
Abstract
RNA modification, a prominent epigenetic mechanism, has been implicated in regulating RNA function, stability, processing, and interactions, including pseudouridylation, acetylation, and methylation. Recent evidence highlights that 5-methylcytosine (m5C) influences key cellular processes such as proliferation, differentiation, apoptosis, and stress responses by modulating RNA stability, translation, transcription, nuclear export, and cleavage. This review consolidates current insights into the role and mechanisms of m5C methylation across various tumor types, underscoring its pivotal involvement in post-transcriptional regulation and its profound effects on gene expression, cellular dynamics, and tumor biology. The mechanisms through which m5C methylation impacts tumor progression, including modulation of glucose and iron metabolism, as well as resistance to therapeutic agents, are also discussed. Finally, the review identifies critical future research avenues, focusing on elucidating the underlying mechanisms, developing targeted therapies, and advancing personalized medicine approaches to leverage m5C methylation in cancer treatment.
Similar content being viewed by others

Introduction
Epigenetics has transformed our understanding of gene regulation and cellular identity by focusing on modifications beyond the DNA sequence itself [1, 2]. These modifications regulate gene transcription, splicing, stability, and chromatin structure, influencing both physiological and pathological cellular processes. Common epigenetic modifications include DNA methylation, histone modifications, non-coding RNA modifications, and RNA modifications [3,4,5]. Among these, DNA methylation has been extensively studied due to its critical role in gene expression and chromatin dynamics. However, recent attention has increasingly turned to RNA methylation, which plays a pivotal role in post-transcriptional regulation and gene expression [6,7,8].
RNA methylation refers to a chemical modification process involving the addition of methyl groups to RNA molecules [6,7,8,9,10,11,12]. This modification regulates gene expression, maintains RNA stability, and plays critical roles in biological development, disease pathogenesis, and progression [13, 14]. Among these modifications, N6-methyladenosine (m6A) is the most prevalent and extensively studied RNA methylation in eukaryotes. It is distributed across diverse RNA types, including mRNA, long non-coding RNA (lncRNA), and circular RNA (circRNA). 5-methylcytosine (m5C) modifications are commonly found in tRNA, ribosomal RNA (rRNA), and specific regions of mRNA, where they influence RNA folding, stability, and protein interactions. In contrast, N1-methyladenosine (m1A) is a structurally distinct modification predominantly localized to tRNA and rRNA, where it alters RNA secondary structure and functional properties [14,15,16,17].
Among these modifications, m5C has emerged as a critical regulator of gene expression. Accumulating evidence indicates that m5C modulates RNA stability, translation, transcription, nuclear export, and cleavage, thereby influencing cellular proliferation, differentiation, apoptosis, stress responses, and other biological functions [18,19,20,21,22]. For example, one study demonstrated that Aly/REF (ALYREF) supports colorectal cancer (CRC) growth and migration by promoting RNA m5C recognition and nuclear export through recruitment of ELAVL1 [23]. Another study showed that enhancing RNA m5C modification via the NSUN2/ALYREF pathway promotes hexavalent chromium [Cr(VI)]-induced malignant transformation and lung cancer by accelerating metabolic reprogramming [24]. Furthermore, Xing et al. reported that NSUN2 regulates the Wnt signaling pathway through m5C modification, thereby promoting hepatocellular carcinoma (HCC) progression [25]. Building on these foundational findings, this review aims to consolidate current insights into the roles and mechanisms of m5C methylation across various tumor types. By synthesizing the latest research, the review seeks to provide a comprehensive understanding of how m5C methylation contributes to tumor development and progression, as well as its potential as a therapeutic target.
Three regulators of RNA m5C; “readers”, “writers” and “erasers”
RNA methylation is mediated by three classes of proteins: “writers,” which catalyze the addition of methyl groups; “readers,” which recognize these modifications; and “erasers,” which remove them (Table 1) [11, 26,27,28,29]. Each class of proteins operates through distinct mechanisms (Fig. 1). These proteins regulate various RNA types and associated signaling pathways, including mRNA, tRNA, lncRNA, and small RNAs (sRNAs) [15, 16, 29]. The expression levels of these regulators across different tumor types, along with their roles in tumor progression and impact on prognosis, are summarized in Table 2.

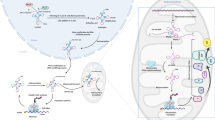
A “Writers” are methyltransferase enzymes that catalyze the addition of m5C methylation modifications. “Erasers” are demethylases responsible for removing these modifications. “Readers” recognize and bind specific m5C methylations, influencing RNA export, stabilization, homologous recombination, and pre-rRNA processing. B The role of m5C in various cancer types. C Common types of RNA involved in m5C methylation modification in cells. D Key Biological Processes involved in m5C methylation modification in tumors. Created with BioRender.com.
Writers
RNA cytosine methyltransferases (RCMTs) utilize S-adenosylmethionine (SAM) as a methyl donor to catalyze the methylation of the fifth carbon atom of cytosine, forming m5C. The “writers” of m5C methylation primarily consist of the NSUN family (NOP2/Sun RNA methyltransferase family members 1–7) and DNA methyltransferase 2 (DNMT2), which recognize specific RNA sequences or structures to catalyze m5C modifications.
NSUN1 (also known as NOP2, NOL1, or p120) primarily catalyzes m5C modification at position 4447 of 28S rRNA and participates in ribosome biogenesis. By forming non-catalytic complexes with box C/D small nucleolar RNAs (snoRNAs), NSUN1 regulates pre-rRNA processing, promotes the recruitment of U3 and U8 snoRNAs to pre-90S ribosomal particles, and maintains the stable assembly of snoRNP complexes [30].
NSUN2, one of the most extensively studied m5C methyltransferases, catalyzes m5C modifications on mRNA and non-coding RNAs such as tRNA and ribosomal RNA (rRNA) [31,32,33]. NSUN2 enhances mRNA stability and promotes nuclear export through m5C modifications. For example, NSUN2 stabilizes circular RNA 505 (circRNA505) via m5C modification and facilitates its nuclear transport through interaction with the binding protein ALY export/import factor (ALYREF) [33]. NSUN2-mediated tRNA modifications regulate tRNA conformation and function, improving mitochondrial tRNA translation efficiency [32].
NSUN3 specifically targets mitochondrial tRNA (mt-tRNA) for cytosine methylation, maintaining mitochondrial translation and oxidative phosphorylation (OXPHOS) [34].
NSUN4 modifies mitochondrial RNA (mtRNA), particularly the terminal m5C of light-strand lncRNA, recruiting polynucleotide phosphorylase (PNPase) via the C1QBP protein to regulate mitochondrial double-stranded RNA (mt-dsRNA) expression [35].
NSUN5 is a conserved rRNA methyltransferase that catalyzes m5C modification at position C3782 of human 28S rRNA and C3438 of mouse 28S rRNA [36]. In glioblastoma (GBM), NSUN5 knockdown significantly reduces protein synthesis, whereas overexpression enhances synthesis, suggesting that it directly regulates translational efficiency through rRNA methylation [37].
Previous studies have demonstrated that NSUN6 influences tRNA stability and function by methylating specific regions of tRNA, such as the variable loop or anticodon loop. This modification regulates tRNA folding and its interaction with ribosomes, thereby impacting translation efficiency [38]. Recent studies have revealed that NSUN6 not only targets tRNA but also specifically binds to the 3’ untranslated region (3’UTR) of mRNA, where it regulates mRNA stability and translation efficiency through m5C modification [39]. For example, in lung cancer, NSUN6 upregulates NM23-H1 expression by methylating m5C sites in the 3’UTR of NM23-H1 mRNA, thereby suppressing tumor cell proliferation, migration, and epithelial-mesenchymal transition (EMT) [40]. In cervical cancer, NSUN6 mediates m5C modification of the 3’UTR of NDRG1 mRNA, enhancing its binding to the m5C reader protein ALYREF. This interaction stabilizes NDRG1 mRNA, promoting homologous recombination repair and conferring resistance to radiotherapy [41]. In osteosarcoma (OS), NSUN6 maintains the stability of EEF1A2 mRNA in an m5C-dependent manner, driving tumor progression by activating the Akt/mTOR signaling pathway [42].
NSUN7 was initially identified as an enhancer RNA (eRNA) m5C methyltransferase [43]. In hepatocyte models, Aza-IP-seq combined with RNA immunoprecipitation quantitative PCR (RIP-qPCR) confirmed that NSUN7 specifically catalyzes m5C modifications on eRNAs associated with key genes such as PFK1, SIRT5, IDH3B, and HMOX2. Methylamp RNA bisulfite sequencing further demonstrated that NSUN7-mediated methylation significantly enhances the stability of these eRNAs.
DNMT2/TRDMT1, a member of the DNMT family, exhibits dual substrate specificity, methylating both DNA and RNA [44]. This suggests that other DNMT family members (e.g., DNMT1, DNMT3A, DNMT3B) may also possess RNA methylation activity, though their specific substrates and mechanisms require validation [45]. The NSUN family and DNMT2 regulate RNA metabolism and function by modifying distinct RNA molecules (mRNA, tRNA, rRNA), thereby influencing critical biological processes such as cell proliferation, mitochondrial energy metabolism, and DNA repair. Dysregulation of these enzymes is closely linked to cancer, neurodegenerative diseases, and metabolic disorders, highlighting their potential as therapeutic targets.
Readers
Readers recognize m5C-modified sites via specific domains to regulate RNA stability, localization, or translation. Core readers include ALYREF and Y-box binding protein 1 (YBX1).
ALYREF facilitates mRNA nucleocytoplasmic transport by recognizing m5C-modified mRNA. In nasopharyngeal carcinoma (NPC), ALYREF binds m5C sites on NOTCH1 mRNA, enhancing its stability and nuclear export [46]. Similar mechanisms are observed in esophageal cancer (EC), where ALYREF stabilizes TBL1XR1 and KMT2E mRNA by binding their m5C sites [47].
YBX1, a multifunctional m5C reader, plays key roles in cancer progression, viral replication, autophagy regulation, and bone metabolism. YBX1 stabilizes target mRNA by directly binding m5C sites. For example, in esophageal squamous cell carcinoma (ESCC), YBX1 stabilizes SMOX mRNA in an m5C-dependent manner, promoting tumor progression [48].
Serine/arginine-rich splicing factor 2 (SRSF2) is a validated m5C reader involved in RNA splicing and leukemogenesis. Leukemia-associated SRSF2 mutations (e.g., P95H) impair its binding to m5C-modified mRNA, reducing interactions with leukemia-related transcripts and potentially driving disease progression [49].
C1QBP (complement component 1Q binding protein) is identified as a reader of m5C-modified mitochondrial double-stranded RNA (mt-dsRNA). It recognizes NSUN4-catalyzed m5C sites (e.g., light-strand lncRNA termini) and recruits polynucleotide phosphorylase (PNPT1) to maintain mtRNA stability and mitochondrial gene expression [50].
In zebrafish, methyl-CpG binding domain protein 5 (MBD5) directly binds m5C-modified RNA, interacting with the Polycomb repressive deubiquitination (PR-DUB) complex to link RNA methylation with histone modifications (e.g., H2A-K119 ubiquitination) and transcriptional silencing [51].
Fragile X mental retardation protein (FMRP), a cytoplasmic RNA-binding protein, coordinates with the m5C writer TRDMT1 and eraser ten-eleven translocation 1 (TET1) to regulate mRNA-dependent DNA repair and cancer cell survival, revealing its role as a novel m5C reader [52, 53].
ALYREF and YBX1 are central readers in cancer, modulating epitranscriptomic regulation, chromatin interactions, and signaling pathways [54]. Their expression levels and functional states correlate with disease progression and treatment response, making them potential diagnostic markers and therapeutic targets [55].
Erasers
Erasers dynamically and reversibly regulate RNA methylation by removing m5C modifications. Key erasers include AlkB homolog 1 (ALKBH1) and the ten-eleven translocation (TET) family (TET1, TET2, TET3) [56, 57].
ALKBH1, a key RNA m5C demethylase, oxidizes m5C to 5-hydroxymethylcytosine (hm5C) and 5-formylcytosine (f5C) via its α-ketoglutarate- and Fe(II)-dependent dioxygenase activity [58]. 5-ethynylcytidine (5-EC) labeling revealed ALKBH1’s broad activity on tRNA and mRNA, particularly in ribosomal RNA processing and translational regulation [59].
TET proteins, initially known for oxidizing DNA 5-methylcytosine (5mC), also catalyze RNA m5C oxidation [60]. For example, TET2 binds chromatin-associated RNA (caRNA) to oxidize m5C to 5hmC, independent of its DNA methyltransferase activity [61]. TET2 dysfunction in gliomas reduces RNA m5C oxidation, potentially promoting tumor progression by altering oncogenic RNA stability [62].
Expression, regulation, and physiological functions of m5C machinery
The expression patterns of m5C regulators are not uniform and exhibit distinct tissue specificity, which underlies their context-dependent roles in oncogenesis. For instance, NSUN2 is highly expressed in tissues with inherently high regenerative and proliferative capacity, such as the testes, intestinal crypts, and skin [63]. This baseline expression profile predisposes these tissues to oncogenic transformation when NSUN2’s pro-growth functions are further amplified. Conversely, the aberrant de novo activation or overexpression of such regulators in tissues where they are normally silent, such as NSUN2 in the lung or liver, can unleash potent oncogenic activity, driving aggressive tumor phenotypes. This paradigm of tissue-specific vulnerability, governed by the physiological expression landscape of the m5C regulatory machinery, provides a critical framework for understanding why certain cancers differentially depend on specific epitranscriptomic pathways.
The expression and activity of m5C regulatory factors are governed by a multi-layered regulatory system operating at transcriptional, post-transcriptional, and post-translational levels [64, 65]. At the transcriptional level, their genes are regulated by oncogenic and tumor-suppressive signaling pathways; for example, TET2 transcription is modulated by diverse cellular signals, and its promoter methylation status is frequently altered in cancer. At the post-transcriptional level, miRNAs and lncRNAs fine-tune mRNA stability and translation efficiency. Post-translational modifications (PTMs), meanwhile, provide a rapid and reversible mechanism to precisely regulate protein activity, stability, and subcellular localization. NSUN2 can be post-translationally modified through both lysyl oxidation by lactate and SUMOylation [37, 66, 67]. The reader protein ALYREF is subject to ubiquitination. Recent studies by Huang et al. have demonstrated that RNF31-mediated ubiquitination of ALYREF represents a promising therapeutic target for reversing paclitaxel resistance in triple-negative breast cancer [68]. Collectively, these examples highlight that PTMs represent a widespread and essential regulatory mechanism across all functional classes of the m5C machinery.
Loss-of-function studies underscore the critical physiological importance of m5C regulators. Genetic deletion of key writers such as NSUN2 or NSUN3, or readers like YBX1, in murine models leads to embryonic lethality or severe postnatal phenotypes, including growth retardation and neurological deficits [69,70,71]. These outcomes demonstrate that m5C regulators perform non-redundant functions in fundamental biological processes, including protein synthesis, mitochondrial metabolism, and embryonic development. Therefore, their dysregulation in cancer typically does not confer novel oncogenic activities but rather reflects the aberrant activation or amplification of pre-existing physiological programs.
Moreover, the genes encoding m5C regulators are themselves frequent targets of genetic alterations in cancer. Somatic mutations lead to inactivation of erasers such as TET2 in myeloid leukemias, while copy number alterations result in amplification and overexpression of writers like NSUN2 and readers such as YBX1 across various solid tumors. These recurrent genomic changes provide strong evidence that m5C regulators act as bona fide drivers of tumorigenesis and are central to understanding the pathophysiological consequences of m5C epitranscriptomic dysregulation.
Role of m5C in various cancer types
The dysregulation of m5C methylation is a hallmark of numerous cancers, driven by aberrant expression, mutations, or altered activity of its regulatory machinery [72]. Although the specific target RNAs and downstream pathways differ across cancer types, recurring mechanistic themes have emerged. m5C modifications primarily promote tumorigenesis by enhancing the stability and translational efficiency of oncogenic transcripts, facilitating their nuclear export, and optimizing the translation of proteins critical for cell proliferation, survival, and metabolic reprogramming [6, 18, 73]. The following sections will examine these roles in a tissue-specific manner, highlighting both context-specific adaptations and conserved molecular principles that define the contribution of m5C to cancer biology (Table 2) (Figs. 2 and 3).

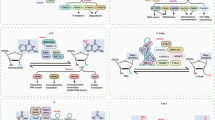
In Esophageal squamous cell carcinoma, YBX1 promotes ESCC progression via m5C-dependent SMOX mRNA stabilization. NSUN2-mediated RNA m5C promotes cancer progression via LIN28B-dependent GRB2 mRNA stabilization.NSUN6 mediated tRNA m5C modifications selectively enhance the translation efficiency of CDH1 mRNA in a codon dependent manner. In hepatocellular carcinoma, ALYREF-mediated RNA m5C modification promotes cancer progression via stabilizing EGFR mRNA and pSTAT3 activation. In addition, LINC00618 promotes the growth and metastasis of hepatocellular carcinoma by promoting NSUN2-mediated SREBP2 m5C modification and by enhancing cholesterol synthesis. These examples highlight the complex role of RNA m5C methylation in regulating the development of various gastrointestinal cancers. Created with BioRender.com.

THOC3 promotes the progression of lung squamous cell carcinoma by modifying PFKFB4 mRNA and interacting with YBX1. The m5C modification in PKM2 mRNA in the HIF-1α/ALYREF/PKM2 axis promotes the glucose metabolism of bladder cancer. In breast cancer, NSUN2/YBX1 enhances the stability of HGH1 mRNA through m5C methylation, thus promoting cancer progression. In head and neck squamous cell carcinoma, NSUN3 promotes tumor progression by regulating the immune invasion of the cancer. These examples highlight the complex role of RNA m5C methylation in regulating the development of other types of cancer. Created with BioRender.com.
Respiratory system
m5C methylation exerts multidimensional roles in the initiation and progression of lung cancer by regulating RNA stability, metabolic reprogramming, and activation of key signaling pathways. Its dynamic regulatory network involves the coordinated actions of methyltransferases, recognition proteins, and downstream effector molecules, exhibiting high heterogeneity across different lung cancer subtypes.
As a core Writer protein, NSUN2 drives lung cancer progression by targeting m5C modifications of key oncogenes. For instance, NSUN2 activates the PI3K-AKT signaling pathway by stabilizing PIK3R2 mRNA, promoting malignant transformation in LUAD [74]. Simultaneously, its synergistic interaction with ALYREF regulates YAP mRNA methylation, enhancing Hippo pathway activation mediated by YAP expression and inducing AZD9291 resistance via exosome secretion [75]. Notably, other NSUN family members exhibit differential regulation in lung cancer: NSUN4 remodels mitochondrial energy metabolism through the circERI3-DDB1-PGC-1α axis to promote tumorigenesis [76], while NSUN6 suppresses lung cancer cell proliferation and EMT by modifying the 3’ UTR of NM23-H1 mRNA, suggesting dual tumor-suppressive and oncogenic functions within this family [40]. The oncogenic effects of m5C modifications depend on Reader proteins for target RNA recognition and stabilization. For example, ALYREF enhances YAP1 mRNA stability by binding LINC02159, activating the Hippo/β-catenin signaling axis to drive NSCLC progression [77], while its collaboration with NOP2 stabilizes EZH2 mRNA to promote EMT [78]. Another critical Reader protein, YBX1, promotes glycolysis and tumor growth in LUSC by recognizing the 3’ UTR m5C site of PFKFB4 mRNA [79]. Additionally, the m5C-YBX1 regulatory axis formed by YBX1 and NSUN2 sustains NRF2 and QSOX1 expression, conferring resistance to ferroptosis and gefitinib in NSCLC cells [80,81,82], highlighting the close link between m5C modifications and oxidative stress or targeted therapy resistance. m5C methylation profoundly influences metabolic adaptability in lung cancer cells by regulating the stability of metabolic enzyme-encoding RNAs. For instance, THOC3 synergizes with YBX1 to facilitate PFKFB4 mRNA nucleocytoplasmic transport and stabilization, enhancing glycolytic capacity in LUSC [79], while NSUN4-mediated transcriptional activation of mitochondrial PGC-1α drives oxidative phosphorylation reprogramming [76]. Notably, m5C modifications also enable intercellular regulation via exosomes. NSUN2/ALYREF-regulated YAP mRNA transmitted via exosomes to neighboring cells may remodel the tumor immune microenvironment and promote drug-resistant clone expansion [75], underscoring the transcellular regulatory functions of m5C modifications in the lung cancer ecosystem.
Digestive system
In ESCC, m5C modifications drive tumor progression through a multidimensional regulatory network. Studies reveal that YBX1 stabilizes SMOX mRNA in an m5C-dependent manner, activating polyamine metabolism to promote ESCC development [48]. Notably, this process is not isolated; NSUN2, as a key methyltransferase, enhances GRB2 mRNA stability via a LIN28B-mediated mechanism, forming a positive feedback loop in growth signaling pathways [83]. Further research demonstrates that m5C regulation extends beyond mRNA stability: NSUN6-mediated tRNA m5C modifications significantly enhance CDH1 mRNA translation efficiency through codon preference selection, uncovering a novel dimension of epigenetic regulation at the protein translation level [84]. Importantly, NSUN2 genetic variants (cis-eQTL) not only promote ESCC progression via mRNA methylation but also correlate with radiotherapy resistance, providing an epigenetic explanation for clinical treatment resistance mechanisms [85].
In gastric cancer (GC), the m5C regulatory network exhibits complex hierarchical interactions. The HCP5-132aa protein bridges YBX1 and ELAVL1 to form a ternary complex that specifically recognizes m5C sites on SLC7A11 and G6PD mRNA. This synergy stabilizes ferroptosis-related transcripts and drives tumor progression via metabolic reprogramming [86]. Meanwhile, NSUN2 participates in gastric carcinogenesis through dual mechanisms: suppressing CDKN1C expression to relieve cell cycle arrest [81, 87] and modulating the Bcl-2/Bax balance to influence chemotherapy sensitivity [88]. Notably, m5C regulation extends to non-coding RNAs-methylated lncRNA NR_033928 remodels glutamine metabolism by stabilizing GLS mRNA, revealing crosstalk between metabolic reprogramming and epigenetic regulation [89]. In the tumor microenvironment, peritoneal adipocytes promote GC peritoneal metastasis via the AMPK/NSUN2/ORAI2 axis, connecting energy-sensing mechanisms with m5C networks [90]. Remarkably, the interaction between DIAPH2-AS1 and NSUN2 stabilizes NTN1 mRNA via ubiquitination regulation, offering new molecular insights into neural invasion mechanisms [91].
In CRC, the m5C regulatory network exhibits unique positive feedback features. NSUN6 not only catalyzes METTL3 m5C modifications to form an oncogenic loop [92], but its synergy with ALYREF enhances RPS6KB2/RPTOR mRNA export via nucleocytoplasmic transport mechanisms, highlighting the role of nuclear transport systems in epigenetic regulation [23]. More intricately, the NSUN2/YBX1/m5C-ENO1 signaling loop establishes a self-sustaining mechanism through histone H3K18 lactylation, dynamically coupling metabolic products with epigenetic modifications [93]. This multi-tiered regulation is further evidenced in SKIL mRNA stability control, confirming the central role of m5C networks in maintaining oncogenic transcripts [94]. Notably, m5C modifications on NXPH4 mRNA influence SQSTM1-mediated RNA autophagy, opening new research directions at the intersection of epigenetic regulation and autophagy [95].
In HCC, m5C modifications exhibit multi-target regulatory properties. ALYREF stabilizes EGFR mRNA to activate the STAT3 pathway, establishing an epigenetic-signal transduction axis [96], while NOP2-mediated XPD mRNA methylation reveals potential links between DNA repair mechanisms and epigenetic regulation [97]. At the metabolic level, the LINC00618/NSUN2 axis drives cholesterol synthesis by stabilizing SREBP2 mRNA, integrating non-coding RNA regulation with lipid metabolism reprogramming [98]. Importantly, NSUN2 modulates key Ras pathway nodes to influence sorafenib sensitivity, offering new strategies for targeted therapy [99]. Additionally, m5C modifications on H19 lncRNA drive tumor progression via the G3BP1/MYC axis, underscoring the pivotal role of non-coding RNA epigenetic modifications in oncogenic networks [100].
In other digestive tumors, m5C networks also play critical roles. In cholangiocarcinoma (CCA), NSUN2 promotes YBX1 binding by modifying NKILA, with its expression significantly correlating with clinical stage and metastasis [101]. In pancreatic cancer (PC), the NSUN2-TIAM2 axis and PIAT/YBX1-mediated neural invasion mechanisms form a complex progression network [102, 103]. Notably, ALYREF alters tumor microenvironment amino acid metabolism via the m5C-dependent JunD/SLC7A5 axis, revealing a novel mechanism by which epigenetic regulation influences immune microenvironments [104].
Urinary system
The study by Tian et al. first elucidated that NOP2 drives clear cell renal cell carcinoma (ccRCC) progression by stabilizing APOL1 mRNA via m5C modifications, activating the PI3K-Akt signaling axis and revealing interactions between epigenetic regulation and classical oncogenic pathways [105]. Notably, this regulatory mechanism exhibits multi-target specificity in ccRCC. The Yang team further discovered that PEBP1P2 downregulation promotes tumor metastasis by stabilizing PEBP1/KLF13 mRNA, suggesting that m5C modifications in ccRCC may form a hierarchical regulatory network through spatiotemporal-specific control of different targets [106]. Together, these studies outline dual dimensions of m5C regulation in ccRCC: directly activating signaling pathways to drive progression while modulating metastasis-related genes to influence disease evolution.
In bladder cancer research, the m5C network displays unique metabolic intervention properties. Wang et al. demonstrated that ALYREF-mediated PKM2 mRNA methylation enhances glycolysis to promote tumor proliferation, a process closely linked to the synergistic action of the YBX1/ELAVL1 complex [107]. Correspondingly, the Chen team revealed that the NSUN2/YBX1 axis targets the 3’ UTR m5C site of HDGF mRNA, constructing an epigenetic-clinical prognostic association network in urothelial carcinoma of the bladder (UCB). High expression of the triple marker (NSUN2/YBX1/HDGF) correlates significantly with poor patient survival [108]. These studies collectively delineate dual regulatory dimensions of m5C in bladder cancer: driving malignant phenotypes via metabolic reprogramming and influencing clinical outcomes through oncogenic transcript stabilization.
Prostate cancer research reveals self-reinforcing features of m5C modifications. Zhu et al. found that NSUN2 establishes a positive feedback loop by clustering m5C modifications on AR mRNA, dynamically coupling androgen signaling with epigenetic regulation [109]. Building on this, the Zhang team further dissected metabolic regulation: CDK13-mediated NSUN5 phosphorylation enhances lipid synthesis via ACC1 mRNA methylation, with ALYREF-dependent nuclear export playing a critical role [110]. This “kinase-methyltransferase-transporter” triad not only clarifies the epigenetic basis of lipid metabolic dysregulation in prostate cancer but also provides a theoretical foundation for combination therapies targeting the m5C network.
Female reproductive system
The role of m5C modifications in female reproductive cancers extends across multiple molecular pathways and cancer subtypes. For instance, NSUN2-YBX1 protein interactions have been identified as critical drivers of breast cancer progression, where they modulate HGH1 expression through site-specific m5C modifications [111]. Expanding on the oncogenic role of YBX1 in hormone-related cancers, subsequent studies revealed that SAT1 (spermidine/arginine N1-acetyltransferase 1) acts as a pivotal regulator in triple-negative breast cancer (TNBC) invasion by engaging the SAT1/YBX1/mTOR axis, highlighting its potential as a therapeutic target [112]. Beyond breast cancer, m5C-mediated RNA stabilization mechanisms also dominate in cervical cancer pathogenesis. Yu et al. demonstrated that NSUN6 induces m5C modifications in NDRG1 mRNA, enabling ALYREF binding to enhance mRNA stability, which promotes homologous recombination-mediated DNA repair and radioresistance [41]. This aligns with findings by Chen et al., who reported that NSUN2 upregulates LRRC8A protein levels via m5C modifications, with YBX1 further stabilizing LRRC8A RNA. Depleting NSUN2 suppressed cervical cancer proliferation and metastasis, underscoring the clinical relevance of the NSUN2-LRRC8A axis [113]. In endometrial cancer, m5C modifications take on a distinct role in apoptosis resistance. Chen et al. linked NSUN2-mediated m5C modifications of SLC7A11 mRNA to iron-induced apoptosis evasion, revealing an epigenetic mechanism underlying therapeutic resistance [114]. Similarly, in ovarian cancer, Meng et al. demonstrated that YBX1 recognizes m5C-modified CHD3 mRNA, stabilizing it via PABPC1 recruitment to enhance chromatin accessibility and homologous recombination repair. This process enables ovarian cancer cells to resist platinum-induced apoptosis, offering a mechanistic basis for overcoming chemoresistance [115]. Further emphasizing the interplay between m5C writers and transcription factors, Liu et al. uncovered a self-reinforcing loop in ovarian cancer: NSUN2 stabilizes E2F1 mRNA via m5C modifications, while E2F1 reciprocally activates NSUN2 transcription, accelerating tumor progression [116].
Nervous system
m5C modifications also play context-dependent roles in gliomas by regulating angiogenesis and immune evasion. A notable example involves glioblastoma endothelial cells (GECs), where Pan et al. observed elevated NSUN2 and LINC00324 levels. NSUN2 stabilizes LINC00324 through m5C modifications, which in turn protects CBX3 mRNA from degradation by competing with AUH binding. CBX3 then activates VEGFR2 transcription to drive angiogenesis, positioning this axis as a therapeutic target [117]. Conversely, in glioma immune regulation, Wu’s group revealed that NSUN5 suppresses tumor growth by promoting CTNNB1 (β-catenin) mRNA degradation. NSUN5-mediated m5C modifications enable TET2 to oxidize m5C to 5hmC on caRNA, which recruits RBFOX2 to degrade these transcripts. This NSUN5/TET2/RBFOX2 network enhances phagocytosis by tumor-associated macrophages (TAMs), suggesting a dual role for m5C in both tumor promotion and immune activation [60].
Blood system
In hematological malignancies, m5C modifications influence both tumor cell behavior and microenvironmental interactions. For instance, Yu et al. demonstrated that NSUN2 and YBX1 upregulate LncRNA MALAT1 via m5C in multiple myeloma (MM) cells. MALAT1 is shuttled to osteoclasts via exosomes, exacerbating bone destruction—a key clinical complication in MM [118]. Beyond RNA stability, m5C also regulates chromatin dynamics in leukemia. Zou et al. discovered that m5C-modified anti-transposon RNA recruits MBD6 to deubiquitinate H2AK119ub, enhancing chromatin accessibility. TET2 counteracts this by oxidizing m5C, and its loss in leukemias creates dependency on MBD6-mediated gene activation. Targeting MBD6 selectively inhibits TET2-mutated leukemia growth, offering a precision therapy strategy [119].
The critical role of TET2 mutations in leukemia highlights the broader pathophysiological consequences of genetic alterations in m5C regulators. Somatic mutations in the m5C eraser TET2 are among the most frequent epigenetic alterations in hematological malignancies, particularly in myeloproliferative neoplasms (MPNs) and acute myeloid leukemia (AML). These loss-of-function mutations disrupt normal hematopoietic differentiation and are recognized as early drivers of leukemogenesis. While the role of TET2 in DNA demethylation is well established, its function as an RNA m5C eraser adds a crucial layer to its tumor-suppressive activity. The consequent loss of TET2-mediated RNA demethylation contributes to the stabilization of oncogenic transcripts and creates a therapeutic vulnerability—such as dependency on the MBD6 pathway—as described above [119]. This paradigm underscores that beyond the expression levels of the m5C machinery, the genetic alteration status of these regulators (e.g., mutations in TET2 or SRSF2 49) is central to understanding disease etiology and to developing targeted therapies for specific molecular subtypes of leukemia.
Other cancer types
The oncogenic impact of m5C extends to diverse solid tumors through metabolic and translational reprogramming. In osteosarcoma, Yang et al. found that NSUN2 stabilizes FABP5 mRNA via m5C, enhancing fatty acid metabolism to fuel tumor progression [81, 120]. Similarly, in retinoblastoma, Zuo et al. linked NSUN2-mediated PFAS mRNA methylation to increased stability and tumor aggressiveness [121]. Uveal melanoma provides another example, where NSUN2 knockdown reduces m5C levels, impairing migration and proliferation by destabilizing β-catenin (CTNNB1) mRNA [122]. In thyroid cancer, NSUN2 supports anaplastic thyroid cancer (ATC) progression by stabilizing tRNAs, which sustain pro-oncogenic translation programs (e.g., c-Myc, BCL2). NSUN2 depletion sensitizes ATC to genotoxic drugs, highlighting its role in therapeutic resistance [21, 123]. Finally, in nasopharyngeal and hypopharyngeal cancers, ALYREF and NSUN2 drive metastasis by stabilizing NOTCH1 RNA and upregulating TEAD1, respectively, through m5C-dependent mechanisms [105, 124].
In summary, despite the vast heterogeneity of cancer types and tissue origins, the oncogenic roles of m5C methylation converge on several fundamental mechanisms that transcend cellular context. A central theme is the pervasive function of m5C in enhancing the stability and translational efficiency of a diverse repertoire of oncogenic transcripts, including messenger RNAs, long non-coding RNAs, and circular RNAs. This epitranscriptomic enhancement fuels core hallmarks of cancer, most notably sustained proliferative signaling, metabolic reprogramming such as enhanced glycolysis, glutaminolysis, and lipid synthesis, and activation of key oncogenic pathways such as the PI3K-Akt, Hippo-YAP, and Wnt-β-catenin pathways. Furthermore, m5C modifications are intricately linked to therapy resistance, mediating evasion from ferroptosis and apoptosis, as well as resistance to targeted therapies including EGFR tyrosine kinase inhibitors and platinum-based chemotherapy. The functional output is orchestrated by a limited set of highly conserved regulator axes, particularly the NSUN2-ALYREF and NSUN2-YBX1 complexes, which emerge as central nodes coordinating RNA nuclear export, stability, and translation. Importantly, the context-dependent consequences of m5C dysregulation are dictated by the specific target RNAs methylated in different tissues, explaining how a common mechanism can drive a wide spectrum of cancer phenotypes. This synthesis underscores that m5C methylation is not merely a passive marker but a dynamic and powerful effector of tumorigenesis, positioning the m5C machinery as a compelling target for the development of novel epigenetic therapies across cancer types.
Different mechanisms of action of m5C affecting tumor progression
Emerging research suggests that RNA m5C modifications can influence tumor formation and progression through various complex mechanisms [20, 125]. This compendium highlights recent studies categorized by their mechanisms of action (Fig. 4), which include influencing tumor cell proliferation, migration, invasion, metabolic reprogramming, EMT, ferroptosis, resistance to therapy, and other regulatory pathways (Table 3).

“Writers”, “Readers” and “Erasers” are widely involved in cancer initiation and development by mediating m5C methylation of target RNA and its regulatory mechanisms include cell proliferation, cell migration and invasion, epithelial-mesenchymal transformation (EMT), iron death, apoptosis, autophagy, angiogenesis, and metabolic reprogramming. Created with BioRender.com.
Cell proliferation, migration, and invasion
Cell proliferation refers to the process by which cells divide to produce new cells [126]. Cell migration is the movement of cells triggered by migration signals or the sensing of a substance gradient [126,127,128,129,130]. Cell invasion involves the ability of a cell to migrate through the extracellular matrix from one area to another [131]. For example, LINC02159 knockdown in NSCLC suppresses tumor growth by inhibiting proliferation, migration, and invasion while inducing apoptosis and cell cycle arrest, whereas its overexpression exacerbates these malignant behaviors [77]. Beyond NSCLC, m5C-mediated mechanisms also drive aggressiveness in colorectal cancer, where ALYREF recruits ELAVL1 to stabilize RPS6KB2 and RPTOR transcripts via m5C recognition, fueling tumor growth and migration [23]. Similarly, in esophageal squamous cell carcinoma (ESCC), YBX1 promotes proliferation and metastasis by stabilizing oncogenic transcripts, underscoring its role as a universal m5C reader across cancer types [48]. This theme extends to renal cell carcinoma, where NOP2 stabilizes APOL1 mRNA in an m5C-dependent manner, activating PI3K-Akt signaling to enhance proliferation, migration, and invasion in ccRCC [111]. Intriguingly, NSUN2 exhibits dual roles in renal and gastric cancers: it suppresses p57Kip2 via m5C to drive ccRCC progression [87] and stabilizes DIAPH2-AS1 in gastric cancer by masking NSUN2 degradation sites, thereby promoting migration and invasion [113]. Finally, in hypopharyngeal squamous cell carcinoma (HPSCC), NSUN2 upregulates TEAD1 via m5C modifications, with knockdown experiments confirming its critical role in sustaining proliferation and invasion both in vitro and in vivo [124].
Metabolic reprogramming
Metabolic reprogramming refers to the adaptive changes in cellular metabolism in response to various stimuli and stresses [132,133,134,135,136,137]. It is a common phenomenon in many diseases, involving alterations in metabolic pathways such as glucose, lipid, and amino acid metabolism. These changes are closely linked to disease progression and development [138]. There is a complex bidirectional regulatory relationship between m5C methylation and metabolic reprogramming, which mainly affects tumorigenesis and development by regulating RNA stability, gene expression and metabolic enzyme activities. For instance, ALYREF stabilizes JunD mRNA via m5C recognition in pancreatic ductal adenocarcinoma (PDAC), enabling JunD to activate SLC7A5 and mTORC1 signaling. This enhances amino acid uptake in tumor cells while impairing CD8+ T cell function, linking m5C to immune-metabolic crosstalk [104]. In addition to amino acid metabolism, m5C modifications regulate glucose utilization. Zhang et al. demonstrated that NSUN2 stabilizes ME1, GLUT3, and CDK2 mRNAs to induce metabolic reprogramming and cell cycle progression [24], while Chen et al. revealed that NSUN2 enhances glucose metabolism in CRC by stabilizing ENO1 mRNA. Lactate produced via this pathway further activates NSUN2 transcription through histone acetylation, creating a feedforward loop that sustains tumor growth [93]. Glycolytic reprogramming also underpins bladder cancer aggressiveness, where ALYREF stabilizes PKM2 mRNA via m5C, accelerating glycolysis and proliferation [107]. Notably, NSUN2 itself acts as a glucose sensor, promoting TREX2 expression and suppressing the cGAS/STING pathway to drive tumorigenesis and immunotherapy resistance. Disrupting glucose-NSUN2 interactions reverses these effects, highlighting its therapeutic potential [139].
EMT
EMT is a key process for tumor cells to acquire migration and invasion abilities, and m5C methylation plays an important role in EMT by regulating the RNA stability and translation efficiency of key genes. For example, ALYREF stabilizes EGFR mRNA via m5C in liver hepatocellular carcinoma (LIHC), activating STAT3 signaling to promote EMT, migration, and tumor formation [96]. This mechanism is not limited to LIHC: the NOP2/ALYREF/EZH2 axis induces EMT in lung cancer by stabilizing transcripts that drive mesenchymal transformation, further linking m5C readers to metastatic progression [78].
Ferroptosis
Ferroptosis is an iron-dependent, non-apoptotic form of regulated cell death. Its core mechanism involves the excessive accumulation of lipid peroxides within cells, leading to oxidative damage to cellular membrane systems [140,141,142,143]. m5C methylation directly or indirectly modulates ferroptosis-related pathways by regulating the RNA stability and translation efficiency of key genes. In endometrial cancer, NSUN2-mediated m5C modifications stabilize SLC7A11 mRNA, elevating its protein levels to inhibit ferroptosis and promote tumor growth. Targeting this axis restores ferroptosis sensitivity, offering a therapeutic strategy [114]. Similarly, NSUN2 enhances antioxidant capacity in other cancers by stabilizing NRF2 mRNA via YBX1 interaction. Reduced NRF2 expression reverses NSUN2-driven proliferation and ferroptosis resistance, underscoring the centrality of this axis [82]. Expanding beyond transcriptional regulation, Zhang et al. showed that NSUN2 stabilizes GPX4 mRNA via 3’UTR m5C modifications. NSUN2 knockdown impairs GPX4 synthesis, triggering ferroptosis in hepatocytes—a mechanism reversible by NSUN2 reexpression [144].
Therapeutic resistance
Drug resistance in cancer cells significantly diminishes treatment efficacy, contributing to recurrence and metastasis. A major factor in this resistance is the epigenetic modification of gene expression through RNA modifications [11, 145, 146]. These modifications play a pivotal role in regulating RNA splicing, translation, transport, degradation, and stability [11, 145, 147]. In cervical cancer, NSUN6-mediated m5C modifications stabilize NDRG1 mRNA, enhancing homologous recombination repair and conferring radioresistance [41]. Beyond radioresistance, m5C modifications drive resistance to targeted therapies: NSUN2 hypermethylates QSOX1 mRNA in EGFR-TKI-resistant NSCLC, stabilizing it via YBX1 to sustain gefitinib resistance. NSUN2 depletion restores drug sensitivity, linking m5C to adaptive therapeutic evasion [80]. Genomic studies further support this role, as NSUN2-driven m5C hypermethylation upregulates oncogenes in ESCC, promoting tumor progression and radioresistance [85].
Other mechanisms
Exosomes are small, single-membrane organelles ranging from 30 to 200 nm in diameter, exhibiting the same topology as their parent cells. These vesicles contain a variety of biomolecules, including nucleic acids, lipids, and proteins, which can be transferred between cells, influencing the behavior of recipient cells [148,149,150,151]. Exosomes play critical roles in numerous biological processes, such as development, immunity, tissue homeostasis, cancer, and neurodegenerative diseases [152,153,154]. Research has shown that NSUN2 and YBX1-induced m5C methylation enhances the expression of the long non-coding RNA MALAT1 in MM cells. This upregulation results in the transfer of MALAT1 to osteoclasts via exosomes, exacerbating bone destruction [118]. Additionally, m5C modification of YAP has been shown to promote the secretion of exosomes, contributing to the aggressive behavior and resistance to AZD9291, a third-generation EGFR tyrosine kinase inhibitor, in LUAD cells [75].
Autophagy is a lysosome-dependent degradation process that recycles damaged cellular components, organelles, and aggregation-prone proteins [155,156,157]. Tian et al. demonstrated that SAT1 accumulation effectively suppresses autophagy by stabilizing mTOR mRNA through YBX1-mediated m5C modification [112].
Wu et al. found that the circular RNA circERI3 interacts with the DNA-binding protein DDB1, modulating its ubiquitination and stabilizing it. This stabilization enhances the transcription of PGC-1α, a key coactivator of peroxisome proliferator-activated receptor gamma (PPARγ), affecting mitochondrial function and energy metabolism, and contributing to lung cancer progression [76]. Furthermore, Meng et al. revealed that the ALYREF-JunD-SLC7A5 axis drives epitranscriptomic metabolic reprogramming and immune evasion strategies, promoting pancreatic ductal adenocarcinoma progression [104].
Conclusion
This review explores the multifaceted roles of m5C methylation in gene regulation and its profound impact on tumor biology. The evidence presented emphasizes the critical role of m5C modification in regulating key biological processes, including RNA stability, translation, and nuclear export, which, in turn, influence cellular proliferation, differentiation, apoptosis, and stress responses.
The mechanisms underlying m5C methylation, involving its “readers,” “writers,” and “erasers,” have been carefully examined, along with the specific RNA types they regulate. Notably, the NSUN2/ALYREF pathway and YBX1-mediated recognition of m5C modifications have emerged as critical regulators in the context of cancer progression.
In tumor biology, m5C methylation has been implicated in the development and progression of various cancer types, including those of the respiratory, digestive, urinary, female reproductive, nervous, and blood systems. The mechanistic roles of m5C in tumor progression—such as in glucose metabolism, ferroptosis, and resistance to therapeutic agents—have been elucidated, offering valuable insights for potential therapeutic targets.
Furthermore, the expression of m5C machinery is not uniform across normal tissues but is instead developmentally regulated and tissue-restricted. This baseline expression pattern likely contributes to tissue-specific vulnerabilities. Cancers may arise in organs where these regulators are normally highly expressed (e.g., NSUN2 in regenerative tissues) by further amplifying their pro-growth functions. Conversely, the cancer-associated reprogramming of these factors in tissues where they are normally quiescent could unleash new oncogenic potential. The embryonic lethality observed upon knockout of key m5C regulators underscores their fundamental role in development and suggests that therapeutic targeting will need to carefully balance efficacy against essential physiological functions.
Further investigation is needed to unravel the precise mechanisms by which m5C modifications influence gene expression and tumor biology. This includes identifying novel m5C modification sites and understanding their interactions with other epigenetic modifications. Moreover, developing therapies that modulate m5C methylation presents a promising strategy for cancer treatment. Targeting m5C “writers” or “erasers,” or enhancing the function of m5C “readers,” could offer alternative approaches to combat cancer drug resistance and promote tumor regression. Given the varying role of m5C methylation across different cancer types, personalized medicine strategies based on m5C profiles could enhance patient prognosis by tailoring treatments to the unique characteristics of individual tumors. A comprehensive understanding of how m5C methylation interacts with other epigenetic modifications, such as DNA methylation and histone modifications, is essential to fully grasp the epigenetic regulation of cancer. In conclusion, the study of m5C methylation in cancer biology holds significant potential for advancing our understanding of tumorigenesis and progression, while also offering new avenues for therapeutic intervention. As research in this field continues to progress, the insights gained will pave the way for more effective and personalized cancer therapies.
References
Chuang JC, Jones PA. Epigenetics and microRNAs. Pediatr Res. 2007;61:24R–9R.
Chen Y, Hong T, Wang S, Mo J, Tian T, Zhou X. Epigenetic modification of nucleic acids: from basic studies to medical applications. Chem Soc Rev. 2017;46:2844–72.
Hogg SJ, Beavis PA, Dawson MA, Johnstone RW. Targeting the epigenetic regulation of antitumour immunity. Nat Rev Drug Discov. 2020;19:776–800.
Wu Y, Zhang S, Gong X, Tam S, Xiao D, Liu S, et al. The epigenetic regulators and metabolic changes in ferroptosis-associated cancer progression. Mol Cancer. 2020;19:39.
Zhang Y, Cao W, Wang S, Zhang L, Li X, Zhang Z, et al. Epigenetic modification of hepatitis B virus infection and related hepatocellular carcinoma. Virulence. 2024;15:2421231.
Li Y, Jin H, Li Q, Shi L, Mao Y, Zhao L. The role of RNA methylation in tumor immunity and its potential in immunotherapy. Mol Cancer. 2024;23:130.
Han M, Sun H, Zhou Q, Liu J, Hu J, Yuan W, et al. Effects of RNA methylation on Tumor angiogenesis and cancer progression. Mol Cancer. 2023;22:198.
An Y, Duan H. The role of m6A RNA methylation in cancer metabolism. Mol Cancer. 2022;21:14.
Jaafar C, Aguiar RCT. Dynamic multilayered control of m(6)A RNA demethylase activity. Proc Natl Acad Sci USA. 2024;121:e2317847121.
Boulias K, Greer EL. Biological roles of adenine methylation in RNA. Nat Rev Genet. 2023;24:143–60.
Chen D, Gu X, Nurzat Y, Xu L, Li X, Wu L, et al. Writers, readers, and erasers RNA modifications and drug resistance in cancer. Mol Cancer. 2024;23:178.
Lou N, Gu X, Fu L, Li J, Xue C. Significant roles of RNA 5-methylcytosine methylation in cancer. Cell Signal. 2025;126:111529.
Ma L, Zuo J, Bai C, Fu A, Wang Q, Zhou Z, et al. The dynamic N(1)-methyladenosine RNA methylation provides insights into the tomato fruit ripening. Plant J. 2024;120:2014–30.
Cui L, Ma R, Cai J, Guo C, Chen Z, Yao L, et al. RNA modifications: importance in immune cell biology and related diseases. Signal Transduct Target Ther. 2022;7:334.
Barbieri I, Kouzarides T. Role of RNA modifications in cancer. Nat Rev Cancer. 2020;20:303–22.
Han D, Xu MM. RNA modification in the immune system. Annu Rev Immunol. 2023;41:73–98.
Chen Y, Jiang Z, Yang Y, Zhang C, Liu H, Wan J. The functions and mechanisms of post-translational modification in protein regulators of RNA methylation: current status and future perspectives. Int J Biol Macromol. 2023;253:126773.
Li M, Tao Z, Zhao Y, Li L, Zheng J, Li Z, et al. 5-methylcytosine RNA methyltransferases and their potential roles in cancer. J Transl Med. 2022;20:214.
Ma J, Song B, Wei Z, Huang D, Zhang Y, Su J, et al. m5C-Atlas: a comprehensive database for decoding and annotating the 5-methylcytosine (m5C) epitranscriptome. Nucleic Acids Res. 2022;50:D196–203.
Xue C, Chu Q, Zheng Q, Jiang S, Bao Z, Su Y, et al. Role of main RNA modifications in cancer: N(6)-methyladenosine, 5-methylcytosine, and pseudouridine. Signal Transduct Target Ther. 2022;7:142.
Yu L, Xu H, Xiong H, Yang C, Wu Y, Zhang Q. The role of m5C RNA modification in cancer development and therapy. Heliyon. 2024;10:e38660.
Yang X, Yang F, Lan L, Wen N, Li H, Sun X. Diagnostic and prognostic value of m5C regulatory genes in hepatocellular carcinoma. Front Genet. 2022;13:972043.
Zhong L, Wu J, Zhou B, Kang J, Wang X, Ye F, et al. ALYREF recruits ELAVL1 to promote colorectal tumorigenesis via facilitating RNA m5C recognition and nuclear export. NPJ Precis Oncol. 2024;8:243.
Zhang RK, Li Y, Sun FL, Zhou ZH, Xie YX, Liu WJ, et al. RNA methyltransferase NSUN2-mediated m5C methylation promotes Cr(VI)-induced malignant transformation and lung cancer by accelerating metabolism reprogramming. Environ Int. 2024;192:109055.
Xing H, Gu X, Liu Y, Xu L, He Y, Xue C. NSUN2 regulates Wnt signaling pathway depending on the m5C RNA modification to promote the progression of hepatocellular carcinoma. Oncogene. 2024;43:3469–82.
Zhang Q, Liu F, Chen W, Miao H, Liang H, Liao Z, et al. The role of RNA m(5)C modification in cancer metastasis. Int J Biol Sci. 2021;17:3369–80.
Flamand MN, Tegowski M, Meyer KD. The proteins of mRNA Modification: Writers, Readers, And Erasers. Annu Rev Biochem. 2023;92:145–73.
Esteve-Puig R, Bueno-Costa A, Esteller M. Writers, readers and erasers of RNA modifications in cancer. Cancer Lett. 2020;474:127–37.
Shi H, Wei J, He C. Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol Cell. 2019;74:640–50.
Liao H, Gaur A, McConie H, Shekar A, Wang K, Chang JT, et al. Human NOP2/NSUN1 regulates ribosome biogenesis through non-catalytic complex formation with box C/D snoRNPs. Nucleic Acids Res. 2022;50:10695–716.
Li D, Liu Y, Yang G, He M, Lu L. Recent insights into RNA m5C methylation modification in hepatocellular carcinoma. Biochim Biophys Acta Rev Cancer. 2024;1879:189223.
Knight HM, Demirbugen Oz M, PerezGrovas-Saltijeral A. Dysregulation of RNA modification systems in clinical populations with neurocognitive disorders. Neural Regen Res. 2024;19:1256–61.
Yao H, Song M, Zhang H, Li Y, Chen D, Li Y, et al. NSUN2/ALYREF-medicated m5C-modified circRNA505 regulates the proliferation, differentiation, and glycolysis of antler chondrocytes via the miRNA-127/p53 axis and LDHA. Int J Biol Macromol. 2025;309:142527.
Zhang Y, Zhang LS, Dai Q, Chen P, Lu M, Kairis EL, et al. 5-methylcytosine (m(5)C) RNA modification controls the innate immune response to virus infection by regulating type I interferons. Proc Natl Acad Sci USA. 2022;119:e2123338119.
Kim S, Tan S, Ku J, Widowati TA, Ku D, Lee K, et al. RNA 5-methylcytosine marks mitochondrial double-stranded RNAs for degradation and cytosolic release. Mol Cell. 2025;85:857.
Zhou J, Kong YS, Vincent KM, Dieters-Castator D, Bukhari AB, Glubrecht D, et al. RNA cytosine methyltransferase NSUN5 promotes protein synthesis and tumorigenic phenotypes in glioblastoma. Mol Oncol. 2023;17:1763–83.
Janin M, Ortiz-Barahona V, de Moura MC, Martinez-Cardus A, Llinas-Arias P, Soler M, et al. Epigenetic loss of RNA-methyltransferase NSUN5 in glioma targets ribosomes to drive a stress adaptive translational program. Acta Neuropathol. 2019;138:1053–74.
Guarnacci M, Zhang PH, Kanchi M, Hung YT, Lin H, Shirokikh NE, et al. Substrate diversity of NSUN enzymes and links of 5-methylcytosine to mRNA translation and turnover. Life Sci Alliance. 2024;7:e202402613.
Selmi T, Hussain S, Dietmann S, Heiß M, Borland K, Flad S, et al. Sequence- and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 2021;49:1006–22.
Lu Z, Liu B, Kong D, Zhou X, Pei D, Liu D. NSUN6 regulates NM23-H1 expression in an m5C manner to affect epithelial-mesenchymal transition in lung cancer. Med Princ Pr. 2024;33:56–65.
Yu M, Ni M, Xu F, Liu C, Chen L, Li J, et al. NSUN6-mediated 5-methylcytosine modification of NDRG1 mRNA promotes radioresistance in cervical cancer. Mol Cancer. 2024;23:139.
Hu S, Yang M, Xiao K, Yang Z, Cai L, Xie Y, et al. Loss of NSUN6 inhibits osteosarcoma progression by downregulating EEF1A2 expression and activation of Akt/mTOR signaling pathway via m(5)C methylation. Exp Therapeutic Med. 2023;26:457.
Aguilo F, Li S, Balasubramaniyan N, Sancho A, Benko S, Zhang F, et al. Deposition of 5-methylcytosine on enhancer RNAs enables the coactivator function of PGC-1α. Cell Rep. 2016;14:479–92.
Li H, Liu H, Zhu D, Dou C, Gang B, Zhang M, et al. Biological function molecular pathways and druggability of DNMT2/TRDMT1. Pharmacol Res. 2024;205:107222.
Vilkaitis G, Masevičius V, Kriukienė E, Klimašauskas S. Chemical expansion of the methyltransferase reaction: tools for DNA labeling and epigenome analysis. Acc Chem Res. 2023;56:3188–97.
Jin Y, Yao J, Fu J, Huang Q, Luo Y, You Y, et al. ALYREF promotes the metastasis of nasopharyngeal carcinoma by increasing the stability of NOTCH1 mRNA. Cell Death Dis. 2024;15:578.
Hu J, Liu Q, Feng B, Lu Y, Chen K. ALYREF-mediated regulation of TBL1XR1 and KMT2E synergistically upregulates APOC1, contributing to oxaliplatin resistance in esophageal cancer. Cancer Res. Treat. 2025;57:1064–89.
Liu L, Chen Y, Zhang T, Cui G, Wang W, Zhang G, et al. YBX1 Promotes Esophageal Squamous Cell Carcinoma Progression via m5C‐Dependent SMOX mRNA Stabilization. Adv Sci. 2024;11:e2302379.
Ma HL, Bizet M, Soares Da Costa C, Murisier F, de Bony EJ, Wang MK, et al. SRSF2 plays an unexpected role as reader of m(5)C on mRNA, linking epitranscriptomics to cancer. Mol Cell. 2023;83:4239–4254.e4210.
Kim S, Tan S, Ku J, Widowati TA, Ku D, Lee K, et al. RNA 5-methylcytosine marks mitochondrial double-stranded RNAs for degradation and cytosolic release. Mol Cell. 2024;84:2935–2948.e2937.
Guo J, Zou Z, Dou X, Zhao X, Wang Y, Wei L, et al. Zebrafish Mbd5 binds to RNA m5C and regulates histone deubiquitylation and gene expression in development metabolism and behavior. Nucleic Acids Res. 2024;52:4257–75.
Yang H, Wang Y, Xiang Y, Yadav T, Ouyang J, Phoon L, et al. FMRP promotes transcription-coupled homologous recombination via facilitating TET1-mediated m5C RNA modification demethylation. Proc Natl Acad Sci USA. 2022;119:e2116251119.
Lu Y, Yang L, Feng Q, Liu Y, Sun X, Liu D, et al. RNA 5-methylcytosine modification: regulatory molecules, biological functions, and human diseases. Genomics Proteomics Bioinformatics 2024;22:qzae063.
Xu R, Wang Y, Kuang Y. Multi-omic analyses of m5C readers reveal their characteristics and immunotherapeutic proficiency. Sci Rep. 2024;14:1651.
Kuwano M, Shibata T, Watari K, Ono M. Oncogenic Y-box binding protein-1 as an effective therapeutic target in drug-resistant cancer. Cancer Sci. 2019;110:1536–43.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–7.
Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29.
Arguello AE, Li A, Sun X, Eggert TW, Mairhofer E, Kleiner RE. Reactivity-dependent profiling of RNA 5-methylcytidine dioxygenases. Nat Commun. 2022;13:4176.
Abbas Z, Rehman MU, Tayara H, Lee SW, Chong KT. m5C-Seq: Machine learning-enhanced profiling of RNA 5-methylcytosine modifications. Comput Biol Med. 2024;182:109087.
Wu R, Sun C, Chen X, Yang R, Luan Y, Zhao X, et al. NSUN5/TET2-directed chromatin-associated RNA modification of 5-methylcytosine to 5-hydroxymethylcytosine governs glioma immune evasion. Proc Natl Acad Sci USA. 2024;121:e2321611121.
Zhang X, Zhang Y, Wang C, Wang X. TET (Ten-eleven translocation) family proteins: structure, biological functions and applications. Signal Transduct Target Ther. 2023;8:297.
Zhao Z, Zhou Y, Lv P, Zhou T, Liu H, Xie Y, et al. NSUN4 mediated RNA 5-methylcytosine promotes the malignant progression of glioma through improving the CDC42 mRNA stabilization. Cancer Lett. 2024;597:217059.
Yang X, Yang Y, Sun BF, Chen YS, Xu JW, Lai WY, et al. 5-methylcytosine promotes mRNA export - NSUN2 as the methyltransferase and ALYREF as an m(5)C reader. Cell Res. 2017;27:606–25.
Xue C, Gu X, Zheng Q, Shi Q, Yuan X, Su Y, et al. ALYREF mediates RNA m(5)C modification to promote hepatocellular carcinoma progression. Signal Transduct Target Ther. 2023;8:130.
Xue C, Zhao Y, Li G, Li L. Multi-omic analyses of the m(5)C regulator ALYREF reveal its essential roles in hepatocellular carcinoma. Front Oncol. 2021;11:633415.
Niu K, Chen Z, Li M, Ma G, Deng Y, Zhang J, et al. NSUN2 lactylation drives cancer cell resistance to ferroptosis through enhancing GCLC-dependent glutathione synthesis. Redox Biol. 2025;79:103479.
Hu Y, Chen C, Tong X, Chen S, Hu X, Pan B, et al. NSUN2 modified by SUMO-2/3 promotes gastric cancer progression and regulates mRNA m5C methylation. Cell Death Dis. 2021;12:842.
Huang S, Shi D, Dai S, Jiang X, Wang R, Yang M, et al. RNF31 induces paclitaxel resistance by sustaining ALYREF cytoplasmic-nuclear shuttling in human triple-negative breast cancer. Clin Transl Med. 2025;15:e70203.
Murakami Y, Wei FY, Kawamura Y, Horiguchi H, Kadomatsu T, Miyata K, et al. NSUN3-mediated mitochondrial tRNA 5-formylcytidine modification is essential for embryonic development and respiratory complexes in mice. Commun Biol. 2023;6:307.
Flores JV, Cordero-Espinoza L, Oeztuerk-Winder F, Andersson-Rolf A, Selmi T, Blanco S, et al. Cytosine-5 RNA methylation regulates neural stem cell differentiation and motility. Stem Cell Rep. 2017;8:112–24.
Zhang T, Chen P, Li W, Sha S, Wang Y, Yuan Z, et al. Cognitive deficits in mice lacking Nsun5, a cytosine-5 RNA methyltransferase, with impairment of oligodendrocyte precursor cells. Glia. 2019;67:688–702.
Gu X, Li P, Gao X, Ru Y, Xue C, Zhang S, et al. RNA 5-methylcytosine writer NSUN5 promotes hepatocellular carcinoma cell proliferation via a ZBED3-dependent mechanism. Oncogene. 2024;43:624–35.
Xue C, Zhao Y, Li L. Advances in RNA cytosine-5 methylation: detection, regulatory mechanisms, biological functions and links to cancer. Biomark Res. 2020;8:43.
Du X, Cheng C, Yang Y, Fan B, Wang P, Xia H, et al. NSUN2 promotes lung adenocarcinoma progression through stabilizing PIK3R2 mRNA in an m(5)C-dependent manner. Mol Carcinog. 2024;63:962–76.
Yu W, Zhang C, Wang Y, Tian X, Miao Y, Meng F, et al. YAP 5-methylcytosine modification increases its mRNA stability and promotes the transcription of exosome secretion-related genes in lung adenocarcinoma. Cancer Gene Ther. 2023;30:149–62.
Wu J, Zhao Q, Chen S, Xu H, Zhang R, Cai D, et al. NSUN4-mediated m5C modification of circERI3 promotes lung cancer development by altering mitochondrial energy metabolism. Cancer Lett. 2024;605:217266.
Yang Q, Wang M, Xu J, Yu D, Li Y, Chen Y, et al. LINC02159 promotes non-small cell lung cancer progression via ALYREF/YAP1 signaling. Mol Cancer. 2023;22:122.
Yang Y, Fan H, Liu H, Lou X, Xiao N, Zhang C, et al. NOP2 facilitates EZH2-mediated epithelial-mesenchymal transition by enhancing EZH2 mRNA stability via m5C methylation in lung cancer progression. Cell Death Dis. 2024;15:506.
Yu T, Zhang Q, Yu SK, Nie FQ, Zhang ML, Wang Q, et al. THOC3 interacts with YBX1 to promote lung squamous cell carcinoma progression through PFKFB4 mRNA modification. Cell Death Dis. 2023;14:475.
Wang Y, Wei J, Feng L, Li O, Huang L, Zhou S, et al. Aberrant m5C hypermethylation mediates intrinsic resistance to gefitinib through NSUN2/YBX1/QSOX1 axis in EGFR-mutant non-small-cell lung cancer. Mol Cancer. 2023;22:81.
Li P, Huang D. NSUN2-mediated RNA methylation: molecular mechanisms and clinical relevance in cancer. Cell Signal. 2024;123:111375.
Chen Y, Jiang Z, Zhang C, Zhang L, Chen H, Xiao N, et al. 5-Methylcytosine transferase NSUN2 drives NRF2-mediated ferroptosis resistance in non-small cell lung cancer. J Biol Chem. 2024;300:106793.
Su J, Wu G, Ye Y, Zhang J, Zeng L, Huang X, et al. NSUN2-mediated RNA 5-methylcytosine promotes esophageal squamous cell carcinoma progression via LIN28B-dependent GRB2 mRNA stabilization. Oncogene. 2021;40:5814–28.
Han H, Sun Y, Wei W, Huang Z, Cheng M, Qiu H, et al. RNA modification-related genes illuminate prognostic signature and mechanism in esophageal squamous cell carcinoma. iScience. 2024;27:109327.
Niu X, Peng L, Liu W, Miao C, Chen X, Chu J, et al. A cis-eQTL in NSUN2 promotes esophageal squamous-cell carcinoma progression and radiochemotherapy resistance by mRNA-m(5)C methylation. Signal Transduct Target Ther. 2022;7:267.
Li Q, Guo G, Chen Y, Lu L, Li H, Zhou Z, et al. HCP5 derived novel microprotein triggers progression of gastric cancer through regulating ferroptosis. Adv Sci. 2024;11:e2407012.
Mei L, Shen C, Miao R, Wang JZ, Cao MD, Zhang YS, et al. RNA methyltransferase NSUN2 promotes gastric cancer cell proliferation by repressing p57(Kip2) by an m(5)C-dependent manner. Cell Death Dis. 2020;11:270.
Shen X, Sun H, Shu S, Tang W, Yuan Y, Su H, et al. Suppression of NSUN2 enhances the sensitivity to chemosensitivity and inhibits proliferation by mediating cell apoptosis in gastric cancer. Pathol Res Pr. 2024;253:154986.
Fang L, Huang H, Lv J, Chen Z, Lu C, Jiang T, et al. m5C-methylated lncRNA NR_033928 promotes gastric cancer proliferation by stabilizing GLS mRNA to promote glutamine metabolism reprogramming. Cell Death Dis. 2023;14:520.
Liu K, Xu P, Lv J, Ge H, Yan Z, Huang S, et al. Peritoneal high-fat environment promotes peritoneal metastasis of gastric cancer cells through activation of NSUN2-mediated ORAI2 m5C modification. Oncogene. 2023;42:1980–93.
Li Y, Xia Y, Jiang T, Chen Z, Shen Y, Lin J, et al. Long noncoding RNA DIAPH2-AS1 promotes neural invasion of gastric cancer via stabilizing NSUN2 to enhance the m5C modification of NTN1. Cell Death Dis. 2023;14:260.
Cui Y, Lv P, Zhang C. NSUN6 mediates 5-methylcytosine modification of METTL3 and promotes colon adenocarcinoma progression. J Biochem Mol Toxicol. 2024;38:e23749.
Chen B, Deng Y, Hong Y, Fan L, Zhai X, Hu H, et al. Metabolic recoding of NSUN2-mediated m(5)C modification promotes the progression of colorectal cancer via the NSUN2/YBX1/m(5)C-ENO1 positive feedback loop. Adv Sci. 2024;11:e2309840.
Zou S, Huang Y, Yang Z, Zhang J, Meng M, Zhang Y, et al. NSUN2 promotes colorectal cancer progression by enhancing SKIL mRNA stabilization. Clin Transl Med. 2024;14:e1621.
Yang L, Shi J, Zhong M, Sun P, Zhang X, Lian Z, et al. NXPH4 mediated by m(5)C contributes to the malignant characteristics of colorectal cancer via inhibiting HIF1A degradation. Cell Mol Biol Lett. 2024;29:111.
Nulali J, Zhang K, Long M, Wan Y, Liu Y, Zhang Q, et al. ALYREF-mediated RNA 5-methylcytosine modification promotes hepatocellular carcinoma progression via stabilizing EGFR mRNA and pSTAT3 activation. Int J Biol Sci. 2024;20:331–46.
Sun GF, Ding H. NOP2-mediated m5C methylation of XPD is associated with hepatocellular carcinoma progression. Neoplasma. 2023;70:340–9.
Li R, Li S, Shen L, Li J, Zhang D, Yu J, et al. LINC00618 facilitates growth and metastasis of hepatocellular carcinoma via elevating cholesterol synthesis by promoting NSUN2-mediated SREBP2 m5C modification. Ecotoxicol Environ Saf. 2024;285:117064.
Song D, An K, Zhai W, Feng L, Xu Y, Sun R, et al. NSUN2-mediated mRNA m(5)C modification regulates the progression of hepatocellular carcinoma. Genomics Proteom Bioinforma. 2023;21:823–33.
Sun Z, Xue S, Zhang M, Xu H, Hu X, Chen S, et al. Aberrant NSUN2-mediated m(5)C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene. 2020;39:6906–19.
Zheng H, Zhu M, Li W, Zhou Z, Wan X. m(5) C and m(6) A modification of long noncoding NKILA accelerates cholangiocarcinoma progression via the miR-582-3p-YAP1 axis. Liver Int. 2022;42:1144–57.
Zhang G, Liu L, Li J, Chen Y, Wang Y, Zhang Y, et al. NSUN2 stimulates tumor progression via enhancing TIAM2 mRNA stability in pancreatic cancer. Cell Death Discov. 2023;9:219.
Zheng S, Hu C, Lin Q, Li T, Li G, Tian Q, et al. Extracellular vesicle-packaged PIAT from cancer-associated fibroblasts drives neural remodeling by mediating m5C modification in pancreatic cancer mouse models. Sci Transl Med. 2024;16:eadi0178.
Meng Q, Xie Y, Sun K, He L, Wu H, Zhang Q, et al. ALYREF-JunD-SLC7A5 axis promotes pancreatic ductal adenocarcinoma progression through epitranscriptome-metabolism reprogramming and immune evasion. Cell Death Discov. 2024;10:97.
Tian J, Gao J, Cheng C, Xu Z, Chen X, Wu Y, et al. NOP2-mediated 5-methylcytosine modification of APOL1 messenger RNA activates PI3K-Akt and facilitates clear cell renal cell carcinoma progression. Int J Biol Sci. 2024;20:4853–71.
Yang L, Yin H, Chen Y, Pan C, Hang H, Lu Y, et al. Low expression of PEBP1P2 promotes metastasis of clear cell renal cell carcinoma by post-transcriptional regulation of PEBP1 and KLF13 mRNA. Exp Hematol Oncol. 2022;11:87.
Wang JZ, Zhu W, Han J, Yang X, Zhou R, Lu HC, et al. The role of the HIF-1alpha/ALYREF/PKM2 axis in glycolysis and tumorigenesis of bladder cancer. Cancer Commun. 2021;41:560–75.
Chen X, Li A, Sun BF, Yang Y, Han YN, Yuan X, et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat Cell Biol. 2019;21:978–90.
Zhu W, Wan F, Xu W, Liu Z, Wang J, Zhang H, et al. Positive epigenetic regulation loop between AR and NSUN2 promotes prostate cancer progression. Clin Transl Med. 2022;12:e1028.
Zhang Y, Chen XN, Zhang H, Wen JK, Gao HT, Shi B, et al. CDK13 promotes lipid deposition and prostate cancer progression by stimulating NSUN5-mediated m5C modification of ACC1 mRNA. Cell Death Differ. 2023;30:2462–76.
Zhang X, An K, Ge X, Sun Y, Wei J, Ren W, et al. NSUN2/YBX1 promotes the progression of breast cancer by enhancing HGH1 mRNA stability through m(5)C methylation. Breast Cancer Res. 2024;26:94.
Tian W, Zhu L, Luo Y, Tang Y, Tan Q, Zou Y, et al. Autophagy deficiency induced by SAT1 potentiates tumor progression in triple-negative breast cancer. Adv Sci. 2024;11:e2309903.
Chen Y, Zuo X, Wei Q, Xu J, Liu X, Liu S, et al. Upregulation of LRRC8A by m(5)C modification-mediated mRNA stability suppresses apoptosis and facilitates tumorigenesis in cervical cancer. Int J Biol Sci. 2023;19:691–704.
Chen SJ, Zhang J, Zhou T, Rao SS, Li Q, Xiao LY, et al. Epigenetically upregulated NSUN2 confers ferroptosis resistance in endometrial cancer via m(5)C modification of SLC7A11 mRNA. Redox Biol. 2024;69:102975.
Meng H, Miao H, Zhang Y, Chen T, Yuan L, Wan Y, et al. YBX1 promotes homologous recombination and resistance to platinum-induced stress in ovarian cancer by recognizing m5C modification. Cancer Lett. 2024;597:217064.
Liu X, Wei Q, Yang C, Zhao H, Xu J, Mobet Y, et al. RNA m(5)C modification upregulates E2F1 expression in a manner dependent on YBX1 phase separation and promotes tumor progression in ovarian cancer. Exp Mol Med. 2024;56:600–15.
Pan A, Xue Y, Ruan X, Dong W, Wang D, Liu Y, et al. m5C modification of LINC00324 promotes angiogenesis in glioma through CBX3/VEGFR2 pathway. Int J Biol Macromol. 2024;257:128409.
Yu M, Cai Z, Zhang J, Zhang Y, Fu J, Cui X. Aberrant NSUN2-mediated m5C modification of exosomal LncRNA MALAT1 induced RANKL-mediated bone destruction in multiple myeloma. Commun Biol. 2024;7:1249.
Zou Z, Dou X, Li Y, Zhang Z, Wang J, Gao B, et al. RNA m(5)C oxidation by TET2 regulates chromatin state and leukaemogenesis. Nature. 2024;634:986–94.
Yang M, Wei R, Zhang S, Hu S, Liang X, Yang Z, et al. NSUN2 promotes osteosarcoma progression by enhancing the stability of FABP5 mRNA via m(5)C methylation. Cell Death Dis. 2023;14:125.
Zuo S, Li L, Wen X, Gu X, Zhuang A, Li R, et al. NSUN2-mediated m(5) C RNA methylation dictates retinoblastoma progression through promoting PFAS mRNA stability and expression. Clin Transl Med. 2023;13:e1273.
Luo G, Xu W, Chen X, Wang S, Wang J, Dong F, et al. NSUN2-mediated RNA m(5)C modification modulates uveal melanoma cell proliferation and migration. Epigenetics. 2022;17:922–33.
Li P, Wang W, Zhou R, Ding Y, Li X. The m(5) C methyltransferase NSUN2 promotes codon-dependent oncogenic translation by stabilising tRNA in anaplastic thyroid cancer. Clin Transl Med. 2023;13:e1466.
Chen L, Ding J, Wang B, Chen X, Ying X, Yu Z, et al. RNA methyltransferase NSUN2 promotes hypopharyngeal squamous cell carcinoma proliferation and migration by enhancing TEAD1 expression in an m(5)C-dependent manner. Exp Cell Res. 2021;404:112664.
Shi H, Chai P, Jia R, Fan X. Novel insight into the regulatory roles of diverse RNA modifications: re-defining the bridge between transcription and translation. Mol Cancer. 2020;19:78.
Entschladen F, TLt Drell, Lang K, Joseph J, Zaenker KS. Tumour-cell migration, invasion, and metastasis: navigation by neurotransmitters. Lancet Oncol. 2004;5:254–8.
Friedl P, Wolf K. Tumour-cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer. 2003;3:362–74.
Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100.
Weiss F, Lauffenburger D, Friedl P. Towards targeting of shared mechanisms of cancer metastasis and therapy resistance. Nat Rev Cancer. 2022;22:157–73.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
Olson MF. Mechanisms of tumour cell invasion and metastasis. J Mol Med. 2007;85:543–4.
Zhang F, Guo J, Yu S, Zheng Y, Duan M, Zhao L, et al. Cellular senescence and metabolic reprogramming: unraveling the intricate crosstalk in the immunosuppressive tumor microenvironment. Cancer Commun. 2024;44:929–66.
Payne KK. Cellular stress responses and metabolic reprogramming in cancer progression and dormancy. Semin Cancer Biol. 2022;78:45–48.
Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science 2020;368:eaaw5473.
Horn P, Tacke F. Metabolic reprogramming in liver fibrosis. Cell Metab. 2024;36:1439–55.
Raggi C, Taddei ML, Rae C, Braconi C, Marra F. Metabolic reprogramming in cholangiocarcinoma. J Hepatol. 2022;77:849–64.
Lin J, Rao D, Zhang M, Gao Q. Metabolic reprogramming in the tumor microenvironment of liver cancer. J Hematol Oncol. 2024;17:6.
Ritterhoff J, Tian R. Metabolic mechanisms in physiological and pathological cardiac hypertrophy: new paradigms and challenges. Nat Rev Cardiol. 2023;20:812–29.
Chen T, Xu ZG, Luo J, Manne RK, Wang Z, Hsu CC, et al. NSUN2 is a glucose sensor suppressing cGAS/STING to maintain tumorigenesis and immunotherapy resistance. Cell Metab. 2023;35:1782–98.e1788.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Djulbegovic MB, Uversky VN. Ferroptosis - An iron- and disorder-dependent programmed cell death. Int J Biol Macromol. 2019;135:1052–69.
Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25:424–42.
Li S, Huang Y. Ferroptosis: an iron-dependent cell death form linking metabolism, diseases, immune cell and targeted therapy. Clin Transl Oncol. 2022;24:1–12.
Zhang X, Zhang Y, Li R, Li Y, Wang Q, Wang Y, et al. STUB1-mediated ubiquitination and degradation of NSUN2 promotes hepatocyte ferroptosis by decreasing m(5)C methylation of Gpx4 mRNA. Cell Rep. 2024;43:114885.
Wood S, Willbanks A, Cheng JX. The role of RNA modifications and RNA-modifying proteins in cancer therapy and drug resistance. Curr Cancer Drug Targets. 2021;21:326–52.
Karati D, Kumar D. Molecular insight into the apoptotic mechanism of cancer cells: an explicative review. Curr Mol Pharm. 2024;17:e18761429273223.
Tang L, Tian H, Min Q, You H, Yin M, Yang L, et al. Decoding the epitranscriptome: a new frontier for cancer therapy and drug resistance. Cell Commun Signal. 2024;22:513.
Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514.
Li C, Teixeira AF, Zhu HJ, Ten Dijke P. Cancer associated-fibroblast-derived exosomes in cancer progression. Mol Cancer. 2021;20:154.
Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977.
Yu D, Li Y, Wang M, Gu J, Xu W, Cai H, et al. Exosomes as a new frontier of cancer liquid biopsy. Mol Cancer. 2022;21:56.
Feng W, Dean DC, Hornicek FJ, Shi H, Duan Z. Exosomes promote pre-metastatic niche formation in ovarian cancer. Mol Cancer. 2019;18:124.
Batista IA, Machado JC, Melo SA. Advances in exosomes utilization for clinical applications in cancer. Trends Cancer. 2024;10:947–68.
Ye L, Li Y, Zhang S, Wang J, Lei B. Exosomes-regulated lipid metabolism in tumorigenesis and cancer progression. Cytokine Growth Factor Rev. 2023;73:27–39.
Nixon RA, Rubinsztein DC. Mechanisms of autophagy-lysosome dysfunction in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2024;25:926–46.
Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24:560–75.
Xia H, Green DR, Zou W. Autophagy in tumour immunity and therapy. Nat Rev Cancer. 2021;21:281–97.
Liu S, Liu Y, Zhou Y, Xia G, Liu H, Zeng Y, et al. NSUN5 promotes tumorigenic phenotypes through the WNT signaling pathway and immunosuppression of CD8+ T cells in gastric cancer. Cell Signal. 2024;124:111475.
Jiang Z, Li S, Han MJ, Hu GM, Cheng P. High expression of NSUN5 promotes cell proliferation via cell cycle regulation in colorectal cancer. Am J Transl Res. 2020;12:3858–70.
Yan D, Xie Y, Huang L, Zhang Y, Gu R, Xie H, et al. RNA m5C methylation orchestrates BLCA progression via macrophage reprogramming. J Cell Mol Med. 2023;27:2398–411.
Li Y, Xue M, Deng X, Dong L, Nguyen LXT, Ren L, et al. TET2-mediated mRNA demethylation regulates leukemia stem cell homing and self-renewal. Cell Stem Cell. 2023;30:1072–90.e1010.
Jin S, Li J, Shen Y, Wu Y, Zhang Z, Ma H. RNA 5-Methylcytosine Regulator NSUN3 promotes tumor progression through regulating immune infiltration in head and neck squamous cell carcinoma. Oral Dis. 2024;30:313–28.
Acknowledgements
The authors would like to express their gratitude to all participants and relevant personnel for their contribution to this study.
Funding
This work was funded by Leading Talents of Zhongyuan Science and Technology Innovation (214200510027), Science and technology department of Henan Province (222102310365), Henan Medical Science and Technology Joint Building Program (LHGJ20210324), Gandan Xiangzhao Research Fund (GDXZ2023001), Henan Provincial Medical Science and Technology Research Plan (SBGJ2018002 and SBGJ202102117), and Outstanding Foreign Scientist Studio in Henan Province (GZS2020004).
Author information
Authors and Affiliations
Contributions
Wenzhi Guo and Yuting He designed the manuscript. Zhenyu Guan and Wendong Li wrote the manuscript. Zhenyu Guan helped with reference collection and draw the figures. Wenzhi Guo and Yuting He revised the manuscript. All authors contributed to the article and approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Guan, Z., Li, W., He, Y. et al. RNA m5C methylation in cancer: mechanisms and biological impact. Oncogenesis 14, 44 (2025). https://doi.org/10.1038/s41389-025-00587-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41389-025-00587-w
