate on cerebral hemodynamics in ADHD children using fNIRS. Clin Neurophysiol. 2012;123:1147–57.
Dipasquale O, Martins D, Sethi A, Veronese M, Hesse S, Rullmann M, et al. Unravelling the effects of methylphenidate on the dopaminergic and noradrenergic functional circuits. Neuropsychopharmacology. 2020;45:1482–9.
Leijenaar JF, Groeneveld GJ, Klaassen ES, Leeuwis AE, Scheltens P, Weinstein HC, et al. Methylphenidate and galantamine in patients with vascular cognitive impairment-the proof-of-principle study STREAM-VCI. Alzheimers Res Ther. 2020;12:10.
Schrantee A, Mutsaerts H, Bouziane C, Tamminga H, Bottelier MA, Reneman L. The age-dependent effects of a single-dose methylphenidate challenge on cerebral perfusion in patients with attention-deficit/hyperactivity disorder. Neuroimage Clin. 2017;13:123–9.
Carrier M, Guilbert J, Lévesque JP, Tremblay M, Desjardins M. Structural and functional features of developing brain capillaries, and their alteration in schizophrenia. Front Cell Neurosci. 2020;14:595002.
Greene C, Kealy J, Humphries MM, Gong Y, Hou J, Hudson N, et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol Psychiatry. 2018;23:2156–66.
Pillai A, Howell KR, Ahmed AO, Weinberg D, Allen KM, Bruggemann J, et al. Association of serum VEGF levels with prefrontal cortex volume in schizophrenia. Mol Psychiatry. 2016;21:686–92.
Casas BS, Vitória G, do Costa MN, Madeiro da Costa R, Trindade P, Maciel R, et al. hiPSC-derived neural stem cells from patients with schizophrenia induce an impaired angiogenesis. Transl Psychiatry. 2018;8:48.
Stankovic I, Notaras M, Wolujewicz P, Lu T, Lis R, Ross ME, et al. Schizophrenia endothelial cells exhibit higher permeability and altered angiogenesis patterns in patient-derived organoids. Transl Psychiatry. 2024;14:53.
Argaw AT, Gurfein BT, Zhang Y, Zameer A, John GR. VEGF-mediated disruption of endothelial CLN-5 promotes blood-brain barrier breakdown. Proc Natl Acad Sci USA. 2009;106:1977–82.
Trindade P, Nascimento JM, Casas BS, Monteverde T, Gasparotto J, Ribeiro CT, et al. Induced pluripotent stem cell-derived astrocytes from patients with schizophrenia exhibit an inflammatory phenotype that affects vascularization. Mol Psychiatry. 2023;28:871–82.
Katsel P, Roussos P, Pletnikov M, Haroutunian V. Microvascular anomaly conditions in psychiatric disease. Schizophrenia - angiogenesis connection. Neurosci Biobehav Rev. 2017;77:327–39.
Rampino A, Annese T, Torretta S, Tamma R, Falcone RM, Ribatti D. Involvement of vascular endothelial growth factor in schizophrenia. Neurosci Lett. 2021;760:136093.
Cantu Gutierrez ME, Hill MC, Largoza GE, Gillespie WB 3rd, Martin JF, Wythe JD. Mapping the transcriptional and epigenetic landscape of organotypic endothelial diversity in the developing and adult mouse. Nat Cardiovasc Res. 2025;473–495. https://doi.org/10.1038/s44161-025-00618-0.
Sabbagh MF, Heng JS, Luo C, Castanon RG, Nery JR, Rattner A, et al. Transcriptional and epigenomic landscapes of CNS and non-CNS vascular endothelial cells. Elife. 2018;7:e36187.
Wakabayashi T, Naito H. Cellular heterogeneity and stem cells of vascular endothelial cells in blood vessel formation and homeostasis: insights from single-cell RNA sequencing. Front Cell Dev Biol. 2023;11:1146399.
Yu M, Nie Y, Yang J, Yang S, Li R, Rao V, et al. Integrative multi-omic profiling of adult mouse brain endothelial cells and potential implications in Alzheimer’s disease. Cell Rep. 2023;42:113392.
Sadanandan J, Thomas S, Mathew IE, Huang Z, Blackburn SL, Tandon N, et al. Key epigenetic and signaling factors in the formation and maintenance of the blood-brain barrier. Elife. 2024;12:RP86978. https://doi.org/10.7554/eLife.86978.
Jeong HW, Diéguez-Hurtado R, Arf H, Song J, Park H, Kruse K, et al. Single-cell transcriptomics reveals functionally specialized vascular endothelium in brain. Elife. 2022;11:e57520.
Jia C, Keasey MP, Malone HM, Lovins C, Sante RR, Razskazovskiy V, et al. Vitronectin from brain pericytes promotes adult forebrain neurogenesis by stimulating CNTF. Exp Neurol. 2019;312:20–32.
Nikolakopoulou AM, Montagne A, Kisler K, Dai Z, Wang Y, Huuskonen MT, et al. Pericyte loss leads to circulatory failure and pleiotrophin depletion causing neuron loss. Nat Neurosci. 2019;22:1089–98.
Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci. 2017;20:406–16.
Ambrosino S, de Zeeuw P, Wierenga LM, van Dijk S, Durston S. What can cortical development in attention-deficit/hyperactivity disorder teach us about the early developmental mechanisms involved? Cereb Cortex. 2017;27:4624–34.
Fujimoto A, Enoki H, Niimi K, Nozaki T, Baba S, Shibamoto I, et al. Epilepsy in patients with focal cortical dysplasia may be associated with autism spectrum disorder. Epilepsy Behav. 2021;120:107990.
Guerrini R, Dobyns WB. Malformations of cortical development: clinical features and genetic causes. Lancet Neurol. 2014;13:710–26.
Leventer RJ, Guerrini R, Dobyns WB. Malformations of cortical development and epilepsy. Dialogues Clin Neurosci. 2008;10:47–62.
Shaw P, Gilliam M, Liverpool M, Weddle C, Malek M, Sharp W, et al. Cortical development in typically developing children with symptoms of hyperactivity and impulsivity: support for a dimensional view of attention deficit hyperactivity disorder. Am J Psychiatry. 2011;168:143–51.
Garcia-Forn M, Boitnott A, Akpinar Z, De Rubeis S. Linking autism risk genes to disruption of cortical development. Cells. 2020;9:2500.
Lindvall O, Kokaia Z. Neurogenesis following stroke affecting the adult brain. Cold Spring Harb Perspect Biol. 2015;7:a019034.
Naseer S, Abelleira-Hervas L, Savani D, de Burgh R, Aleksynas R, Donat CK, et al. Traumatic brain injury leads to alterations in contusional cortical miRNAs involved in dementia. Biomolecules. 2022;12:1457.
Romito-DiGiacomo RR, Menegay H, Cicero SA, Herrup K. Effects of Alzheimer’s disease on different cortical layers: the role of intrinsic differences in Abeta susceptibility. J Neurosci. 2007;27:8496–504.
Jenny B, Kanemitsu M, Tsupykov O, Potter G, Salmon P, Zgraggen E, et al. Fibroblast growth factor-2 overexpression in transplanted neural progenitors promotes perivascular cluster formation with a neurogenic potential. Stem Cell. 2009;27:1309–17.
Lassiter CM, Gal JS, Becker S, Hartman NW, Grabel L. Embryonic stem cell-derived neural progenitors transplanted to the hippocampus migrate on host vasculature. Stem Cell Res. 2016;16:579–88.
Hill J, Cave J. Targeting the vasculature to improve neural progenitor transplant survival. Transl Neurosci. 2015;6:162–7.
Acknowledgements
PKT acknowledges support from NIH grants R01 (R01NS121339) and UT Health startup funds. Figures were generated using the website Biorender.com under a license to UT Health Houston.
Author information
Authors and Affiliations
Contributions
PKT conceived the study. PKT and LKA wrote the manuscript. DWM and SLB read and provided comments to improve the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ahirwar, L.K., Blackburn, S.L., McBride, D.W. et al. Reviewing vascular influences on neuronal migration, cortical development, and neurodevelopmental disorders: focus on autism, ADHD and schizophrenia. Mol Psychiatry 30, 5953–5966 (2025). https://doi.org/10.1038/s41380-025-03200-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41380-025-03200-z
