
Abstract
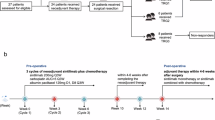
Esophageal squamous cell carcinoma (ESCC) presents significant clinical and therapeutic challenges due to its aggressive nature and generally poor prognosis. We initiated a Phase II clinical trial (ChiCTR1900027160) to assess the efficacy of a pioneering neoadjuvant chemo-immunotherapy regimen comprising programmed death-1 (PD-1) blockade (Toripalimab), nanoparticle albumin-bound paclitaxel (nab-paclitaxel), and the oral fluoropyrimidine derivative S-1, in patients with locally advanced ESCC. This study uniquely integrates clinical outcomes with advanced spatial proteomic profiling using Imaging Mass Cytometry (IMC) to elucidate the dynamics within the tumor microenvironment (TME), focusing on the mechanistic interplay of resistance and response. Sixty patients participated, receiving the combination therapy prior to surgical resection. Our findings demonstrated a major pathological response (MPR) in 62% of patients and a pathological complete response (pCR) in 29%. The IMC analysis provided a detailed regional assessment, revealing that the spatial arrangement of immune cells, particularly CD8+ T cells and B cells within tertiary lymphoid structures (TLS), and S100A9+ inflammatory macrophages in fibrotic regions are predictive of therapeutic outcomes. Employing machine learning approaches, such as support vector machine (SVM) and random forest (RF) analysis, we identified critical spatial features linked to drug resistance and developed predictive models for drug response, achieving an area under the curve (AUC) of 97%. These insights underscore the vital role of integrating spatial proteomics into clinical trials to dissect TME dynamics thoroughly, paving the way for personalized and precise cancer treatment strategies in ESCC. This holistic approach not only enhances our understanding of the mechanistic basis behind drug resistance but also sets a robust foundation for optimizing therapeutic interventions in ESCC.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on SpringerLink
- Instant access to the full article PDF.
USD 39.95
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others


Data availability
The IMC data can be accessed via OMIX/OMIX006370.
Code availability
Please note that the IMC analysis in this study does not involve the original code. To access the original codes used, please refer to the “Methods” section and the respective references provided. If you require any additional information to reanalyze the data presented in this paper, please contact the corresponding author directly.
References
Uhlenhopp DJ, Then EO, Sunkara T, Gaduputi V. Epidemiology of esophageal cancer: update in global trends, etiology and risk factors. Clin J Gastroenterol. 2020;13:1010–21.
Liu CQ, Ma YL, Qin Q, Wang PH, Luo Y, Xu PF, et al. Epidemiology of esophageal cancer in 2020 and projections to 2030 and 2040. Thorac Cancer. 2023;14:3–11.
Li J, Xu J, Zheng Y, Gao Y, He S, Li H, et al. Esophageal cancer: epidemiology, risk factors and screening. Chin J Cancer Res. 2021;33:535–47.
Deybasso HA, Roba KT, Nega B, Belachew T. Dietary and environmental determinants of oesophageal cancer in arsi zone, oromia, central ethiopia: a case-control study. Cancer Manag Res. 2021;13:2071–82.
Gao YB, Chen ZL, Li JG, Hu XD, Shi XJ, Sun ZM, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097–102.
Ajani JA, D’Amico TA, Bentrem DJ, Cooke D, Corvera C, Das P, et al. Esophageal and esophagogastric junction cancers, version 2.2023, NCCN clinical practice guidelines in oncology. J Natl Compr Canc Netw. 2023;21:393–422.
Kitagawa Y, Uno T, Oyama T, Kato K, Kato H, Kawakubo H, et al. Esophageal cancer practice guidelines 2017 edited by the Japan Esophageal Society: part 1. Esophagus. 2019;16:1–24.
Lordick F, Hölscher AH, Haustermans K, Wittekind C. Multimodal treatment of esophageal cancer. Langenbecks Arch Surg. 2013;398:177–87.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: Globocan estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Topalian SL, Taube JM, Pardoll DM. Neoadjuvant checkpoint blockade for cancer immunotherapy. Science. 2020;367.
Jiang Y, Chen M, Nie H, Yuan Y. Pd-1 and pd-l1 in cancer immunotherapy: clinical implications and future considerations. Hum Vaccin Immunother. 2019;15:1111–22.
Chen Y, Liu R, Li C, Song Y, Liu G, Huang Q, et al. Nab-paclitaxel promotes the cancer-immunity cycle as a potential immunomodulator. Am J Cancer Res. 2021;11:3445–60.
Saif MW, Syrigos KN, Katirtzoglou NA. S-1: a promising new oral fluoropyrimidine derivative. Expert Opin Investig Drugs. 2009;18:335–48.
Kozai H, Ogino H, Mitsuhashi A, Nguyen NT, Tsukazaki Y, Yabuki Y, et al. Potential of fluoropyrimidine to be an immunologically optimal partner of immune checkpoint inhibitors through inducing immunogenic cell death for thoracic malignancies. Thorac Cancer. 2024;15:369–78.
Zou G, Anwar J, Pizzi MP, Abdelhakeem A. Unraveling the intricacies of neoadjuvant immune checkpoint blockade in esophageal squamous cell carcinoma: a comprehensive single-cell perspective. J Thorac Dis. 2024;16:826–8.
Pei L, Liu Y, Liu L, Gao S, Gao X, Feng Y, et al. Roles of cancer-associated fibroblasts (cafs) in anti- pd-1/pd-l1 immunotherapy for solid cancers. Mol Cancer. 2023;22:29.
Fang J, Lu Y, Zheng J, Jiang X, Shen H, Shang X, et al. Exploring the crosstalk between endothelial cells, immune cells, and immune checkpoints in the tumor microenvironment: new insights and therapeutic implications. Cell Death Dis. 2023;14:586.
Di Martino JS, Akhter T, Bravo-Cordero JJ. Remodeling the ECM: implications for metastasis and tumor dormancy. Cancers. 2021;13.
Che G, Yin J, Wang W, Luo Y, Chen Y, Yu X, et al. Circumventing drug resistance in gastric cancer: a spatial multi-omics exploration of chemo and immuno-therapeutic response dynamics. Drug Resist Updat. 2024;74:101080.
Bao X, Li Q, Chen D, Dai X, Liu C, Tian W, et al. A multiomics analysis-assisted deep learning model identifies a macrophage-oriented module as a potential therapeutic target in colorectal cancer. Cell Rep. Med. 2024;5:101399.
Kiessling P, Kuppe C. Spatial multi-omics: novel tools to study the complexity of cardiovascular diseases. Genome Med. 2024;16:14.
Baharlou H, Canete NP, Cunningham AL, Harman AN, Patrick E. Mass cytometry imaging for the study of human diseases-applications and data analysis strategies. Front Immunol. 2019;10:2657.
Ji Y, Sun D, Zhao Y, Tang J, Tang J, Song J, et al. A high-throughput mass cytometry barcoding platform recapitulating the immune features for HCC detection. Nano Today. 2023;52:101940.
Rörby E, Adolfsson J, Hultin E, Gustafsson T, Lotfi K, Cammenga J, et al. Multiplexed single‐cell mass cytometry reveals distinct inhibitory effects on intracellular phosphoproteins by midostaurin in combination with chemotherapy in AML cells. Exp Hematol Oncol. 2021;10:7.
Du J, Zhang J, Wang L, Wang X, Zhao Y, Lu J, et al. Selective oxidative protection leads to tissue topological changes orchestrated by macrophage during ulcerative colitis. Nat Commun. 2023;14:3675.
Sheng J, Zhang J, Wang L, Tano V, Tang J, Wang X, et al. Topological analysis of hepatocellular carcinoma tumour microenvironment based on imaging mass cytometry reveals cellular neighbourhood regulated reversely by macrophages with different ontogeny. Gut. 2022;71:1176–91.
Shao W, Shi H, Liu J, Zuo Y, Sun L, Xia T, et al. Multi-instance multi-task learning for joint clinical outcome and genomic profile predictions from the histopathological images. IEEE Trans Med Imaging. 2024;43:2266–78.
Shao W, Zuo Y, Shi Y, Wu Y, Tang J, Zhao J, et al. Characterizing the survival-associated interactions between tumor-infiltrating lymphocytes and tumors from pathological images and multi-omics data. IEEE Trans Med Imaging. 2023;42:3025–35.
Shao W, Liu J, Zuo Y, Qi S, Hong H, Sheng J, et al. Fam3l: feature-aware multi-modal metric learning for integrative survival analysis of human cancers. IEEE Trans Med Imaging. 2023;42:2552–65.
Hamamoto R, Koyama T, Kouno N, Yasuda T, Yui S, Sudo K. et al. Introducing AI to the molecular tumor board: One direction toward the establishment of precision medicine using large-scale cancer clinical and biological information. Exp Hematol Oncology. 2022;11:82.
Health Commission Of The People’s Republic Of China N. National guidelines for diagnosis and treatment of esophageal carcinoma 2022 in China (English version). Chin J Cancer Res. 2022;34:309–34.
Bottlaender L, Amini-Adle M, Maucort-Boulch D, Robinson P, Thomas L, Dalle S. Cutaneous adverse events: a predictor of tumour response under anti-pd-1 therapy for metastatic melanoma, a cohort analysis of 189 patients. J Eur Acad Dermatol Venereol. 2020;34:2096–105.
Chaw S, Majeed AA, Dalley A, Chan A, Stein S, Farah C. Epithelial to mesenchymal transition (EMT) biomarkers–e-cadherin, beta-catenin, APC and vimentin–in oral squamous cell carcinogenesis and transformation. Oral Oncol. 2012;48:997–1006.
Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LM, et al. Ido2 is critical for ido1-mediated t-cell regulation and exerts a non-redundant function in inflammation. Int Immunol. 2014;26:357–67.
Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100a8/a9 in inflammation. Front Immunol. 2018;9:1298.
Paver EC, Cooper WA, Colebatch AJ, Ferguson PM, Hill SK, Lum T, et al. Programmed death ligand-1 (pd-l1) as a predictive marker for immunotherapy in solid tumours: a guide to immunohistochemistry implementation and interpretation. Pathology. 2021;53:141–56.
Yang M, Lu J, Zhang G, Wang Y, He M, Xu Q, et al. Cxcl13 shapes immunoactive tumor microenvironment and enhances the efficacy of PD-1 checkpoint blockade in high-grade serous ovarian cancer. J Immunother Cancer. 2021;9.
Feng X, Zhang L, Acharya C, An G, Wen K, Qiu L, et al. Targeting cd38 suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23:4290–4300.
Revel M, Sautès-Fridman C, Fridman W-H, Roumenina LT. C1q+ macrophages: passengers or drivers of cancer progression. Trends Cancer. 2022;8:517–26.
Zhao YX, Song JY, Bao XW, Zhang JL, Wu JC, Wang LY, et al. Single-cell RNA sequencing-guided fate-mapping toolkit delineates the contribution of yolk sac erythro-myeloid progenitors. Cell Rep. 2023;42:113364.
Guo G, Sun L, Yang L, Xu H. Ido1 depletion induces an anti-inflammatory response in macrophages in mice with chronic viral myocarditis. Cell Cycle. 2019;18:2598–613.
Bill R, Wirapati P, Messemaker M, Roh W, Zitti B, Duval F, et al. Cxcl9: Spp1 macrophage polarity identifies a network of cellular programs that control human cancers. Science. 2023;381:515–24.
Zhu S, Yi M, Wu Y, Dong B, Wu K. Roles of tumor-associated macrophages in tumor progression: implications on therapeutic strategies. Exp Hematol Oncol. 2021;10:60.
Wang C, Chen L, Fu D, Liu W, Puri A, Kellis M, et al. Antigen presenting cells in cancer immunity and mediation of immune checkpoint blockade. Clin Exp Metastasis. 2024:1–17.
Zhang Q, Wu S. Tertiary lymphoid structures are critical for cancer prognosis and therapeutic response. Front Immunol. 2023;13:1063711.
Hoch T, Schulz D, Eling N, Gómez JM, Levesque MP, Bodenmiller B. Multiplexed imaging mass cytometry of the chemokine milieus in melanoma characterizes features of the response to immunotherapy. Sci Immunol. 2022;7:eabk1692.
Lu C, Liu Y, Ali NM, Zhang B, Cui X. The role of innate immune cells in the tumor microenvironment and research progress in anti-tumor therapy. Front Immunol. 2023;13:1039260.
Hou J, Xie S, Gao J, Jiang T, Zhu E, Yang X, et al. NK cell transfer overcomes resistance to PD-(l)1 therapy in aged mice. Exp Hematol Oncol 2024;13:48.
Mentlik James A, Cohen AD, Campbell KS. Combination immune therapies to enhance anti-tumor responses by NK cells. Front Immunol. 2013;4:481.
Risom T, Glass DR, Averbukh I, Liu CC, Baranski A, Kagel A, et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell. 2022;185:299–310.e218.
Speiser JL, Miller ME, Tooze J, Ip E. A comparison of random forest variable selection methods for classification prediction modeling. Expert Syst Appl. 2019;134:93–101.
Dobson AJ, Barnett AG. An introduction to generalized linear models. 3rd ed. Boca Raton, FL: Chapman and Hall/CRC; 2018.
Zhang Z, Zhang Z. Artificial Neural Network. In: Multivariate Time Series Analysis in Climate and Environmental Research. Cham, Switzerland: Springer; 2018. pp. 1–35.
Latha R, Venkatachalam K, Al-Amri JF, Abouhawwash M. Shrinkage linear with quadratic Gaussian discriminant analysis for big data classification. Intell Autom Soft Comput. 2022;34:1803–1818.
Zheng Y, Chen Z, Han Y, Han L, Zou X, Zhou B, et al. Immune suppressive landscape in the human esophageal squamous cell carcinoma microenvironment. Nat Commun. 2020;11:6268.
Lazăr DC, Avram MF, Romoșan I, Cornianu M, Tăban S, Goldiș A. Prognostic significance of tumor immune microenvironment and immunotherapy: novel insights and future perspectives in gastric cancer. World J Gastroenterol. 2018;24:3583.
Tang J, Sheng J, Zhang Q, Ji Y, Wang X, Zhang J, et al. Runx3-overexpression cooperates with ex vivo akt inhibition to generate receptor-engineered t cells with better persistence, tumor-residency, and antitumor ability. J Immunother Cancer. 2023;11.
Sheng J, Wu J, Yin X, Sun Z, Wang X, Zhang J, et al. Synergetic treatment of oxygen microcapsules and lenvatinib for enhanced therapy of HCC by alleviating hypoxia condition and activating anti-tumor immunity. Chin Chem Lett. 2023;34:107738.
Ling Y, Zhong J, Weng Z, Lin G, Liu C, Pan C, et al. The prognostic value and molecular properties of tertiary lymphoid structures in oesophageal squamous cell carcinoma. Clin Transl Med. 2022;12:e1074.
Huang T-C, Liang C-W, Li Y-I, Guo J-C, Lin C-C, Lee J-M, et al. The prognostic role of tertiary lymphoid structure (TLS) in locally advanced esophageal squamous cell carcinoma (ESCC). Cancer Res. 2023;83:2211–2211.
Li R, Huang X, Yang W, Wang J, Liang Y, Zhang T, et al. Tertiary lymphoid structures favor outcome in resected esophageal squamous cell carcinoma. J Pathol: Clin Res. 2022;8:422–35.
Zhang T, Ren Y, Yang P, Wang J, Zhou H. Cancer-associated fibroblasts in pancreatic ductal adenocarcinoma. Cell Death Dis. 2022;13:897.
Toledo B, Zhu Chen L, Paniagua-Sancho M, Marchal JA, Perán M, Giovannetti E. Deciphering the performance of macrophages in tumour microenvironment: a call for precision immunotherapy. J Hematol Oncol. 2024;17:44.
Liu Y, Zugazagoitia J, Ahmed FS, Henick BS, Gettinger SN, Herbst RS, et al. Immune cell PD-l1 colocalizes with macrophages and is associated with outcome in PD-1 pathway blockade therapy. Clin Cancer Res. 2020;26:970–7.
Kazemi MH, Sadri M, Najafi A, Rahimi A, Baghernejadan Z, Khorramdelazad H, et al. Tumor-infiltrating lymphocytes for treatment of solid tumors: It takes two to tango? Front Immunol. 2022;13:1018962.
Chen Y, Kim J, Yang S, Wang H, Wu CJ, Sugimoto H, et al. Type I collagen deletion in αSMA(+) myofibroblasts augments immune suppression and accelerates progression of pancreatic cancer. Cancer Cell. 2021;39:548–565.e546.
Monteran L, Erez N. The dark side of fibroblasts: cancer-associated fibroblasts as mediators of immunosuppression in the tumor microenvironment. Front Immunol. 2019;10:1835.
Qi J, Sun H, Zhang Y, Wang Z, Xun Z, Li Z, et al. Single-cell and spatial analysis reveal interaction of FAP+ fibroblasts and SPP1+ macrophages in colorectal cancer. Nat Commun. 2022;13:1742.
Sojka DK, Huang YH, Fowell DJ. Mechanisms of regulatory T-cell suppression - a diverse arsenal for a moving target. Immunology. 2008;124:13–22.
Xin S, Liu X, Li Z, Sun X, Wang R, Zhang Z, et al. ScRNA-seq revealed an immunosuppression state and tumor microenvironment heterogeneity related to lymph node metastasis in prostate cancer. Experimental Hematology &. Exp. Hematol Oncol. 2023;12:49.
Bao X, Li Q, Chen J, Chen D, Ye C, Dai X, et al. Molecular subgroups of intrahepatic cholangiocarcinoma discovered by single-cell RNA sequencing–assisted multiomics analysis. Cancer Immunol Res. 2022;10:811–28.
Tharp KM, Kersten K, Maller O, Timblin GA, Stashko C, Canale FP, et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat Cancer. 2024. https://doi.org/10.1038/s43018-024-00775-4.
Funding
This work was supported by the National Key Research and Development Program of China (grant 2019YFA0803000 to JS), the Excellent Youth Foundation of Zhejiang Scientific (grant R22H1610037 to JS), the National Natural Science Foundation of China (grant 82173078 to JS), the Natural Science Foundation of Zhejiang Province (grant 2022C03037 to JS). Supported by the Henan Provincial Department of Science and Technology, No. 212102310047. Supported by the Henan Provincial Health Commission, No. SBGJ202003005.
Author information
Authors and Affiliations
Contributions
CW and GZ conceptualized the study, designed the experiments, and drafted the manuscript. LW conducted data analysis and figure preparation. All three authors contributed equally to this work. ZJ and HT performed the experimental work. QL and JL performed the RF analysis. YH and XH organized the clinical trial and obtained the clinical samples. GZ assisted in coordinating the clinical trials. WZ and JS contributed to proofreading the manuscript and reviewed the manuscript. YH and XH supervised the project. JH helped with the manuscript review. JS secured funding.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41388_2024_3123_MOESM1_ESM.pdf (download PDF )
Correlation between skin rashes and treatment outcomes. Distribution of patients with and without rashes among those exhibiting good (0–1) and poor (2–3) responses.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, C., Zhang, G., Wang, L. et al. Spatial proteomic profiling elucidates immune determinants of neoadjuvant chemo-immunotherapy in esophageal squamous cell carcinoma. Oncogene 43, 2751–2767 (2024). https://doi.org/10.1038/s41388-024-03123-z
Received:
Revised:
Accepted:
Published:
Version of record:
Issue date:
DOI: https://doi.org/10.1038/s41388-024-03123-z
This article is cited by
-
Tertiary lymphoid structures in cancer: spatiotemporal heterogeneity, immune orchestration, and translational opportunities
Journal of Hematology & Oncology (2025)
-
AI-based neoadjuvant immunotherapy response prediction across pan-cancer: a comprehensive review
Cancer Cell International (2025)
-
Neoadjuvant serplulimab combined with chemotherapy for resectable oesophageal squamous cell carcinoma: a single-arm, phase 2 trial
Nature Communications (2025)
-
Spatial proteomics for investigating solid tumor resistance mechanisms
Cancer and Metastasis Reviews (2025)
