
Abstract
The dynamic interplay between neoplastic cells and the host has been increasingly recognized as important players in the pathogenesis of cancer cachexia, a syndrome affecting ~50–80% of cancer patients with various incidences of different types of malignancies. Despite its prevalence, a comprehensive understanding of cancer cachexia progression, with a holistic view at the cross-organismal, cellular and molecular levels, remains elusive. In this review, we undertake an in-depth exploration of the relevant target organs and their regulatory roles in cancer cachexia, with a particular focus on macroenvironmental interactions via various organismal crosstalk axes. Moreover, we highlight how systemic metabolic remodeling, a hallmark of cancer cachexia, plays essential roles in modulating the inflammatory responses of immune and stromal cells in the tumor microenvironment (TME). These cellular responses, in turn, disrupt energy metabolism in distant organs and perturb organismal homeostasis by secreting a variety of mediators that activate specific signaling pathways, thereby fostering a vicious cycle that exacerbates cancer cachexia. We comprehensively summarize these complex cellular and molecular networks that constitute reciprocally regulatory dynamics between systemic metabolic reprogramming and inflammatory cascades. Notably, targeting the multifaceted interplay of organismal metabolic remodeling and cancer-associated inflammation holds great promise for clinical translation, as illustrated by a series of innovative therapeutic strategies and ongoing clinical trials aimed at mitigating cachexia in cancer patients.
Similar content being viewed by others


Introduction
Cancer cachexia is a systemic inflammatory response that is commonly characterized by body weight loss and atrophy of muscle and adipose tissues during the progression of cancer. These features stem primarily from increased energy expenditure, hypermetabolism, and anorexia.1,2 Clinical criteria have been proposed for individuals with cancer cachexia,3 including (1) ≥ 5% weight loss within six months; (2) ≥ 2% weight loss in patients with a body mass index (BMI) < 20 kg/m²; and (3) ≥ 2% weight loss in sarcopenic patients. Some studies have revealed that diagnostic sensitivity differs from cancer type to cancer type. Unsettlingly, the clinical diagnosis of cancer cachexia in obese individuals may be delayed because their high BMI can obscure fat loss and skeletal muscle atrophy,4 preventing them from meeting the diagnostic criteria for cachexia. This is particularly concerning in patients with skeletal muscle depletion, which is an independent risk factor for mortality.5 Therefore, refining BMI into specifics of body composition, such as lean and fat mass, can be important for achieving more accurate diagnoses. Cancer cachexia negatively affects patients’ quality of life, exacerbates treatment-related toxicity, and significantly increases the mortality rate of cancer by 20–30%.1,6 The incidence of cancer cachexia varies among tumor types, with pancreatic, gastrointestinal, and lung cancers being more prominently associated with cachexia, accounting for ~40–70% of all cases.1,2 In patients with these tumors, the deterioration of cachexia is accompanied by the progression of cancer, which may be attributed to reduced food intake and deranged digestion, influenced by the tumor in the upper digestive tract.2
Although cancer cachexia has been documented in humans for centuries, our comprehensive understanding of this disease has emerged only in the last few decades. The term ‘cachexia’ has etymological roots in the ancient Greek lexemes kakós (bad) and hexis (habit), and it refers to the loss of appetite and a general wasting condition. Hippocrates (~460–377 BC) described it as linked to conditions such as hydropsy (edema or fluid retention).7 Owing to the relentless efforts and significant research advancements made by numerous scientific researchers, we now have a deeper understanding of cancer cachexia (Fig. 1). Currently, cancer cachexia is recognized as a systemic metabolic syndrome that involves multiple tissues and organs, including musculoskeletal, skeletal, adipose, neurological, gastrointestinal, and hepatic tissues.8 This raises the following question: How do tumors affect distant organs? Recently, some studies reported that tumors can communicate with distant organs through neural, blood, and lymphatic networks via metabolites and inflammatory factors.9,10 Consequently, both inflammatory cytokines and metabolites may play regulatory roles in mediating cross-organ crosstalk. We hypothesize that catabolism activation and anabolic suppression are important features in cachexia patients and that metabolic remodeling potentially triggers an inflammatory response in various cells. The altered immune and stromal cells then disseminate from primary organs via the circulatory system and secrete various inflammatory factors, such as tumor necrosis factor α (TNF-α), interleukin 6 (IL-6) and members of the transforming growth factor β (TGF-β) family, which induce skeletal muscle and adipose tissue catabolism,11,12,13 forming a regulatory loop. Thus, multiorgan interactions or cellular crosstalk and, consequently, inflammatory factor-mediated systemic perturbations may drive cachexia. In this review, we highlight the importance of crosstalk among distinct organs and metabolite-inflammatory factors in the cachexia macroenvironment and microenvironment.

Timeline and milestones in the study of cancer cachexia. The figure presents a comprehensive timeline depicting the significant milestones achieved in cancer cachexia research. Early studies identified the clinical manifestations, underlying mechanisms, animal models, and novel therapeutic strategies. The figure was generated with BioRender (https://biorender.com)
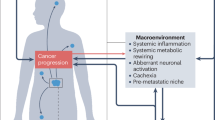
The tumor microenvironment (TME) is an intricate ecosystem in which cancer cells thrive and can influence tumor growth, metastatic spread, and response to treatment. It comprises mesenchymal elements such as immune cells, cancer-associated fibroblasts (CAFs), endothelial cells (ECs), pericytes, and tissue-specific cells such as adipocytes and neurons, as well as noncellular constituents such as the extracellular matrix (ECM), extracellular vesicles, and soluble factors.14 The cancer cachexia microenvironment focuses on the local tissue and cellular milieu where cachexia-related changes occur, including the impact of metabolic products and inflammatory imbalances on cellular signaling pathways and functional states. However, changes in the tumor host body extend beyond the TME. They involve the crosstalk between multiple distal compartments at places beyond tumor beds through hormonal signals, inflammatory mediators, and circulating immune cells,15 which also serve as critical contributors to the pathophysiology of cancer cachexia. Understanding the interactions between the microenvironment and macroenvironment offers novel avenues to enhance clinical outcomes in cancer cachexia patients.
Clinical manifestations and prevalence
Clinical diagnosis
The diagnostic criteria for cancer cachexia have undergone continuous refinement. The progression in these standards facilitated earlier intervention and management for patients with cachexia. In 2008, a consensus conference on cachexia was held in Washington, D.C., USA, and preliminary diagnostic criteria for this condition were proposed.16 Currently, the most widely used diagnostic criteria for cachexia are the European Palliative Care Research Collaboration (EPCRC), upon which cancer cachexia management guidelines have been developed3 (Fig. 1). Nevertheless, these criteria incorporate thresholds based on Western populations, which may not accurately reflect the situation for Asians due to variations in body composition, dietary patterns, lifestyles, and metabolic characteristics. To address this issue, the Asian Working Group for Cachexia (AWGC) established the Asian criteria for cachexia: patients with chronic wasting disease and a BMI < 21 kg/m2 or weight loss >2% over the preceding 3–6 months, coupled with anorexia, diminished handgrip strength (<28 kg for males and <18 kg for females), or C-reactive protein (CRP) > 0.5 mg/dL.17 The differences between the AWGC and EPCRC criteria were the addition of anorexia, grip strength, and CRP level. Xie et al. established the utility of the AWGC2023 criteria in predicting survival and medical burden among Chinese cancer patients.18 In a study comparing both the AWGC and EPCRC criteria in lung cancer patients, the AWGC criteria were found to be more effective in diagnosing cancer cachexia in the Asian population and provided superior prognostic indicators.19 These findings underscore the pivotal role of racial characteristics in the diagnosis of cachexia and underscore the potential of the AWGC criteria for improved patient assessment and management in Asian populations.
The international consensus group delineated cachexia as a continuous process encompassing three clinically relevant stages: precachexia, cachexia and refractory cachexia3 (Fig. 1). On the basis of these stages, Vigano et al. assessed the outcomes of 207 patients with advanced non-small cell lung cancer (NSCLC) or gastrointestinal cancer and reported that precachexia and cachectic patients presented similar outcomes but were significantly different from noncachectic and refractory cachexia patients.20 These findings imply that the clinical application of this staging system, such as treatment planning and prognosis assessment, has certain limitations. Notably, there are emerging strategies for staging cachexia. For example, the Glasgow prognostic score, which integrates CRP and albumin levels, provides a straightforward and objective framework for assessing and managing cancer cachexia.21 A novel cachexia classification system based on the modified Glasgow prognostic score was used to classify cancer cachexia into four different stages: no cachexia, undernourishment, precachexia, and refractory cachexia. These stages exhibited robust correlations with poor clinical outcomes and demonstrated the capacity to predict overall survival.22 Recently, Jin et al. identified cancer cachexia by characterizing longitudinal body composition trajectories and categorized cancer patients into three phases. This classification can also effectively predict survival prognosis and the occurrence of adverse events.23 However, the clinical application of these staging systems requires further research.
In summary, the diagnostic criteria for cancer cachexia remain controversial, posing considerable challenges for clinical practice and driving the continuous exploration of novel diagnostic techniques. The diversity and complexity observed in cancer cachexia patients may be attributable to variations across tumor types, ethnicities, and geographical regions.
Prevalence of cancer cachexia among various geographical regions and cancer types
The prevalence of cancer cachexia significantly varies among different tumor types and demonstrates geographical disparities across countries and regions (Fig. 2). In 2019, the prevalence of cancer cachexia in the USA and European Union was documented on the basis of 21 studies published between 1980 and 2017 involving 31,047 cancer patients.24 A study in China enrolled 47,604 patients with 16 common cancers from June 2012 to December 2020 to investigate the prevalence of cancer cachexia (Fig. 1). These findings indicate that, irrespective of the cancer site, advanced TNM stages are linked to a notably higher incidence of cachexia among the general cancer patient population. Furthermore, compared with younger individuals and males, elderly individuals and males are more likely to develop cachexia.25

Epidemiological statistics of cancer cachexia patients based on studies published in the past 5 years. The prevalence of cancer cachexia varies by region and tumor type. a The prevalence of cachexia among pancreatic cancer patients is high in various countries, and a significantly lower prevalence has been reported in breast cancer patients. In the chart, the blue column highlights the tumor type with the lowest cachexia incidence rate, whereas the yellow column denotes the highest incidence. b Digestive system and lung cancer have a high prevalence, whereas breast cancer and melanoma have the lowest prevalence on the basis of available data. The figure was generated with BioRender (https://biorender.com)
To obtain more accurate epidemiological data on cancer cachexia, we incorporated dozens of additional studies published in the past five years,19,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45 which provide comprehensive information for the 14 selected cancer entities analyzed (liver cancer, pancreatic cancer, gastric cancer, esophageal cancer, esophagogastric cancer, colorectal cancer, lung cancer, thyroid cancer, head and neck cancer, gynecological tumors, urogenital cancer, hematologic malignancies, breast cancer, melanoma and others) (Fig. 2). These data, unweighted for patient origin, age, tumor stage and treatment strategy, offer a realistic midpoint reflecting clinical practice scenarios. In these studies, the cachexia prevalence in patients with pancreatic cancer was the highest in numerous countries, including China (63%),25 France (67%),24 Germany (41%), Italy (74%), Japan (60%)27 and the Netherlands (71%).26 Conversely, the incidence of cachexia in breast cancer patients was lower, with rates of 15% reported in China,25 12% in France,24 and 14% reported in both Italy and the USA (Fig. 2a).
In terms of cancer type, the incidence of cachexia was markedly greater in patients diagnosed with pancreatic, gastrointestinal, or lung cancer than in those with various types of cancer. Conversely, the cachexia incidence rate was notably lower in those with breast cancer and melanoma (Fig. 2b). For example, clinical studies reported that cachexia incidence rates in pancreatic cancer patients range from 22% to 84%,28,29 whereas melanoma patients presented significantly lower rates, varying between 14% and 30%.24 The variations in diagnostic criteria, patient staging, and treatment phases across studies significantly contribute to discrepancies in cachexia prevalence rates. Furthermore, publication biases, particularly the tendency to publish positive results (indicating high prevalence), may introduce limitations in data interpretation. Collectively, these observations underscore the importance of implementing targeted interventions to address cachexia in high-risk cancer patients.
Patients with gastrointestinal cancer are more prone to cachexia, which is potentially attributed to tumor blocking or anatomical changes after esophagogastrostomy, exacerbating the imbalance between inadequate food intake and increased tumor metabolism. Notably, one study evaluated the impact of demographic and socioeconomic factors on cachexia in gastrointestinal tract cancer patients and revealed an elevated risk among socioeconomically disadvantaged and uninsured patients.41 Liver cancer, particularly hepatocellular carcinoma (HCC), predominantly arises from cirrhosis, leading to liver dysfunction, such as protein synthesis disorders, and often results in muscle wasting and physical debility in advanced stages. This may explain the high prevalence of cancer cachexia within the liver cancer patient population. Importantly, Rich et al. reported that cachexia and precachexia are prevalent across all HCC stages, including patients with early-stage tumors.45 Similarly, cachexia in pancreatic cancer patients is highly prevalent and clinically relevant, primarily because of alterations in both exocrine and endocrine pancreatic functions, as well as potential impacts on the structure of other digestive organs or indirect modifications to gut physiology.46
Despite the scarcity of research on thyroid cancer cachexia, the available data revealed a strikingly high prevalence of thyroid cancer cachexia among at-risk patients, with 39.9% and even 69% in one cohort study.24 This elevated incidence is partly due to the adverse effects caused by the administration of multikinase inhibitors, which include weight loss, nausea, and diarrhea.47 Furthermore, the anatomical location of the tumor, for instance, head and neck cancer, may have an impact on nutritional intake. Malignancy and its treatment frequently compromise essential physiological functions critical for chewing, swallowing, and saliva production, thereby exacerbating the risk of cachexia in patients with thyroid cancer.48
The relatively high prevalence of cachexia in lung cancer patients is potentially due to anorexia, cytokines, and metabolic abnormalities.49 Interestingly, patients with epidermal growth factor receptor (EGFR)-mutated lung cancers have a lower risk of developing cachexia, possibly because these cancer cells tend to progress more slowly than those without EGFR mutations.30 However, comprehensive elucidation of the mechanisms driving cachexia in lung cancer patients may facilitate the development of targeted interventions and better outcomes for this vulnerable population.
Breast cancer patients exhibit the lowest incidence of cancer cachexia and often avoid weight loss after diagnosis. In contrast, they face a risk of weight gain. This may be attributed to reduced metabolism caused by treatment-related ovarian failure and premature menopause, coupled with decreased physical activity after diagnosis.50 These findings highlight the necessity of implementing tailored weight management strategies for patients with distinct types of cancer.
In summary, the prevalence of cachexia in these studies was between 11% (endometrial cancer)24 and 84% (pancreatic cancer).28 Notably, the lack of standardized definitions across the studies precluded precise conclusions. However, we can deduce that the incidence of cancer cachexia is potentially influenced by the unique characteristics of the primary tumor. Therefore, when devising treatment plans, it is imperative to consider the differences among various tumor types.
Prognostic impacts on clinical outcomes
Cancer cachexia frequently results in adverse prognoses and elevated mortality rates among patients. The 12-month mortality rate for patients with cancer cachexia has reached 30.2%.51 After adjusting for various factors, such as age, sex, race/ethnicity, stage, and treatment, cachexia remained an independent predictor of poorer survival across diverse cancer patient populations.45 Despite the unclear causal relationship, there is a notable connection between cancer treatment and cachexia. While chemotherapy is effective in improving the 5-year survival probability, it leads to various adverse events, including skeletal muscle deconditioning.52 Several studies have indicated that approximately half of patients with advanced colorectal cancer develop cachexia within 24 weeks after receiving first-line systemic chemotherapy.53 Importantly, cancer cachexia and its metabolic alterations may impair patients’ tolerance to cancer therapies. Patients with cachexia experience more severe appetite loss and fatigue, potentially disrupting their ability to continue therapy and adversely affecting their well-being and quality of life.30,31 These findings underscore the importance of continuous monitoring of cachexia during cancer treatment.
Given the detrimental impacts of cancer cachexia on clinical outcomes, increasing attention has been given to elucidating its pathogenesis. Therefore, our review aims to consolidate and analyze existing research to offer comprehensive insight into the underlying mechanisms of cancer cachexia. However, as mentioned above, the characteristics of cachexia vary among different types of tumors and are closely tied to the primary tumor. Considering the complexity, our discussion will focus primarily on the common mechanisms for most types of cancer cachexia.
Regulatory roles and interactions of multiple organs in cancer cachexia
Over 3500 years ago, ancient biblical accounts documented a monarch affected by primary carcinoma of the prostate or kidney with subsequent metastases to bones. The textual descriptions—including “I forgot to eat my bread” (anorexia), “My knees are weak through fasting, and my flesh has lost its fatness” (asthenia and adipose depletion), “My strength has failed… and my bones are consumed” (osteolysis), “My bones wasted away through my anguished roaring all day long” (chronic pain-induced demineralization), and “I am feeble and depressed” (affective disturbances)—collectively suggest a clinical presentation congruent with cancer-related cachexia.54 It is evident that cancer cachexia, arising from uncontrolled and disrupted interactions among multiple organs or tissues, frequently manifests as systemic metabolic disturbances and augmented inflammatory responses.1,2,6 However, the complex mechanism by which diverse organs or tissues drive cancer cachexia through the excretion of various molecular signals and jointly regulating the metabolic and immune landscapes within organs is still poorly understood (Fig. 3). A systematic review of multifactorial alterations, along with their underlying molecular mechanisms, may offer crucial insights for developing therapeutic interventions and optimizing the efficacy of pharmacological treatment for cachexia management in oncology.

Overview of the interactions between systemic metabolic remodeling and the cellular inflammatory response in cancer cachexia. Crosstalk among various tissues and organs contributes to the complex clinical manifestations observed in cancer cachexia, which is characterized by metabolic disturbances. Systemic metabolic remodeling is potentially associated with the cellular inflammatory response. Subsequently, the cells that undergo functional and quantitative alterations, along with the cachexia-associated factors they secrete, penetrate the vascular system and influence remote target organs/tissues. These inflammatory alterations can lead to a shift toward a catabolic state in terms of systemic metabolism, thereby exacerbating the progression of cancer cachexia. The network diagram above illustrates the intricate interplay between organs and tissues. The left chart below depicts the metabolic reprogramming that occurs in the state of cachexia, whereas the right chart below reveals the specific impacts of these metabolites on various cell types, which in turn affect target organs, further exacerbating the systemic metabolic imbalance. This figure was created with BioRender (https://biorender.com)
Involvement and regulatory roles of multiple organs
Cachexia involves distinct clinical stages, and each stage can display different clinical symptoms. In the initial stage, patients may experience anorexia and metabolic changes, with insignificant weight loss but gradual adipose tissue depletion. As cachexia progresses, patients experience unconscious weight loss exceeding 5% within six months, accompanied by skeletal muscle atrophy.3 Metabolic changes in the liver, such as bile acid, may occur before cachexia, which is attributed to gut microbial dysbiosis.55 Therefore, we may conclude that in some cases, gut microbiota disorders may occur before liver metabolic disorders. Petruzzelli et al. identified a phenomenon termed white adipose tissue (WAT) browning, which takes place in the initial stages of cancer cachexia before skeletal muscle atrophy.56 A study of 68 pancreatic cancer patients via CT revealed that the loss of subcutaneous adipose tissue was first observed in cancer cachexia patients, followed by skeletal muscle and visceral adipose tissue loss.57 This finding was corroborated in a larger cohort of 1690 patients with pancreatic ductal adenocarcinoma (PDAC).58 However, another study employing age-, sex-, and race-standardized tissue measurements reported that skeletal muscle decline precedes adipose tissue loss, occurring 18 months and 6 months before pancreatic cancer diagnosis, respectively.59 These inconsistencies could be ascribed to differences in evaluation methodologies and sample sizes. Notably, both muscle and bone volume loss become evident in the late stage of cancer cachexia.58 In this review, we succinctly outline the crucial roles of target organs in cancer cachexia in the sequence of the nervous system, gut, liver, adipose tissue, skeletal muscle, and bone. However, the sequence of organ involvement may differ on the basis of individual variations and research approaches, necessitating further exploration in future studies to derive more definitive conclusions.
Central nervous system dysfunction
The effects of the neuroendocrine system on anorexia and appetite control have garnered considerable attention in cancer cachexia research.60 Appetite regulation is involved in many neural circuits and regions within the central nervous system (CNS). Neurons stimulate appetite by secreting neuropeptide Y (NPY) and agouti-related protein (AgRP). In contrast, proopiomelanocortin (POMC) and cocaine- and amphetamine-regulated transcript (CART) neurons exert opposite effects.61 POMC and AgRP neurons residing in the hypothalamic arcuate nucleus serve as critical regulators of melanocortin signaling in the CNS, which is associated with cachexia-related appetite dysfunction through POMC activation and AgRP suppression.62,63 There are five melanocortin receptors (MCRs) known to exist. Among them, MC3R and MC4R are expressed primarily in the brain. α-MSH is considered an anorexigenic agonist, whereas AgRP is considered an orexigenic antagonist/inverse agonist. The activation of MC4R by α-melanocyte-stimulating hormone (α-MSH) released from POMC neurons inhibits appetite and food intake.64,65 Conversely, MC4R antagonists mitigate cancer cachexia through antagonizing central melanocortin signaling, stimulating appetite, and promoting anabolism.62,63 Recent research highlighted the anorectic effects of diverse biopeptides, especially lipocalin 2 (LCN2)66,67 and glucagon-like peptide-1 (GLP-1),68 which directly or indirectly modulate the melanocortin system to suppress appetite. In addition to the melanocortin system, the area postrema and nucleus of the solitary tract (AP/NTS) have also emerged as targets for cancer anorexia-cachexia syndrome. A study demonstrated that pharmacological blockade of brainstem GLP-1 signaling attenuates cachexia in rats.69 Similarly, the knockdown of brainstem prolactin-releasing peptide reduced weight reduction and appetite suppression in tumor-bearing rats, which might be involved in hypothalamic‒pituitary‒adrenal (HPA) axis inhibition.70 Circulating growth differentiation factor 15 (GDF15) traverses the blood‒brain barrier (BBB) to activate the GDNF family receptor alpha-like and Ret proto-oncogene (GFRAL‒RET) receptors in the AP/NTS of the hindbrain, initiating a neural cascade culminating in anorexia through parabrachial nucleus activation.71
The current scientific consensus supports a strong correlation between chronic inflammatory processes and anorexia. The hypothalamus, the key regulator of appetite, contains numerous neurons that recognize multiple inflammatory signals, which trigger skeletal muscle atrophy, anorexia, and weight loss.72 TIR-domain-containing adaptor-inducing interferon-β (TRIF) has been reported to induce the production of several cytokines and chemokines in the hypothalamus, as well as microglial activation and neutrophil mobilization into the brain. The activation of these inflammatory signals induces anorexia and gastrocnemius catabolism.73 Dilp8/INSL3 derived from tumor tissue activates the Lgr3 receptor in the hypothalamus, which increases the expression of anorexigenic nucleobinding 1 and decreases the expression of the orexigenic neuropeptides short neuropeptide F (sNPF) and NPF, triggering cachexia-associated anorexia.74 In addition to peripheral tissue-derived inflammatory molecules, the neuroimmune axis has recently been implicated in cachexia in mouse models and human specimens. For example, the C-C motif chemokine ligand 2/C-C motif chemokine receptor 2 (CCL2/CCR2) axis recruits neutrophils to the brain to induce anorexia and sarcopenia in patients with pancreatic cancer.75 Since inflammation plays a pivotal role in cachexia-associated anorexia, elevated intestinal permeability may represent a potential mechanism underlying central inflammation.
Gut microbiota dysbiosis and intestinal barrier disruption
The presence of cancer located outside the gut can disturb the delicate balance of intestinal homeostasis and modify the gut microbiome. By adjusting the gut microbiota composition and fortifying gut barrier function in mice with cachexia, alleviating the distinctive physiological characteristics associated with this condition may be feasible.76 For a long period, chemotherapy was previously believed to disrupt intestinal barrier function and the gut microbiota composition among cancer patients, at least in part, by killing intestinal stem cells and the microbiome to affect the mucus layer, epithelium, and immune system.77 However, gut barrier dysfunction is also observed in mouse models of cancer cachexia without chemotherapy.78 The gut microbiota, as a component of the intestinal microenvironment, regulates host–tumor interactions through the inflammatory response and metabolism regulation. Not surprisingly, gut dysbiosis impairs host immune function by decreasing immune activation markers, including CD11b (innate immune system), CD11c (proinflammatory macrophage), CD3γ (T lymphocytes), Tbet (Th1 lymphocytes), and IL-17A (Th17 lymphocytes).76 Therefore, enhancing gut microbial diversity effectively alleviates cachexia-related symptoms partly through regulating the expression of T-cell differentiation-related genes and inflammation-related genes in the colonic mucosa and inhibiting the abundance of FoxP3+ regulatory T cells (Tregs) and Th17 cells in mesenteric lymph nodes.79 Additionally, the gut microbiota produces short-chain fatty acids (SCFAs) and other metabolites that support barrier function and energy metabolism.80 Given the importance of the intestinal barrier in intestinal homeostasis, an increase in its permeability often leads to systemic inflammation and metabolic disturbances.
Hepatic metabolic reprogramming
The liver, as the central organ of metabolism and energy homeostasis, coordinates the intricate balance of energy and nutrient demands by mediating energy status and glucose, amino acid, and lipid metabolism.81,82,83,84 Rats with cancer cachexia induced by peritoneal carcinosis exhibit impaired hepatic mitochondrial oxidative phosphorylation efficiency, which is attributed to elevated fatty acid incorporation into hepatic mitochondrial cardiolipin, suggesting that there is an increased energy demand for adenosine‒triphosphate (ATP) synthesis.81 Reduced glycogen, glucose, and lactate levels, coupled with downregulated glucokinase expression, indicate impaired hepatic glycolysis in patients with cancer cachexia. Despite an overall increase in hepatic amino acid levels, the downregulation of the gluconeogenic enzymes phosphoenolpyruvate kinase and glucose-6-phosphatase in the liver suggests that amino acids are mainly used for biosynthesis of acute-phase reactants rather than gluconeogenesis or tricarboxylic acid (TCA) cycle activity.82 Cachexia model mice with colon carcinoma 26 (C26) exhibit severe hepatic steatosis, which is potentially attributed to reduced carnitine levels and hepatic phosphatidylcholine synthesis. The decrease in carnitine levels impairs β-oxidation in mitochondria, making them unable to provide the necessary energy. Moreover, reduced levels of phosphatidylcholine promote triglyceride accumulation in the liver by impeding very low-density lipoprotein-dependent triglyceride export.82 Another study reported that the transcription factor TGF-β1-stimulated clone (TSC) 22D4 is increased in the hepatic tissue of individuals with cancer cachexia, which is associated with hepatic lipid accumulation and decreased serum triglyceride levels via the inhibition of hepatic very low-density lipoprotein release and lipogenic gene expression.83 Consistent with these findings, hepatic dysfunction is observed in cancer patients with cachexia, as decreased B vitamin-related liver enzymes are detected in blood samples from these patients.84 These findings suggest that cachexia is accompanied by a spectrum of metabolic disturbances within the liver. Addressing these metabolic abnormalities may hold potential for both preventive and therapeutic interventions.
Fat mobilization and browning
Fat tissue, which is composed of WAT and brown adipose tissue (BAT), serves as a vital metabolic and secretory organ in cancer cachexia. In higher vertebrates, WAT and BAT typically exhibit opposing physiological functions. WAT primarily serves as an energy reservoir, storing triglycerides within white adipocytes. During periods of nutrient scarcity, triglycerides are catalyzed into fatty acids through a lipolytic cascade involving hormone-sensitive lipase (HSL), adipose triglyceride lipase (ATGL), and monoacylglycerol lipase (MGL).85 Suppressing lipolysis through targeted genetic inactivation of Atgl or Hsl can ameliorate certain symptoms of cancer cachexia.86 Conversely, BAT dissipates energy as heat during nonshivering thermogenesis, featuring multilocular lipid droplets, abundant mitochondria, and high expression of uncoupling protein 1 (UCP1), contributing to its high metabolic activity.87 WAT cells can reversibly transform into BAT cells to adapt to the environment, including cold exposure or some pharmacological agents, such as β3-adrenergic receptor agonists and thiazolidinediones, as well as various peptides and hormones, which is often called “WAT browning”.56,88 Locally activated thermogenic adipocytes in the TME not only accelerate cancer progression by providing a fuel source, potentially leading to chemotherapy resistance but also result in weight loss.89 Researchers discovered that the complex formed by glucose-regulated protein 75 and adenine nucleotide translocase 2 serves as a critical determinant of UCP1 transcriptional upregulation, ultimately facilitating the browning of adipocytes in cancer cachexia.90 However, a previous study demonstrated that neither Ucp1 knockout nor thermoneutral housing conditions prevented the loss of fat, sparking a debate regarding the importance of Ucp1-mediated thermogenesis in cancer cachexia.91 While WAT browning positively impacts health by favorably influencing energy expenditure, obesity-related metabolic disorders, insulin resistance, and hyperlipidemia, it has also been implicated as a potential driver of cancer cachexia.56,88 The process of WAT browning in cancer cachexia can be triggered by proinflammatory mediators56 and hormones (parathyroid hormone/parathyroid hormone-related protein, PTH/PTHrP).92,93 Furthermore, adipose tissue is now recognized not only as a fuel reservoir or fat depot but also as an endocrine organ that releases hormones such as adiponectin and leptin, regulating energy balance and inflammation.94 In conclusion, adipose tissue loss in cachexia patients involves multiple mechanisms spanning neurological, endocrine, and metabolic domains.
Myofiber catabolism induced skeletal muscle atrophy
Skeletal muscle atrophy is the most prominent characteristic of cancer cachexia and is attributed to muscle protein degradation involving multiple molecular pathways.95 Notably, the ubiquitin‒proteasome system (UPS) and the autophagy‒lysosome pathway (ALP) play pivotal roles in this process, with calcium (Ca²⁺) serving as a significant modulator.96 The UPS is a conserved and dynamic cascade process involving ubiquitin, ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes (E2), ubiquitin ligases (E3), and the proteasome. The UPS begins with the ubiquitination of target proteins, after which E3 ligases recognize the degraded protein and ligate ubiquitin to the substrate for degradation.97 Two pivotal E3 ligases, i.e., muscle RING-finger protein-1 (MuRF1) and muscle atrophy F-box (MAFbx/Atrogin-1), were shown to regulate skeletal muscle atrophy by triggering the degradation of different muscle proteins. MuRF1 selectively targets sarcomeric proteins such as actin, myosin heavy chain (MHC), and troponin, whereas MAFbx/Atrogin-1 mainly degrades regulatory proteins such as myogenic differentiation (MyoD).98 Furthermore, the autophagic machinery orchestrates skeletal muscle atrophy by encapsulating proteins and organelles destined for degradation within autophagosomes. The upregulation of autophagy-related genes such as Atg5, Atg7, Beclin1, and Lc3b, along with an increase in the number of autophagosomes observed in cachectic muscle, supports the role of autophagy in atrophy.99,100 A clear link was found between alterations in Ca2+ homeostasis and decreases in muscle performance. For example, imbalances between calpains and their inhibitors and reduced 130 kDa Ca²+-ATPase activity were observed in skeletal muscle or heart tissue in a cancer cachexia model, which suggests that Ca2+-dependent proteolysis was activated in tumor-bearing animals.101
Cancer-associated skeletal muscle atrophy directly fuels tumor progression by serving as a metabolic substrate for cancer cell proliferation. Specifically, Zhou et al. reported that acetyl-coenzyme A synthetase short-chain family member 2 promotes muscle wasting in pancreatic cancer patients through the GSK3β/TRAIL signaling pathway and augments tumor cell macropinocytosis, a vital mechanism for amino acid supply.102 This dual mechanism creates a vicious cycle: muscle wasting supplies tumors with essential nutrients via paracrine signaling and direct metabolic coupling, while tumor-secreted factors further exacerbate muscle catabolism. Consequently, therapeutic strategies targeting muscle atrophy may not only preserve lean body mass but also disrupt tumor metabolic dependency.
Bone resorption, marrow adiposity, and hematopoietic failure
Patients with cancer cachexia exhibit aberrant bone metabolism, which is attributed to primary tumors and bone metastases.103 Additionally, chemotherapeutic agents such as carboplatin and cisplatin can inflict varying degrees of bone loss.104,105 Bone abnormalities are always accompanied by muscle atrophy in various cachexia mouse models.106 Cancer cell-derived factors increase the expression of osteoclast markers such as Acp5, CtsK, Atp6v0d2, and Mmp13 in IDG-SW3 osteocytes, which causes weight loss and muscle atrophy.107 Receptor activator of nuclear factor-κB (NF-κb) ligand (RANKL)-induced inflammation is responsible for osteoclast differentiation and bone resorption. Tumor-derived RANKL promotes bone loss and skeletal muscle atrophy in a cachexia mouse model of ovarian cancer.108 In addition to peripheral bone destruction, the bone marrow microenvironment may also suffer dysfunction in cancer cachexia. The differentiation of bone marrow hematopoietic stem cells (HSCs) into various immune cells is crucial for systemic immune responses and is tightly regulated by the microenvironment, which includes various cells, such as mesenchymal stem cells (MSCs). MSCs possess multilineage differentiation potential, enabling them to differentiate into various types of tissue cells, encompassing adipogenic, chondrogenic, and osteogenic lineages.109 Some inflammatory factors, such as IL-6, can regulate the differentiation trajectory of MSCs, such as toward an adipogenic state, via the Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway and glucocorticoid signaling.110 Osteoclast-releasing factors such as TGF-β can also disrupt the ecological niche of HSCs, leading to cancer-related anemia.111 In conclusion, skeletal tissue plays a pivotal role in metabolic and endocrine homeostasis and coordinates the differentiation of various cell types.
Complex organismal crosstalk via diverse axes in cancer cachexia
Cancer cachexia is a multisystem disorder driven by systemic inflammation, metabolic derangements, and neuroendocrine alterations, whose pathological progression is amplified through multiorgan crosstalk. For example, disturbances in gut epithelial tight junction barriers may involve intact bacteria, lipopolysaccharides (LPS), and other bacterial components and digestive enzymes, which further lead to bacterial translocation, immune activation, and systemic inflammation. Notably, inflammation resulting from intestinal dysbiosis often impacts multiple organs throughout the body, including the gut‒brain axis, gut‒liver axis, and gut‒muscle axis, thereby eliciting a cascade effect. Skeletal muscle is the predominant tissue that undergoes catabolic remodeling in cancer cachexia.96 Skeletal muscle is no longer considered to be just a motor or energy organ but rather an endocrine organ that produces myokines,112 allowing for crosstalk between the muscle and other organs, such as the muscle‒adipose axis and the muscle‒bone axis. Furthermore, the role of the nervous system in influencing peripheral tissues has become increasingly clear in cancer cachexia (Fig. 4).

Cross-organismal interactions in cancer cachexia. Cancer cachexia involves multiple organs/tissues, including the nervous system, liver, gut and microbiota, adipose tissue, skeletal muscle, and bone. Notably, the intricate interaction among these organs and tissues exacerbates the abnormal physiological balance of the body, involving the gut‒brain axis, gut‒liver axis, gut‒muscle axis, muscle‒adipose axis, muscle‒bone axis, and dysregulated communication between peripheral tissues and the autonomic nervous system. Furthermore, the interplay of metabolic products and inflammatory signaling pathways within and among these organs creates a complex network, which contributes to the overall decline in body weight and functional capacity observed in cancer cachexia patients. The arrow line represents activation, and the blunt-headed arrow represents inhibition. Abbreviations: OXPHOS oxidative phosphorylation, LPL lipoprotein lipase, FAA free amino acid, BAIBA β-aminoisobutyric acid, OPG osteoprotegerin. This figure was created with BioRender (https://biorender.com)
Although we focused on dissecting the contributions of the gut and skeletal muscle to crosstalk, the importance of other organs in the cachexia process should not be ignored. Focusing on the multiorgan axes of these two organs enables a clearer elucidation of the various axes involved in cancer cachexia.
The gut‒brain axis
Multiple lines of evidence implicate gut–brain axis dysfunction as a central mediator in the pathological process of cancer cachexia (Fig. 4). The axis often modulates intricate biological mechanisms via bidirectional communication between the enteric nervous system (ENS) and the CNS, which is facilitated by proinflammatory cytokines, neuropeptides and neurotransmitters, vagal afferents, stress hormones, immune-mediators, SCFAs and other microbiome metabolites.113 When intestinal barrier permeability increases, bacterial LPS stimulates the secretion of IL-6 in the hypothalamus, which can be amplified by tumor-derived prostaglandin E2 (PGE2).114 Furthermore, increased plasma levels of LPS are related to hypothalamic inflammatory status, where decreased expression of the immune checkpoint receptors programmed death receptor-1 (PD-1) and CD112R, as well as increased levels of corticosterone and LCN2, occurs.115 These results indicate that intestinal barrier dysfunction ascribed to gut microbiota alterations may leak LPS into the plasma during cachexia, leading to central inflammation and triggering cachectic phenotypes. Notably, gut dysbiosis not only affects the intestinal barrier but also directly increases BBB permeability. Therapeutic interventions with Lactobacillus and sodium butyrate can reduce BBB permeability, upregulate tight junction protein expression, and reinstate endothelial barrier integrity.116 Increased BBB permeability may facilitate the penetration of circulating factors, such as GDF15, LCN2, TNF-α, and IL-6, thereby reducing food intake.
Hormones such as ghrelin, cholecystokinin (CCK), GLP-1, and peptide YY (PYY), which are derived from the gastrointestinal tract, have been shown to interact in the modulation of food consumption and body weight. For example, ghrelin participates in cancer cachexia mainly through three mechanisms: (1) stimulation of numerous hypothalamic and brainstem neurons, as well as gastric acid secretion and gastrointestinal motility; (2) increased secretion of growth hormone and reduced energy expenditure; and (3) decreased expression of anorectic cytokines.117 Elevated levels of CCK were observed in cachexia models associated with chronic heart failure, leading to cardiac cachexia, which is characterized by significant weight loss and muscle wasting.118 Choi et al. reported that Sipjeondaebo-tang, a traditional Chinese medicine, improves cancer-related anorexia and anemia by modulating cytokines (IL-6 and CCL2) and hormones (GLP-1 and PYY) in cachectic tumor-bearing mice, further emphasizing the essential regulatory role of gastrointestinal hormones in cancer cachexia.119 The nervous system controls intestinal function, whereas both inflammatory mediators resulting from gut microbiota dysbiosis and gastrointestinal hormones can affect central appetite regulation. These findings underscore the complex bidirectional communication along the gut‒brain axis during cachexia pathogenesis.
The gut–liver axis
The gut‒liver axis comprises the biliary tract, portal vein, and systemic circulation. The liver regulates nutrient absorption, metabolism, the intestinal microbiota, and gut wall permeability by releasing bile acids and other bioactive mediators into the intestine. Reciprocally, gut-derived dietary and microbial metabolites modulate bile acid biosynthesis and glucose‒lipid metabolism in the liver via the portal and systemic circulation. During cancer cachexia, the equilibrium of the gut‒liver axis, including the gut microbiota ecology, hepatic function, metabolic processes and immune responses, is frequently perturbed (Fig. 4). Li et al. reported that progressive damage to the intestinal structure during tumor progression in a liver cancer zebrafish model manifested as villus injury, intestinal wall thinning, increased goblet cell numbers, eosinophil infiltration and disrupted intestinal epithelial cell renewal, suggesting that intestinal inflammation may be a promising therapeutic target in cachexia management.120 It is conceivable that gut dysbiosis increases intestinal permeability, allowing harmful factors to traverse the gut barrier into the bloodstream and subsequently activate Kupffer cells to release proinflammatory cytokines, thereby initiating hepatic inflammatory responses. Studies have shown that CD68+ macrophage infiltration and hepatic inflammasome pathway activation trigger systemic chronic inflammation and cachectic phenotypes by increasing IL-1β activity in liver tissue.121
Enterohepatic circulation, as a bridge between the gut and liver, has emerged as an important driver of the progression of cachexia. Farnesoid X receptor (FXR) is widely expressed in the liver and intestine. Activated by bile acid in the gut, FXR induces the expression of fibroblast growth factor 15 (FGF15), which is secreted into the liver via the portal system to activate liver FXR, thus regulating the expression of genes governing enterohepatic bile acid homeostasis.122,123 Notably, oral administration of tauroursodeoxycholic acid alleviates cancer cachexia and reverses cholestasis by increasing bile acid secretion in a mouse model.123 A recent study similarly highlighted the contribution of gut microbial dysbiosis to perturbing bile acid dysmetabolism in individuals with cancer cachexia, especially the reduction in the levels of several secondary bile acids, mainly taurodeoxycholic bile acids, which occur before the development of cachexia.55 These findings underscore the therapeutic potential of taurodeoxycholic acid for hepatic cholesterol homeostasis in cachexia. Abnormal bile secretion may further impact the gut environment, increasing the accumulation of harmful hepatic metabolites and perpetuating the cycle of bile acid production and inflammation. However, Thibaut et al. reported that ursodeoxycholic acid-induced bile acid secretion fails to alleviate liver inflammation in cachectic mice but exacerbates muscle wasting due to diminished Takeda G protein-coupled receptor 5 (TGR5) activity, which can promote muscle differentiation and hypertrophy.124 The differences in the structural and functional characteristics of bile acids should be further considered.
Intestinal barrier disruption and bile acid metabolism disturbances damage liver function; however, certain factors may safeguard liver functions. Aryl hydrocarbon receptor (AHR), a ligand-dependent transcription factor, senses tryptophan metabolites and promotes intestinal barrier integrity through the upregulation of tight junction proteins and decreased levels of inflammatory factors,125 suggesting that the gut‒liver axis is essential for maintaining liver functions. Dolly et al. revealed that, compared with those in normal mice, several indole derivatives and AHR agonists are much lower in the feces of cachectic mice. The authors further reported that the AHR target gene is downregulated in the liver by the IL-6/STAT3/hypoxia-inducible factor 1α pathway. Despite failing to reverse alterations in intestinal permeability or barrier function, AHR agonists ameliorate liver inflammation and glycemic disturbances.126
In conclusion, gut microbiota dysbiosis and abnormal bile acid secretion can modify the intestinal milieu, facilitating the translocation of harmful metabolites across the intestinal barrier into the bloodstream. Consequently, immune cells are activated to release proinflammatory cytokines, sparking hepatic inflammatory responses and augmenting the accumulation of deleterious metabolites in the liver. Addressing intestinal permeability or abnormal bile acid secretion may confer protection against cancer cachexia.
The gut–muscle axis
Recent studies highlighted the interaction between the gut microbiota and skeletal muscle in cancer cachexia through metabolic and inflammatory regulation (Fig. 4). For example, reduced levels of Lactobacillus species were observed in mouse models of cancer cachexia, and Lactobacillus supplementation attenuated muscle wasting in these models, suggesting that Lactobacillus supplementation may serve as a potential therapeutic avenue for cachexia-related muscle atrophy.76 They also maintain gut barrier integrity by reducing intestinal permeability and upregulating antimicrobial proteins,76 emphasizing the profound impact of the gut microbiota on skeletal muscle health. Similarly, cisplatin and docetaxel chemotherapy induce cachexia-related muscle wasting in mice by reducing Ruminococcaceae and Bacteroides.127 These studies indicate the presence of widespread gut microbial dysbiosis across various malignancies.
Mechanistically, the gut microbiota plays dual regulatory roles in maintaining the homeostasis of skeletal muscle: (1) modulating nutrient digestion, nutrient absorption128 and systemic metabolism through bioactive metabolites80 and (2) stimulating host inflammatory responses. Metabolism and inflammation regulation frequently do not function as independent entities but rather interconnect with each other. Fiber supplementation obviously reinforces the colonic mucus layer and reduces circulating LPS-binding protein and IL-6 levels in cachexia model mice, suggesting that the modification of the colonic mucus barrier is a major contributor to the alleviation of systemic inflammation. Furthermore, dietary fiber reverses skeletal muscle wasting by inhibiting Atrogin-1, MuRF1, and autophagy markers such as LC3 and Bnip3.129 Conversely, antibiotic-mediated gut dysbiosis and aberrant bile acid metabolism induce muscle atrophy through the repression of skeletal muscle protein synthesis, which is mediated by the FXR-FGF15/19-extracellular-signal-regulated protein kinase (ERK)1/2 signaling pathway.130 Under pathological conditions, the endogenous components of the intestinal microbiota, such as LPS and flagella, may mediate inflammatory responses. The release of LPS, which passes through the compromised gut barrier into the circulation, can activate toll-like receptors (TLRs) and subsequently trigger the synthesis of proinflammatory factors, such as IL-6.131 TRIF also orchestrates critical signal transduction cascades in TLR responses to LPS.73 Once recognized by pattern recognition receptors, gut microbiota-derived flagellin promotes the secretion of CCL2 and IL-6 in C26-induced cachexia models, thereby significantly exacerbating myogenesis in C2C12 myoblasts.132 In addition to inflammatory factors, gut barrier disruption promotes the infiltration of eosinophils, M1/M2 macrophages, and fibroblasts, which is accompanied by increased concentrations of IL-13 and TGF-β3 in the colon mucosa, supporting the hypothesis that the intestinal barrier is impaired in cancer cachexia. The depletion of goblet cells in the colon epithelium may further affect the integrity of the mucus barrier.133 In summary, inflammatory signals and metabolic imbalance within the context of gut barrier impairment and microbiota dysbiosis exert a long-range effect on skeletal muscle homeostasis, exacerbating their negative nitrogen balance, wherein the synthesis of skeletal muscle proteins falls below their breakdown rates.
The muscle‒adipose axis
Skeletal muscle and adipose tissue function as dual-role organs with respect to metabolic and endocrine roles. Muscle‒adipose tissue crosstalk maintains the response to nutritional and physical stimuli. Dysregulation of this crosstalk contributes to the pathogenesis of various diseases, including cancer cachexia (Fig. 4). IL-15, a highly expressed cytokine in muscle tissue, regulates muscle‒adipose interactions by reducing visceral fat mass.134 However, Molanouri et al. reported that aerobic interval training and antioxidant therapy prevent muscle atrophy and preserve adipose mass through increased IL-15 expression within the skeletal muscle of cachexia-bearing mice,135 highlighting the complexity of IL-15 in regulating adipose tissue. Another myokine, myostatin (MSTN), can cause muscle atrophy, but plasma Mstn concentrations negatively correlate with the development of the cachexia phenotype in colorectal or lung cancer patients.136 Consistently, patients with cachexia presented the highest FGF21 levels.137 Fu et al. revealed that ablation of FUNDC1 in skeletal muscle, a mitophagy mediator, promotes adipose tissue thermogenesis by increasing FGF21 levels,138 suggesting that FGF21-mediated energy metabolism modulates muscle‒adipose interactions. Although this regulatory relationship has not been definitively confirmed in tumors, the possibility cannot be ruled out. In addition to skeletal muscle-derived myokines, adipose tissue also secretes adipokines with potential effects on cachexia (Fig. 4). Adipocyte-derived LCN2 promotes lipolysis and muscle atrophy through transcriptional activation of MuRF-1 and Atrogin-1 expression during pancreatic cancer cachexia.139 Some factors, such as FGF21, can be secreted by both skeletal muscle and adipocytes. Under cold conditions, FGF21 derived from BAT serves as a mediator of β3-adrenergic-dependent GDF15 gene transcription and is released in BAT, which suppresses proinflammatory gene expression and decreases TNF and CCL2 secretion in macrophages.140 However, GDF15 appears to exert a proinflammatory effect in cancer cachexia,141 implying that the same factor can have various effects depending on the physiological or pathological context. Recent studies have demonstrated that tumor epithelial cells not only increase IL-6 production in adipocytes but also increase IL-6R levels in myocytes and subsequently sIL-6R levels in plasma, which induce myotube wasting and adipocyte lipolysis. It highlights the complex crosstalk between tumors, adipose tissue, and muscle.142 Another study reported that adipocyte-derived IL-6 promotes adipose tissue inflammation, whereas IL-6 derived from muscle inhibits macrophage infiltration in adipose tissue, triggering an anti-inflammatory response.143 These findings indicate that the source of IL6 influences the type of inflammatory response. Overall, energy metabolism disruption and endocrine dysregulation within adipose tissues and muscle are particularly important for the development of many diseases, including cancer cachexia.
The muscle‒bone axis
The intricate interplay between bone and skeletal muscle is underscored by their anatomical proximity and reciprocal signaling mediated through bone- and muscle-derived factors (Fig. 4). Research has demonstrated that cisplatin-treated bone-conditioned media elicits profound atrophy in muscle tubules, indicating that bone-derived soluble factors induce muscle wasting. Phosphonates mitigate bone damage and ameliorate muscle atrophy by inhibiting osteoclast-mediated bone resorption.105 Within the pathogenic milieu of bone metastases, bone-derived TGF-β contributes to muscle weakness by decreasing Ca2+-induced muscle contractility.144 Collectively, these findings underscore the implications of bone-derived factors for muscle function, emphasizing that interventions aimed at preventing bone destruction may effectively preserve skeletal muscle mass and safeguard contractile performance.
Notably, the role of muscle‒bone interactions in other physiological and pathological processes may reveal potential roles in cancer cachexia. Osteoblast/osteocyte-derived Connexin43 modulates muscle growth and function, potentially via an endocrine effect of the undercarboxylated isoform of osteocalcin.145 Additionally, RANKL inhibitors and osteoprotegerin antagonists synergistically regulate muscle insulin sensitivity and glucose uptake.146 These findings should inspire further investigations to explore treatments for muscle diseases characterized by elevated muscle wasting, including cancer cachexia. Conversely, skeletal muscle releases regulatory factors that act on bone. For example, skeletal muscle secretes MSTN to induce RANKL-mediated osteoclast formation in vitro through Smad2-dependent regulation of the nuclear factor of activated T cells.147 The exercise-induced muscle factor β-aminoisobutyric acid prevents bone loss and muscle dysfunction by counteracting ROS-mediated mitochondrial breakdown and osteocyte apoptosis.148 The regulation of muscle‒bone interactions in cancer cachexia remains elusive, and new investigations should be pursued to reveal the complex mechanisms involved.
Regulatory role of the SNS and PNS in several tissues
The CNS, along with the sympathetic nervous system (SNS) and parasympathetic nervous system (PNS), collaboratively maintains body homeostasis by integrating body information, triggering “fight or flight” responses, and promoting rest and digestion, respectively. SNS and the PNS can influence multiple distant organs, especially the CNS, muscles, and adipose tissues (Fig. 4).
Hypothalamic POMC neurons can regulate energy metabolism through SNS activation. This pathway suggests a potential mechanistic link between POMC-mediated SNS hyperactivity and the pathogenesis of cachexia. For example, cannabinoid 1 receptors in the hypothalamus and in the limbic system mediate orexigenic effects. Notably, the conditional knockout of cannabinoid 1 receptors in sympathetic neurons led to sympathetic activation, which resulted in a lean phenotype.149 These results demonstrate the importance of SNS activity in cancer cachexia. The vagus nerve, an important component of the PNS, can sense tumor growth potentially via proinflammatory cytokines. This sensory information may subsequently be conveyed to brainstem areas and ultimately to the hypothalamus, triggering the activation of the melanocortin system to decrease food intake.150 Indeed, subdiaphragmatic vagotomy can prevent the cancer cachexia phenotype,151 which further implies a potential contribution of the vagal PNS to the pathogenesis of cancer cachexia. However, recent research revealed that subdiaphragmatic vagal deafferentation failed to alleviate anorexia or prevent body weight loss, suggesting that vagal afferents may not be obligatory for mediating cancer cachexia syndrome in this experimental model.152 These findings collectively underscore the potential interactions with several orexigenic and anorexigenic neuropeptides and hormones, the CNS, the SNS, and the PNS in cancer cachexia.
Some studies have demonstrated that β-2 adrenergic agonists can effectively counteract the accelerated muscle protein catabolism observed in cancer cachexia models, including myocardium and skeletal muscles,153,154 suggesting the potential role of SNS activity in modulating skeletal muscle wasting during cancer cachexia. Consistently, a study demonstrated that the use of a neutralizing antibody targeting GDF15 signaling or sympathectomy alone prevented cancer cachexia by reducing excessive β-oxidation in adipose tissue.155 Interestingly, Xie et al. reported that increased peripheral sympathetic activity may be induced by intraadipose macrophages. Specifically, IL-4 receptor deficiency hindered the alternative activation of these macrophages, resulting in decreased sympathetic activity and inhibited WAT browning.156 These findings suggest that the SNS contributes to the lipolytic response in cancer cachexia. In general, the SNS and PNS, as the two primary constituents of the autonomic nervous system, are extensively distributed across various organs and tissues. They regulate the functional activities of these organs by releasing distinct neurotransmitters, thereby potentially modulating the progression of cachexia.
Other cross-organ interactions
In addition to the aforementioned crosstalk, other tissues have intricate interconnections. The heart, as the pivotal organ of the circulatory system, is responsible for circulating blood throughout systemic tissues and organs. Myocardial atrophy, ventricular remodeling, and decreased cardiac function have been consistently documented in murine models of cancer cachexia. These cardiac alterations may be attributed, at least in part, to oxidative stress-mediated pathways.157 Compromised cardiac function may lead to inadequate blood supply, thereby exacerbating tissue hypoxia and nutrient deprivation in peripheral organs. Simultaneously, cachexia-induced metabolic reprogramming drives aberrant interorgan communication: the augmented Cori cycle exemplifies this cross-talk, where lactate shuttled from cachectic skeletal muscle to the liver fuels compensatory gluconeogenesis. This diverted metabolic flux not only supports tumor growth but also perpetuates a vicious cycle, which further strains cardiac function through energy-consuming futile cycles and accelerates cachexia progression.158
Owing to space limitations, we focus more on elucidating the primary roles of select organs and their interactions in cancer cachexia. Specifically, we focused on molecules beneficial to tissues and organs under other conditions but unreported in cancer cachexia. Furthermore, we observed that the same molecules may exhibit opposite effects in different contexts, on which unraveling the underlying mechanisms offers new insights into the pathology of cachexia. In summary, intricate tissue‒organ interactions are paramount in cancer cachexia; therefore, interfering with these interactions could contribute to the treatment of cachexia.
Molecular and cellular mechanisms: metabolic alterations, inflammatory responses, key mediators, and signaling pathways
The extensively reported organ interactions involved in cancer cachexia, such as the gut‒brain axis, gut‒liver axis, gut‒muscle axis, muscle‒adipose axis, and muscle‒bone axis, and the regulatory role of the SNS and PNS in several tissues have been thoroughly described in recent decades. However, inflammation and metabolic disorders are intertwined in the crosstalk of these organs/tissues, which raises the chicken-and-egg question of whether it is a disturbance of metabolism or inflammation that initiates cancer cachexia. Metabolic disorders, characterized by disruptions in carbohydrates, lipids, and proteins, play essential roles in cancer cachexia.159 The notable abnormalities in host metabolism diminish nutrient and oxygen availability within the TME and lead to the accumulation of metabolic byproducts, which undoubtedly pose additional threats to cellular health and function. The increased metabolic burden subsequently accelerates cellular damage and dysfunction, resulting in inflammatory responses among immune cells and stromal cells (Fig. 5). In this scenario, the involved cells release certain cachexia-inducing factors (Fig. 6), which disrupt homeostasis by activating relevant signaling pathways (Fig. 7) and ultimately drive the aggressive progression of cachexia. The formation of the feedback loop underscores the intricate interplay between cellular functional transformation and dynamic metabolic‒inflammatory interactions during cancer cachexia progression.

Metabolic remodeling in cancer cachexia and its regulatory effects on cellular functions. a The network diagram above illustrates the profound metabolic dysregulation associated with cancer cachexia, such as decreased hepatic ketogenesis, impaired lactate metabolism, elevated hepatic triglyceride accumulation, reduced gut microbiota-derived SCFA production, and suppressed protein synthesis. In glucose metabolism and the TCA cycle, an increase in energy consumption accompanied by an abnormal shift toward anaerobic glycolysis exacerbates the imbalance between energy supply and demand. In fat metabolism, accelerated fat breakdown coexists with WAT browning, resulting in rapid depletion of body fat reserves. In protein metabolism, intensified protein degradation coupled with weakened synthetic capacity leads to significant muscle wasting. b The effects of metabolites on immune and stromal cells include phagocytosis and polarization of macrophages, proliferation and effector functions of T cells, ECM production by fibroblasts, and angiogenesis in endothelial cells. The arrow line represents activation, and the blunt-headed arrow represents inhibition. Abbreviations: BAs bile acid, CoA coenzyme A, 3PG 3-phosphoglycerate, GLUT glucose transporter type, GSSG oxidized glutathione, KB ketone body, Pyr pyruvate, α-KG α-ketoglutarate, MPC mitochondrial pyruvate carrier, APP acute phase protein, FAs fatty acids, VLDL very low-density lipoprotein, TSC22D4 transforming growth factor β1-stimulated clone 22D4, Arg arginine, Leu leucine, Met methionine, Ser serine, Gly glycine, Cys cysteine, Glu glutamate, SAM S-adenosylmethionine, TCF7 transcription factor 7, MMP9 matrix metalloproteinases-9, IFP interstitial fluid pressure. This figure was created with BioRender (https://biorender.com)

Tumor-associated inflammation alters organismal metabolic status in cancer cachexia. Cancer cachexia is closely associated with the dysfunction of multiple cell types, including TAMs, TANs, TILs, MDSCs, CAFs, and ECs, all of which are currently the subject of extensive research interest. The metabolic burden on these cells fosters a macroenvironment that is more conducive to tumor growth and the progression of cachexia. Mechanistically, they reach distant organs/tissues through the circulatory system to exert their effects or exert a broader influence by releasing cachexia-related factors. The arrow line represents activation, and the blunt-headed arrow represents inhibition. Abbreviations: ETS1 V-et erythroblastosis virus E26 oncogene homolog 1, NDP neutrophil-derived proteases, FAA free amino acid, CB2 cannabinoid 2, LPL lipoprotein lipase, mTEC medullary thymic epithelial cell, ONOO- peroxynitrite, eIF2α eukaryotic translation initiation factor 2α, MyoF myofibroblast, RA retinoic acid, ICAM1 intercellular cell adhesion molecule-1. This figure was created with BioRender (https://biorender.com)

Inflammatory ligand-activated signaling pathways in cachexia-related muscle and fat atrophy. In patients with cachexia, elevated cytokine levels activate or inhibit multiple signaling pathways, leading to muscle and fat wasting. IL-1, IL-6, and TNF-α stimulate the release of CRH in the hypothalamus, which promotes the secretion of glucocorticoids through the HPA axis. This process inhibits the Akt signaling pathway while upregulating the expression of MuRF1, MAFbx, and autophagy-related genes. IL-1 and TNF-α recruit TRAF2/6 to activate the MAPK and NF-κB signaling pathways, facilitating the nuclear translocation of p38 MAPK and NF-κB, respectively, which upregulates the expression of MuRF1 and MAFbx. The TGF-β family engages its receptors, recruiting TRAF4/6 and Smad2/3 to activate the NF-κB and Smad signaling pathways. This promotes the nuclear translocation of the NF-κB and Smad2/3/4 complexes, increasing the expression of MuRF1 and MAFbx. The IL-6 family binds to its receptors to activate the JAK/STAT signaling pathway, leading to the nuclear translocation of STAT3 and the subsequent upregulation of MuRF1 and MAFbx expression. Moreover, the nuclear translocation of p38 MAPK and STAT3 induced by IL-1, TNF-α, and the IL-6 family can also upregulate UCP-1 expression, contributing to fat wasting through mitochondrial involvement. Additionally, the IL-1-, TNF-α-, and IL-6-mediated FoxO and STAT3 pathways directly participate in ATGL- and HSL-dependent lipolysis. This figure was created with BioRender (https://biorender.com)
Alterations in metabolites and their potential impacts on immune and stromal cells
Metabolic imbalance in cancer cachexia cannot be overlooked, as it may reshape cellular function and exacerbate the progression of cachexia. Macroenvironmental metabolites are crucial for maintaining homeostasis and serve as essential constituents of energy sources. In addition to their classic roles via the allosteric regulation of enzymes involved in metabolism, some metabolites also convey a specific signaling message through protein receptors.160 Furthermore, metabolites function as substrates or cofactors for chromatin-modifying enzymes, and they regulate gene expression through both transcriptional and epigenetic mechanisms.161 Importantly, the cellular inflammatory response is considered a key hallmark of cancer progression and can be induced by abnormal metabolism. Given the vast array of metabolites, we classified them into three main categories: glucose metabolism and the TCA cycle, amino acid metabolism, and fatty acid metabolism. Variations in the dynamic levels of these components can result in modifications to the immune response and stromal cell functionality, thereby fostering a microenvironment conducive to tumorigenesis (Table 1). Thus, metabolic imbalance and inflammatory responses in cancer cachexia play pivotal roles in shaping disease progression.
Glucose metabolism and the TCA cycle
Metabolites constitute an extensive and intricate system that is indispensable for understanding numerous diseases, ranging from traditional metabolic disorders to cancers. During the metabolic process, they not only release energy to serve as a power source but also modulate enzyme activity through intricate feedback mechanisms governing key metabolic pathways to influence the metabolic rate. Crucially, cancer metabolites can affect epigenetic modifications, posttranscriptional modifications, and signal transduction processes.162 In this discussion, we focus on lactate, pyruvate, alpha-ketoglutarate, and ketone bodies, which are frequently reported in the context of cancer cachexia.
Lactate
Organisms harness energy from glucose via fermentation and respiration, both of which initiate glycolysis to yield pyruvate. During fermentation, lactic dehydrogenase (LDH) reduces pyruvate to lactate, which is then expelled into the cytoplasm. The oxidative respiration channel nicotinamide adenine dinucleotide reduces (NADH) and pyruvate to the mitochondria, which is facilitated by pyruvate dehydrogenase (PDH).163 Warburg’s seminal work in the 1920s highlighted that cancer cells preferentially convert glycolytic pyruvate to lactate, even under aerobic conditions.164
During cancer cachexia progression, abnormal metabolism of lactate can occur in various tissues, including skeletal muscle, adipose tissue, the liver, and the CNS (Fig. 5). LDH reportedly correlates with systemic inflammation and survival rates in advanced cancer patients.165 The decreased activity of PDH and succinate dehydrogenase in cachectic skeletal muscle indicates a reduced flux of glycolytic pyruvate into the TCA cycle.166 Under conditions of metabolic stress, such as nutrient deprivation and hypoxia, a reduction in PDH activity is anticipated. This reduction occurs due to the inactivation of PDH by PDH kinase (PDK) 4, which is upregulated in cancer cachexia. This upregulation can subsequently lead to the shrinkage of C2C12 myotubes and the induction of muscle atrophy in vivo.167 Increased glucose uptake, decreased oxygen consumption, upregulated LDH expression, and enhanced lactate production in cachectic myotubes and adipose tissue were identified. Inhibition of LDH can alleviate the cancer cachexia phenotype.168 Monocarboxylate transporters (MCTs) 1--4 are proton-dependent membrane proteins that serve as critical mediators of transmembrane flux for glycolysis-derived metabolites (i.e., lactate and pyruvate) and ketone bodies (i.e., acetoacetate and β-hydroxybutyrate).169 Lactate exerts metabolic effects and activates signaling cascades in neurons through transmembrane transport via MCT2 or by acting on specific receptors,170 implicating the critical modulator of lactate in the nervous system. The infusion of lactate into mice via the hepatic portal vein or the vena cava augments postprandial satiety and reduces feeding, implying that increased peripheral lactate may induce neurogenic anorexia, promoting weight loss and tissue wasting.171
The multifaceted roles beyond the energy supply of lactate have been well elucidated. For example, lactate-induced G protein-coupled receptor (GPR) 81-mediated cachexia in adipose tissue occurs via the Gi-Gβγ-RhoA/ROCK1-p38 signaling cascade. This cascade enhances adipose browning and lipolysis and ultimately leads to muscle wasting and systemic hypercatabolism.172 Lactate and other metabolites may also modulate autophagy through reactive oxygen species (ROS)-mediated signaling cascades involving ERK1/2/mammalian target of rapamycin (mTOR)/p70S6K.173
The increased levels of lactate in the circulatory system are closely related to the deterioration of cachexia. Furthermore, at the cellular level, increased lactate profoundly influences diverse cellular states and functionalities (Table 1). At relatively high concentrations, lactate enhances histone H3 lysine 27 acetylation (H3K27ac) at Tcf7 superenhancer sites by inhibiting histone deacetylase activity, thereby increasing Tcf7 gene expression in CD8+ T cells to increase their stemness and improve the efficacy of anti-PD-1 therapy.174 The addition of lactate to glucose-starved mouse bone marrow-derived macrophages (BMDMs) increases H3K9ac at promoter regions of typical M2-associated genes, indicating an epigenetic role of lactate in promoting macrophage polarization and immunosuppressive function.175 Furthermore, lactylation, identified as a novel epigenetic modification of histone lysine residues, can also directly stimulate gene transcription in mouse and human cells, such as arginase 1 expression in mouse BMDMs.176 Lactate also inhibits tumor surveillance by suppressing the function of natural killer (NK) cells.177 Linares et al. demonstrated that lactate impairs poly(ADP‒ribose) polymerase 1 (PARP-1) activity by reducing the NAD+/NADH ratio. PARP-1 inhibition blocks the poly(ADP-ribosyl)ation of c-FOS and c-JUN, leading to p62 downregulation, which is associated with the induction of the CAF phenotype.178 Furthermore, several studies have shown that lactic acidosis in hypoxic tumor tissue can activate TGF-β signaling, which subsequently stimulates neovascularization by regulating the expression of vascular endothelial growth factor (VEGF) and matrix metalloproteinases. This process facilitates tumor cell invasion and progression.179
Elevated lactate levels in cancer cachexia can induce skeletal muscle atrophy, anorexia nervosa, hepatic energy deficiency, and WAT browning. Various studies have demonstrated that lactate in the microenvironment impacts multiple cellular functions, including but not limited to T cells, NK cells, macrophages, fibroblasts, and ECs. However, these findings have yet to be confirmed in cancer cachexia patients and necessitate further investigation.
Pyruvate
Pyruvate, along with its corresponding metabolic enzymes, can restore energy balance by entering the TCA cycle and therefore regulate genes associated with skeletal muscle differentiation and degradation, ultimately impacting cancer cachexia (Fig. 5). Myotubes treated with conditioned media containing sodium pyruvate from CT26 colon carcinoma cells presented normalized lactic acid production, oxygen consumption, and PDH activity. Notably, the mitochondrial pyruvate carrier serves as the sole entry point for the entry of pyruvate into mitochondria. Inhibition of the mitochondrial pyruvate carrier in myotubes recapitulates metabolic perturbations and cachectic traits in addition to those observed with conditioned media from CT26 cells, thereby emphasizing the importance of the mitochondrial pyruvate content in mitigating cachexia.180 A previous study revealed that PDHB knockout causes lactate accumulation by reducing pyruvate metabolism and suppressing genes related to muscle differentiation. Ariadne RBR E3 ubiquitin protein ligase 2 is a pivotal downstream mediator in this process and is involved in aging-associated muscle degeneration and ubiquitin-dependent modification in skeletal muscle.181 PDK4 promotes gluconeogenesis through the preservation of PDH substrates such as pyruvate, lactate, and alanine. However, recent experimental evidence has demonstrated that virus-mediated PDK4 overexpression in myotube cultures is sufficient to promote myofiber shrinkage, which is consistent with increased protein catabolism and mitochondrial abnormalities.167
Multiple studies have highlighted the effects of pyruvate metabolism on functional phenotypes across diverse cellular populations (Table 1), such as macrophage polarization, mitochondrial dynamics, and the formation of mitochondria-associated membranes, all of which are indispensable for macrophage function. T-cell differentiation is also linked to pyruvate metabolism, with PDK inhibiting pyruvate oxidation and promoting proinflammatory T-cell polarization.182 In cytotoxic CD8+ T cells, pyruvate fuels the TCA cycle via pyruvate carboxylase and PDH. Pyruvate carboxylase, a pivotal gluconeogenic enzyme, converts pyruvate to mitochondrial oxaloacetate, which maintains succinate secretion and initiates autocrine signaling through succinate receptor 1, triggering the production of cytotoxic molecules in T cells to augment their tumoricidal capacity. Conversely, tumor-derived lactate perturbs pyruvate flux within the TCA cycle, favoring PDH-mediated reactions over pyruvate carboxylase activity, highlighting the metabolic orchestration of the TME.183 Similarly, pyruvate carboxylase inhibition in TAMs induced by a hypoxic tumor environment has immunosuppressive effects on TAMs, which is mainly mediated by CD8+ T cells and macrophages.184 Intriguingly, studies have shown that inhibiting mitochondrial pyruvate import in CD8+ T cells promotes glutamine and fatty acid oxidation, leading to elevated CoA levels, particularly H3K27ac, which enhances histone acetylation and chromatin accessibility. This shift facilitates CD8+ T-cell differentiation toward memory T cells, partly through the transcriptional regulation of RUNX1.185
Pyruvate metabolism dysfunction in cancer cachexia involves multiple enzymes, such as PDH, PDK and pyruvate carboxylase, which affect metabolite levels along the metabolic axis and orchestrate critical processes in myogenic differentiation and muscle proteolysis. Further exploration of the cellular functions of pyruvate in the progression of cancer cachexia is necessary.
Alpha( α )-ketoglutarate
α-ketoglutarate, a crucial intermediate in the TCA cycle and a product of glutamine deamination, inversely correlates with glucose levels in the gastrocnemius muscle of cachectic mice.186 α-ketoglutarate plays a protective role in muscle development, such as promoting C2C12 myoblast proliferation, alleviating glucose deprivation-induced myotube atrophy, and maintaining the antioxidant capacity of cells.186 Additionally, Ruiz et al. discovered that α-ketoglutarate, as a cofactor for histone demethylases, inhibits TNF family member Tnfrsf12a/Fn14 expression-induced cachectic manifestations by reversing the glutamine deficiency-induced tri-methylation of lysine 4 on histone H3 (H3K4me3) enrichment at the Tnfrsf12a promoter, indicating the protective role of dimethyl α-ketoglutarate187 (Fig. 5).
Dimethyl-α-ketoglutarate reshapes the tumor immune microenvironment by enhancing radiation therapy-induced tumor cell apoptosis and immunogenic cell death, as well as increasing CD8+ T-cell infiltration.188 However, contrary findings suggest that α-ketoglutarate production activates mTORC1 signaling and sustains an immunosuppressive M2-like macrophage phenotype.189 Moreover, α-ketoglutarate significantly alters the DNA methylation profile of naïve CD4+ T cells, attenuating the differentiation of Tregs and enhancing the production of inflammatory cytokines and antitumor cytotoxicity.190 This multifaceted role of α-ketoglutarate underscores its complexity in regulating immune responses and metabolic pathways in diverse biological contexts (Table 1).
Ketone bodies
Ketone bodies, consisting of β-hydroxybutyrate, acetoacetate, and acetone, are intermediary products of fatty acid oxidation and decomposition. Studies have revealed decreased hepatic ketogenesis in cachectic mice compared with healthy food-restricted or starved counterparts, indicating their roles in cancer cachexia (Fig. 5). Indeed, 2-deoxy-D-glucose supplementation enhances hepatic ketogenesis and promotes ketone metabolism in skeletal muscle, which elevates ATP synthesis efficiency and blocks aberrant Cori cycling, thereby effectively preventing muscle atrophy.158 Tumor-secreted IL-6 inhibits peroxisome proliferator-activated receptor (PPAR) α, a transcriptional regulator of ketogenesis, diminishing hepatic ketogenic capacity in precachectic mice. This metabolic reprogramming and energy deficit impair the ability of the host to provide alternative endogenous energy sources. More importantly, it exacerbates the stress response, leading to elevated glucocorticoids and potentially undermining the effectiveness of antitumor immunotherapy.191 In contrast, a PPARα agonist restores ketone production, mitigating skeletal muscle and body weight loss.192 Moreover, research in gastrointestinal cancer patients revealed upregulated serum ketone bodies, potentially driven by increased lipolysis and amino acid breakdown.193 Increased β-hydroxybutyrate in the liver, portal vein, and systemic circulation of cachectic mice is accompanied by a reduction in hepatic ketogenesis transcripts, suggesting the activation of alternative ketogenesis pathways such as kidney ketogenesis.82 Brain glucose utilization is decreased in tumor-bearing animals, potentially substituted by lactate and 3-hydroxybutyrate, implying a shift to ketone bodies as primary brain energy sources.194
Research on ketogenic diets in cachexia patients underscores the transition of energy supply from glucose to fatty acids, which provides therapeutic benefits for cancer cachexia through the targeting of ketone bodies. Supplementation of CT26 cell cultures with 3-hydroxybutyrate antagonizes tumor-induced C2C12 myotube atrophy by increasing TCA cycle activity, protein synthesis, and metabolic balance while attenuating proteolysis and inflammation.195 Cumulative evidence indicates that diets fostering ketone body production and anti-inflammatory strategies may serve as promising preventive measures for cancer cachexia patients.196 Nevertheless, the effects of ketone bodies are not uniformly beneficial. Ketogenic diet accelerates cachexia onset and shortens survival in IL-6-related cancer cachexia mouse models. This effect may be attributed to increased lipid hydroperoxide (LOOH) production, which saturates the glutathione (GSH) pathway, leading to ferroptosis in cancer cells. Additionally, redox imbalance and NADPH depletion impair the biosynthesis of cortisol, a vital metabolic stress regulator in the adrenal glands, causing relative adrenal insufficiency and metabolic maladaptation in ketogenic diet-fed mice.197
Ketone bodies can regulate the functions of different cells. β-hydroxybutyrate levels are reduced in human CRC, whereas increased ketogenesis inhibits KLF5 expression in CAFs by suppressing histone deacetylase 1, causing the downregulation of C-X-C motif chemokine ligand 12 (CXCL12). This mechanism, in turn, inhibits M2 macrophage accumulation, promotes NK cell and cytotoxic T-cell infiltration, and augments the efficacy of anti-PD1 therapy.198 Similarly, ketone bodies are essential fuels that support CD8+ T-cell metabolism and effector function by enhancing mitochondrial respiratory capacity and TCA cycle synthesis. Furthermore, β-hydroxybutyrate serves as the primary source of acetyl-CoA in T cells, facilitating H3K27ac at promoters of effector gene loci (e.g., Ifng, Gzmb and Tbx21), thereby augmenting cytokine production in CD8+ T cells (e.g., IFN-γ, TNF-α) and modulating cellular immune functions.199 These findings stress the importance of ketone body supplementation in enhancing antitumor immunity (Table 1).
Decreased hepatic ketogenesis in cancer cachexia patients results in an energy deficit and impairs antitumor immunotherapy efficacy, which aligns with reports that ketones enhance antitumor immunity. While some conflicting results exist, the clinical utility of ketogenic therapy for treating cancer cachexia has been demonstrated.
Amino acid metabolism
In addition to intermediate metabolites, amino acids and their derivatives play pivotal roles in metabolic reprogramming, and their metabolic disorders are associated with numerous pathological conditions. Tumor cells exhibit a unique capacity to redirect carbohydrate intermediates from oxidative phosphorylation to various anabolic pathways, a phenomenon known as the Warburg effect,164 but emerging evidence underscores the crucial involvement of amino acids, the fundamental building blocks of proteins, in the Warburg effect.200 Numerous strategies targeting amino acid depletion have been proposed to repress cancer progression by modulating amino acid transporters, inhibiting amino acid biosynthesis, or consuming amino acids.201 While amino acid depletion therapies exhibit broad applicability in cancer treatment, intriguing findings suggest that, in certain contexts, specific amino acids may possess antitumor properties.202 The metabolic profiling of cachectic patients revealed significant perturbations in amino acid and protein metabolism.203 Targeted amino acid supplementation strategies have the potential to preserve energy homeostasis and alleviate chemotherapy-induced adverse effects in cancer patients.204
Emerging evidence suggests that the intricate relationship between altered amino acid metabolism and pivotal tumor characteristics, including growth, metastasis, and therapeutic recalcitrance, is mediated through the orchestration of immune cell fates (Table 1). Numerous amino acids reportedly modulate immune cellular function.202 For example, extracellular L-arginine concentrations regulate T-cell antigen receptor ζ-chain expression, thereby influencing the normal function of T cells.205 Glutamine enhances GSH de novo synthesis via catabolism, providing essential precursors for glutathione biosynthesis, and inhibits the production of ROS by generating GSH and NADPH, which are required during Th17 cell differentiation to maintain redox balance and support effector function.206 Glutamine deprivation inhibits M2-like macrophage differentiation and CCL22 chemokine production,207 which may be induced by the inhibited generation and recruitment of MDSCs.208 Methionine provides essential methyl groups for DNA and RNA methylation, thereby promoting T-cell differentiation and proliferation by mediating mTORC1 activity.209
In conclusion, the metabolic reprogramming of amino acids has been characterized extensively across various tumor types, modulating infiltrating cell functions within the tumor macroenvironment. Further elucidation of the dysregulated amino acid metabolism associated with cancer cachexia could lead to the development of novel immunotherapy strategies.
Essential amino acids
In animals, essential amino acids (EAAs) include valine, isoleucine, leucine, phenylalanine, tryptophan, methionine, lysine, threonine, and histidine, whose carbon frameworks cannot be biosynthesized de novo by animal cells and therefore must depend on an exogenous dietary supply to sustain growth and nitrogen homeostasis.210 EAA/leucine supplementation in advanced lung cancer patients elicits anabolic responses, which favorably impact patient prognosis.211 Among EAAs, BCAA, including valine, leucine, and isoleucine, have garnered extensive attention in cachexia research. Metabolomic analyses linked BCAA degradation to sarcopenia in liver cirrhosis patients.212 Accelerated onset of cancer cachexia is observed in tumor-bearing mice with conditional knockout of muscle-specific branched-chain α-ketoacid dehydrogenase kinase due to increased protein ubiquitination, degradation and compromised synthesis. Importantly, supplementation with BCAAs can ameliorate these effects.213 Modulating the muscle BCAA transporter LAT1 has emerged as a promising therapeutic approach for cachexia.214
Baek et al. identified the C-terminal unique attached sequence motif domain of leucyl-tRNA synthetase as an activating domain of mTORC1, suggesting its potential as a muscle-enhancing mTOR-targeting protein.215 However, another study reported that a leucine-rich diet improved muscle function in cachectic tumor-bearing rats without altering mTOR activity, implying that this dietary intervention may exert beneficial effects through an mTOR-independent pathway.216 Furthermore, a leucine-rich diet inhibits tumor-induced cardiac injury, myocardial proteolysis, and apoptosis.217 Interestingly, maternal leucine nutritional supplementation improved muscle protein balance in cancer cachexia-induced muscle damage in adult offspring rats.218 When combined with exercise, a leucine-rich diet improves protein turnover.219 Similarly, β-hydroxy β-methyl butyrate, a leucine metabolite, exhibits protective properties.220 However, leucine supplementation in a murine model of pancreatic cancer accelerated tumor progression in both lean and overweight phenotypes, albeit with different biological outcomes.221 In lean mice, leucine enhances mTOR phosphorylation and subsequent S6 ribosomal protein activation, whereas in overweight mice, it disrupts glucose homeostasis by impairing clearance mechanisms, thereby increasing circulating glucose availability for tumor cell utilization.221 Schrems et al. reported that leucine potentially exacerbates mortality in male mice but not in female mice,222 and the mechanisms underlying sex-specific disparities in the impact of leucine on cancer cachexia need further study.
Elevated levels of the ketogenic amino acids phenylalanine and tryptophan were observed in cancer cachexia mouse models, suggesting their potential as biomarkers for skeletal muscle loss and reduced protein synthesis.223 However, mice subjected to a tryptophan-deficient diet presented a greater fiber diameter in the anterior tibialis than did those fed a standard diet. The reduction in glycolysis due to tryptophan deficiency may suppress mTOR signaling, while the increased levels of amino acids seem to compensate for the diminished pyruvate-derived energy production in skeletal muscle.224 Additionally, lysine was shown to serve as a diagnostic biomarker for cancer cachexia.225 In summary, EAAs play a multifaceted role in the pathological process of cancer cachexia (Fig. 5). Although supplementation with these amino acids can stimulate protein anabolism, it is important to recognize that certain amino acids may also exacerbate cachexia by modulating inflammation, immunity, and energy metabolism.
Nonessential amino acids
In most mammals (e.g., humans, rats, and pigs), the traditionally categorized nonessential amino acids (NEAAs) include alanine, arginine, asparagine, aspartic acid, cysteine, glutamic acid, glutamine, glycine, proline, serine and tyrosine, which can be synthesized through their own metabolic pathways to fulfill normal physiological needs, even if these amino acids are absent from the diet.210 Recent investigations underscore their crucial role in cancer cachexia (Fig. 5). For example, elevated levels of methylarginine metabolites in tumor-bearing mice inhibit protein synthesis and mitochondrial protein quality, thereby contributing to muscle wasting in cachexia.226 The cationic amino acid transporter, which is an essential factor for the transport of L-arginine into mitochondria, is significantly downregulated in the mitochondrial membranes of cachectic mouse muscles. These findings suggest that mitochondrial dysfunction may impact amino acid metabolism in cachectic skeletal muscles.227
Research indicates that gastrointestinal cancer patients receiving cysteine-supplemented parenteral nutrition exhibit reduced overall survival compared with those without cysteine supplementation. In vitro and in vivo mouse experiments confirmed that cystine increases cancer cell proliferation by activating mTORC1 via the GCN2-ATF4-SESN2 axis. Additionally, cystine confers resistance to chemotherapy by quenching chemotherapy-induced oxidative stress through GSH synthesis.228 However, other evidence suggests that coadministration of cystine and theanine can significantly prevent weight loss and adipose and skeletal muscle depletion. Therefore, the underlying mechanisms behind this phenomenon remain to be further investigated.229
Inhibition of the initial rate-limiting enzyme in serine biosynthesis drastically reduces myofibrillar protein synthesis, thereby emphasizing the crucial role of serine production in maintaining muscle size.230 Low serum levels of glutamine, histidine, alanine, and glycine correlate with poor survival among cancer patients, supporting their important roles in cancer progression.231 Supplementation with glycine232 and β-alanine233 reduces oxidative and inflammatory burdens, thereby protecting skeletal muscle from cancer-induced atrophy and functional loss. These studies demonstrate that these amino acids cannot be dismissed as unimportant or completely neglected in the human body. Moderate consumption of NEAAs is necessary for maintaining normal physiological functions. Furthermore, in specific circumstances such as cancer cachexia, the body’s requirement for NEAAs may increase, making it advantageous to moderately increase the intake of relevant foods.
Amino acid derivatives
Amino acid derivatives are compounds that possess novel properties and functions, arising from a series of chemical reactions involving amino acids, and play diverse and crucial roles within organisms. Importantly, multiple studies have indicated that supplementation with specific amino acid derivatives can be advantageous for alleviating the phenotypic manifestations of cachexia. Patients suffering from cachexia exhibit low basal circulatory levels of glutamine.234 Further studies confirmed that L-glutamine supplementation curtails tumor growth and tumor-induced cachexia.235 Tumor progression often elicits oxidative stress, which is linked to stromal neuropathy and intestinal atrophy and is partially attributed to depletion of GSH, the primary endogenous antioxidant. L-GSH supplementation elevates GSH levels and therefore minimizes the overexpression of nNOS and nitric oxide (NO) production in the gut, thereby preventing intestinal neurotoxicity mediated by peroxynitrite formation. This, in turn, helps to preserve the ileal neuronal count and morphology, alleviating cachexia-related gastrointestinal motility impairments.236
Taurine, a cysteine derivative, protects myoblasts against cisplatin-mediated cytotoxicity by enhancing ROS scavenging and GSH synthesis. Moreover, it regulates myotube differentiation by modulating the expression of myogenic markers and attenuates myotube atrophy.237
Creatine, an amino acid derivative synthesized from L-arginine, glycine, and L-methionine, has been extensively investigated in the context of tumor progression. Creatine supplementation effectively prevents tumor-induced increases in plasma homocysteine levels and hepatic oxidative stress, thereby alleviating tumor growth and weight loss.238 Compared with tumor-bearing rats, creatine intake diminishes plasma TNF-α and IL-6 levels and alleviates splenic morphological alterations, such as reduced white pulp and lymph follicle sizes, indicating that creatine may prevent skeletal muscle atrophy by modulating the tumor-induced proinflammatory milieu.239
L-carnitine, a derivative of lysine, has been shown to alleviate fatigue, stimulate appetite, and increase lean body mass in advanced cancer patients following supplementation.240 L-carnitine can induce the restoration of hepatic lipid metabolic dysfunction, accompanied by a reduction in the circulating concentrations of TNF-α and IL-6.241,242
Generally, amino acid metabolites, including glutamine, GHS, taurine, creatine, and L-carnitine, mitigate cachexia phenotypes by mitigating inflammatory reactions, alleviating oxidative stress, increasing energy expenditure, and modulating metabolism.
Fatty acid metabolism
Increasing evidence suggests that fatty acids play crucial roles in cancer cachexia via diverse mechanisms, including energy provision, metabolic modulation, anti-inflammatory and immune regulation and intestinal barrier maintenance (Fig. 5). According to their carbon chain length, fatty acids are classified into short-chain (SCFAs, <6 carbons), mid-chain (MCFAs, 6–12 carbons) and long-chain (LCFAs, >12 carbons) fatty acids. Furthermore, fatty acids can be dichotomized on the basis of saturation status into saturated (SFAs) and unsaturated (UFAs) fatty acids, with UFAs further subdivided into monounsaturated (MUFAs) and polyunsaturated (PUFAs).243 PUFAs are categorized into Omega (ω)-3 and ω-6 families on the basis of the location of the first double bond from the methyl end. ω-3 PUFAs include alpha-linolenic acid (ALA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), whereas the ω-6 series includes linoleic acid (LA), gamma-linolenic acid, conjugated linoleic acid, and arachidonic acid.243 Elucidating their potential roles in cancer cachexia may promote the development of enhanced nutritional and metabolic interventions.
Long-chain fatty acids
During the occurrence and progression of cancer cachexia, alterations in long-chain fatty acid composition, both intracellularly and in plasma, may occur. An investigation utilizing a rodent model of cancer cachexia revealed increased palmitic acid and decreased LA levels in WAT, associated with downregulated expression of ELOVL6, a gene encoding an enzyme crucial for fatty acid elongation. These findings suggest that the modulation of adipocyte lipid composition and content may inhibit excessive lipogenesis.244 The level of oleic acid, a type of MUFA, was significantly greater in the plasma of PDAC patients with cachexia, potentially reflecting compensatory mechanisms in response to malnutrition or impaired tissue uptake of oleic acid. Furthermore, LA, an ω-6 PUFA, is positively correlated with albumin and hemoglobin levels in cachexia patients, suggesting potential nutritional support from increased dietary LA.245 However, Wang et al. reported elevated LA levels and decreased LA-rich cardiolipins in the hearts of cancer cachexia patients. The degradation of LA-rich cardiolipins may underlie the increased LA levels, which are implicated in inflammation, apoptosis, and atrophy of heart tissues in cancer cachexia. This finding aligns with an elevated ω-6/ω-3 PUFA ratio in heart tissues and indicates that targeting disturbances in glycerophospholipid and fatty acid metabolism in cancer cachexia hearts may be helpful.246 Therefore, further research is needed to identify the role of ω-6 PUFAs in cancer cachexia.
The role of ω-3 PUFAs in cachexia has been extensively reported. Among these, EPA and DHA are the most studied because of their potent anti-inflammatory and antioxidant properties.247 The plasma level of ω-3 fatty acids is decreased during chemotherapy for lung cancer patients, which may exacerbate muscle wasting.248 Additionally, a meta-analysis suggested that low-dose ω-3 FA supplementation stabilizes weight, appetite, and quality of life in cancer patients, especially those with tumors of the upper digestive tract and pancreas.249 However, fish oil therapy enriched with ω-3 FAs could have potential gastrointestinal side effects such as nausea, emesis, and diarrhea.250 Low-dose marine phospholipid supplementation offers comparable benefits to those of fish oil, with better tolerability and fewer side effects, effectively improving weight and appetite in cancer patients.251
In tumor-bearing mice, EPA antagonizes the loss of skeletal muscle proteins in cancer cachexia by suppressing ATP-dependent proteolysis and the expression of 20S proteasome alpha-subunits and the p42 regulator,252 which can also prevent NF-κB nuclear accumulation, thereby dampening proteolysis-inducing factor-triggered ubiquitin‒proteasome proteolysis.253 DHA-phospholipid and EPA-phospholipid act against TNF-α-triggered lipolysis in adipocytes.254 The combination of free fatty acid receptor FFA1/FFA4 agonists (GW9508 and TUG891) with ALA/DHA reduces tumor weight in LLC mice. Furthermore, these findings suggest that ALA and DHA may exert their therapeutic effects on cancer cachexia, at least in part, through the activation of FFA1/FFA4 receptors.255 Collectively, ω-6 and ω-3 PUFAs appear to exert opposing effects on cachexia, underscoring the need for further investigation into their mechanistic interplay and the identification of optimal ω-6/ω-3 ratios for therapeutic benefit in cachexia patients.
Accumulating evidence highlights the intricate link between PUFAs and the function of immune cells, which may provide a possible explanation for the anti-inflammatory changes observed after exogenous administration of PUFAs during the progression of cancer cachexia (Table 1). ω-3 PUFAs enhance anti-inflammatory responses by augmenting IL-10 release from macrophages, fostering Treg induction and inhibiting excessive Th17 cell differentiation. They also reduce oxidative stress and NF-κB-mediated inflammation in immune cells.256 GPR120/FFA4, a G protein-coupled receptor for long-chain fatty acids, is potently activated by ALA.257 Once FFA4 is activated by ALAs, macrophages are polarized toward the M2 phenotype to modulate inflammation in adipose tissues. For example, FFA4 activation reduces TNF-α and IL-6 secretion in mouse macrophages.257 ω-3 PUFAs also modulate NK cell function. DHA inhibits tumor growth by increasing peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1α-mediated mitochondrial oxidative phosphorylation activity in NK cells and increasing IFN-γ production.258 However, in certain contexts, PUFAs may exert immunosuppressive effects. Tumor-derived alpha-fetoprotein binds more PUFAs than does umbilical blood-derived alpha-fetoprotein, which induces a glycolytic phenotype in dendritic cells. Therefore, ω-3 PUFAs enhance glucose uptake and lactate secretion, leading to suppression of the antitumor capabilities of dendritic cells.259 Apart from immune cells, ω-3 PUFAs are crucial in regulating stromal cell functions (Table 1). Interstitial fluid pressure, a significant barrier to anticancer drug diffusion, is elevated by NO-mediated tumor angiogenesis. ω-3 PUFAs diminish endothelial NO synthase activation, thereby lowering interstitial fluid pressure and enhancing anticancer drug delivery efficiency.260
LCFAs, particularly ω-3 PUFAs, have been extensively investigated in cancer cachexia, presumably because of their potential immunomodulatory and redox-regulatory capacities. Notably, several studies have indicated their crucial roles in diverse immune and stromal cell functions, warranting further research in the cachexia field.
Mid-chain fatty acids
MCFAs include both even-carbon MCFAs (including hexanoic acid, octanoic acid, and decanoic acid and, although controversial, lauric acid due to its twelve carbons) and odd-carbon MCFAs (such as heptanoic acid, nonanoic acid, and undecanoic acid). MCFAs, notably lauric acid, are rapidly assimilated into mitochondria, stimulating oxidative phosphorylation and resulting in the apoptosis of tumor cells. This effect is tumor specific, sparing skeletal muscle and other nontumor cells. The consumption of lauric acid elevates skeletal muscle protein levels and halts tumor growth through a tumor-selective oxidative stress response, mitigating cancer-induced skeletal muscle atrophy.261 Furthermore, dietary intervention combining lauric acid and glucose for cancer cachexia may ameliorate cancer-associated myocardial damage by increasing mitochondrial respiration and ATP synthesis without exacerbating oxidative stress.157 A study indicated that caprylic acid has a greater capacity than capric acid and lauric acid for improving mitochondrial quality and promoting skeletal muscle maturation. These findings support the therapeutic role of caprylic acid in cancer cachexia.262 Diets with medium-chain triglycerides consisting of glycerine and MCFAs also offer an optimal ketogenic regimen for reversing weight loss in cancer cachexia patients, accompanied by a reduction in tumor size.263 In the immune microenvironment, the dysregulation of MCFA may affect the functional modulation of immune effector cells. For example, endogenous MCFAs can activate GPR84 to enhance inflammatory responses and macrophage phagocytosis.264 Disorders of MCFAs in cancer cachexia may disrupt macrophage function.
Short-chain fatty acids
SCFAs are metabolic byproducts generated via microbial fermentation of complex carbohydrates in the gut, which emerge as pivotal players in cancer cachexia, especially butyrate and acetate.80,82 The decrease in butyrate and acetate is closely related to the reduction in the abundance of members of the Ruminococcaceae and Lachnospiraceae families in cachectic cancer mice, thereby exacerbating intestinal inflammation through the impairment of intestinal barrier integrity.82 Mechanistically, butyrate not only minimizes endotoxin translocation and oxidative stress but also promotes polarization toward the M2 phenotype, inhibiting inflammation and macrophage infiltration into muscles. Furthermore, it counteracts cachexia-induced muscle atrophy by modulating pathways such as the Akt/mTOR/Foxo3a and Fbox32/Trim63 pathways.265 Microbiome-derived SCFAs can induce IL-22 production in CD4+ T cells and innate lymphoid cells by enhancing HIF-1α binding to the Il-22 promoter via histone modification, thereby maintaining intestinal homeostasis.266
In addition to preserving intestinal homeostasis, fatty acids play critical roles in orchestrating host immune responses and modulating systemic inflammation, thereby having the potential to attenuate cachexia progression. It is necessary to further understand their importance, and individualized treatments for patients should be developed.
Other metabolites
Nevertheless, numerous other metabolites are associated with the progression of cachexia. For example, vitamin D supplementation effectively ameliorates the disruption of UCP1 and ATP levels in adipose tissue and muscle. It modulates biomarkers of beige adipocytes and browning and normalizes the levels of the entire Tlr/NF-κB pathway in inguinal WAT. It also inhibits muscle catabolic signaling and enhances muscle regeneration and the myogenesis process.267 Furthermore, supplementation with 25(OH)D₃ may confer more pronounced improvements in muscle atrophy and adipose browning in cachectic mice than supplementation with 1,25(OH)₂D₃.268
Decreased ATP concentrations and total adenine nucleotide pools represent frequent features of atrophic muscles in human diseases. In cancer cachexia, energy metabolism is frequently disrupted by tumor proliferation and increased catabolism, resulting in reduced ATP and ADP levels in muscle tissue that normalize after tumor resection.269 Xanthine oxidase inhibitors hold promise as potential therapeutic agents to mitigate muscle wasting and cardiac dysfunction in cancer cachexia.270 In lung cancer patients, upregulated AMPK contributes to alleviating cancer-induced metabolic perturbations by preventing glucose intolerance and insulin resistance and decreasing glucose disposal in skeletal muscle and WAT. The AMPK-dependent upregulation of PDKs may represent an adaptive metabolic reprogramming strategy to maintain essential metabolic functions within the muscle, emphasizing the metabolic underpinnings of cachexia.271
Cells generate several active byproducts of metabolic pathways, such as ROS, NAD + /NADH, and NADP + /NADPH. The concurrent impairment of NAD+ and downregulation of the NAD+ biosynthetic enzyme nicotinamide riboside kinase 2 were identified as common features of severe cachexia in both preclinical murine models and cancer patients. The supplementation of vitamin B3 niacin enhances NAD+ levels and increases mitochondrial biogenesis to ameliorate muscle mass loss and metabolic disorders.272 Preemptive intake of nicotinamide riboside, a substrate for NAD+, significantly reduces the levels of the cachexia-inducing cytokines TNF-α and IL-6 in a mouse model, decreases muscle-specific ubiquitin‒proteasome ligase activity, and inhibits lipolysis.273
Oxidative stress, intricately linked to cachexia, results from an imbalance between oxidation and antioxidation. Oxidative stress generates excessive ROS, such as superoxide anions, hydrogen peroxide, and hydroxyl radicals, which are harmful to cellular proteins, DNA, and lipids.274 Elevated oxidative stress in skeletal muscle disrupts mitochondrial function, energy metabolism and ATP production, leading to mitochondrial autophagy-related muscle damage.275 This mitochondrial dysfunction, in turn, leads to increased oxidative stress. Furthermore, oxidative stress can activate the UPS276 and calpains277 in skeletal muscle to accelerate protein degradation and modulate Akt phosphorylation to affect protein synthesis, such as the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt) pathway,278 ultimately exacerbating muscle atrophy.
In conclusion, these metabolites are closely associated with cancer cachexia. These aberrant alterations not only reflect tumor metabolic signatures but also impact host cell function via intricate metabolic networks and signaling cascades, thereby intensifying catabolic processes and cachexia manifestations.
Regulatory effects of immune and stromal cells on systemic metabolism
These profound and aberrant metabolic transformations are coupled with the progression of cancer cachexia. Importantly, these metabolic alterations strongly impact the internal architecture, functional properties, and dynamic quantity of immune cells and stromal cells. As a result, immune cells such as TAMs, tumor-associated neutrophils (TANs), tumor-infiltrating T lymphocytes (TILs) and MDSCs can modulate cancer cachexia via the following mechanisms: (1) they impact key target organs such as skeletal muscle, adipose tissue and even the nervous system, inducing a cascade of pathological effects within these tissues through signal transduction mechanisms; and (2) they exert a broader influence by secreting a range of bioactive molecules, termed “cachectic factors”, which further propagate their effects (Fig. 6). Upon dissemination through the circulatory system, cachectic factors act as potent signaling molecules, promoting cachexia progression across tissues and organs. By binding to receptors on target cells, they elicit downstream signaling cascades that augment inflammation, accelerate tissue catabolism, and inhibit anabolic processes, thereby exacerbating cachexia. The reconfiguration of stromal cellular phenotypes plays a significant role. CAFs can produce cytokines, secrete extracellular vesicles, and synthesize ECM, all of which influence tumor progression. The destruction of ECs leads to microvascular circulation disorders in various tissues, such as skeletal muscle and adipose tissue, ultimately resulting in malnutrition (Fig. 6). In summary, these cellular alterations exacerbate clinical symptoms such as weight loss, muscle atrophy and fat depletion, severely compromising immune defenses and posing formidable obstacles to disease management and prognosis.
Tumor-associated macrophages
TAMs constitute up to 50% of the TME in certain solid tumors and play a pivotal role in immune cell infiltration. Upon receiving signals from the TME, macrophages are differentiated into either proinflammatory or anti-inflammatory phenotypes on the basis of environmental cues. Although macrophages can be classified into two subsets, i.e., M1-like macrophages or M2-like macrophages, they are collectively designated TAMs.279 Accumulating evidence underscores the critical role of TAMs in mediating cancer cachexia (Fig. 6). In pancreatic cancer mouse models, macrophage depletion restores muscle and epididymal fat weight, accompanied by increased grip strength and mobility.280
Macrophages are major immune cells in adipose tissue. They support the nutrient storage function of adipocytes in the absence of metabolic dysfunction and then release inflammatory factors to promote insulin resistance during the progression of metabolic syndrome.281 The activation of beige adipose thermogenesis is orchestrated by both the SNS and adipose tissue macrophages (ATMs), but a detailed mechanism for this process is lacking. In BAT, the minimal fundamental thermogenic unit is the brown adipocyte, with norepinephrine serving as the paramount regulatory factor.87 Ajay’s group reported that cold stimuli can recruit macrophages to adipose tissue regions in a CCR2-dependent manner. Moreover, eosinophils induce catecholamine production in macrophages via the IL-4/13-IL-4Rα-STAT6 signaling axis, triggering thermogenic gene expression in BAT and promoting lipolysis in WAT.282,283 Furthermore, in a cancer cachexia mouse model, researchers reported an increase in the number of IL-4-induced M2 macrophages, which express neurotrophic factors that promote a neuroprotective milieu, within the inguinal WAT. These factors facilitate catecholamine production by peripheral sympathetic nerves and then orchestrate lipid metabolism, WAT browning, and adipose tissue atrophy in cancer cachexia. These findings highlight the potential role of ATM-SNS crosstalk in cancer cachexia.156 However, another study reported that macrophages significantly promote beige adipose thermogenesis independently of the SNS after inducing sympathetic neuronal apoptosis with 6-hydroxydopamine in mice.284 M2 macrophage-derived TGF-β induces senescence in adipose progenitor cells and inhibits adipogenesis in aged mice. However, the role of this mechanism in cancer cachexia remains unclear and warrants further investigation.285 In the fibrotic regions of subcutaneous adipose tissue from gastrointestinal tumor patients, infiltrated CD68+ macrophages and CD3-Ly have been identified, suggesting their potential roles in cachexia-related adipose tissue remodeling.286 Inactivation of hypoxia-inducible factor 1α in myeloid cells severely impairs energy generation, leading to robustly compromised cellular function and defective myeloid cell-mediated inflammation. However, reduced levels of proinflammatory cytokines unexpectedly increase cachexia-associated fat loss coupled with reduced ATMs. This finding suggests that ATMs may play a protective role in cancer cachexia-related adipose loss.287
In addition to facilitating adipose tissue catabolism, macrophages influence skeletal muscle primarily through the secretion of inflammatory cytokines. For example, the coculture of macrophages and tumor cells accelerates myotube atrophy via macrophage-derived IL-6 and IL-1α.288 Another experiment further solidified the intimate connection between macrophages and tumor cells. An increased percentage of CD68+ macrophages was observed in tumor tissues from cachectic patients compared with noncachectic individuals. Notably, the transcriptional upregulation of ZXDC in tumor cells facilitates macrophage recruitment through the CCL2/CCR2 signaling axis. These macrophages subsequently secrete CCL5, which activates the NF-κB signaling cascade, leading to the upregulation of TWEAK in tumor cells.289 Elevated TWEAK subsequently initiates muscle remodeling and promotes muscle atrophy via MuRF1 activation.289,290 However, macrophages can also facilitate muscle recovery and regeneration via insulin-like growth factor 1 (IGF-1) autocrine signaling, which shifts polarization toward the M2 phenotype.291 Recent research by Pryce et al. may explain the contrary role of macrophages in cachexia. They identified distinct cellular subtypes of macrophages that are induced by NF-κB. Proinflammatory macrophages promote atrophy by inhibiting muscle regeneration, whereas anti-inflammatory macrophages, to some extent, contribute to maintaining myofiber size.292
Tumor-associated neutrophils
Neutrophils, some of the most abundant immune cells in the human circulation, perform pivotal functions in combating inflammation caused by infections and injuries. Accumulating evidence indicates that neutrophils also participate in various aspects of tumor biology, including metastasis, drug resistance, and poor prognosis.279 Compared with noncachectic patients, cachectic patients exhibit a marked elevation in absolute neutrophil count coupled with an elevated neutrophil‒lymphocyte ratio (NLR),51,293,294,295,296 accompanied by the upregulation of angiotensin II and neutrophil-derived proteases.293 Currently, several studies have reported the important role of neutrophil-related indicators in evaluating inflammatory status, such as the NLR-to-handgrip strength ratio,294 the neutrophil-to-albumin ratio,296 and the advanced lung cancer inflammation index, which is calculated as BMI (kg/m2) × albumin (g/dl)/NLR.295 Additionally, the modified advanced lung cancer inflammation index, which is defined as the appendicular skeletal muscle index × serum albumin/NLR, was also introduced.297 These indices were confirmed to be negatively correlated with survival rates in cancer cachexia patients, suggesting the prognostic significance of neutrophil-mediated processes in cancer cachexia patients (Fig. 6).
As tumors proliferate, tissues gradually undergo damage, necrosis, and hypoxia, triggering the release of numerous chemokines, such as TNF-α, which facilitate the recruitment and activation of neutrophils and macrophages.298 During the precachexia stage, neutrophils are elevated in tissues such as the liver and lungs and increase in number in the circulatory system.299 Upon recruitment, activated neutrophils unleash neutrophil-derived proteases and angiotensin II. Elevated plasma levels of angiotensin II are associated with increased production of inflammatory cytokines such as IL-6, IL-8, and TGF-β1, which contribute to the inflammatory state and cause the activation of proteolytic pathways and/or disruption of protein synthesis, thus promoting cancer cachexia.293 Recently, researchers defined a subset of neutrophil-like monocytes, termed cachexia-inducible monocytes, which express CD38+ and induce muscle atrophy by producing IL-36G, thereby exacerbating cachexia phenotypes in advanced cancer models.300
In murine models of cancer cachexia, neutrophils are increased in various tissues and secrete cytotoxic proteins such as LCNs, which can suppress appetite or promote ferroptosis-related inflammation.67,301 Anorexia, a common symptom in cachexia patients, is driven by cerebral inflammation. Studies have revealed that neutrophils can be recruited to the brain through TRIF-dependent mechanisms.73 Afterward, neutrophils accumulate in a CCR2-rich meningeal region of the velum interpositum, which is adjacent to the hippocampus and habenula areas that are crucial for appetite regulation. Here, neutrophils contribute to anorexia and muscle catabolism through neuroimmune circuits.75
Neutrophils contribute to skeletal muscle atrophy and anorexia nervosa through the release of substantial amounts of inflammatory cytokines and cytotoxic proteins, highlighting the importance of preventing their detrimental effects on cancer cachexia.
Tumor-infiltrating T lymphocytes
Emerging evidence underscores the pivotal role of adaptive immune cells, particularly T cells, in the pathology of cachexia. Studies have revealed a positive relationship between muscle strength and the frequencies of CD8+ naïve T cells, CD8+ effector T cells, and CD197+CD45RA+ Tregs. Conversely, CD8+ memory T cells and CD95+CD8+ T cells inversely correlate with muscle function, suggesting that the infusion of specific T-cell subsets may restore a balanced T-cell profile, thereby mitigating muscle atrophy and alleviating cachexia in cancer-bearing mice.302 Nevertheless, the intricate nature of the immune system implies that multiple factors concurrently influence muscle function and mass, resulting in inconsistencies in research findings and thereby obscuring the precise role of T cells in cachexia (Fig. 6). The thymus and spleen, which are crucial for T-cell progenitor generation and differentiation, exhibit atrophy and quantitative reductions in Tlymphocyte populations in cachexia-bearing mice. Notably, upregulated expression of immune checkpoint markers, such as cytotoxic T-lymphocyte-associated antigen 4 (CTLA-4) and PD-1, was found within CD4+ T cells, highlighting the involvement of immunodeficiency in cancer cachexia.303 The engineered rimiducid-inducible MyD88- and CD40-driven chimeric antigen receptor (CAR)-T cells elicited antitumor responses, but these CAR-T cells also caused cachexia in mice through dose-dependent weight loss. The authors further demonstrated that CD4+ T helper cells are the primary source of cachexia-related proinflammatory cytokines such as TNF-α. Conversely, the purification of CD8+ T cells enhances antitumor efficacy while minimizing cytokine-related toxicity.304 However, other studies have shown that CD4+ T cells play a protective role in cachexia. For example, cachectic mice display notable depletion of CD44low CD4+ T cells, a type of quiescent naïve cell, in lymph nodes and spleens compared with noncachectic diabetic tumor-bearing mice. Intriguingly, infusion of these CD44low CD4 + T cells attenuated body weight reduction and preserved skeletal muscle mass in tumor-bearing mice.305
The role of CD8+ T cells in cachexia is equally complex. A study demonstrated that the transcriptional signature of CD8+ T cells is negatively correlated with genes involved in muscle wasting pathways, the ubiquitin proteasome, and apoptosis/autophagy, suggesting a potential protective role of CD8+ T cells in cachexia.306 In a mouse model of PDAC, a TLR7/8 agonist showed an antitumor response and improved cachexia by increasing CD8+ T-cell infiltration and decreasing the number of Tregs.307 Interestingly, CD8+ T cells trigger cachexia through antigen-specific activation in a reversible cachexia mouse model induced by chronic viral infection.308 Mechanistically, CD8+ T cells modulate adipose tissue lipid metabolism in a type I IFN-dependent manner but not the levels of the cytokines IL-6, TNF, and IFN-γ, which often mediate cancer cachexia.308 These findings suggest that the mechanisms causing cachexia may differ among various diseases, suggesting that when studying and treating cachexia, we need to consider the specific pathological background and avoid generalization.
Notably, the abnormal metabolism associated with cancer cachexia can reciprocally influence the function of immune cells. In advanced lung patients, cachexia status inversely correlates with immunotherapy response. Mechanistically, nutrient intake and energy utilization are largely impaired by chronic inflammation, metabolic disturbances, anemia, and anorexia, which result in defective T-cell activation and exhaustion, thereby hindering antitumour activity and immunotherapy responsiveness.309 These findings further support the bidirectional crosstalk between metabolic and inflammatory regulation in the cancer cachexia process.
Myeloid-derived suppressor cells
MDSCs, which originate from bone marrow progenitors and immature myeloid cells, possess the ability to differentiate along myeloid lineages, including dendritic cells, macrophages and granulocytes, thereby exhibiting a remarkable capacity to suppress immune cell responses, thereby accelerating the progression of cachexia (Fig. 6). During the development of cancer cachexia, a marked increase in MDSC counts is observed in the circulation of cachectic cancer patients, indicating the importance of MDSCs in cancer cachexia.310 Chronic inflammation mediated by IL-17 may contribute to immunosuppression or cachexia by promoting MDSC infiltration in cancer patients.311 Synergistic blockade of MEK and PI3K signaling reduces splenic MDSC counts, effectively alleviating weight loss and skeletal muscle atrophy.312 In 4T1-bearing animals, MDSC expansion increases susceptibility to inflammation-induced organ injury and death due to increased energy expenditure, adipose tissue depletion, and increased expression of acute-phase hepatic proteins.313
Although MDSCs alone may not be sufficient to induce cachexia, they can exaggerate cancer cachexia by suppressing adaptive immunity. For example, MDSCs inhibit T-cell activation and function, allowing tumor cells to release cachexia-related factors.314 A study demonstrated that partial inhibition of PARP-1 is sufficient to block the suppressive activity of MDSCs, such as impairing their ability to inhibit T-cell proliferation. Therefore, this treatment synergizes with anti-PD-1 immunotherapy and other immune checkpoint inhibitors to enhance CD8+ T-cell infiltration, offering protection against cachexia in a colon cancer mouse model.315
MDSCs frequently represent a pivotal source of cachexia-associated factors that regulate cachexia. In cachectic mice, Ly6G+CD244+ polymorphonuclear MDSCs can secrete activin A in skeletal and cardiac muscles, leading to muscle atrophy via the upregulation of E3 ubiquitin ligases.316 Notably, therapeutic intervention with anti-Ly6G antibodies targeting MDSCs and neutrophils reduces weight loss and muscular atrophy in both skeletal and cardiac tissues.316,317
However, another study reported that increased infiltration of CD11b+Ly6g+ cells is observed in skeletal muscle and adipose tissue in a colon cancer mouse model. When 5-FU chemotherapy reduces this infiltration, cachectic phenotypes are not alleviated, indicating that MDSC cells may not be the primary driver of tumor- and chemotherapy-induced cachexia.318 Similarly, in a preclinical cancer cachexia model, soluble ACVR2B can alleviate the cachectic phenotype by enhancing hepatic protein synthesis and reducing splenomegaly. However, the levels of circulating inflammatory cytokines and MDSCs in the spleen remain elevated. Thus, the changes in MDSCs do not explain the cachectic phenotypes in these contexts.319
Cancer-associated fibroblasts
CAFs are prevalent in various tumor tissues and exert detrimental effects by promoting tumor growth, angiogenesis, and metastasis. Recent studies increasingly implicate the role of CAFs in cancer cachexia (Fig. 6). Hypoxia within cachectic tumor regions activates the TGF-β signaling cascade via noncanonical routes such as MAPK signaling to trigger fibroblast-to-myofibroblast transdifferentiation. This transition increases the levels of ECM components and inflammatory and angiogenic factors, thereby modulating tumor aggressiveness.320
CAFs can also secrete various proinflammatory factors to modulate cachectic phenotypes by influencing ECM components within the TME. For example, in lung cancer, CAFs secrete IL-6 via the circNOX4/miR-329-5p/FAP axis.321 In breast cancer, infiltrating TAMs secrete CCL18 to induce the production of IL-6 and IL-8 by breast fibroblasts through the NF-κB signaling pathway.322 A prior study indicated that CAFs can secrete GDF15, supporting the role of CAFs in regulating cachectic phenotypes.323
Fibroblast activation protein (FAP), also known as F19, a serine protease involved in ECM remodeling, is highly expressed in CAFs but virtually absent in normal fibroblasts or other normal tissues.324 Moreover, FAPα+ fibroblasts are implicated in immune suppression. For example, FAPα+ fibroblasts impair the responsiveness to the T-cell-activating immune checkpoint inhibitors α-CTLA-4 and α-PD-L1 via the secretion of CXCL12.325 Moreover, the secretion of CCL2 by FAP+ fibroblasts promotes the recruitment of MDSCs,326 indicating the importance of FAP in modulating the deleterious biological behaviors of CAFs.327 However, recent studies have suggested that inhibiting FAP-positive CAFs can induce cancer cachexia. Tran et al. reported that the adoptive transfer of T cells genetically engineered with FAP-reactive CARs into mice with subcutaneous tumors markedly induced cachexia and bone marrow toxicity. Further investigation revealed that FAP5-CAR-transduced T cells might target FAP-positive multipotent bone marrow stromal cells, which could explain the observed bone marrow toxicity.328 Similarly, Roberts et al. reported that the depletion of FAP+ stromal cells results in muscle wasting and altered hematopoiesis.329 Collectively, these observations implicate fibroblasts as critical regulators of muscle homeostasis, and the absence of fibroblasts in skeletal muscle can contribute to muscle atrophy in cancer cachexia.
Tumor-associated endothelium
ECs, which constitute the luminal layer of nascent blood vessels, play critical roles in maintaining vascular integrity and function across diverse tissues, including the tumor stroma, skeletal muscles, and CNS. Within the TME, these vessels serve as nutrient suppliers for the tumor and therefore regulate tumor progression and metastatic potential via intricate vascular endocrine signaling mechanisms.330 Elevated VEGF levels and increased CD34+ microvessel density are widely observed in cancer patients and indicate neoangiogenesis during the progression of malignant tumors.331 Additionally, the matricellular protein SPARC, a novel tumor-derived vascular permeability factor, disrupts the endothelial barrier through vascular cell adhesion molecule 1 and p38 MAPK-mediated signaling, thereby promoting the metastatic competence of tumor cells.332 The high expression of the oncoprotein BRAFV600E in undifferentiated thyroid carcinoma tumor cells promotes in vitro angiogenesis and facilitates the secretion of factors such as VEGFA, VEGFC, and IL-6 by tumor cells, which contributes to cachexia and metabolic alterations in advanced thyroid cancer.333 Similarly, the delicate balance between anabolic protein synthesis and catabolic proteolytic breakdown of skeletal muscle partly depends on the integrity of the blood microcirculation. Healthy skeletal muscles are abundantly supplied with capillaries. When ECs are dysfunctional, the nutrient supply within muscles is impaired. Therefore, protein synthesis is reduced, impairing muscle regeneration and leading to fibrosis and compromised contractility.334 In KPC mice with cachexia, muscle tissue is characterized by increased capillary leakage and infiltration of immune cells and inflammatory factors such as IL-1β, IL-10, and IL-6. Notably, Activin-A inhibits PGC1α expression in ECs within muscle tissue, which disrupts vascular integrity, leading to vascular leakage and muscle loss.335 This finding is consistent with studies showing that PGC1α overexpression in myocytes can partially inhibit age-related muscle atrophy by activating mitochondrial oxidative metabolism and neovascularization.336
Moreover, the BBB serves as a crucial interface for communication between the CNS and peripheral tissues, facilitating the transport and interaction of appetite-regulating cytokines with brain ECs and thereby modulating the release of appetite-altering substances into the brain interstitial fluid.337 Endothelial peroxynitrite generation by cisplatin mediates neurotoxicity through neuronal caspase-1, which participates in abnormal neuronal γ oscillations in the hypothalamic arcuate nucleus, causing decreased food intake and weight loss in mice.338 ECs play pivotal roles not only in angiogenesis but also in regulating immune cell infiltration and inflammatory responses (Fig. 6). P-selectin, which is expressed on platelets and ECs, facilitates leukocyte recruitment by binding with PSGL-1, enabling leukocyte capture and rolling on stimulated ECs. This mechanism increases the aggregation of leukocytes and activated platelets and the secretion of inflammatory cytokines, which promote the manifestation of cachectic phenotypes.339 TNF-α and IL-1β can induce the expression of the notch ligand JAG1, and tumor-derived exosomes may also transfer DLL4 to activate notch receptors on distant ECs. This activation increases the synthesis of retinoic acid and IL-33 in ECs. Secreted IL-33, in turn, stimulates excessive production of retinoic acid in adjacent adipocytes and macrophages, which contributes to maintaining catecholamine-induced WAT reduction via increased UCP1 levels. Additionally, retinoic acid enhances the expression of IGFBP3, which promotes antiadipogenesis and apoptosis, thereby influencing lipid storage and adipocyte turnover.340 Tumor ECs release chemerin in response to chemotherapy, which can increase NK cell recruitment and alleviate WAT lipolysis and proteolytic effects on skeletal muscle. Targeting VEGF-A in myeloid cells can promote the release of chemerin and ameliorate cachexia.341
ECs are critical cellular components that form the vascular lumen, and their structural integrity and functional status are essential for nutrient delivery to peripheral tissues. Their destruction not only results in energy deficiency but also disrupts the blood vessel barrier, potentially promoting tumor cell metastasis, immune cell mislocalization, and the accumulation of inflammatory factors, ultimately accelerating cachexia progression.
Regulatory roles of other cell types in the TME
The development of cachexia is intricately intertwined with various cellular components and their secreted cytokines. It is well documented that distinct types of cells within the TME, including TAMs, TANs, TILs, MDSCs, CAFs, and ECs, modulate metabolic and immunologic responses. However, other cellular components within the TME should not be overlooked, as they may also have potential roles in cachexia. For example, in a lung cancer mouse model of cachexia, a significant increase in activated and degranulated mast cells was observed in skeletal muscle. Consistently, exposure of C2C12 myotubes to conditioned media from tumor-activated mast cells results in a marked reduction in myotube diameter, suggesting that tumor-activated mast cells secrete certain factors to mediate skeletal muscle atrophy.342 NK cells may alleviate cachexia by eradicating senescent tumor cells,341 and they may also contribute to cachexia pathogenesis in specific mouse models. For example, upregulated NK cell activity was identified in a peritonitis-induced sepsis model.343 Furthermore, NK cells swiftly activate the immune system by producing cytokines such as IFN-γ, TNF-α, IL-10, GM-CSF, and chemokines, which are canonical cachexia-associated factors.344 While the direct contribution of B lymphocytes to cachexia is less understood, the antibodies produced by B lymphocytes can modulate cancer cachexia progression by regulating immune responses and metabolic states.
In conclusion, the onset and progression of cachexia are intricately governed by diverse cellular components. Elucidating the intricate interplay between cachexia and cells within the TME is paramount for understanding its pathogenesis and identifying effective therapeutic interventions.
Cachexia-associated inflammatory cytokines regulate metabolic reprogramming
As mentioned earlier, cancer cachexia is often accompanied by dysregulation of inflammation regulation. Therefore, in the past few decades, various studies have focused extensively on the role of proinflammatory cytokines in cachexia syndrome. A series of factors, including members of the interleukin family (IL-1, IL-4, IL-6, and IL-20), interferon (IFN-γ), TNF-α, and growth factors such as TGF-β, oncostatin M (OSM), leukemia inhibitory factor (LIF) and GDF15, as well as chemokines (LCN2), were identified. All of these factors are significantly elevated in cachexia patients.345 Metabolic abnormalities in various organs or tissues, such as the gut, muscles, and fat, can lead to elevated levels of cytokines. Prolonged high levels of inflammatory factors can further disrupt muscle and fat metabolism, exacerbating cachexia. However, numerous studies have indicated that anti-inflammatory treatments targeting these factors have not yielded satisfactory results, suggesting that a deep understanding of the pathology of cancer cachexia is needed. Recently, several new cachexia-related factors, such as the growth factor GDF15 and the chemokine LCN2, which participate in cachexia through mechanisms that amplify inflammatory signals beyond traditional pathways, have been reported (Table 2).
Interleukin family
Interleukin-1
In advanced cancer patients, immune cells secrete large amounts of proinflammatory cytokines such as IL-1 and IL-6, which further exacerbates the progression of cachexia.346 Mechanistically, IL-1 triggers the release of α-MSH from POMC neurons347 and has an inhibitory effect on NPY neurons,348 which affects appetite by regulating MC4R expression in neurons. These phenotypes can be alleviated by a melanocortin receptor antagonist.349 IL-1 can also stimulate the release of corticotropin-releasing hormone (CRH) from hypothalamic neurons, promoting the secretion of adrenocorticotropic hormone and cortisol, thereby mediating the catabolic effects observed in cachexia.350 Following adrenalectomy, muscle catabolism is alleviated in mice, suggesting that the HPA axis may serve as a central pathway to mediate the impact of IL-1 on muscle degradation.351 IL-1 can be further classified into IL-1α and IL-1β on the basis of different coding genes. Compared with IL-1α, IL-1β has more pronounced proinflammatory effects.352 IL-1β facilitates the release of various inflammatory factors through central inflammation, resulting in the dysregulation of POMC and AgRP, which subsequently triggers protein hydrolysis and lipolysis.353
Interleukin-6
IL-6 was initially identified for its roles in B-cell differentiation and the acute phase of the immune response.354,355 Indeed, IL-6 is a crucial cytokine that regulates both innate and adaptive immunity and plays significant roles in various physiological processes, including inflammation, metabolism, apoptosis, cell differentiation, bone homeostasis, and angiogenesis.356,357 Although IL-6 exerts its anti-inflammatory and regenerative effects by regulating the JAK/STAT pathway, it also participates in proinflammatory responses through trans-signaling.358 Despite its dual function in inflammation, increased levels of IL-6 are detected in patients with cancer cachexia, supporting its role in promoting cancer cachexia.359 Indeed, short-term activation of IL-6 signaling can stimulate muscle growth. However, owing to its proinflammatory role, prolonged activation can cause muscle atrophy by stimulating muscle degradation and impairing oxidative capacity.357,360 Similarly, administration of recombinant IL-6 and IL-6 overexpression results in decreased muscle mass in a cancer cachexia mouse model,359 which can be alleviated by injecting IL-6 receptor antibodies.361 Not surprisingly, IL-6 can decrease the myofibrillar protein content and increase muscle wasting. Mechanistically, IL-6 inhibits the activity of the mTOR signaling pathway by reducing the phosphorylation of mTOR, ribosomal S6 kinase and eukaryotic translation initiation factor 4E-binding protein 1.362,363 In addition, IL-6 and PTHrP can reduce adipose tissue mass by promoting adipose tissue browning.56,92 In mouse models of cachexia, treatment with IL-6 receptor antibodies can suppress lipolysis and prevent browning of WAT.364 Recent studies have indicated that peripheral circulating IL-6 can enter the brain and activate neurons in the area postrema and its associated neural networks, leading to brain dysfunction and subsequent cachexia.365
In addition to IL-6, IL-4 and IL-8 also play regulatory roles in the process of cachexia. IL-4 is a pleiotropic cytokine that can promote the differentiation of fibro-/adipogenic progenitors into fibroblasts, enhancing the phagocytosis of cellular debris by activating the STAT6 signaling pathway. This process facilitates muscle regeneration. However, fibro-/adipogenic progenitors can also differentiate into adipocytes, which may promote muscle growth.366,367 Additionally, treatment with the proinflammatory cytokine IL-20, an anti-IL-20, can improve weight loss and prevent a reduction in adipose tissue mass.368
Tumor necrosis factor
Tumor necrosis factor α
TNF-α is a key regulatory factor in cancer cachexia, as its concentration is positively correlated with the degree of weight loss and muscle wasting. TNF-α influences the development of cachexia through multiple mechanisms, including lipid metabolism, protein homeostasis, and neural regulation. On the one hand, TNF-α inhibits the expression of lipoprotein lipase in adipocytes, resulting in reduced lipid uptake.369 On the other hand, TNF-α inhibits G0S2 while promoting the activity of ATGL, thereby enhancing ATGL-mediated lipolysis.370 Reduced intake and enhanced degradation collectively shift the lipid metabolic balance in a detrimental direction. In vitro studies using myogenic cell cultures revealed that TNF-α suppresses myoblast differentiation and induces protein loss via the NF-κB signaling pathway.371 TNF-α can also stimulate oxidative stress by promoting the production of ROS, which can further activate the NF-κB pathway, thereby increasing muscle protein degradation.372 Other studies reported that TNF-α stimulates protein degradation in muscle cells by activating the UPS and upregulating the expression of ubiquitin ligases (such as MuRF-1 and Atrogin-1). Moreover, TNF-α inhibits mTOR signaling by activating AMPK and suppressing the PI3K/Akt pathway, which leads to decreased muscle protein synthesis.373 Recent studies have suggested that TNF-α suppresses appetite and increases feelings of satiety by influencing the secretion of appetite-regulating neuropeptides in the hypothalamus, including both appetite suppressants and stimulants.374
Growth factors
Transforming growth factor-β
The TGF-β superfamily encompasses a variety of growth factors, including TGF-β itself, growth differentiation factors, bone morphogenetic proteins, activins and inhibins, which play physiological and pathological roles in different tissues. TGF-β primarily impacts skeletal muscle metabolism, exacerbating the occurrence of cachexia by inhibiting muscle protein synthesis and promoting muscle protein degradation. In a mouse model of Marfan syndrome characterized by fibrillin-1 deficiency, the administration of TGF-β antagonists significantly improved muscle regeneration, indicating that TGF-β is critical for maintaining muscle balance.375 In the context of muscle injury, the upregulation of TGF-β promotes muscle tissue fibrosis, further impairing muscle function and contributing to muscle wasting.376 The binding of TGF-β to its type I (TGF-βR1) and type II (TGF-βR2) receptors results in the formation of a heterotetrameric complex,377 which subsequently induces the formation of the Smad2/3/4 complex, facilitating its translocation into the nucleus. Activated TGF-β signaling, in turn, regulates the proliferation and differentiation of skeletal muscle precursor cells and the transcription of genes associated with protein degradation pathways in fibroblasts.378,379 Furthermore, recent studies have shown that TGF-β influences lipid metabolism. TGF-β derived from M2 macrophages in aged mice induces DNA damage through the production of reactive ROS, thereby leading to the senescence of adipose progenitor cells and the inhibition of adipogenesis. However, although elevated p16 expression and adipose tissue atrophy are observed in a cancer cachexia model, blocking TGF-β does not affect the number of p16-high adipose progenitor cells.285
Oncostatin M
Oncostatin M is involved in various physiological functions, including hematopoiesis, liver regeneration, wound healing, inflammation and metabolism. By binding to the glycoprotein gp130, OSM activates the downstream JAK/STAT signaling pathway and stimulates the expression of STAT-dependent genes by recruiting either the LIF receptor β or the OSM receptor β.380 Recent studies have established a link between OSM and cancer cachexia. The upregulated expression of OSM target genes, such as OSMR, is detected in the muscles of mice with tumor implantation. The introduction of recombinant OSM protein into myotubes promotes cellular atrophy by inducing the expression of several muscle atrophy-related genes, including Atrogin-1, which is mediated by the JAK/STAT signaling pathway.381,382,383 While IL-6, LIF, and OSM induce muscle atrophy via the JAK/STAT pathway, OSM appears to exert a more pronounced effect on the cellular transcriptome of myotubes, thus resulting in a stronger atrophic response. Recently, Bilgic et al. reported that OSM can increase the levels of EDA2R in muscle tissues, leading to increased skeletal muscle atrophy, suppressed adipocyte differentiation, and increased adipocyte degradation.382 Furthermore, depletion of OSMR in muscle fibers protects mice from weight loss and muscle wasting, which, at least in part, is attributed to suppression of the upregulation of Eda2r induced by tumors.382 In other words, in patients with cachexia, OSM has a significant effect on both muscle and fat metabolism.
Leukemia inhibitory factor
LIF plays a significant role in immune system activation and tumorigenesis, functioning as a pleiotropic cytokine. It affects muscle and fat metabolism and is involved in the CNS’s regulation of appetite, particularly in patients with cancer cachexia. LIF primarily binds to the surface heterodimeric glycoprotein complex gp130/LIF receptor, thereby activating downstream signaling pathways, including the JAK/STAT3, PI3K/Akt/mTOR, and MAPK signaling pathways.384 LIF can promote lipolysis and induce cachexia. Experimental findings indicate that LIF activates the JAK/STAT signaling pathway, which allows ATGL to hydrolyze triglycerides into free fatty acids while reducing lipoprotein lipase activity, further accelerating lipolysis.385,386,387 Moreover, LIF serves as an inducer of anorexia. Recent research has demonstrated that LIF expressed in POMC neurons located in the arcuate nucleus of the hypothalamus stimulates the release of α-MSH, thereby inducing anorexia.353 LIF secreted from tumor cells can interact with various neuropeptides, leading to the onset of anorexia.388 LIF also promotes anorexia by increasing the expression of leptin during acute inflammatory responses.389 Additionally, patients with muscle wasting show excessive expression of LIF, suggesting a potential link between LIF and muscle wasting, possibly related to the activation of signaling pathways such as the JAK/STAT, ERK1, and MAPK pathways.390 Furthermore, treatment with LIF antibodies such as PIAS3 and ruxolitinib effectively alleviated muscle wasting.390
Growth differentiation factor 15
GDF15 is a stress-induced cytokine and a new member of the TGF-β family that was first discovered in activated macrophages. Research has demonstrated that GDF15 expression is significantly elevated in various cancers, including prostate cancer, colorectal cancer, and breast cancer, and its serum levels in patients are nearly twice as high as those in healthy controls.141,391 Like TGF-β, GDF15 can activate the Smad signaling pathway, the PI3K/Akt signaling pathway, and the NF-κB signaling pathway.392 Johnen et al. reported that overexpression of GDF15 in tumor-bearing mice significantly reduced food intake, which indirectly caused decreased fat reserves and atrophy in the tibialis and gastrocnemius muscles.393 GDF15 can also bind to the specific receptor GFRAL to activate the Ret proto-oncogene, which promotes ERK and Akt phosphorylation, resulting in decreased appetite. It activates the HPA axis to promote the secretion of CRH and glucocorticoids.155,394 In cachectic mice, the activation of the GFRAL/RET signaling pathway upregulates the expression of iodothyronine deiodinase 2 and β3-adrenergic receptors (β3-AR), causing fat reduction.155 GDF15 is also an important factor for inducing muscle wasting. In vitro experiments have shown that GDF15 can promote the expression of MuRF1 and MAFbx/Atrogin-1, leading to a reduction in muscle fiber diameter.395 Lerner et al. confirmed that GDF15 facilitates the reduction of the gastrocnemius and soleus muscles by activating mitogen-activated protein kinase 11.396 Furthermore, GDF15 enhances the expression of the lipid-mobilizing factor ZAG through the JPA axis and stimulates lipolysis via β3-AR.397 Similarly, another study demonstrated that GDF15 promotes the expression of differentiation and thermogenic genes in brown adipocytes. Additionally, GDF15 promotes osteoclast differentiation and inhibits osteoblast differentiation both in vivo and in vitro, leading to bone metabolism disorders.398 These findings support the crucial role of GDF15 in regulating distinct cancer cachexia phenotypes via different mechanisms.
Interferon
Interferon-γ
IFN-γ, also known as immune interferon, is classified as a type II interferon that is produced primarily by activated Th1 cells, NK cells and CD8+ cytotoxic T lymphocytes. IFN-γ exacerbates systemic inflammatory responses and promotes protein breakdown, making it a key factor leading to the progression of cachexia. Moreover, IFN-γ is considered a crucial element in the progression of cachexia. Research has demonstrated that IFN-γ promotes the upregulation of muscle-specific E3 ubiquitin ligases, such as MuRF1 and Atrogin-1, by activating the JAK/STAT signaling pathway, which accelerates protein degradation.399 The elevation of IFN-γ is closely associated with the release of proinflammatory signaling molecules (such as TNF-α and IL-6), which collectively enhance systemic inflammatory responses and worsen the symptoms of cachexia.
Chemokines
Lipocalin 2
LCN2, which is synthesized primarily by the immune system, is secreted into the circulation in various diseases associated with cachexia.400 Recent studies have indicated that LCN2 has an appetite-inhibiting effect. When LCN2 is injected intraperitoneally into Lcn2-deficient mice, it specifically binds to MC4R in the paraventricular and ventromedial neurons of the hypothalamus and activates an MC4R-dependent anorexigenic pathway.66 Similarly, the absence of LCN2 mitigates the loss of lean and fat mass associated with cachexia-induced anorexia, whereas the restoration of Lcn2 expression in the bone marrow rescues the anorexia feature of cachexia.67 Moreover, the increase in food intake due to LCN2 blockade is unrelated to systemic inflammatory status or immune activation.67 These findings suggest that LCN2 plays a key role in reducing appetite in cancer cachexia patients.
In summary, various cytokines secreted by different cells in the microenvironment play significant roles in the development of cancer cachexia. These cytokines include classical inflammatory factors such as TNF-α, IL-1, IL-6, and IFN-γ. Upon release into the circulation, these cytokines exert diverse effects on various tissues, such as the brain, muscle, and adipose tissues, leading to appetite suppression, suppression of muscular protein production, and promotion of lipolysis, which ultimately lead to the progression of cachexia. Furthermore, TNF-α and IL-1 can enhance the activation of the IL-6 signaling pathway, further exacerbating the progression of cachexia. Therefore, targeting cachexia-inducing factors may provide new avenues for managing cancer cachexia.
Regulatory roles of cell signaling pathways
Cytokines constitute pivotal components in the pathogenesis of cachexia, primarily regulating skeletal muscle wasting and fat depletion via receptor-mediated signal transduction cascades. Therefore, various signaling pathways activated by proinflammatory cytokines or inflammation-related ligands are crucial for muscle atrophy through the regulation of muscle protein turnover. Additionally, various signaling pathways exacerbate fat depletion by regulating the activity of lipid metabolism-related enzymes and accelerating the browning of WAT. Thus, signaling pathways serve as the key hubs between cytokines and skeletal muscle wasting, as well as lipid metabolic dysregulation. A deeper investigation into these signaling pathways will enhance our understanding of the underlying pathological mechanisms of related diseases.
PI3K/Akt signaling pathway
The PI3K/Akt signaling pathway is involved in numerous physiological and pathological processes, including the regulation of cell proliferation and apoptosis, angiogenic responses, metabolic homeostasis, and the generation of muscle and fat tissue.401 This pathway is modulated by multiple signals, and in patients with cachexia, the PI3K/Akt pathway is modulated primarily by the HPA axis. The axis is a crucial component of the neuroendocrine system, integrating signals from the brain to peripheral systems and coordinating the endocrine system, nervous system, and immune system. The HPA axis consists of three main parts: the hypothalamic paraventricular nucleus, the anterior pituitary, and the adrenal cortex402 (Fig. 7). The paraventricular nucleus secretes CRH, which can suppress appetite by modulating leptin levels.403 The adrenal cortex synthesizes glucocorticoids that participate in various bodily functions, including digestion, reproductive behaviors, and energy storage and expenditure. Research has indicated that glucocorticoids induce muscle atrophy by suppressing the PI3K/Akt signaling pathway, leading to the upregulation of FoxO transcriptional regulators (FoxO3a and FoxO1). This activation increases the transcription of MAFbx and MuRF1, enhancing the proteolytic activity of the UPS. Concurrently, glucocorticoids can increase skeletal muscle resistance to anabolic regulators, such as IGF-1, promoting protein degradation while inhibiting protein synthesis, ultimately causing muscle atrophy.404 Importantly, the HPA axis is also modulated by inflammatory cytokines such as IL-1, IL-6, and TNF-α. These cytokines may individually or synergistically stimulate CRH secretion, ultimately leading to increased glucocorticoid release, appetite suppression, and exacerbation of muscle wasting in patients.405,406
NF-κB signaling pathway
The NF-κB signaling pathway is important for diverse biological activities, including inflammatory responses, immune responses, cell apoptosis, and stress responses. Hyperactivation of the NF-κB pathway has been linked to numerous human diseases, particularly tumorigenesis. Abnormalities in the NF-κB signaling pathway lead to dysregulation of cell proliferation, viability, motility, and invasion, thereby promoting tumor development.407,408 In cachexia patients, circulating concentrations of inflammatory factors (such as IL-1 and TNF-α) are significantly elevated. These bioactive molecules activate their respective receptors, recruiting adaptor proteins to activate I-kappa-B kinase, which in turn triggers the nuclear translocation of NF-κB. Among these, TRAFs serve as intracellular adaptors that interact with surface receptors such as TNFR-1, TNFR-2, TLR4, and IL-1R, integrating upstream inflammatory signals and activating the downstream NF-κB pathway.409,410 This activation upregulates the expression of MAFbx and MuRF1 (Fig. 7). The activation of NF-κB is considered a critical step in inflammation-mediated skeletal muscle atrophy. In models of systemic inflammation, the inhibition of muscle NF-κB signaling in genetically modified mice alleviated muscle wasting.411 This signaling pathway, particularly in response to IL-1 and TNF-α, serves as a significant mechanism for inducing muscle wasting. In vitro studies revealed that C2C12 myoblasts exhibit increased NF-κB pathway activation following TNF-α treatment. Moreover, the use of the NF-κB inhibitor PDTC effectively suppressed the upregulation of MuRF1 mediated by TNF-α intervention.412 IL-1, a proinflammatory cytokine with effects similar to those of TNF-α, is also significantly elevated in cachexia patients. Parallel experiments indicate that IL-1 activates NF-κB signaling, resulting in the upregulation of MuRF1.413 Additionally, NF-κB reduces the abundance and activity of MyoD and Myf-5 proteins, decreases MyoD mRNA expression, and inhibits Akt-mediated dephosphorylation of FoxO3a, facilitating its nuclear translocation and inducing the transcription of the target genes MAFbx and MuRF-1. This cascade ultimately impairs postnatal myogenesis.412,414
JAK-STAT signaling pathway
The JAK-STAT signaling pathway is a crucial inflammatory signaling cascade that plays significant roles in regulating cell development, proliferation, metabolism, inflammation, and cancer. Approximately 60 cytokines, including IFNs, colony-stimulating factors, interleukins, and growth factors, are known to activate the JAK/STAT signaling pathway.415 Upon cytokine binding to its receptor, JAKs are recruited and phosphorylated. The phosphorylated JAKs then activate STAT proteins, which dimerize and translocate to the nucleus, where they attach to target DNA sequences and modulate gene expression415 (Fig. 7). The JAK family consists of four nonreceptor tyrosine kinases, namely, JAK1, JAK2, JAK3, and tyrosine kinase 2 (TYK2), which interact noncovalently with the cytoplasmic domains of cytokine receptors.415,416 The STAT family includes STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Once phosphorylated, STAT proteins form dimers that act as transcription factors, altering chromatin accessibility and inducing gene transcription.417 In cachexia patients, the regulation of the JAK/STAT pathway by the IL-6 family of receptors (including IL-6, OSM, and LIF) is particularly pronounced. After binding to their receptor complexes, IL-6 family factors activate STAT transcription, promoting the expression of MSTN, MAFbx, MuRF1, and caspase-3 in muscle fibers,418,419 which leads to muscle protein catabolism. Studies have shown that IL-6 levels and the STAT3 signaling pathway are markedly increased in the skeletal muscle of cachexia mice induced by C26 cancer cells and that the inhibition of STAT3 alleviated muscle wasting. Similar results were observed through the inhibition of STAT3 signaling in C2C12 cells in vitro.359 Similarly, the administration of IL-6 receptor antibodies to C26 cachexia mice mitigated muscle loss.420 Hashimoto et al. demonstrated that recombinant IL-6 treatment reduced the diameter of C2C12 myotubes while increasing STAT3 phosphorylation and Atrogin-1 transcription.421 Overall, IL-6 phosphorylates STAT3 via the JAK/STAT pathway, leading to the overexpression of MAFbx and subsequent muscle wasting. Additionally, the STAT transcription factor family can be regulated by other kinases, such as mTOR and MAPK, which can phosphorylate STAT3.422 STAT transcription factors also interact with FoxO and NF-κB to regulate gene transcription.423,424 In addition to the IL-6 family, IFN-γ also utilizes the JAK/STAT signaling pathway to upregulate MuRF1 and MAFbx, accelerating protein degradation.399 Recent studies have shown that IL-6 can promote the expression of UCP-1 via the JAK/STAT signaling cascade, thereby facilitating WAT browning, and can also directly enhance ATGL-dependent lipolysis.425,426,427
TGF-β/SMAD signaling pathway
The SMAD signaling pathway involves an intracellular signaling mechanism that modulates cell growth, differentiation, migration, and invasion. The SMAD proteins are categorized into three classes: the first class comprises receptor-regulated SMADs (R-Smads), which are downstream signaling molecules of the TGF-β receptor complex and include mainly Smad1, Smad2, Smad3, Smad5, Smad8, and Smad9; the second class comprises comediator SMADs, of which only Smad4 has been identified in mammals; and the third class consists of inhibitory SMADs (I-Smads), which include Smad6 and Smad7. The SMAD signaling pathway is regulated by various ligands, with members of the TGF-β family playing a significant role in modulating SMAD signaling and regulating skeletal muscle generation and metabolism.428 In cachexia patients, TGF-β family members (such as MSTN, TGF-β, and GDF15) bind to receptor complexes (TGFβRI, TGFβRII, and ActRIIB), leading to the phosphorylation of Smad2 and Smad3, which then combine with Smad4. This complex stimulates FoxO-dependent transcription (Fig. 7), thereby regulating the expression of genes linked to the proliferation and differentiation of skeletal muscle progenitor cells, as well as protein breakdown in mature muscle fibers.429,430,431 Furthermore, phosphorylated Smad2/3 can inhibit Akt, leading to the activation of FoxO, which results in increased protein catabolism.432 Costelli et al. demonstrated that MSTN can induce cachexia in mice via ActRIIB.433 Conversely, blocking the ActRIIB receptor can prevent cachexia in mice with C26 tumors, with no significant alterations detected in the levels of IL-6, TNF-α, or IL-1β.434 Additionally, TGF-β can activate noncanonical signaling cascades, such as the MAPK pathway, to influence the development of cachexia.
MAPK signaling pathway
The MAPK signaling pathway is one of the oldest evolutionary signaling pathways, mediating various intercellular interactions and receiving various extracellular stimuli, including cytokines and environmental changes, to regulate cellular responses. The MAPK signaling cascade consists of three tiers: MAPK, MAPK/ERK kinase (MEK or MKK), and MAPK kinase kinase (MEKK or MKKK). Moreover, the MAPK pathway is subdivided into four distinct branches: ERK1/2, p38 MAPK, c-Jun N-terminal kinase (JNK), and ERK5.435 In skeletal muscle cells, the MAPK signaling pathway regulates myocyte growth and stress responses, particularly in cachexia patients, where MAPK activation promotes protein catabolism.436 This process is primarily mediated by IL-1 or TNF-α. When TNF-α binds to its receptors TNFR1/2, it activates a TRAF2-mediated JNK-dependent kinase cascade, sequentially activating MKKs, MAPKs, and ultimately p38 MAPK. After nuclear translocation, p38 MAPK upregulates the transcription of MuRF1 and Atrogin-1, leading to increased protein degradation437 (Fig. 7). Similarly, IL-1 was shown to upregulate Atrogin-1 expression through the phosphorylation of p38 MAPK.413 Additionally, JNK can inhibit muscle formation by phosphorylating MRF4, which suppresses the expression of late-selective myogenic genes, thus adversely affecting myogenesis.438 These findings underscore the crucial function of the MAPK signaling pathway in the manifestation of cancer cachexia.
Clinical trials and therapeutic approaches
Despite considerable attention being given to the fundamental mechanisms of cancer cachexia, the complicated pathogenesis of cancer cachexia across different cancer types remains incompletely understood, leading to a lack of effective treatment options for different cancer types. To date, most interventional studies have concentrated on dietary and appetite-enhancing measures, anabolic stimulation, and anticytokine therapies. In this section, we summarize the results of clinical trials conducted over the past several years (Table 3) and discuss the clinical trials and promising agents used in preclinical models aimed at delineating cancer cachexia across different cancer types.
Gastrointestinal cancers
Gastrointestinal cancer affects many individuals with lesions from the esophagus to the stomach and down to the rectum. Gastrointestinal cancer patients are more susceptible to cancer cachexia, as direct mechanical or digestive issues related to the tumor burden often impair their appetite and oral intake, and the systemic inflammation and metabolic disorders caused by the tumor also contribute to this risk.439 Multiple trials have evaluated treatment methods for cachexia in patients with gastrointestinal cancer and have shown encouraging outcomes.
Considering the widespread accessibility and economic cost of dietary supplements, they undoubtedly hold significance in the management of gastrointestinal cancer. For example, fish oil, which is rich in ω-3 fatty acids, EPA, DHA, and other PUFAs, can lower the concentration of proinflammatory mediators.439,440 The use of fish oil may benefit patients with gastrointestinal cancers through increased skeletal and lean muscle mass caused by decreased CRP levels.441 However, another study indicated that fish oil can lead to decreased global quality of life and increased appetite loss.442 Thus, larger trials are needed in gastrointestinal cancer patients to determine the efficacy of fish oil and to determine which composition of fish oil strongly contributes to its efficacy. Creatine, an amino acid derivative, has no effect on cancer anorexia syndrome in patients with gastrointestinal cancers but may augment muscle strength and improve strength in CRC patients.443 Some dietary supplements, such as fish oil and guarana, have favorable effects in the treatment of cachexia, whereas others have not. Therefore, more extensive clinical trials are needed to explore the influence of dietary supplements on cancer cachexia.
Appetite stimulants, including steroids, progestational agents, cyproheptadine, and cannabinoids, were first used to treat cachexia.444 For example, Megestrol acetate, a synthetic progestin, is commonly employed to stimulate appetite by acting on NPY present in the ventromedial hypothalamus or by decreasing the synthesis and liberation of proinflammatory cytokines.445 Consistently, a randomized, double-blind clinical trial in which Megestrol acetate 320 mg/day was used demonstrated significant improvements in appetite, body weight, grip strength, and quality of life in gastrointestinal cancer patients.446 Ghrelin is a neuropeptide that is currently undergoing clinical trials for cancer cachexia. Ghrelin was significantly correlated with the BMI loss ratio and Glasgow prognostic score in gastric cancer patients and demonstrated efficacy in treating cancer cachexia.447 Another trial revealed that there were no statistically significant differences in nutritional intake or eating-related symptoms between the groups receiving ghrelin and those receiving a placebo.448 Anamorelin and macimorelin are ghrelin receptor agonists that can promote ghrelin secretion by activating the ghrelin receptor, thereby increasing appetite, weight, and muscle mass.449,450 In addition, clinical trials have shown their ability to treat cachexia in patients with gastrointestinal tumors.450,451,452 Cannabis, which mainly contains Δ9-tetrahydrocannabinol and cannabidiol, has a notable effect on stimulating appetite and enhancing body weight among HIV-positive individuals and cancer patients. Additionally, clinical trials in gastrointestinal cancer patients confirmed this finding.453 In a clinical trial, olanzapine, an antipsychotic medication with antagonistic effects on dopamine and serotonin receptors, was found to enhance appetite and result in weight gain in patients with gastric cancer.454 Mirtazapine, a widely used tetracyclic antidepressant with serotonergic and noradrenergic action, has potential as a promising treatment option because of its ability to increase body weight and appetite.455,456
Chronic inflammation is one of the main hallmarks of cancer cachexia, and targeting inflammatory cytokines may alleviate the symptoms of cancer cachexia.13 MABp1, an antibody targeting IL-1α, resulted in a significant reduction in systemic inflammation and thrombocytosis in CRC patients but did not improve cancer cachexia symptoms. Therefore, larger clinical trials should be conducted to evaluate the treatment efficacy of MABp1 in patients with cancer cachexia.457 Curcumin, a polyphenolic compound obtained from turmeric, attenuated proteolysis and muscle wasting and suppressed adipose wasting in mouse models bearing MAC16 and C26 tumors.458 Nevertheless, it failed to show superiority over placebo in enhancing body composition among patients with gastrointestinal cancers; thus, additional research is warranted.459 An increasing amount of evidence has demonstrated that celecoxib, an inhibitor of cyclooxygenase 2 (COX-2), alleviates cachexia in preclinical models and patients with gastrointestinal cancer through the downregulation of the serum levels of IL-6, VEGF, or TNF-α.446,460,461 However, etanercept (a TNF-decoy receptor) does not seem to alleviate cancer anorexia syndrome in patients.462 As an immunomodulatory and anti-inflammatory agent, thalidomide has been shown to prevent weight loss, improve appetite, and enhance quality of life in patients with some types of cancer, including gastrointestinal cancer, largely because of its ability to reduce the generation of TNF-α, degrade TNF-α mRNA, and suppress NF-κB pathway activation.463,464,465 Similarly, OHR/AVR118, another immunomodulatory agent, significantly improved anorexia, dyspepsia, and strength and achieved weight stabilization or gain in patients with gastric cancer and CRC.466 N-acetylcysteine enhances muscular performance and decreases TNF-α levels in elderly individuals.467 Furthermore, a clinical trial demonstrated that N-acetylcysteine decreased TNF-α levels in gastrointestinal cancer patients, thereby alleviating cancer cachexia (NCT00196885).
Cancer cachexia is also driven by metabolic alterations, including increased energy expenditure, elevated plasma glucose, and insulin resistance. Insulin is an important hormone that maintains organ vitality and regulates the metabolism of glucose, proteins, and lipids. Decreased insulin sensitivity leads to reduced glucose absorption by organs and leads to atrophy and a reduction in skeletal muscle and adipose tissue.468 Insulin treatment increased carbohydrate intake, lowered serum-free fatty acids, and increased body fat, mainly in the trunk and legs, without affecting lean tissue mass. More importantly, it enhances metabolic efficiency during exercise and improves survival in patients without affecting tumor growth.468
GDF15, a member of the TGF-β superfamily, modulates food consumption, the energy metabolism rate, and body weight.469 Elevated GDF15 concentrations are correlated with weight loss and poor outcomes in patients with gastrointestinal cancer.470,471 Ponsegromab is a potent and highly selective humanized monoclonal antibody designed to bind to circulating GDF15, which effectively blocks the interaction between GDF15 and the GFRAL receptor.472,473 A phase Ib study with 10 patients, including 4 CRC patients, revealed that ponsegromab led to improvements in weight, appetite, and physical activity by reducing serum GDF15 levels.472 Soon after, a larger trial involving 187 cancer patients reported that inhibiting GDF15 with ponsegromab resulted in increased weight gain, improved overall activity levels, and reduced cachexia symptoms.473 These findings support the use of ponsegromab as a therapeutic option for cachexia.
Accumulating evidence suggests that elevated levels of MSTN and its analog activin A lead to the progression of atrophy and cachexia.474 ActRIIB, a high-affinity activin type 2 receptor, plays a crucial role in mediating signaling pathways activated by a particular group of TGF-β family ligands, including MSTN, activin, and GDF11. Among these ligands, activins are the most potent negative regulators of muscle mass.474 Treatment with sActRIIB, an ActRIIB decoy receptor, not only reversed the decrease in skeletal muscle mass and cancer-induced cardiac atrophy but also prolonged the survival of C26 tumor-bearing mice.434 Unfortunately, clinical trials involving decoy ActRIIB led to treatment-related bleeding problems in volunteers, causing their discontinuation as a potential treatment for cachexia (NCT01099761).
Hepatopancreatic biliary cancers
Hepatopancreato-biliary (HPB) cancers are characterized by poor prognosis and surgically challenging management. Older patients diagnosed with HPB malignancies often suffer from malnutrition and cancer cachexia, resulting in accelerated loss of muscle mass and function beyond the typical age-related decline.475 In patients with HPB malignancy, the causes of malnutrition and weight loss are diverse and complex; thus, treatment strategies aimed at maintaining weight and muscle indices are necessary to improve postoperative outcomes.
Systemic inflammation resulting from elevated levels of CRP, an increased neutrophil-to-lymphocyte ratio, and heightened levels of proinflammatory cytokines, including TNF-α, IL-1α, and IL6, is correlated with pancreatic cancer cachexia, poor treatment response, and adverse prognoses.476 Thalidomide, an inhibitor of TNF-α, has good tolerability and effectiveness in reducing the loss of weight and lean body mass in patients with cachexia owing to advanced PDAC.477 However, infliximab, a monoclonal antibody that targets TNF-α, failed to increase lean body mass.478 L-carnitine can regulate inflammatory response mechanisms and improve nutritional status (including body mass and body fat), along with parameters related to quality of life, while also increasing overall survival rates in patients with PDAC.479 As mentioned previously, insulin and ponsegromab treatment could alleviate the symptoms of cachexia in patients with gastrointestinal tumors, and the same effect was observed in patients with PDAC.468,473 Although patients with BTC generally have a poor prognosis, a recent study demonstrated that the MAPK signaling pathway could play a crucial role in these patients.480,481 For example, patients treated with selumetinib, a MEK1/2 inhibitor, experienced an average increase in nonfluid weight and enhanced muscle gain, and another MEK1/2 inhibitor, binimetinib, also demonstrated muscle gain in patients with BTC.481,482,483
For liver cancer, one clinical study, VT-122 (NCT01265576), is under investigation to evaluate the therapeutic effects of clinical drugs for cachexia. However, many preclinical drugs have been reported to alleviate cachexia in mouse models bearing AH-130 Yoshida ascites ascites hepatoma, a cachectic rat tumor. In the Yoshida rat model, the atypical β-blocker S-oxprenolol dose-dependently decreased both weight and lean mass.484 In a cancer cachexia clinical trial, MT-102 led to great improvements in lean mass and handgrip strength.485 Similarly, in the Yoshida rat model, MT-102 prevented the progressive decrease in fat mass, lean mass, and body weight by decreasing overall protein degradation and stimulating protein synthesis.485,486 L-carnitine treatment also ameliorated atrophy of both slow- and fast-twitch muscle fibers, reversed muscle structural abnormalities, and reduced oxidative stress, proteolytic activity, and signaling markers, thereby relieving symptoms in Yoshida tumors in mice.479 Therefore, more drugs that have proven effective in preclinical models of liver cancer can be expeditiously transitioned into clinical studies to ascertain their effectiveness and safety in combating cachexia in patients with liver cancer.
Lung cancer
Lung cancer is the leading contributor to cancer death globally, with NSCLC accounting for the majority (85%) of cases.487 Patients with advanced cancer have a heightened risk of developing cancer cachexia. However, approximately 20% of individuals diagnosed with early-stage NSCLC exhibit signs of cachexia, even without any changes in caloric intake.488,489 In addition, ~40% of patients with metastatic NSCLC develop cancer cachexia, which is associated with shorter survival.489 Therefore, early identification and intervention of cancer cachexia should be considered a top priority and a vital component in the management of NSCLC.
Among the drugs discussed earlier, some can also alleviate cachexia symptoms in lung cancer patients. For example, among patients with NSCLC, mirtazapine led to a significant increase in the intake of energy, including protein, carbohydrate, and fat, as well as a reduction in the proportion of patients who experienced sarcopenia.490 Nabilone, a cannabinoid, has been demonstrated to be a safe and adequate therapeutic compound for increasing caloric intake and enhancing the quality of life of lung cancer patients diagnosed with anorexia.491 As mentioned in gastrointestinal cancer, ponsegromab enhances body mass and increases appetite among cancer patients, including those with NSCLC.472,473
Selective androgen receptor modulators (SARMs) have the potential to increase lean body mass and muscle mass without adverse effects. Enobosarm, a nonsteroidal SARM, is currently under clinical development as a therapy for preventing and treating muscle wasting in cancer patients, as demonstrated in the POWER 1 and 2 trials.492 Upon activation, it elicits conformational shifts in the androgen receptor, selectively modulating its interaction with coactivator and corepressor proteins present in various tissues, thereby altering the receptor’s capacity to modulate gene expression.493 The enobosarm was granted fast-track designation by the U.S. Food and Drug Administration (FDA), and the outcomes of the phase III trials will be instrumental in determining its approval for clinical use in preventing and treating muscle wasting in patients with NSCLC.492 Nevertheless, a completed trial involving patients with NSCLC and CRC revealed the effectiveness of the enobosarm in enhancing lean body mass without exhibiting the toxic impacts typically correlated with androgenic and progestational agents.494
β2-adrenergic agonists serve as potent muscle growth accelerators, whereas nonselective β receptor blockade can decrease catabolism. Additionally, antagonism of the central 5-HT1a receptor can lead to reduced fatigue and thermogenesis.485,495 Espindolol, a novel nonselective β-blocker, has multiple effects on the aforementioned three pharmacological targets.485 It markedly reversed weight loss, enhanced fat-free mass, preserved fat mass, and augmented handgrip strength in advanced NSCLC patients and CRC-related cachexia patients in an ACT-ONE trial.485,496 VT-122, another β2-adrenergic agonist for NSCLC, is in progress (NCT00527319).
The most extensively studied anticachexia drug in clinical trials for treating lung cancer is anamorelin. It is a novel selective ghrelin receptor agonist that enhances anabolic processes and stimulates appetite by promoting growth hormone secretion.497 In a murine xenograft model of lung cancer, anamorelin significantly increased body weight without promoting tumor growth.498 Since then, numerous clinical trials have reported the therapeutic efficacy of anamorelin in patients with NSCLC.499,500,501,502 For example, in phase 3, the international, randomized, double-blind, placebo-controlled trials ROMANA 1 (323 to anamorelin, 161 to placebo) and ROMANA 2 (330 to anamorelin, 165 to placebo), anamorelin was well tolerated and achieved an increase in lean body mass after 12 weeks, without significant effects on handgrip strength.499 ROMANA 3 served as a safety extension of ROMANA 1 and 2, with a total of 513 patients enrolled, including 345 patients receiving anamorelin and 168 patients receiving placebo. Throughout the 24-week treatment period, anamorelin was easily tolerated and significantly increased body weight compared with the placebo.500 Concurrently, two randomized phase 2 trials comprising Japanese patients with NSCLC demonstrated that anamorelin treatment resulted in improvements in lean body mass, body weight, appetite, and quality of life without any tolerability concerns.501,502 On the basis of the aforementioned clinical study conducted in Japanese cancer cachexia patients, anamorelin (Adlumiz), the first drug used to treat cancer cachexia, was approved for the treatment of cancer cachexia in Japan on January 22, 2021.503
Head and neck cancer
Head and neck cancer (HNC), which directly involves the aerodigestive tract, leads to swallowing dysfunction due to local tumor infiltration and treatment toxicity, resulting in varying degrees of malnutrition in ~50–70% of HNC patients.504 Among patients with HNC, those with head and neck squamous cell carcinoma (HNSCC) are particularly prone to developing malnutrition. This is because HNSCC not only promotes the development of cachexia due to its direct physiological effects (leading to dysphagia, odynophagia, and restricted dietary intake) but also exacerbates this process through the secretion of inflammatory cytokines and other cachexia-inducing mediators.505
Several pharmacological treatments have been employed in the therapeutic approach for cachexia among patients with HNC. A phase III, placebo-controlled, double-blind randomized study revealed that taking 800 mg of Megestrol acetate per day for 12 weeks helped patients with HNC maintain their weight during curative radiotherapy and enhanced their quality of life.506 An Ethanwell/Ethanzyme regimen enriched with ω-3 fatty acids, micronutrients, and probiotics was found to increase body weight, as well as serum albumin and prealbumin levels, in patients with HNC.507 However, echium oil, a plant source of ω-3 fatty acids, fails to protect against weight loss or improve nutritional parameters.508 Studies conducted both in vitro and in vivo have demonstrated that curcuminoids, extracts derived from curcumin, have an inhibitory effect on NF-κB via intracellular phosphorylation.458 Notably, patients treated with curcumin exhibited an increase in muscle mass, along with a decrease in handgrip muscle strength and absolute lymphocyte count.509 Cachectic patients with HNC receiving celecoxib can gain weight, experience increased BMI, and improve quality of life without adverse events.460,461 By binding to muscle-specific androgen receptors, testosterone represents a well-established approach for addressing skeletal muscle loss.510 Indeed, compared with placebo, adjunct testosterone was found to increase lean body mass, improve quality of life, and enhance physical activity in patients with HNSCC.511 Immunomodulatory agents, such as thalidomide and OHR/AVR118, also have potential in combating cachexia associated with many cancer types, including HNC; however, standalone cohort studies specifically focused on this unique cancer are lacking.463,464
Other types of cancers
Breast cancer is a life-threatening disease for females and the primary cause of death among women. Some studies have shown that breast cancer patients often have increased serum levels of PGE2 and proinflammatory cytokines, including TNF-α, IL-1β, IL-6, and CRP, which exacerbates the progression of cancer cachexia.512 In a randomized, double-blind, controlled study, the plasma fatty acid composition of breast cancer patients who received EPA and DHA significantly altered, and stable CD4+ T-cell counts and serum hsCRP levels were maintained, indicating a potential beneficial impact on the immune system and reduced inflammatory activity.513 Owing to the increase in proinflammatory cytokines among breast cancer patients, anti-inflammatory drugs may be beneficial for these patients.
HT1080 tumor-bearing mice, which serve as a fibrosarcoma mouse model, can secrete high levels of GDF15 into mouse serum, mimicking the condition of cancer cachexia.155,396,514 The relevant signaling complex, which comprises GFRAL and RET in brainstem neurons, was shown to mediate the weight loss induced by GDF15 in mice.515,516 A preclinical study demonstrated that 3P10, a therapeutic antagonistic monoclonal antibody targeting GFRAL, can mitigate high-level lipid oxidation and prevent cancer cachexia in tumor-bearing mice by inhibiting RET signaling by blocking the GDF15-induced interaction between RET and GFRAL on the cell surface, even when the mice are under calorie-restricted conditions.155 Another RET-selective inhibitor, selpercatinib (LOXO-292), notably increased food intake, enhanced skeletal muscle mass and strength, and promoted adipose tissue while simultaneously reducing body weight loss without exerting a significant effect on tumor growth in tumor-bearing animals. Compared with traditional antibodies, nanobodies, considered next-generation antibody-derived tools, were developed and evaluated in different stages of clinical trials for cancer treatment.517 GB18-06, a nanobody specifically directed against GDF15, effectively blocks the GDF15-GFRAL signaling pathway in vitro and reduces weight loss (>20%) in HT1080 tumor-bearing mice.518 Bringing these drugs into clinical research as soon as possible will greatly benefit patients with cachexia.
Although several clinical studies have investigated genitourinary, hematologic, and urogenital cancers, the small number of patients involved necessitates further clinical and preclinical studies to identify effective pharmacological agents for these specific cancer types.
Conclusion and future perspectives
In recent decades, investigative efforts in cancer cachexia pathophysiology have focused on skeletal muscle wasting and adipose tissue depletion. Currently, cancer cachexia is recognized as a systemic disease. The progression of cancer cachexia involves other abnormalities in the gut, brain, bone, and heart, highlighting the significant contributions of metabolism, inflammation, and immunology. In this review, we elucidate the intricate crosstalk between various organs/tissues involved in cachexia, which is characterized by metabolic disorders. However, many interaction axes have not been fully characterized, and their roles remain unclear. Research has indicated that altered gut microbiota composition in obese individuals can lead to increased release of gut-derived inflammatory factors into both portal and lymphatic circulations.519 Importantly, these factors can induce adipose tissue inflammation by directly infiltrating mesenteric WAT. Therefore, the gut-adipose-liver axis is crucial in obesity and potentially in cancer cachexia. Recently, the liver‒brain axis has also received widespread attention. On the one hand, the regulation of eating behavior within the CNS is influenced by stimuli originating from the liver. On the other hand, neural signals emanating from the CNS influence glucose, lipid, and protein metabolism in the liver.520 Additionally, several studies have underscored the importance of the gut‒bone axis521 and the bone‒adipose axis in metabolic diseases,522 hinting their potential relevance in cachexia. Elucidating the molecular underpinnings of organ crosstalk in cachexia could provide new insights into its pathogenesis and facilitate the development of novel therapeutic strategies.
Various cancer-promoting metabolites circulate in the bloodstream and can elicit inflammatory responses by either directly infiltrating or releasing cachexia-related metabolites and cytokines into secondary sites. Inflammatory responses exacerbate metabolic abnormalities and form a vicious cycle. However, few studies have directly investigated the role of metabolites in cellular responses in cancer cachexia progression. This research gap may represent a pivotal breakthrough in the etiology of cancer cachexia. By delving into how metabolites influence and reshape cellular structure and function, we can gain a deeper understanding of the underlying causes of these cellular alterations in cancer cachexia. This exploration not only offers a novel perspective on the pathophysiology of cachexia but also provides crucial clues and evidence for the development of innovative therapies targeting these cellular alterations.
The role of immune and stromal cells in cachexia is particularly complex. Some cells can mediate both protective and pathogenic responses in cancer cachexia. The inconsistencies observed in research findings within this domain can be attributed to several contributing factors. First, the immune system is highly dynamic and responsive to an extensive range of stimuli, thereby posing significant challenges in isolating the specific contributions of cells in cachexia. Second, the complex interplay between diverse immune cell types and their interactions with other biological systems, such as the nervous and endocrine systems, further enhances the complexity of elucidating the role of certain cells in cancer cachexia. Furthermore, cachexia itself is a multifaceted condition with a plethora of underlying causes, which can further complicate the interpretation of research outcomes. These intricate interactions can influence muscle function and mass in manners that are yet to be fully understood.
Intriguingly, cachexia arising from diverse pathological origins may involve distinct underlying mechanisms. Specifically, macrophage-derived TGF-β1 has been implicated in mediating the senescence of adipocyte progenitors in aged mice, yet it appears to be nonfunctional in the context of cancer cachexia.285 Analogously, cytokines that are pertinent to cancer cachexia do not significantly vary with infection-related cachexia.308 Furthermore, the functional role of a particular molecule can differ under different pathological scenarios. In response to cold exposure, GDF15 secreted by BAT targets macrophages to suppress the expression of proinflammatory genes.140 Conversely, in the context of cancer cachexia, GDF15 is frequently associated with proinflammatory activities. These observations imply that, despite the clinical similarities among these metabolic disorders, their underlying mechanisms cannot be generalized. Investigating the underlying causes of these discrepancies may provide deeper insights into the fundamental mechanisms governing these diseases.
Notably, the incidence rate of cancer cachexia significantly varies among different tumor types, likely because of primary tumor characteristics and specific therapeutic interventions. This variability highlights the importance of the precise elucidation of the distinct molecular pathogenesis characterizing each tumor subtype when developing targeted therapeutic strategies. Our analysis also revealed a significant discrepancy between preclinical and clinical outcomes for certain treatments. While these treatments have shown promise in preclinical models, their efficacy has been markedly reduced in clinical settings. This suggests that current preclinical models may lack the complexity required to fully recapitulate human diseases, limiting the translation of preclinical findings to clinical practice. Therefore, there is an urgent need to develop more sophisticated preclinical models that better mimic human physiopathology. This could involve the use of advanced tissue engineering techniques, such as organoids or patient-derived xenografts, to create more physiologically relevant models. Additionally, incorporating immune components and the TME into these models may further increase their value for clinical translation.
Current treatment strategies for cancer cachexia primarily focus on a multifaceted approach that includes nutritional support, anti-inflammatory therapies, immunotherapy, exercise therapy, and psychological interventions. Studies have revealed that metabolic dysregulation in cancer cachexia patients is closely associated with epigenetic mechanisms such as DNA methylation and histone modifications.523 Specific epigenetic changes may lead to an imbalance in muscle synthesis and degradation.524 Recent studies have reported that ncRNAs, such as microRNAs, long noncoding RNAs (lncRNAs), and circular RNAs (circRNAs), play a significant role in cancer cachexia.525,526 For example, Shi et al. reported that circANAPC7, via the CREB-miR-373-PHLPP2 axis, suppressed tumor growth and muscle wasting in pancreatic cancer.526 Therefore, the development of targeted drugs and therapies aimed at epigenetic markers, particularly demethylation agents, holds promise for breakthroughs in the treatment of cancer cachexia.
Overall, the detailed mechanisms of cachexia are complex. Although several causes have been identified and related drugs have been developed, only a very limited number of emerging drugs and other intervention strategies for cancer cachexia exist. Since research aimed at identifying the systematic mechanisms of cancer cachexia is far from saturated, additional studies on this topic are likely to contribute to a more comprehensive understanding of cancer cachexia progression. Overall, a more systematic integration of immunologic and metabolomics approaches, together with the utilization of optimal animal models to mimic the cachectic state, is necessary to uncover novel targets of cancer cachexia.
References
Argilés, J. M., Busquets, S., Stemmler, B. & López-Soriano, F. J. Cancer cachexia: understanding the molecular basis. Nat. Rev. Cancer 14, 754–762 (2014).
Baracos, V. E. et al. Cancer-associated cachexia. Nat. Rev. Dis. Prim. 4, 17105 (2018).
Fearon, K. et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol. 12, 489–495 (2011).
Prado, C. M., Cushen, S. J., Orsso, C. E. & Ryan, A. M. Sarcopenia and cachexia in the era of obesity: clinical and nutritional impact. Proc. Nutr. Soc. 75, 188–198 (2016).
Martin, L. et al. Cancer cachexia in the age of obesity: skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol. 31, 1539–1547 (2013).
Argilés, J. M., López-Soriano, F. J., Stemmler, B. & Busquets, S. Cancer-associated cachexia - understanding the tumor macroenvironment and microenvironment to improve management. Nat. Rev. Clin. Oncol. 20, 250–264 (2023).
Bennani-Baiti, N. & Walsh, D. What is cancer anorexia-cachexia syndrome? A historical perspective. J. R. Coll. Physicians Edinb. 39, 257–262 (2009).
Schmidt, S. F., Rohm, M., Herzig, S. & Berriel Diaz, M. Cancer Cachexia: more than skeletal muscle wasting. Trends Cancer 4, 849–860 (2018).
Elinav, E. et al. Inflammation-induced cancer: crosstalk between tumors, immune cells and microorganisms. Nat. Rev. Cancer 13, 759–771 (2013).
Ganguly, K. & Kimmelman, A. C. Reprogramming of tissue metabolism during cancer metastasis. Trends Cancer 9, 461–471 (2023).
Fearon, K. C., Glass, D. J. & Guttridge, D. C. Cancer cachexia: mediators, signaling, and metabolic pathways. Cell Metab. 16, 153–166 (2012).
Strassmann, G., Fong, M., Kenney, J. S. & Jacob, C. O. Evidence for the involvement of interleukin 6 in experimental cancer cachexia. J. Clin. Investig. 89, 1681–1684 (1992).
Oliff, A. et al. Tumors secreting human TNF/cachectin induce cachexia in mice. Cell 50, 555–563 (1987).
de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: from cancer initiation to metastatic outgrowth. Cancer Cell 41, 374–403 (2023).
Mottini, C. et al. The cross-talk between the macro and micro-environment in precursor lesions of pancreatic cancer leads to new and promising circulating biomarkers. J. Exp. Clin. Cancer Res. 43, 198 (2024).
Evans, W. J. et al. Cachexia: a new definition. Clin. Nutr. 27, 793–799 (2008).
Arai, H. et al. Diagnosis and outcomes of cachexia in Asia: working Consensus Report from the Asian Working Group for Cachexia. J. Cachexia Sarcopenia Muscle 14, 1949–1958 (2023).
Xie, H. et al. AWGC2023 cachexia consensus as a valuable tool for predicting prognosis and burden in Chinese patients with cancer. J. Cachexia Sarcopenia Muscle 15, 2084–2093 (2024).
Zhang, F. M. et al. Characteristics and prognostic impact of cancer cachexia defined by the Asian Working Group for Cachexia consensus in patients with curable gastric cancer. Clin. Nutr. 43, 1524–1531 (2024).
Vigano, A., Del Fabbro, E., Bruera, E. & Borod, M. The cachexia clinic: from staging to managing nutritional and functional problems in advanced cancer patients. Crit. Rev. Oncog. 17, 293–303 (2012).
Douglas, E. & McMillan, D. C. Towards a simple objective framework for the investigation and treatment of cancer cachexia: the Glasgow Prognostic Score. Cancer Treat. Rev. 40, 685–691 (2014).
Silva, G. A. D., Wiegert, E. V. M., Calixto-Lima, L. & Oliveira, L. C. Clinical utility of the modified Glasgow Prognostic Score to classify cachexia in patients with advanced cancer in palliative care. Clin. Nutr. 39, 1587–1592 (2020).
Jin, Z. C. et al. Longitudinal body composition identifies hepatocellular carcinoma with cachexia following combined immunotherapy and target therapy (CHANCE2213). J. Cachexia Sarcopenia Muscle 15, 2705–2716 (2024).
Anker, M. S. et al. Orphan disease status of cancer cachexia in the USA and in the European Union: a systematic review. J. Cachexia Sarcopenia Muscle 10, 22–34 (2019).
Li, X. et al. Cancer cachexia statistics in China. Precision Nutrition. 1, https://doi.org/10.1097/PN1099.0000000000000008 (2022).
Latenstein, A. E. J. et al. Cachexia, dietetic consultation, and survival in patients with pancreatic and periampullary cancer: a multicenter cohort study. Cancer Med. 9, 9385–9395 (2020).
Takeda, T. et al. The impact of cachexia and sarcopenia in elderly pancreatic cancer patients receiving palliative chemotherapy. Int. J. Clin. Oncol. 26, 1293–1303 (2021).
Hou, Y. C. et al. The differential clinical impacts of cachexia and sarcopenia on the prognosis of advanced pancreatic cancer. Cancers. 14, 3137 (2022).
Furuse, J. et al. Effect of cancer cachexia on first-line chemotherapy in patients with advanced pancreatic cancer: a claims database study in Japan. Int. J. Clin. Oncol. 29, 456–463 (2024).
Shukuya, T. et al. Epidemiology, risk factors and impact of cachexia on patient outcome: results from the Japanese Lung Cancer Registry Study. J. Cachexia Sarcopenia Muscle 14, 1274–1285 (2023).
Roch, B. et al. Cachexia - sarcopenia as a determinant of disease control rate and survival in non-small lung cancer patients receiving immune-checkpoint inhibitors. Lung Cancer 143, 19–26 (2020).
Liu, C. A. et al. Nutrition impact symptoms: noteworthy prognostic indicators for lung cancer. Clin. Nutr. 42, 550–558 (2023).
Miura, K. et al. Impact of cachexia and first-line systemic therapy for previously untreated advanced non-small cell lung cancer: NEJ050A. J. Cachexia Sarcopenia Muscle 15, 2618–2628 (2024).
Zhang, J. et al. Cancer cachexia as a predictor of adverse outcomes in patients with non-small cell lung cancer: a meta-analysis. Clin. Nutr. 43, 1618–1625 (2024).
Kawachi, H. et al. Clinical impact of cancer cachexia on the outcome of patients with non-small cell lung cancer with PD-L1 tumor proportion scores of ≥50% receiving pembrolizumab monotherapy versus immune checkpoint inhibitor with chemotherapy. Oncoimmunology 14, 2442116 (2025).
White, R. et al. Determining the prevalence and severity of cancer cachexia in advanced non-small cell lung cancer and its relationship with chemotherapy outcomes. Support. Care Cancer 28, 4373–4380 (2020).
Li, H. et al. Effect of longitudinal changes of cachexia on the efficacy and toxicity of immune checkpoint inhibitors in esophageal squamous cell cancer (ESCC) patients. Nutrition 124, 112462 (2024).
Dijksterhuis, W. P. M. et al. Cachexia and dietetic interventions in patients with esophagogastric cancer: a multicenter cohort study. J. Natl Compr. Canc. Netw. 19, 144–152 (2021).
Brown, L. R. et al. Comparison between single- and multi-slice computed tomography body composition analysis in patients with oesophagogastric cancer. J. Cachexia Sarcopenia Muscle 16, e13673 (2025).
Brown, L. R. et al. The prognostic impact of pre-treatment cachexia in resectional surgery for oesophagogastric cancer: a meta-analysis and meta-regression. Br. J. Surg. 110, 1703–1711 (2023).
Olaechea, S. et al. Race, ethnicity, and socioeconomic factors as determinants of cachexia incidence and outcomes in a retrospective cohort of patients with gastrointestinal tract cancer. JCO Oncol. Pract. 19, 493–500 (2023).
Ruan, G. T. et al. Prognostic value of systemic inflammation and for patients with colorectal cancer cachexia. J. Cachexia Sarcopenia Muscle 14, 2813–2823 (2023).
Molfino, A. et al. Histomorphological and inflammatory changes of white adipose tissue in gastrointestinal cancer patients with and without cachexia. J. Cachexia Sarcopenia Muscle 13, 333–342 (2022).
Davis, E. W. et al. The association of body composition phenotypes before chemotherapy with epithelial ovarian cancer mortality. J. Natl. Cancer Inst. 116, 1513–1524 (2024).
Rich, N. E. et al. Cachexia is prevalent in patients with hepatocellular carcinoma and associated with worse prognosis. Clin. Gastroenterol. Hepatol. 20, e1157–e1169 (2022).
Kordes, M., Larsson, L., Engstrand, L. & Löhr, J. M. Pancreatic cancer cachexia: three dimensions of a complex syndrome. Br. J. Cancer 124, 1623–1636 (2021).
Agate, L. et al. Nutrition in advanced thyroid cancer patients. Nutrients. 14, 1298 (2022).
Mäkitie, A. A. et al. Managing cachexia in head and neck cancer: a systematic scoping review. Adv. Ther. 39, 1502–1523 (2022).
Zhu, R. et al. Updates on the pathogenesis of advanced lung cancer-induced cachexia. Thorac. Cancer 10, 8–16 (2019).
Demark-Wahnefried, W., Campbell, K. L. & Hayes, S. C. Weight management and its role in breast cancer rehabilitation. Cancer 118, 2277–2287 (2012).
Zhang, Q. et al. Association of systemic inflammation with survival in patients with cancer cachexia: results from a multicenter cohort study. J. Cachexia Sarcopenia Muscle 12, 1466–1476 (2021).
Mallard, J. et al. A single chemotherapy administration induces muscle atrophy, mitochondrial alterations and apoptosis in breast cancer patients. J. Cachexia Sarcopenia Muscle 15, 292–305 (2024).
Shibata, M. et al. A retrospective cohort study to investigate the incidence of cachexia during chemotherapy in patients with colorectal cancer. Adv. Ther. 37, 5010–5022 (2020).
Ben-Noun, L. L. The disease that caused weight loss in King David the Great. J. Gerontol. A Biol. Sci. Med. Sci. 59, 143–145 (2004).
Thibaut, M. M. et al. The microbiota-derived bile acid taurodeoxycholic acid improves hepatic cholesterol levels in mice with cancer cachexia. Gut Microbes 17, 2449586 (2025).
Petruzzelli, M. et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 20, 433–447 (2014).
Sah, R. P. et al. Phases of metabolic and soft tissue changes in months preceding a diagnosis of pancreatic Ductal Adenocarcinoma. Gastroenterology 156, 1742–1752 (2019).
Klatte, D. C. F. et al. Temporal trends in body composition and metabolic markers prior to diagnosis of pancreatic Ductal Adenocarcinoma. Clin. Gastroenterol. Hepatol. 22, 1830–1838.e1839 (2024).
Babic, A. et al. Adipose tissue and skeletal muscle wasting precede clinical diagnosis of pancreatic cancer. Nat. Commun. 14, 4317 (2023).
Garattini, S. et al. Anorexia and cancer in animals and man. Cancer Treat. Rev. 7, 115–139 (1980).
Qi, Y. et al. Agrp-negative arcuate NPY neurons drive feeding under positive energy balance via altering leptin responsiveness in POMC neurons. Cell Metab. 35, 979–995.e977 (2023).
Dallmann, R. et al. The orally active melanocortin-4 receptor antagonist BL-6020/979: a promising candidate for the treatment of cancer cachexia. J. Cachexia Sarcopenia Muscle 2, 163–174 (2011).
Zhu, X. et al. Melanocortin-4 receptor antagonist TCMCB07 ameliorates cancer- and chronic kidney disease-associated cachexia. J. Clin. Investig. 130, 4921–4934 (2020).
Sweeney, P., Gimenez, L. E., Hernandez, C. C. & Cone, R. D. Targeting the central melanocortin system for the treatment of metabolic disorders. Nat. Rev. Endocrinol. 19, 507–519 (2023).
Marks, D. L., Ling, N. & Cone, R. D. Role of the central melanocortin system in cachexia. Cancer Res. 61, 1432–1438 (2001).
Mosialou, I. et al. MC4R-dependent suppression of appetite by bone-derived lipocalin 2. Nature 543, 385–390 (2017).
Olson, B. et al. Lipocalin 2 mediates appetite suppression during pancreatic cancer cachexia. Nat. Commun. 12, 2057 (2021).
Steward, T. Endocrinology-informed neuroimaging in eating disorders: GLP1, orexins, and psilocybin. Trends Mol. Med. 30, 321–323 (2024).
Borner, T., Liberini, C. G., Lutz, T. A. & Riediger, T. Brainstem GLP-1 signaling contributes to cancer anorexia-cachexia syndrome in the rat. Neuropharmacology 131, 282–290 (2018).
Navarro, I. B. K., Schraner, M. & Riediger, T. Brainstem prolactin-releasing peptide contributes to cancer anorexia-cachexia syndrome in rats. Neuropharmacology 180, 108289 (2020).
Breit, S. N., Brown, D. A. & Tsai, V. W. The GDF15-GFRAL pathway in health and metabolic disease: friend or foe?. Annu. Rev. Physiol. 83, 127–151 (2021).
Braun, T. P. & Marks, D. L. Pathophysiology and treatment of inflammatory anorexia in chronic disease. J. Cachexia Sarcopenia Muscle 1, 135–145 (2010).
Burfeind, K. G. et al. TRIF is a key inflammatory mediator of acute sickness behavior and cancer cachexia. Brain. Behav. Immun. 73, 364–374 (2018).
Yeom, E. et al. Tumor-derived Dilp8/INSL3 induces cancer anorexia by regulating feeding neuropeptides via Lgr3/8 in the brain. Nat. Cell Biol. 23, 172–183 (2021).
Burfeind, K. G. et al. Circulating myeloid cells invade the central nervous system to mediate cachexia during pancreatic cancer. Elife 9, e54095 (2020).
Bindels, L. B. et al. Synbiotic approach restores intestinal homeostasis and prolongs survival in leukemic mice with cachexia. ISME J. 10, 1456–1470 (2016).
Chrysostomou, D., Roberts, L. A., Marchesi, J. R. & Kinross, J. M. Gut microbiota modulation of efficacy and toxicity of cancer chemotherapy and immunotherapy. Gastroenterology 164, 198–213 (2023).
Puppa, M. J. et al. Gut barrier dysfunction in the Apc(Min/+) mouse model of colon cancer cachexia. Biochim. Biophys. Acta 1812, 1601–1606 (2011).
Jia, H. et al. Eggshell membrane modulates gut microbiota to prevent murine pre-cachexia through suppression of T helper cell differentiation. J. Cachexia Sarcopenia Muscle 13, 2088–2101 (2022).
Ubachs, J. et al. Gut microbiota and short-chain fatty acid alterations in cachectic cancer patients. J. Cachexia Sarcopenia Muscle 12, 2007–2021 (2021).
Dumas, J. F. et al. Efficiency of oxidative phosphorylation in liver mitochondria is decreased in a rat model of peritoneal carcinosis. J. Hepatol. 54, 320–327 (2011).
Pötgens, S. A. et al. Multi-compartment metabolomics and metagenomics reveal major hepatic and intestinal disturbances in cancer cachectic mice. J. Cachexia Sarcopenia Muscle 12, 456–475 (2021).
Jones, A. et al. TSC22D4 is a molecular output of hepatic wasting metabolism. EMBO Mol. Med. 5, 294–308 (2013).
Kojima, Y. et al. Decreased liver B vitamin-related enzymes as a metabolic hallmark of cancer cachexia. Nat. Commun. 14, 6246 (2023).
Zechner, R. et al. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 15, 279–291 (2012).
Das, S. K. et al. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science 333, 233–238 (2011).
Cannon, B. & Nedergaard, J. Brown adipose tissue: function and physiological significance. Physiol. Rev. 84, 277–359 (2004).
Tamucci, K. A., Namwanje, M., Fan, L. & Qiang, L. The dark side of browning. Protein Cell 9, 152–163 (2018).
Yin, X. et al. The evolving view of thermogenic fat and its implications in cancer and metabolic diseases. Signal Transduct. Target Ther. 7, 324 (2022).
Chen, X. et al. GRP75 triggers white adipose tissue browning to promote cancer-associated cachexia. Signal Transduct. Target Ther. 9, 253 (2024).
Rohm, M. et al. An AMP-activated protein kinase-stabilizing peptide ameliorates adipose tissue wasting in cancer cachexia in mice. Nat. Med. 22, 1120–1130 (2016).
Kir, S. et al. Tumor-derived PTH-related protein triggers adipose tissue browning and cancer cachexia. Nature 513, 100–104 (2014).
Kir, S. et al. PTH/PTHrP receptor mediates cachexia in models of kidney failure and cancer. Cell Metab. 23, 315–323 (2016).
Kahn, C. R., Wang, G. & Lee, K. Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 129, 3990–4000 (2019).
Llovera, M. et al. Protein turnover in skeletal muscle of tumor-bearing transgenic mice overexpressing the soluble TNF receptor-1. Cancer Lett. 130, 19–27 (1998).
Martin, A., Gallot, Y. S. & Freyssenet, D. Molecular mechanisms of cancer cachexia-related loss of skeletal muscle mass: data analysis from preclinical and clinical studies. J. Cachexia Sarcopenia Muscle 14, 1150–1167 (2023).
Eldridge, A. G. & O’Brien, T. Therapeutic strategies within the ubiquitin proteasome system. Cell Death Differ. 17, 4–13 (2010).
Bodine, S. C. & Baehr, L. M. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1. Am. J. Physiol. Endocrinol. Metab. 307, E469–E484 (2014).
de Castro, G. S. et al. Human cachexia induces changes in mitochondria, autophagy and apoptosis in the skeletal muscle. Cancers 11, 1264 (2019).
Zhang, R. et al. Corylifol A ameliorates muscle atrophy by inhibiting TAOK1/p38-MAPK/FoxO3 pathway in cancer cachexia. J. Cachexia Sarcopenia Muscle 14, 2098–2113 (2023).
Costelli, P., De Tullio, R., Baccino, F. M. & Melloni, E. Activation of Ca(2+)-dependent proteolysis in skeletal muscle and heart in cancer cachexia. Br. J. Cancer 84, 946–950 (2001).
Zhou, Z. et al. Acetyl-Coenzyme A Synthetase 2 Potentiates Macropinocytosis and muscle wasting through metabolic reprogramming in pancreatic cancer. Gastroenterology 163, 1281–1293.e1281 (2022).
Marino, S. et al. Paradoxical effects of JZL184, an inhibitor of monoacylglycerol lipase, on bone remodeling in healthy and cancer-bearing mice. EBioMedicine 44, 452–466 (2019).
Hain, B. A. et al. Zoledronic acid improves muscle function in healthy mice treated with chemotherapy. J. Bone Min. Res. 35, 368–381 (2020).
Essex, A. L. et al. Bisphosphonate treatment ameliorates chemotherapy-induced bone and muscle abnormalities in young mice. Front. Endocrinol.10, 809 (2019).
Bonetto, A. et al. Differential bone loss in mouse models of colon cancer cachexia. Front. Physiol. 7, 679 (2016).
Pin, F. et al. Non-bone metastatic cancers promote osteocyte-induced bone destruction. Cancer Lett. 520, 80–90 (2021).
Pin, F. et al. RANKL blockade reduces cachexia and bone loss induced by non-metastatic ovarian cancer in mice. J. Bone Min. Res. 37, 381–396 (2022).
Pittenger, M. F. et al. Multilineage potential of adult human mesenchymal stem cells. Science 284, 143–147 (1999).
Yu, J. et al. Bone marrow homeostasis is impaired via JAK/STAT and glucocorticoid signaling in cancer cachexia Model. Cancers 13, 1059 (2021).
Wang, B. et al. Inhibition of TGFβ improves hematopoietic stem cell niche and ameliorates cancer-related anemia. Stem Cell. Res. Ther. 12, 65 (2021).
Severinsen, M. C. K. & Pedersen, B. K. Muscle-organ crosstalk: the emerging roles of myokines. Endocr. Rev. 41, 594–609 (2020).
Morais, L. H., Schreiber, H. L. 4th & Mazmanian, S. K. The gut microbiota-brain axis in behavior and brain disorders. Nat. Rev. Microbiol. 19, 241–255 (2021).
Li, X. et al. Lipopolysaccharide-induced hypothalamic inflammation in cancer cachexia-anorexia is amplified by tumor-derived prostaglandin E2. J. Cachexia Sarcopenia Muscle 13, 3014–3027 (2022).
Suda, Y. et al. Peripheral-central network analysis of cancer cachexia status accompanied by the polarization of hypothalamic microglia with low expression of inhibitory immune checkpoint receptors. Mol. Brain. 17, 20 (2024).
Wen, J., Ding, Y., Wang, L. & Xiao, Y. Gut microbiome improves postoperative cognitive function by decreasing permeability of the blood-brain barrier in aged mice. Brain Res. Bull. 164, 249–256 (2020).
Khatib, M. N. et al. Ghrelin for the management of cachexia associated with cancer. Cochrane Database Syst. Rev. 2, Cd012229 (2018).
Wang, C. et al. The relationship of appetite-regulating hormones in the development of cardiac cachexia. Int. Heart J. 60, 384–391 (2019).
Choi, Y. K. et al. Effect of Sipjeondaebo-tang on cancer-induced anorexia and cachexia in CT-26 tumor-bearing mice. Mediators Inflamm. 2014, 736563 (2014).
Li, Y. et al. Systematic characterization of the disruption of intestine during liver tumor progression in the xmrk oncogene transgenic zebrafish model. Cells 11, 1810 (2022).
das Neves, R. X. et al. Cachexia causes time-dependent activation of the inflammasome in the liver. J. Cachexia Sarcopenia Muscle 14, 1621–1630 (2023).
Modica, S. et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 142, 355–365 (2012). e351-354.
Feng, L. et al. Bile acid metabolism dysregulation associates with cancer cachexia: roles of liver and gut microbiome. J. Cachexia Sarcopenia Muscle 12, 1553–1569 (2021).
Thibaut, M. M. et al. Bile acid dysregulation is intrinsically related to cachexia in tumor-bearing mice. Cancers 13, 6389 (2021).
Dong, F. et al. Intestinal microbiota-derived tryptophan metabolites are predictive of Ah receptor activity. Gut Microbes 12, 1–24 (2020).
Dolly, A. et al. Impairment of aryl hydrocarbon receptor signaling promotes hepatic disorders in cancer cachexia. J. Cachexia Sarcopenia Muscle 14, 1569–1582 (2023).
Wu, S. Y. et al. Polysaccharide of ganoderma lucidum ameliorates cachectic myopathy induced by the combination cisplatin plus docetaxel in mice. Microbiol Spectr. 11, e0313022 (2023).
Jumpertz, R. et al. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 94, 58–65 (2011).
Sakakida, T. et al. Water-soluble dietary fiber alleviates cancer-induced muscle wasting through changes in gut microenvironment in mice. Cancer Sci. 113, 1789–1800 (2022).
Qiu, Y. et al. Depletion of gut microbiota induces skeletal muscle atrophy by FXR-FGF15/19 signaling. Ann. Med. 53, 508–522 (2021).
Zhong, W. et al. Gut dysbiosis promotes prostate cancer progression and docetaxel resistance via activating NF-κB-IL6-STAT3 axis. Microbiome 10, 94 (2022).
Pekkala, S. et al. Blocking activin receptor ligands is not sufficient to rescue cancer-associated gut Microbiota-A role for gut microbial flagellin in colorectal cancer and cachexia?. Cancers 11, 1799 (2019).
Costa, R. G. F. et al. Cancer cachexia induces morphological and inflammatory changes in the intestinal mucosa. J. Cachexia Sarcopenia Muscle 10, 1116–1127 (2019).
Quinn, L. S. et al. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am. J. Physiol. Endocrinol. Metab. 296, E191–E202 (2009).
Molanouri Shamsi, M. et al. Combined effect of aerobic interval training and selenium nanoparticles on expression of IL-15 and IL-10/TNF-α ratio in skeletal muscle of 4T1 breast cancer mice with cachexia. Cytokine 90, 100–108 (2017).
Loumaye, A. et al. Role of Activin A and myostatin in human cancer cachexia. J. Clin. Endocrinol. Metab. 100, 2030–2038 (2015).
Franz, K. et al. Higher serum levels of fibroblast growth factor 21 in old patients with cachexia. Nutrition 63-64, 81–86 (2019).
Fu, T. et al. Mitophagy directs muscle-adipose crosstalk to alleviate dietary obesity. Cell Rep. 23, 1357–1372 (2018).
Lemecha, M. et al. Lcn2 mediates adipocyte-muscle-tumor communication and hypothermia in pancreatic cancer cachexia. Mol. Metab. 66, 101612 (2022).
Campderrós, L. et al. Brown adipocytes secrete GDF15 in response to thermogenic activation. Obesity 27, 1606–1616 (2019).
Suzuki, H. et al. Clinical and tumor characteristics of patients with high serum levels of growth differentiation factor 15 in advanced pancreatic cancer. Cancers. 13, 4842 (2021).
Rupert, J. E. et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 218, e20190450 (2021).
Han, M. S. et al. Regulation of adipose tissue inflammation by interleukin 6. Proc. Natl. Acad. Sci. USA 117, 2751–2760 (2020).
Waning, D. L. et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 21, 1262–1271 (2015).
Shen, H., Grimston, S., Civitelli, R. & Thomopoulos, S. Deletion of connexin43 in osteoblasts/osteocytes leads to impaired muscle formation in mice. J. Bone Min. Res. 30, 596–605 (2015).
Bonnet, N. et al. RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J. Clin. Investig. 129, 3214–3223 (2019).
Dankbar, B. et al. Myostatin is a direct regulator of osteoclast differentiation and its inhibition reduces inflammatory joint destruction in mice. Nat. Med. 21, 1085–1090 (2015).
Kitase, Y. et al. β-aminoisobutyric Acid, l-BAIBA, is a muscle-derived osteocyte survival factor. Cell Rep. 22, 1531–1544 (2018).
Engeli, S. Central and peripheral cannabinoid receptors as therapeutic targets in the control of food intake and body weight. Handb. Exp. Pharmacol. 209, 357–381 (2012).
Molfino, A., Gioia, G., Rossi Fanelli, F. & Laviano, A. Contribution of neuroinflammation to the pathogenesis of cancer cachexia. Mediators Inflamm. 2015, 801685 (2015).
Bernstein, I. L. Neutral mediation of food aversions and anorexia induced by tumor necrosis factor and tumors. Neurosci. Biobehav. Rev. 20, 177–181 (1996).
Borner, T. et al. Anorexia-cachexia syndrome in hepatoma tumor-bearing rats requires the area postrema but not vagal afferents and is paralleled by increased MIC-1/GDF15. J. Cachexia Sarcopenia Muscle 8, 417–427 (2017).
Piffar, P. M. et al. Naproxen, clenbuterol and insulin administration ameliorates cancer cachexia and reduce tumor growth in Walker 256 tumor-bearing rats. Cancer Lett. 201, 139–148 (2003).
Busquets, S. et al. Anticachectic effects of formoterol: a drug for potential treatment of muscle wasting. Cancer Res. 64, 6725–6731 (2004).
Suriben, R. et al. Antibody-mediated inhibition of GDF15-GFRAL activity reverses cancer cachexia in mice. Nat. Med. 26, 1264–1270 (2020).
Xie, H. et al. An immune-sympathetic neuron communication axis guides adipose tissue browning in cancer-associated cachexia. Proc. Natl. Acad. Sci. USA 119, e2112840119 (2022).
Nukaga, S. et al. Combined administration of lauric acid and glucose improved cancer-derived cardiac atrophy in a mouse cachexia model. Cancer Sci. 111, 4605–4615 (2020).
Wei, L. et al. 2-Deoxy-D-glucose alleviates cancer cachexia-induced muscle wasting by enhancing ketone metabolism and inhibiting the cori cycle. Cells 11, 2987 (2022).
DeWys, W. D. Pathophysiology of cancer cachexia: current understanding and areas for future research. Cancer Res. 42, 721s–726s (1982).
Baker, S. A. & Rutter, J. Metabolites as signaling molecules. Nat. Rev. Mol. Cell Biol. 24, 355–374 (2023).
Lin, J. G. et al. Metabolic modulation of transcription: the role of one-carbon metabolism. Cell Chem. Biol. 29, (2022).
Nalbantoglu, S. & Karadag, A. Metabolomics bridging proteomics along metabolites/oncometabolites and protein modifications: Paving the way toward integrative multiomics. J. Pharm. Biomed. Anal. 199, 114031 (2021).
Rabinowitz, J. D. & Enerbäck, S. Lactate: the ugly duckling of energy metabolism. Nat. Metab. 2, 566–571 (2020).
Warburg, O. On the origin of cancer cells. Science 123, 309–314 (1956).
McGovern, J. et al. Lactate dehydrogenase: relationship with the diagnostic GLIM criterion for cachexia in patients with advanced cancer. Br. J. Cancer 128, 760–765 (2023).
Pin, F. et al. Cachexia induced by cancer and chemotherapy yield distinct perturbations to energy metabolism. J. Cachexia Sarcopenia Muscle 10, 140–154 (2019).
Pin, F. et al. PDK4 drives metabolic alterations and muscle atrophy in cancer cachexia. FASEB J. 33, 7778–7790 (2019).
Mannelli, M. et al. STAT3 signaling drives LDH Up-regulation and adiponectin down-regulation in cachectic adipocytes. Int. J. Mol. Sci. 24, 16343 (2023).
Felmlee, M. A. et al. Monocarboxylate Transporters (SLC16): function, regulation, and role in health and disease. Pharmacol. Rev. 72, 466–485 (2020).
Magistretti, P. J. & Allaman, I. Lactate in the brain: from metabolic end-product to signaling molecule. Nat. Rev. Neurosci. 19, 235–249 (2018).
Silberbauer, C. J., Surina-Baumgartner, D. M., Arnold, M. & Langhans, W. Prandial lactate infusion inhibits spontaneous feeding in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 278, R646–R653 (2000).
Liu, X. et al. Activation of GPR81 by lactate drives tumor-induced cachexia. Nat. Metab. 6, 708–723 (2024).
Nikooie, R., Moflehi, D. & Zand, S. Lactate regulates autophagy through ROS-mediated activation of ERK1/2/m-TOR/p-70S6K pathway in skeletal muscle. J. Cell Commun. Signal 15, 107–123 (2021).
Feng, Q. et al. Lactate increases stemness of CD8 + T cells to augment anti-tumor immunity. Nat. Commun. 13, 4981 (2022).
Noe, J. T. et al. Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci. Adv. 7, eabi8602 (2021).
Zhang, D. et al. Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580 (2019).
Brand, A. et al. LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK Cells. Cell Metab. 24, 657–671 (2016).
Linares, J. F. et al. The lactate-NAD(+) axis activates cancer-associated fibroblasts by downregulating p62. Cell Rep. 39, 110792 (2022).
Rastogi, S. et al. Lactate acidosis and simultaneous recruitment of TGF-β leads to alter plasticity of hypoxic cancer cells in tumor microenvironment. Pharm. Ther. 250, 108519 (2023).
Mannelli, M. et al. Pyruvate prevents the onset of the cachectic features and metabolic alterations in myotubes downregulating STAT3 signaling. FASEB J. 36, e22598 (2022).
Jiang, X. et al. Pyruvate dehydrogenase B regulates myogenic differentiation via the FoxP1-Arih2 axis. J. Cachexia Sarcopenia Muscle 14, 606–621 (2023).
Kim, M. J. et al. The role of pyruvate metabolism in mitochondrial quality control and inflammation. Mol. Cells 46, 259–267 (2023).
Elia, I. et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab. 34, 1137–1150.e1136 (2022).
Shu, Y. et al. Intervening pyruvate carboxylase stunts tumor growth by strengthening anti-tumor actions of tumor-associated macrophages. Signal Transduct. Target Ther. 7, 34 (2022).
Wenes, M. et al. The mitochondrial pyruvate carrier regulates memory T cell differentiation and antitumor function. Cell Metab. 34, 731–746.e739 (2022).
Cui, P. et al. Metabolic profiling of tumors, sera, and skeletal muscles from an orthotopic murine model of gastric cancer associated-cachexia. J. Proteome Res. 18, 1880–1892 (2019).
Ruiz, B. I. et al. Alpha-Ketoglutarate regulates Tnfrsf12a/Fn14 expression via histone modification and prevents cancer-induced cachexia. Genes. 14, 1818 (2023).
Tan, H. et al. Ketoglutaric acid can reprogram the immunophenotype of triple-negative breast cancer after radiotherapy and improve the therapeutic effect of anti-PD-L1. J. Transl. Med. 21, 462 (2023).
Cai, Z. et al. Targeting PHGDH reverses the immunosuppressive phenotype of tumor-associated macrophages through α-ketoglutarate and mTORC1 signaling. Cell. Mol. Immunol. 21, 448–465 (2024).
Matias, M. I. et al. Regulatory T cell differentiation is controlled by αKG-induced alterations in mitochondrial metabolism and lipid homeostasis. Cell Rep. 37, 109911 (2021).
Flint, T. R. et al. Tumor-Induced IL-6 reprograms host metabolism to suppress anti-tumor immunity. Cell Metab. 24, 672–684 (2016).
Goncalves, M. D. et al. Fenofibrate prevents skeletal muscle loss in mice with lung cancer. Proc. Natl. Acad. Sci. USA 115, E743–e752 (2018).
von Renesse, J. et al. Tumor catabolism independent of malnutrition and inflammation in upper GI cancer patients revealed by longitudinal metabolomics. J. Cachexia Sarcopenia Muscle 14, 298–309 (2023).
Mulligan, H. D. & Tisdale, M. J. Metabolic substrate utilization by tumor and host tissues in cancer cachexia. Biochem. J. 277, 321–326 (1991).
Zhou, Y. et al. Exploring the therapeutic potential of Ethyl 3-hydroxybutyrate in alleviating skeletal muscle wasting in cancer cachexia. Biomolecules. 13, 1330 (2023).
Arneson-Wissink, P. C. et al. Hepatic signal transducer and activator of transcription-3 signaling drives early-stage pancreatic cancer cachexia via suppressed ketogenesis. J. Cachexia Sarcopenia Muscle 15, 975–988 (2024).
Ferrer, M. et al. Ketogenic diet promotes tumor ferroptosis but induces relative corticosterone deficiency that accelerates cachexia. Cell Metab. 35, 1147–1162.e1147 (2023).
Wei, R. et al. Ketogenesis attenuates KLF5-dependent production of CXCL12 to overcome the immunosuppressive tumor microenvironment in colorectal cancer. Cancer Res. 82, 1575–1588 (2022).
Luda, K. M. et al. Ketolysis drives CD8(+) T cell effector function through effects on histone acetylation. Immunity 56, 2021–2035.e2028 (2023).
Hosios, A. M. et al. Amino acids rather than glucose account for the majority of cell mass in proliferating mammalian cells. Dev. Cell 36, 540–549 (2016).
Tabe, Y., Lorenzi, P. L. & Konopleva, M. Amino acid metabolism in hematologic malignancies and the era of targeted therapy. Blood 134, 1014–1023 (2019).
Yang, L. et al. Amino acid metabolism in immune cells: essential regulators of the effector functions, and promising opportunities to enhance cancer immunotherapy. J. Hematol. Oncol. 16, 59 (2023).
Cala, M. P. et al. Multiplatform plasma fingerprinting in cancer cachexia: a pilot observational and translational study. J. Cachexia Sarcopenia Muscle 9, 348–357 (2018).
Bozzetti, F. & Bozzetti, V. Is the intravenous supplementation of amino acid to cancer patients adequate? A critical appraisal of literature. Clin. Nutr. 32, 142–146 (2013).
Zea, A. H. et al. L-Arginine modulates CD3zeta expression and T-cell function in activated human T lymphocytes. Cell. Immunol. 232, 21–31 (2004).
Lian, G. et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T-cell differentiation. Elife. 7, e36158 (2018).
Jha, A. K. et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42, 419–430 (2015).
Oh, M. H. et al. Targeting glutamine metabolism enhances tumor-specific immunity by modulating suppressive myeloid cells. J. Clin. Investig. 130, 3865–3884 (2020).
Sinclair, L. V. et al. Antigen receptor control of methionine metabolism in T cells. Elife 8, e44210 (2019).
Jiménez-Alonso, J. J. & López-Lázaro, M. Dietary manipulation of amino acids for cancer therapy. Nutrients 15, 2879 (2023).
Engelen, M. et al. High anabolic potential of essential amino acid mixtures in advanced non-small cell lung cancer. Ann. Oncol. 26, 1960–1966 (2015).
Aliwa, B. et al. Altered gut microbiome, bile acid composition and metabolome in sarcopenia in liver cirrhosis. J. Cachexia Sarcopenia Muscle 14, 2676–2691 (2023).
Chen, L. et al. Bckdk-mediated branch chain amino acid metabolism reprogramming contributes to muscle atrophy during cancer cachexia. Mol. Nutr. Food Res. 68, e2300577 (2024).
Mora, S. & Adegoke, O. A. J. Maintenance of the branched-chain amino acid transporter LAT1 counteracts myotube atrophy following chemotherapy. Am. J. Physiol. Cell Physiol. 326, C866–c879 (2024).
Baek, M. O. et al. Self-transducible LRS-UNE-L peptide enhances muscle regeneration. J. Cachexia Sarcopenia Muscle 13, 1277–1288 (2022).
Viana, L. R. et al. Leucine-rich diet improved muscle function in cachectic walker 256 tumor-bearing wistar rats. Cells. 10, 3272 (2021).
Toneto, A. T. et al. Nutritional leucine supplementation attenuates cardiac failure in tumor-bearing cachectic animals. J. Cachexia Sarcopenia Muscle 7, 577–586 (2016).
Miyaguti, N., Oliveira, S. C. P. & Gomes-Marcondes, M. C. C. Maternal leucine-rich diet minimizes muscle mass loss in tumor-bearing adult rat offspring by improving the balance of muscle protein synthesis and degradation. Biomolecules. 9, 229 (2019).
Salomão, E. M. & Gomes-Marcondes, M. C. Light aerobic physical exercise in combination with leucine and/or glutamine-rich diet can improve the body composition and muscle protein metabolism in young tumor-bearing rats. J. Physiol. Biochem. 68, 493–501 (2012).
Caperuto, E. C. et al. Beta-hydoxy-beta-methylbutyrate supplementation affects Walker 256 tumor-bearing rats in a time-dependent manner. Clin. Nutr. 26, 117–122 (2007).
Liu, K. A., Lashinger, L. M., Rasmussen, A. J. & Hursting, S. D. Leucine supplementation differentially enhances pancreatic cancer growth in lean and overweight mice. Cancer Metab. 2, 6 (2014).
Schrems, E. R. et al. Leucine supplementation exacerbates morbidity in male but not female mice with colorectal cancer-induced cachexia. Nutrients 15, 4570 (2023).
Lautaoja, J. H. et al. Muscle and serum metabolomes are dysregulated in colon-26 tumor-bearing mice despite amelioration of cachexia with activin receptor type 2B ligand blockade. Am. J. Physiol. Endocrinol. Metab. 316, E852–e865 (2019).
Ninomiya, S. et al. Low levels of serum tryptophan underlie skeletal muscle atrophy. Nutrients. 12, 978 (2020).
Qiu, X. et al. Metabolic signatures and potential biomarkers for the diagnosis and treatment of colon cancer cachexia. Acta Biochim. Biophys. Sin. 55, 1913–1924 (2023).
Kunz, H. E. et al. Methylarginine metabolites are associated with attenuated muscle protein synthesis in cancer-associated muscle wasting. J. Biol. Chem. 295, 17441–17459 (2020).
Kunzke, T. et al. Derangements of amino acids in cachectic skeletal muscle are caused by mitochondrial dysfunction. J. Cachexia Sarcopenia Muscle 11, 226–240 (2020).
Wu, J. et al. Cyst(e)ine in nutrition formulation promotes colon cancer growth and chemoresistance by activating mTORC1 and scavenging ROS. Signal Transduct. Target Ther. 6, 188 (2021).
Kudamatsu, H. et al. Ameliorating effects of cystine and theanine in a cancer cachexia mouse model. J. Pharmacol. Sci. 152, 163–166 (2023).
Baumert, P. et al. Skeletal muscle hypertrophy rewires glucose metabolism: An experimental investigation and systematic review. J. Cachexia Sarcopenia Muscle 15, 989–1002 (2024).
Sirniö, P. et al. Alterations in serum amino-acid profile in the progression of colorectal cancer: associations with systemic inflammation, tumor stage and patient survival. Br. J. Cancer 120, 238–246 (2019).
Ham, D. J. et al. Glycine administration attenuates skeletal muscle wasting in a mouse model of cancer cachexia. Clin. Nutr. 33, 448–458 (2014).
Posa, D. K. et al. Skeletal muscle analysis of cancer patients reveals a potential role for carnosine in muscle wasting. J. Cachexia Sarcopenia Muscle 14, 1802–1814 (2023).
Tůma, P., Hložek, T., Kamišová, J. & Gojda, J. Monitoring of circulating amino acids in patients with pancreatic cancer and cancer cachexia using capillary electrophoresis and contactless conductivity detection. Electrophoresis 42, 1885–1891 (2021).
Fracaro, L. et al. Walker 256 tumor-bearing rats demonstrate altered interstitial cells of Cajal. Effects on ICC in the Walker 256 tumor model. Neurogastroenterol. Motil. 28, 101–115 (2016).
Vicentini, G. E. et al. Does l-glutamine-supplemented diet extenuate NO-mediated damage on myenteric plexus of Walker 256 tumor-bearing rats?. Food Res. Int. 101, 24–34 (2017).
Zhou, L., Lu, R., Huang, C. & Lin, D. Taurine protects C2C12 myoblasts from impaired cell proliferation and myotube differentiation under cisplatin-induced ROS Exposure. Front. Mol. Biosci. 8, 685362 (2021).
Deminice, R. et al. Creatine supplementation prevents hyperhomocysteinemia, oxidative stress and cancer-induced cachexia progression in Walker-256 tumor-bearing rats. Amino Acids 48, 2015–2024 (2016).
Cella, P. S. et al. Creatine supplementation in Walker-256 tumor-bearing rats prevents skeletal muscle atrophy by attenuating systemic inflammation and protein degradation signaling. Eur. J. Nutr. 59, 661–669 (2020).
Gramignano, G. et al. Efficacy of l-carnitine administration on fatigue, nutritional status, oxidative stress, and related quality of life in 12 advanced cancer patients undergoing anticancer therapy. Nutrition 22, 136–145 (2006).
Liu, S. et al. L-carnitine ameliorates cancer cachexia in mice by regulating the expression and activity of carnitine palmityl transferase. Cancer Biol. Ther. 12, 125–130 (2011).
Silvério, R., Laviano, A., Rossi Fanelli, F. & Seelaender, M. L-Carnitine induces recovery of liver lipid metabolism in cancer cachexia. Amino Acids 42, 1783–1792 (2012).
de Carvalho, C. & Caramujo, M. J. The various roles of fatty acids. Molecules. 23, 2583 (2018).
Jin, C. et al. Low expression of ELOVL6 may be involved in fat loss in white adipose tissue of cancer-associated cachexia. Lipids Health Dis. 23, 144 (2024).
Gumpper-Fedus, K. et al. Altered plasma fatty acid abundance is associated with cachexia in treatment-naïve pancreatic cancer. Cells 11, 910 (2022).
Wang, Y. P. et al. The involvement and possible targeting of cardiolipins degradation and disturbed linoleic acid metabolism in cardiac atrophy under cancer cachexia. Eur. J. Pharmacol. 985, 177108 (2024).
Calder, P. C. Omega-3 fatty acids and inflammatory processes: from molecules to man. Biochem. Soc. Trans. 45, 1105–1115 (2017).
Murphy, R. A. et al. Skeletal muscle depletion is associated with reduced plasma (n-3) fatty acids in non-small cell lung cancer patients. J. Nutr. 140, 1602–1606 (2010).
Colomer, R. et al. N-3 fatty acids, cancer and cachexia: a systematic review of the literature. Br. J. Nutr. 97, 823–831 (2007).
Burns, C. P. et al. Phase II study of high-dose fish oil capsules for patients with cancer-related cachexia. Cancer 101, 370–378 (2004).
Werner, K. et al. Dietary supplementation with n-3-fatty acids in patients with pancreatic cancer and cachexia: marine phospholipids versus fish oil - a randomized controlled double-blind trial. Lipids Health Dis. 16, 104 (2017).
Whitehouse, A. S., Smith, H. J., Drake, J. L. & Tisdale, M. J. Mechanism of attenuation of skeletal muscle protein catabolism in cancer cachexia by eicosapentaenoic acid. Cancer Res. 61, 3604–3609 (2001).
Whitehouse, A. S. & Tisdale, M. J. Increased expression of the ubiquitin-proteasome pathway in murine myotubes by proteolysis-inducing factor (PIF) is associated with activation of the transcription factor NF-kappaB. Br. J. Cancer 89, 1116–1122 (2003).
Yang, Y. H. et al. Docosahexaenoic acid-enriched phospholipids and eicosapentaenoic acid-enriched phospholipids inhibit tumor necrosis factor-alpha-induced lipolysis in 3T3-L1 adipocytes by activating sirtuin 1 pathways. Food Funct. 12, 4783–4796 (2021).
Freitas, R. D. S. et al. Targeting FFA1 and FFA4 receptors in cancer-induced cachexia. Am. J. Physiol. Endocrinol. Metab. 319, E877–e892 (2020).
Fu, Y. et al. Associations among dietary omega-3 polyunsaturated fatty acids, the gut microbiota, and intestinal immunity. Mediators Inflamm. 2021, 8879227 (2021).
Im, D. S. Functions of omega-3 fatty acids and FFA4 (GPR120) in macrophages. Eur. J. Pharmacol. 785, 36–43 (2016).
Wu, S. et al. The ω-3 polyunsaturated fatty acid docosahexaenoic acid enhances NK-Cell antitumor effector functions. Cancer Immunol. Res. 12, 744–758 (2024).
Munson, P. V. et al. Polyunsaturated fatty acid-bound α-Fetoprotein promotes immune suppression by altering human dendritic cell metabolism. Cancer Res. 83, 1543–1557 (2023).
Kornfeld, S. et al. Reducing endothelial NOS activation and interstitial fluid pressure with n-3 PUFA offset tumor chemoresistance. Carcinogenesis 33, 260–267 (2012).
Mori, T. et al. Giving combined medium-chain fatty acids and glucose protects against cancer-associated skeletal muscle atrophy. Cancer Sci. 110, 3391–3399 (2019).
Nishida, R. et al. Differential effects of three medium-chain fatty acids on mitochondrial quality control and skeletal muscle maturation. Antioxidants. 13, 821 (2024).
Tisdale, M. J. & Brennan, R. A. A comparison of long-chain triglycerides and medium-chain triglycerides on weight loss and tumor size in a cachexia model. Br. J. Cancer 58, 580–583 (1988).
Zhang, X. et al. Pro-phagocytic function and structural basis of GPR84 signaling. Nat. Commun. 14, 5706 (2023).
Liu, H. et al. The metabolite butyrate produced by gut microbiota inhibits cachexia-associated skeletal muscle atrophy by regulating intestinal barrier function and macrophage polarization. Int. Immunopharmacol. 124, 111001 (2023).
Yang, W. et al. Intestinal microbiota-derived short-chain fatty acids regulation of immune cell IL-22 production and gut immunity. Nat. Commun. 11, 4457 (2020).
Cheung, W. W. et al. Vitamin D repletion ameliorates adipose tissue browning and muscle wasting in infantile nephropathic cystinosis-associated cachexia. J. Cachexia Sarcopenia Muscle 11, 120–134 (2020).
Zhou, P. et al. Metabolic advantage of 25(OH)D(3) versus 1,25(OH)(2)D(3) supplementation in infantile nephropathic cystinosis-associated adipose tissue browning and muscle wasting. Cells. 11, 3264 (2022).
Hochwald, S. N. et al. Depletion of high energy phosphate compouds in the tumor-bearing state and reversal after tumor resection. Surgery 120, 534–541 (1996).
Konishi, M. et al. Febuxostat improves outcome in a rat model of cancer cachexia. J. Cachexia Sarcopenia Muscle 6, 174–180 (2015).
Raun, S. H. et al. Adenosine monophosphate-activated protein kinase is elevated in human cachectic muscle and prevents cancer-induced metabolic dysfunction in mice. J. Cachexia Sarcopenia Muscle 14, 1631–1647 (2023).
Beltrà, M. et al. NAD(+) repletion with niacin counteracts cancer cachexia. Nat. Commun. 14, 1849 (2023).
Park, J. M. et al. Nicotinamide riboside vitamin B3 mitigated C26 adenocarcinoma-induced cancer cachexia. Front. Pharmacol. 12, 665493 (2021).
Le Moal, E. et al. Redox control of skeletal muscle regeneration. Antioxid. Redox Signal. 27, 276–310 (2017).
Puig-Vilanova, E. et al. Oxidative stress, redox signaling pathways, and autophagy in cachectic muscles of male patients with advanced COPD and lung cancer. Free Radic. Biol. Med. 79, 91–108 (2015).
Gomes-Marcondes, M. C. & Tisdale, M. J. Induction of protein catabolism and the ubiquitin‒proteasome pathway by mild oxidative stress. Cancer Lett. 180, 69–74 (2002).
Min, K. et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J. Physiol. 593, 2017–2036 (2015).
Tan, P. L., Shavlakadze, T., Grounds, M. D. & Arthur, P. G. Differential thiol oxidation of the signaling proteins Akt, PTEN or PP2A determines whether Akt phosphorylation is enhanced or inhibited by oxidative stress in C2C12 myotubes derived from skeletal muscle. Int. J. Biochem. Cell Biol. 62, 72–79 (2015).
Kim, J. & Bae, J. S. Tumor-associated macrophages and neutrophils in tumor microenvironment. Mediators Inflamm. 2016, 6058147 (2016).
Markov, S. D., Gonzalez, D. & Mehla, K. Preclinical models for studying the impact of macrophages on cancer cachexia. Curr. Protoc. Pharmacol. 91, e80 (2020).
Odegaard, J. I. & Chawla, A. Type 2 responses at the interface between immunity and fat metabolism. Curr. Opin. Immunol. 36, 67–72 (2015).
Nguyen, K. D. et al. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 480, 104–108 (2011).
Qiu, Y. et al. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 157, 1292–1308 (2014).
Wu, S. et al. M2 macrophages independently promote beige adipogenesis via blocking adipocyte Ets1. Nat. Commun. 15, 1646 (2024).
Zeng, X. et al. M2 macrophage-derived TGF-β induces age-associated loss of adipogenesis through progenitor cell senescence. Mol. Metab. 84, 101943 (2024).
Batista, M. L. Jr. et al. Cachexia-associated adipose tissue morphological rearrangement in gastrointestinal cancer patients. J. Cachexia Sarcopenia Muscle 7, 37–47 (2016).
Erdem, M. et al. Macrophages protect against loss of adipose tissue during cancer cachexia. J. Cachexia Sarcopenia Muscle 10, 1128–1142 (2019).
Shukla, S. K. et al. Macrophages potentiate STAT3 signaling in skeletal muscles and regulate pancreatic cancer cachexia. Cancer Lett. 484, 29–39 (2020).
Liu, M. et al. The crosstalk between macrophages and cancer cells potentiates pancreatic cancer cachexia. Cancer Cell 42, 885–903.e884 (2024).
Johnston, A. J. et al. Targeting of Fn14 prevents cancer-induced cachexia and prolongs survival. Cell 162, 1365–1378 (2015).
Tonkin, J. et al. Monocyte/Macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther. 23, 1189–1200 (2015).
Pryce, B. R. et al. Muscle inflammation is regulated by NF-κB from multiple cells to control distinct states of wasting in cancer cachexia. Cell Rep. 43, 114925 (2024).
Penafuerte, C. A. et al. Identification of neutrophil-derived proteases and angiotensin II as biomarkers of cancer cachexia. Br. J. Cancer 114, 680–687 (2016).
Zhang, K. L. et al. Integrated neutrophil-to-lymphocyte ratio and handgrip strength better predict survival in patients with cancer cachexia. Nutrition 122, 112399 (2024).
Ruan, G. T. et al. Association of systemic inflammation and overall survival in elderly patients with cancer cachexia - results from a multicenter study. J. Inflamm. Res. 14, 5527–5540 (2021).
Xie, H. et al. Evaluation and validation of neutrophil to albumin ratio as a promising prognostic marker for all-cause mortality in patients with cancer: a multicenter cohort study. Nutrition 121, 112365 (2024).
Xie, H. L. et al. The prognostic value of the combination of body composition and systemic inflammation in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 14, 879–890 (2023).
Peterson, J. M., Feeback, K. D., Baas, J. H. & Pizza, F. X. Tumor necrosis factor-alpha promotes the accumulation of neutrophils and macrophages in skeletal muscle. J. Appl Physiol. 101, 1394–1399 (2006).
Petruzzelli, M. et al. Early Neutrophilia marked by aerobic glycolysis sustains host metabolism and delays cancer cachexia. Cancers. 14, 963 (2022).
Hayashi, Y. et al. IL36G-producing neutrophil-like monocytes promote cachexia in cancer. Nat. Commun. 15, 7662 (2024).
Wang, D. et al. LCN2 secreted by tissue-infiltrating neutrophils induces the ferroptosis and wasting of adipose and muscle tissues in lung cancer cachexia. J. Hematol. Oncol. 16, 30 (2023).
Narsale, A. et al. Cancer-driven changes link T-cell frequency to muscle strength in people with cancer: a pilot study. J. Cachexia Sarcopenia Muscle 10, 827–843 (2019).
Ju, J. E. et al. Potential role of immunological factors in early diagnosis of cancer cachexia in C26 tumor-bearing mice. Appl Biol. Chem. 62, 3 (2019).
Collinson-Pautz, M. R. et al. Constitutively active MyD88/CD40 costimulation enhances expansion and efficacy of chimeric antigen receptor T cells targeting hematological malignancies. Leukemia 33, 2195–2207 (2019).
Wang, Z., Zhao, C., Moya, R. & Davies, J. D. A novel role for CD4+ T cells in the control of cachexia. J. Immunol. 181, 4676–4684 (2008).
Anoveros-Barrera, A. et al. Immunohistochemical phenotyping of T cells, granulocytes, and phagocytes in the muscle of cancer patients: association with radiologically defined muscle mass and gene expression. Skelet. Muscle 9, 24 (2019).
Michaelis, K. A. et al. The TLR7/8 agonist R848 remodels tumor and host responses to promote survival in pancreatic cancer. Nat. Commun. 10, 4682 (2019).
Baazim, H. et al. CD8(+) T cells induce cachexia during chronic viral infection. Nat. Immunol. 20, 701–710 (2019).
Macciò, A. & Madeddu, C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology 159, 357–364 (2020).
Gonda, K. et al. Multiple immunological mechanisms of cancer cachexia in patients with solid tumors. J. Clin. Oncol. 34, 667–667 (2016).
Shibata, M. et al. Abstract 1285: Production of interleukin-17 is increased in patients with gastrointestinal cancer and correlates with immune suppression involving MDSC, nutritional impairment, and poor prognosis. Cancer Res. 75, 1285–1285 (2015).
Yang, J. et al. Abstract 4695: dual targeting of MEK and PI3K pathways can act via tumor-intrinsic mechanisms to overcome resistance and tumor-extrinsic mechanisms to modulate immunity and limit cancer cachexia. Cancer Res. 75, 4695–4695 (2015).
Cuenca, A. G. et al. Novel role for tumor-induced expansion of myeloid-derived cells in cancer cachexia. J. Immunol. 192, 6111–6119 (2014).
Winfield, R. D. et al. Myeloid-derived suppressor cells in cancer cachexia syndrome: a new explanation for an old problem. JPEN J. Parenter. Enter. Nutr. 32, 651–655 (2008).
Ghonim, M. A. et al. Targeting PARP-1 with metronomic therapy modulates MDSC suppressive function and enhances anti-PD-1 immunotherapy in colon cancer. J. Immunother. Cancer. 9, e001643 (2021).
Dzierlega, K. et al. Activin A-expressing polymorphonuclear myeloid-derived suppressor cells infiltrate skeletal and cardiac muscle and promote cancer cachexia. J. Immunol. 211, 497–507 (2023).
Deyhle, M. R. et al. Depleting Ly6G positive myeloid cells reduces pancreatic cancer-induced skeletal muscle atrophy. Cells 11, 1893 (2022).
VanderVeen, B. N. et al. The complex heterogeneity of immune cell signatures across wasting tissues with C26 and 5-fluorouracil-induced cachexia. Am. J. Physiol. Cell Physiol. 326, C606–c621 (2024).
Nissinen, T. A. et al. Treating cachexia using soluble ACVR2B improves survival, alters mTOR localization, and attenuates liver and spleen responses. J. Cachexia Sarcopenia Muscle 9, 514–529 (2018).
Lima, J. et al. Tumor-derived transforming growth factor-β signaling contributes to fibrosis in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 10, 1045–1059 (2019).
Zhao, Y. et al. circNOX4 activates an inflammatory fibroblast niche to promote tumor growth and metastasis in NSCLC via FAP/IL-6 axis. Mol. Cancer 23, 47 (2024).
Zeng, W. et al. CCL18 signaling from tumor-associated macrophages activates fibroblasts to adopt a chemoresistance-inducing phenotype. Oncogene 42, 224–237 (2023).
Bruzzese, F. et al. Local and systemic protumorigenic effects of cancer-associated fibroblast-derived GDF15. Cancer Res. 74, 3408–3417 (2014).
Rettig, W. J. et al. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc. Natl. Acad. Sci. USA 85, 3110–3114 (1988).
Feig, C. et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 110, 20212–20217 (2013).
Yang, X. et al. FAP promotes immunosuppression by cancer-associated fibroblasts in the tumor microenvironment via STAT3-CCL2 signaling. Cancer Res. 76, 4124–4135 (2016).
Zhao, L. et al. Antitumor efficacy and potential mechanism of FAP-targeted radioligand therapy combined with immune checkpoint blockade. Signal Transduct. Target Ther. 9, 142 (2024).
Tran, E. et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 210, 1125–1135 (2013).
Roberts, E. W. et al. Depletion of stromal cells expressing fibroblast activation protein-α from skeletal muscle and bone marrow results in cachexia and anemia. J. Exp. Med. 210, 1137–1151 (2013).
Maishi, N. & Hida, K. Tumor endothelial cells accelerate tumor metastasis. Cancer Sci. 108, 1921–1926 (2017).
Inda, A. M. et al. Evaluation of angiogenesis with the expression of VEGF and CD34 in human non-small cell lung cancer. J. Exp. Clin. Cancer Res. 26, 375–378 (2007).
Tichet, M. et al. Tumor-derived SPARC drives vascular permeability and extravasation through endothelial VCAM1 signaling to promote metastasis. Nat. Commun. 6, 6993 (2015).
Husain, A., Hu, N., Sadow, P. M. & Nucera, C. Expression of angiogenic switch, cachexia and inflammation factors at the crossroad in undifferentiated thyroid carcinoma with BRAF(V600E). Cancer Lett. 380, 577–585 (2016).
Hsu, B. G. et al. Association of endothelial dysfunction and peripheral arterial disease with sarcopenia in chronic kidney disease. J. Cachexia Sarcopenia Muscle 15, 1199–1208 (2024).
Kim, Y.-M. et al. Skeletal muscle endothelial dysfunction through the activin A-PGC1α axis drives progression of cancer cachexia. Nat. Cancer 6, 1835–1845 (2025).
Yang, S. et al. Functional effects of muscle PGC-1alpha in aged animals. Skelet. Muscle 10, 14 (2020).
Banks, W. A. Anorectic effects of circulating cytokines: role of the vascular blood‒brain barrier. Nutrition 17, 434–437 (2001).
Sun, M. et al. Endothelial peroxynitrite causes disturbance of neuronal oscillations by targeting caspase-1 in the arcuate nucleus. Redox Biol. 47, 102147 (2021).
Ling, T., Liu, J., Dong, L. & Liu, J. The roles of P-selectin in cancer cachexia. Med. Oncol. 40, 338 (2023).
Taylor, J. et al. Endothelial Notch1 signaling in white adipose tissue promotes cancer cachexia. Nat. Cancer 4, 1544–1560 (2023).
Klose, R. et al. Targeting VEGF-A in myeloid cells enhances natural killer cell responses to chemotherapy and ameliorates cachexia. Nat. Commun. 7, 12528 (2016).
Widner, D. B. et al. Activated mast cells in skeletal muscle can be a potential mediator for cancer-associated cachexia. J. Cachexia Sarcopenia Muscle 12, 1079–1097 (2021).
Cho, D. S. et al. Single-cell deconstruction of post-sepsis skeletal muscle and adipose tissue microenvironments. J. Cachexia Sarcopenia Muscle 11, 1351–1363 (2020).
Morvan, M. G. & Lanier, L. L. NK cells and cancer: you can teach innate cells new tricks. Nat. Rev. Cancer 16, 7–19 (2016).
Baazim, H., Antonio-Herrera, L. & Bergthaler, A. The interplay of immunology and cachexia in infection and cancer. Nat. Rev. Immunol. 22, 309–321 (2022).
Yeh, S. S. & Schuster, M. W. Geriatric cachexia: the role of cytokines. Am. J. Clin. Nutr. 70, 183–197 (1999).
Scarlett, J. M. et al. Regulation of central melanocortin signaling by interleukin-1 beta. Endocrinology 148, 4217–4225 (2007).
Marks, D. L. et al. Differential role of melanocortin receptor subtypes in cachexia. Endocrinology 144, 1513–1523 (2003).
Whitaker, K. W. & Reyes, T. M. Central blockade of melanocortin receptors attenuates the metabolic and locomotor responses to peripheral interleukin-1beta administration. Neuropharmacology 54, 509–520 (2008).
Ericsson, A., Kovács, K. J. & Sawchenko, P. E. A functional anatomical analysis of central pathways subserving the effects of interleukin-1 on stress-related neuroendocrine neurons. J. Neurosci. 14, 897–913 (1994).
Braun, T. P. et al. Central nervous system inflammation induces muscle atrophy via activation of the hypothalamic‒pituitary‒adrenal axis. J. Exp. Med. 208, 2449–2463 (2011).
Krelin, Y. et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res. 67, 1062–1071 (2007).
Grossberg, A. J. et al. Arcuate nucleus proopiomelanocortin neurons mediate the acute anorectic actions of leukemia inhibitory factor via gp130. Endocrinology 151, 606–616 (2010).
Hirano, T. et al. Complementary DNA for a novel human interleukin (BSF-2) that induces B lymphocytes to produce immunoglobulin. Nature 324, 73–76 (1986).
Kopf, M. et al. Impaired immune and acute-phase responses in interleukin-6-deficient mice. Nature 368, 339–342 (1994).
Soler, M. F. et al. New perspectives in cancer immunotherapy: targeting IL-6 cytokine family. J. Immunother. Cancer 11, e007530 (2023).
Hunter, C. A. & Jones, S. A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 16, 448–457 (2015).
Wolf, J., Rose-John, S. & Garbers, C. Interleukin-6 and its receptors: a highly regulated and dynamic system. Cytokine 70, 11–20 (2014).
Bonetto, A. et al. JAK/STAT3 pathway inhibition blocks skeletal muscle wasting downstream of IL-6 and in experimental cancer cachexia. Am. J. Physiol. Endocrinol. Metab. 303, E410–E421 (2012).
Zimmers, T. A., Fishel, M. L. & Bonetto, A. STAT3 in the systemic inflammation of cancer cachexia. Semin. Cell Dev. Biol. 54, 28–41 (2016).
Tsujinaka, T. et al. Interleukin 6 receptor antibody inhibits muscle atrophy and modulates proteolytic systems in interleukin 6 transgenic mice. J. Clin. Investig. 97, 244–249 (1996).
White, J. P. et al. Muscle mTORC1 suppression by IL-6 during cancer cachexia: a role for AMPK. Am. J. Physiol. Endocrinol. Metab. 304, E1042–E1052 (2013).
Magnuson, B., Ekim, B. & Fingar, D. C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signaling networks. Biochem. J. 441, 1–21 (2012).
Mishra, D. et al. Parabrachial interleukin-6 reduces body weight and food intake and increases thermogenesis to regulate energy metabolism. Cell Rep. 26, 3011–3026.e3015 (2019).
Sun, Q. et al. Area postrema neurons mediate interleukin-6 function in cancer cachexia. Nat. Commun. 15, 4682 (2024).
Heredia, J. E. et al. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153, 376–388 (2013).
Meng, J. et al. Accelerated regeneration of the skeletal muscle in RNF13-knockout mice is mediated by macrophage-secreted IL-4/IL-6. Protein Cell 5, 235–247 (2014).
Lu, S. W. et al. IL-20 antagonist suppresses PD-L1 expression and prolongs survival in pancreatic cancer models. Nat. Commun. 11, 4611 (2020).
Beutler, B. et al. Purification of cachectin, a lipoprotein lipase-suppressing hormone secreted by endotoxin-induced RAW 264.7 cells. J. Exp. Med. 161, 984–995 (1985).
Yang, X. et al. Relative contribution of adipose triglyceride lipase and hormone-sensitive lipase to tumor necrosis factor-α (TNF-α)-induced lipolysis in adipocytes. J. Biol. Chem. 286, 40477–40485 (2011).
Li, Y. P. & Reid, M. B. NF-kappaB mediates the protein loss induced by TNF-alpha in differentiated skeletal muscle myotubes. Am. J. Physiol. Regul. Integr. Comp. Physiol. 279, R1165–R1170 (2000).
Yoshida, T. et al. Molecular mechanisms and signaling pathways of angiotensin II-induced muscle wasting: potential therapeutic targets for cardiac cachexia. Int. J. Biochem. Cell Biol. 45, 2322–2332 (2013).
Wang, D. T. et al. Resveratrol prevents TNF-α-induced muscle atrophy via regulation of Akt/mTOR/FoxO1 signaling in C2C12 myotubes. Int. Immunopharmacol. 19, 206–213 (2014).
Patel, H. J. & Patel, B. M. TNF-α and cancer cachexia: molecular insights and clinical implications. Life Sci. 170, 56–63 (2017).
Chen, J. L. et al. Specific targeting of TGF-β family ligands demonstrates distinct roles in the regulation of muscle mass in health and disease. Proc. Natl. Acad. Sci. USA 114, E5266–e5275 (2017).
Walton, K. L., Johnson, K. E. & Harrison, C. A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 8, 461 (2017).
Massagué, J. TGFβ signaling in context. Nat. Rev. Mol. Cell Biol. 13, 616–630 (2012).
Lee, S. J. et al. Functional redundancy of type I and type II receptors in the regulation of skeletal muscle growth by myostatin and activin A. Proc. Natl. Acad. Sci. USA 117, 30907–30917 (2020).
Sartori, R., Romanello, V. & Sandri, M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat. Commun. 12, 330 (2021).
Domaniku, A., Bilgic, S. N. & Kir, S. Muscle wasting: emerging pathways and potential drug targets. Trends Pharmacol. Sci. 44, 705–718 (2023).
Domaniku-Waraich, A. et al. Oncostatin M signaling drives cancer-associated skeletal muscle wasting. Cell Rep. Med. 5, 101498 (2024).
Bilgic, S. N. et al. EDA2R-NIK signaling promotes muscle atrophy linked to cancer cachexia. Nature 617, 827–834 (2023).
Miki, Y. et al. Oncostatin M induces C2C12 myotube atrophy by modulating muscle differentiation and degradation. Biochem. Biophys. Res. Commun. 516, 951–956 (2019).
Viswanadhapalli, S. et al. Targeting LIF/LIFR signaling in cancer. Genes Dis. 9, 973–980 (2022).
Li, X. & Sun, K. Regulation of lipolysis in adipose tissue and clinical significance. Adv. Exp. Med. Biol. 1090, 199–210 (2018).
Basu, D. & Goldberg, I. J. Regulation of lipoprotein lipase-mediated lipolysis of triglycerides. Curr. Opin. Lipidol. 31, 154–160 (2020).
Arora, G. K. et al. Cachexia-associated adipose loss induced by tumor-secreted leukemia inhibitory factor is counterbalanced by decreased leptin. JCI Insight 3, e121221 (2018).
Terawaki, K. et al. New cancer cachexia rat model generated by implantation of a peritoneal dissemination-derived human stomach cancer cell line. Am. J. Physiol. Endocrinol. Metab. 306, E373–E387 (2014).
Sarraf, P. et al. Multiple cytokines and acute inflammation raise mouse leptin levels: potential role in inflammatory anorexia. J. Exp. Med. 185, 171–175 (1997).
Seto, D. N., Kandarian, S. C. & Jackman, R. W. A key role for leukemia inhibitory factor in C26 cancer cachexia. J. Biol. Chem. 290, 19976–19986 (2015).
Wischhusen, J., Melero, I. & Fridman, W. H. Growth/Differentiation Factor-15 (GDF-15): from biomarker to novel targetable immune checkpoint. Front. Immunol. 11, 951 (2020).
Rochette, L. et al. GDF15 and cardiac cells: current concepts and new insights. Int. J. Mol. Sci. 22, 8889 (2021).
Johnen, H. et al. Tumor-induced anorexia and weight loss are mediated by the TGF-β superfamily cytokine MIC-1. Nat. Med. 13, 1333–1340 (2007).
Cimino, I. et al. Activation of the hypothalamic‒pituitary‒adrenal axis by exogenous and endogenous GDF15. Proc. Natl. Acad. Sci. USA 118, e2106868118 (2021).
Garfield, B. E. et al. Growth/differentiation factor 15 causes TGFβ-activated kinase 1-dependent muscle atrophy in pulmonary arterial hypertension. Thorax 74, 164–176 (2019).
Lerner, L. et al. MAP3K11/GDF15 axis is a critical driver of cancer cachexia. J. Cachexia Sarcopenia Muscle 7, 467–482 (2016).
Elattar, S., Dimri, M. & Satyanarayana, A. The tumor secretory factor ZAG promotes white adipose tissue browning and energy wasting. FASEB J. 32, 4727–4743 (2018).
Westhrin, M. et al. Growth differentiation factor 15 (GDF15) promotes osteoclast differentiation and inhibits osteoblast differentiation and high serum GDF15 levels are associated with multiple myeloma bone disease. Hematologica 100, e511–e514 (2015).
Hao, J. et al. IFN-γ induces lipogenesis in mouse mesangial cells via the JAK2/STAT1 pathway. Am. J. Physiol. Cell Physiol. 304, C760–C767 (2013).
Sun, W. Y. et al. Lipocalin-2 derived from adipose tissue mediates aldosterone-induced renal injury. JCI Insight. 3, e120196 (2018).
Glaviano, A. et al. PI3K/AKT/mTOR signal transduction pathway and targeted therapies in cancer. Mol. Cancer 22, 138 (2023).
Tsigos, C. & Chrousos, G. P. Hypothalamic‒pituitary‒adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 53, 865–871 (2002).
Gioldasi, S. et al. Metabolic Association between Leptin and the Corticotropin Releasing Hormone. Endocr. Metab. Immune Disord. Drug Targets 19, 458–466 (2019).
Menconi, M. et al. Role of glucocorticoids in the molecular regulation of muscle wasting. Crit. Care Med. 35, S602–S608 (2007).
Chrousos, G. P. The hypothalamic‒pituitary‒adrenal axis and immune-mediated inflammation. N. Engl. J. Med. 332, 1351–1362 (1995).
Tsigos, C. et al. Dose effects of recombinant human interleukin-6 on pituitary hormone secretion and energy expenditure. Neuroendocrinology 66, 54–62 (1997).
Gupta, R. et al. Multifaceted role of NF-κB in hepatocellular carcinoma therapy: Molecular landscape, therapeutic compounds and nanomaterial approaches. Environ. Res. 228, 115767 (2023).
Shi, P., Xu, J. & Cui, H. The recent research progress of NF-κB signaling on the proliferation, migration, invasion, immune escape and drug resistance of glioblastoma. Int. J. Mol. Sci. 24, (2023).
Miao, C. et al. Pyrrolidine Dithiocarbamate (PDTC) attenuates cancer cachexia by affecting muscle atrophy and fat lipolysis. Front. Pharmacol. 8, 915 (2017).
Paul, P. K. et al. Targeted ablation of TRAF6 inhibits skeletal muscle wasting in mice. J. Cell Biol. 191, 1395–1411 (2010).
Langen, R. C. et al. NF-κB activation is required for the transition of pulmonary inflammation to muscle atrophy. Am. J. Respir. Cell Mol. Biol. 47, 288–297 (2012).
Guttridge, D. C. et al. NF-kappaB-induced loss of MyoD messenger RNA: possible role in muscle decay and cachexia. Science 289, 2363–2366 (2000).
Li, W. et al. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 297, C706–C714 (2009).
Hanna, R. A. et al. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J. Biol. Chem. 287, 19094–19104 (2012).
Philips, R. L. et al. The JAK-STAT pathway at 30: Much learned, much more to do. Cell 185, 3857–3876 (2022).
O’Shea, J. J., Holland, S. M. & Staudt, L. M. JAKs and STATs in immunity, immunodeficiency, and cancer. N. Engl. J. Med. 368, 161–170 (2013).
Samra, S., Bergerson, J. R. E., Freeman, A. F. & Turvey, S. E. JAK-STAT signaling pathway, immunodeficiency, inflammation, immune dysregulation, and inborn errors of immunity. J. Allergy Clin. Immunol. 155, 357–367 (2025).
Zhang, L. et al. Stat3 activation links a C/EBPδ to myostatin pathway to stimulate loss of muscle mass. Cell Metab. 18, 368–379 (2013).
Silva, K. A. et al. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin‒proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 290, 11177–11187 (2015).
Fujita, J. et al. Anti-interleukin-6 receptor antibody prevents muscle atrophy in colon-26 adenocarcinoma-bearing mice with modulation of lysosomal and ATP-ubiquitin-dependent proteolytic pathways. Int. J. Cancer 68, 637–643 (1996).
Hashimoto, S. et al. Exercise-induced vitamin D receptor and androgen receptor mediate inhibition of IL-6 and STAT3 in muscle. Biochem. Biophys. Rep. 37, 101621 (2024).
Testa, M. T. J. et al. Resistance training attenuates activation of STAT3 and muscle atrophy in tumor-bearing mice. Front. Oncol. 12, 880787 (2022).
Oh, H. M. et al. STAT3 protein interacts with Class O Forkhead transcription factors in the cytoplasm and regulates nuclear/cytoplasmic localization of FoxO1 and FoxO3a proteins in CD4(+) T cells. J. Biol. Chem. 287, 30436–30443 (2012).
Yoon, S. et al. NF-κB and STAT3 cooperatively induce IL6 in starved cancer cells. Oncogene 31, 3467–3481 (2012).
Gandhi, A. Y. et al. Cytokine-mediated STAT3 transcription supports ATGL/CGI-58-dependent adipocyte lipolysis in cancer cachexia. Front. Oncol. 12, 841758 (2022).
Abdullahi, A. et al. Browning of white adipose tissue after a burn injury promotes hepatic steatosis and dysfunction. Cell Death Dis. 10, 870 (2019).
Anderson, L. J. et al. Whole-body and adipose tissue metabolic phenotype in cancer patients. J. Cachexia Sarcopenia Muscle 13, 1124–1133 (2022).
Lee, J. H. & Jun, H. S. Role of myokines in regulating skeletal muscle mass and function. Front. Physiol. 10, 42 (2019).
Burks, T. N. & Cohn, R. D. Role of TGF-β signaling in inherited and acquired myopathies. Skelet. Muscle 1, 19 (2011).
Amthor, H. & Hoogaars, W. M. Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy. Curr. Gene Ther. 12, 245–259 (2012).
El Shafey, N. et al. Inhibition of the myostatin/Smad signaling pathway by short decorin-derived peptides. Exp. Cell Res. 341, 187–195 (2016).
Schiaffino, S. et al. Mechanisms regulating skeletal muscle growth and atrophy. FEBS J. 280, 4294–4314 (2013).
Costelli, P. et al. Muscle myostatin signaling is enhanced in experimental cancer cachexia. Eur. J. Clin. Investig. 38, 531–538 (2008).
Zhou, X. et al. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell 142, 531–543 (2010).
Kramer, H. F. & Goodyear, L. J. Exercise, MAPK, and NF-κB signaling in skeletal muscle. J. Appl. Physiol. 103, 388–395 (2007).
Yang, J. et al. ZIP4 promotes muscle wasting and cachexia in mice with orthotopic pancreatic tumors by stimulating RAB27B-Regulated release of extracellular vesicles from cancer cells. Gastroenterology 156, 722–734.e726 (2019).
Chiappalupi, S. et al. Targeting RAGE prevents muscle wasting and prolongs survival in cancer cachexia. J. Cachexia Sarcopenia Muscle 11, 929–946 (2020).
Perdiguero, E. et al. Genetic analysis of p38 MAP kinases in myogenesis: fundamental role of p38alpha in abrogating myoblast proliferation. EMBO J. 26, 1245–1256 (2007).
Yoon, S. L. & Grundmann, O. Relevance of dietary supplement use in gastrointestinal-cancer-associated cachexia. Nutrients. 15, 3391 (2023).
Coniglio, S., Shumskaya, M. & Vassiliou, E. Unsaturated fatty acids and their immunomodulatory properties. Biology. 12, 279 (2023).
Loyala, J. V., Down, B., Wong, E. & Tan, B. Treatment of cachexia in gastric cancer: exploring the use of anti-inflammatory natural products and their derivatives. Nutrients. 16, 1246 (2024).
Mocellin, M. C., Camargo, C. D. Q., Fabre, M. E. D. S. & Trindade, E. B. S. D. M. Fish oil effects on quality of life, body weight and free fat mass change in gastrointestinal cancer patients undergoing chemotherapy: a triple blind, randomized clinical trial. J. Funct. Foods 31, 113–122 (2017).
Jatoi, A. et al. A double-blind, placebo-controlled randomized trial of creatine for the cancer anorexia/weight loss syndrome (N02C4): an Alliance trial. Ann. Oncol. 28, 1957–1963 (2017).
Inui, A. Cancer anorexia-cachexia syndrome: current issues in research and management. CA Cancer J. Clin. 52, 72–91 (2002).
Tchekmedyian, N. S., Tait, N., Moody, M. & Aisner, J. High-dose megestrol acetate. A possible treatment for cachexia. JAMA 257, 1195–1198 (1987).
Kouchaki, B. et al. Randomized double-blind clinical trial of combined treatment with megestrol acetate plus celecoxib versus megestrol acetate alone in cachexia-anorexia syndrome induced by GI cancers. Support. Care Cancer 26, 2479–2489 (2018).
Kerem, M. et al. Adipokines and ghrelin in gastric cancer cachexia. World J. Gastroenterol. 14, 3633–3641 (2008).
Strasser, F. et al. Safety, tolerability and pharmacokinetics of intravenous ghrelin for cancer-related anorexia/cachexia: a randomized, placebo-controlled, double-blind, double-crossover study. Br. J. Cancer 98, 300–308 (2008).
Dev, R., Amano, K., Naito, T. & Del Fabbro, E. Anamorelin for the treatment of cancer anorexia-cachexia syndrome. Curr. Oncol. Rep. 26, 762–772 (2024).
Herodes, M. et al. Pilot clinical trial of macimorelin to assess safety and efficacy in patients with cancer cachexia. J. Cachexia Sarcopenia Muscle 14, 835–846 (2023).
Hamauchi, S. et al. A multicenter, open-label, single-arm study of anamorelin (ONO-7643) in advanced gastrointestinal cancer patients with cancer cachexia. Cancer 125, 4294–4302 (2019).
Iwai, N. et al. Predictors of response to anamorelin in gastrointestinal cancer patients with cachexia: a retrospective study. Support. Care Cancer 31, 115 (2023).
Bar-Sela, G., Zalman, D., Semenysty, V. & Ballan, E. The effects of dosage-controlled cannabis capsules on cancer-related cachexia and anorexia syndrome in advanced cancer patients: pilot study. Integr. Cancer Ther. 18, 1534735419881498 (2019).
Sandhya, L. et al. Randomized double-blind placebo-controlled study of olanzapine for chemotherapy-related anorexia in patients with locally advanced or metastatic gastric, hepatopancreaticobiliary, and lung cancer. J. Clin. Oncol. 41, 2617–2627 (2023).
Riechelmann, R. P. et al. Phase II trial of mirtazapine for cancer-related cachexia and anorexia. Am. J. Hosp. Palliat. Care. 27, 106–110 (2010).
Almeida, O. L. S. et al. Mirtazapine versus megestrol in the treatment of anorexia-cachexia syndrome in patients with advanced cancer: a randomized, double-blind, controlled phase II clinical trial. Cancers. 15, 3588 (2023).
Hickish, T. et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomized, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 18, 192–201 (2017).
Wang, R. et al. Curcumin treatment suppresses cachexia-associated adipose wasting in mice by blocking the cAMP/PKA/CREB signaling pathway. Phytomedicine 109, 154563 (2023).
Chaiworramukkul, A., Seetalarom, K., Saichamchan, S. & Prasongsook, N. A double-blind, placebo-controlled randomized phase IIa study: evaluating the effect of curcumin for treatment of cancer anorexia-cachexia syndrome in solid cancer patients. Asian Pac. J. Cancer Prev. 23, 2333–2340 (2022).
Lai, V. et al. Results of a pilot study of the effects of celecoxib on cancer cachexia in patients with cancer of the head, neck, and gastrointestinal tract. Head. Neck 30, 67–74 (2008).
Mantovani, G. et al. Phase II nonrandomized study of the efficacy and safety of COX-2 inhibitor celecoxib on patients with cancer cachexia. J. Mol. Med. 88, 85–92 (2010).
Jatoi, A. et al. A placebo-controlled double blind trial of etanercept for the cancer anorexia/weight loss syndrome: results from N00C1 from the North Central Cancer Treatment Group. Cancer 110, 1396–1403 (2007).
Davis, M. et al. A Phase II dose titration study of thalidomide for cancer-associated anorexia. J. Pain. Symptom Manag. 43, 78–86 (2012).
Yennurajalingam, S. et al. The role of thalidomide and placebo for the treatment of cancer-related anorexia-cachexia symptoms: results of a double-blind placebo-controlled randomized study. J. Palliat. Med. 15, 1059–1064 (2012).
Wen, H. S. et al. Clinical studies on the treatment of cancer cachexia with megestrol acetate plus thalidomide. Chemotherapy 58, 461–467 (2012).
Chasen, M., Hirschman, S. Z. & Bhargava, R. Phase II study of the novel peptide-nucleic acid OHR118 in the management of cancer-related anorexia/cachexia. J. Am. Med. Dir. Assoc. 12, 62–67 (2011).
Hauer, K. et al. Improvement in muscular performance and decrease in tumor necrosis factor level in old age after antioxidant treatment. J. Mol. Med. 81, 118–125 (2003).
Lundholm, K. et al. Insulin treatment in cancer cachexia: effects on survival, metabolism, and physical functioning. Clin. Cancer Res. 13, 2699–2706 (2007).
Hsu, J. Y. et al. Non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 550, 255–259 (2017).
Breen, D. M. et al. GDF-15 neutralization alleviates platinum-based chemotherapy-induced emesis, anorexia, and weight loss in mice and nonhuman primates. Cell Metab. 32, 938–950.e936 (2020).
Lerner, L. et al. Plasma growth differentiation factor 15 is associated with weight loss and mortality in cancer patients. J. Cachexia Sarcopenia Muscle 6, 317–324 (2015).
Crawford, J. et al. A Phase Ib first-in-patient study assessing the safety, tolerability, pharmacokinetics, and pharmacodynamics of ponsegromab in participants with cancer and cachexia. Clin. Cancer Res. 30, 489–497 (2024).
Groarke, J. D. et al. Ponsegromab for the treatment of cancer cachexia. N. Engl. J. Med. 391, 2291–2303 (2024).
Chen, J. L. et al. Elevated expression of activins promotes muscle wasting and cachexia. FASEB J. 28, 1711–1723 (2014).
Christopher, C. N. et al. Exercise and nutrition interventions for prehabilitation in hepato-pancreato-biliary cancers: a narrative review. Nutrients. 15, 5044 (2023).
Gukovsky, I. et al. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology 144, 1199–1209.e1194 (2013).
Gordon, J. N. et al. Thalidomide in the treatment of cancer cachexia: a randomized placebo controlled trial. Gut 54, 540–545 (2005).
Wiedenmann, B. et al. A multicenter, phase II study of infliximab plus gemcitabine in pancreatic cancer cachexia. J. Support. Oncol. 6, 18–25 (2008).
Kraft, M. et al. L-Carnitine-supplementation in advanced pancreatic cancer (CARPAN)–a randomized multicenter trial. Nutr. J. 11, 52 (2012).
Khan, S. A., Thomas, H. C., Davidson, B. R. & Taylor-Robinson, S. D. Cholangiocarcinoma. Lancet 366, 1303–1314 (2005).
Bekaii-Saab, T. et al. Multi-institutional phase II study of selumetinib in patients with metastatic biliary cancers. J. Clin. Oncol. 29, 2357–2363 (2011).
Prado, C. M. et al. Skeletal muscle anabolism is a side effect of therapy with the MEK inhibitor: selumetinib in patients with cholangiocarcinoma. Br. J. Cancer 106, 1583–1586 (2012).
Finn, R. S. et al. Phase 1b investigation of the MEK inhibitor binimetinib in patients with advanced or metastatic biliary tract cancer. Investig. N. Drugs 36, 1037–1043 (2018).
Yuan, L. et al. The atypical β-blocker S-oxprenolol reduces cachexia and improves survival in a rat cancer cachexia model. J. Cachexia Sarcopenia Muscle 14, 653–660 (2023).
Stewart et al. Espindolol for the treatment and prevention of cachexia in patients with stage III/IV non-small cell lung cancer or colorectal cancer: a randomized, double-blind, placebo-controlled, international multicenter phase II study (the ACT-ONE trial). J. Cachexia Sarcopenia Muscle 7, 355–365 (2016).
Pötsch, M. S. et al. MT-102 prevents tissue wasting and improves survival in a rat model of severe cancer cachexia. J. Cachexia Sarcopenia Muscle 11, 594–605 (2020).
Sung, H. et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 71, 209–249 (2021).
Op den Kamp, C. M. et al. Pre-cachexia in patients with stages I-III non-small cell lung cancer: systemic inflammation and functional impairment without activation of skeletal muscle ubiquitin proteasome system. Lung Cancer 76, 112–117 (2012).
Cortiula, F. et al. Physical exercise at the crossroad between muscle wasting and the immune system: implications for lung cancer cachexia. J. Cachexia Sarcopenia Muscle 13, 55–67 (2022).
Arrieta, O. et al. Mirtazapine as appetite stimulant in patients with non-small cell lung cancer and anorexia: a randomized clinical trial. JAMA Oncol. 10, 305–314 (2024).
Turcott, J. G. et al. The effect of nabilone on appetite, nutritional status, and quality of life in lung cancer patients: a randomized, double-blind clinical trial. Support. Care Cancer 26, 3029–3038 (2018).
Crawford, J. et al. Study design and rationale for the phase 3 clinical development program of enobosarm, a selective androgen receptor modulator, for the prevention and treatment of muscle wasting in cancer patients (POWER Trials). Curr. Oncol. Rep. 18, 37 (2016).
Bohl, C. E. et al. Structural basis for accommodation of nonsteroidal ligands in the androgen receptor. J. Biol. Chem. 280, 37747–37754 (2005).
Dobs, A. S. et al. Effects of enobosarm on muscle wasting and physical function in patients with cancer: a double-blind, randomized controlled phase 2 trial. Lancet Oncol. 14, 335–345 (2013).
Setiawan, T. et al. Cancer cachexia: molecular mechanisms and treatment strategies. J. Hematol. Oncol. 16, 54 (2023).
Stewart et al. The ACT-ONE trial, a multicenter, randomized, double-blind, placebo-controlled, dose-finding study of the anabolic/catabolic transforming agent, MT-102 in subjects with cachexia related to stage III and IV non-small cell lung cancer and colorectal cancer: study design. J. Cachexia Sarcopenia Muscle 2, 201–207 (2011).
Zhang, H. & Garcia, J. M. Anamorelin hydrochloride for the treatment of cancer-anorexia-cachexia in NSCLC. Expert Opin. Pharmacother. 16, 1245–1253 (2015).
Northrup, R. et al. Effect of ghrelin and anamorelin (ONO-7643), a selective ghrelin receptor agonist, on tumor growth in a lung cancer mouse xenograft model. Support. Care Cancer 21, 2409–2415 (2013).
Temel, J. S. et al. Anamorelin in patients with non-small cell lung cancer and cachexia (ROMANA 1 and ROMANA 2): results from two randomized, double-blind, phase 3 trials. Lancet Oncol. 17, 519–531 (2016).
Currow, D. et al. ROMANA 3: a phase 3 safety extension study of anamorelin in advanced non-small cell lung cancer (NSCLC) patients with cachexia. Ann. Oncol. 28, 1949–1956 (2017).
Takayama, K. et al. Anamorelin (ONO-7643) in Japanese patients with non-small cell lung cancer and cachexia: results of a randomized phase 2 trial. Support. Care Cancer 24, 3495–3505 (2016).
Katakami, N. et al. Anamorelin (ONO-7643) for the treatment of patients with non-small cell lung cancer and cachexia: Results from a randomized, double-blind, placebo-controlled, multicenter study of Japanese patients (ONO-7643-04). Cancer 124, 606–616 (2018).
Nakanishi, Y., Higuchi, J., Honda, N. & Komura, N. Pharmacological profile and clinical efficacy of anamorelin HCl (ADLUMIZ(®)Tablets), the first orally available drug for cancer cachexia with ghrelin-like action in Japan]. Nihon Yakurigaku Zasshi 156, 370–381 (2021).
Kwon, M. et al. Prevalence and clinical significance of cancer cachexia based on time from treatment in advanced-stage head and neck squamous cell carcinoma. Head. Neck 39, 716–723 (2017).
Muthanandam, S. & Muthu, J. Understanding cachexia in head and neck cancer. Asia Pac. J. Oncol. Nurs. 8, 527–538 (2021).
McQuellon, R. P. et al. Supportive use of megestrol acetate (Megace) with head/neck and lung cancer patients receiving radiation therapy. Int. J. Radiat. Oncol. Biol. Phys. 52, 1180–1185 (2002).
Yeh, K. Y. et al. Omega-3 fatty acid-, micronutrient-, and probiotic-enriched nutrition helps body weight stabilization in head and neck cancer cachexia. Oral. Surg. Oral. Med Oral. Pathol. Oral. Radio. 116, 41–48 (2013).
Pottel, L. et al. Echium oil is not protective against weight loss in head and neck cancer patients undergoing curative radio(chemo)therapy: a randomized-controlled trial. BMC Complement. Altern. Med. 14, 382 (2014).
Thambamroong, T. et al. Efficacy of curcumin on treating cancer anorexia-cachexia syndrome in locally or advanced head and neck cancer: a double-blind, placebo-controlled randomized phase IIa Trial (CurChexia). J. Nutr. Metab. 2022, 5425619 (2022).
Rossetti, M. L., Steiner, J. L. & Gordon, B. S. Androgen-mediated regulation of skeletal muscle protein balance. Mol. Cell Endocrinol. 447, 35–44 (2017).
Wright, T. J. et al. A randomized trial of adjunct testosterone for cancer-related muscle loss in men and women. J. Cachexia Sarcopenia Muscle 9, 482–496 (2018).
Goldberg, J. E. & Schwertfeger, K. L. Proinflammatory cytokines in breast cancer: mechanisms of action and potential targets for therapeutics. Curr. Drug Targets 11, 1133–1146 (2010).
Paixão, E. et al. The effects of EPA and DHA enriched fish oil on nutritional and immunological markers of treatment naïve breast cancer patients: a randomized double-blind controlled trial. Nutr. J. 16, 71 (2017).
Khatri, U. et al. Selpercatinib mitigates cancer cachexia independent of anti-tumor activity in the HT1080 tumor model. Cancer Lett. 611, 217444 (2025).
Hsu, J. Y. et al. Erratum: non-homeostatic body weight regulation through a brainstem-restricted receptor for GDF15. Nature 551, 398 (2017).
Mullican, S. E. et al. GFRAL is the receptor for GDF15 and the ligand promotes weight loss in mice and nonhuman primates. Nat. Med. 23, 1150–1157 (2017).
Liu, M., Li, L., Jin, D. & Liu, Y. Nanobody-A versatile tool for cancer diagnosis and therapeutics. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 13, e1697 (2021).
Huang, Y. et al. GB18-06, a nanobody targeting GDF15, effectively alleviates weight loss and restores physical function in cachexia models. MAbs 16, 2416453 (2024).
Konrad, D. & Wueest, S. The gut-adipose-liver axis in the metabolic syndrome. Physiol. 29, 304–313 (2014).
Matsubara, Y. et al. Organ and brain crosstalk: the liver-brain axis in gastrointestinal, liver, and pancreatic diseases. Neuropharmacology 205, 108915 (2022).
Zaiss, M. M., Jones, R. M., Schett, G. & Pacifici, R. The gut-bone axis: how bacterial metabolites bridge the distance. J. Clin. Investig. 129, 3018–3028 (2019).
Gómez-Ambrosi, J., Rodríguez, A., Catalán, V. & Frühbeck, G. The bone-adipose axis in obesity and weight loss. Obes. Surg. 18, 1134–1143 (2008).
Gray, J. S., Wani, S. A. & Campbell, M. J. Epigenomic alterations in cancer: mechanisms and therapeutic potential. Clin. Sci. 136, 473–492 (2022).
Tseng, Y. C. et al. Preclinical investigation of the novel histone deacetylase inhibitor AR-42 in the treatment of cancer-induced cachexia. J. Natl Cancer Inst. 107, djv274 (2015).
Kottorou, A., Dimitrakopoulos, F. I. & Tsezou, A. Non-coding RNAs in cancer-associated cachexia: clinical implications and future perspectives. Transl. Oncol. 14, 101101 (2021).
Shi, X. et al. Circular RNA ANAPC7 inhibits tumor growth and muscle wasting via PHLPP2-AKT-TGF-β signaling axis in pancreatic cancer. Gastroenterology 162, 2004–2017.e2002 (2022).
Han, J., Meng, Q., Shen, L. & Wu, G. Interleukin-6 induces fat loss in cancer cachexia by promoting white adipose tissue lipolysis and browning. Lipids Health Dis. 17, 14 (2018).
Bodine, S. C. et al. Identification of ubiquitin ligases required for skeletal muscle atrophy. Science 294, 1704–1708 (2001).
Aiello, D., Patel, K. & Lasagna, E. The myostatin gene: an overview of mechanisms of action and its relevance to livestock animals. Anim. Genet. 49, 505–519 (2018).
Shintaku, J. et al. MyoD regulates skeletal muscle oxidative metabolism cooperatively with alternative NF-κB. Cell Rep. 17, 514–526 (2016).
Bailey, J. L., Hang, H., Boudreau, A. & Elks, C. M. Oncostatin M Induces lipolysis and suppresses insulin response in 3T3-L1 Adipocytes. Int. J. Mol. Sci. 23, 4689 (2022).
Johnen, H. et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 13, 1333–1340 (2007).
Fouladiun, M. et al. Body composition and time course changes in regional distribution of fat and lean tissue in unselected cancer patients on palliative care–correlations with food intake, metabolism, exercise capacity, and hormones. Cancer 103, 2189–2198 (2005).
Iguchi, H. et al. Involvement of parathyroid hormone-related protein in experimental cachexia induced by a human lung cancer-derived cell line established from a bone metastasis specimen. Int. J. Cancer 94, 24–27 (2001).
Cabedo Martinez, A. I. et al. Biochemical and structural characterization of the interaction between the siderocalin NGAL/LCN2 (Neutrophil Gelatinase-associated Lipocalin/Lipocalin 2) and the N-terminal domain of its endocytic receptor SLC22A17. J. Biol. Chem. 291, 2917–2930 (2016).
Garcia, J. M. et al. Anamorelin for patients with cancer cachexia: an integrated analysis of two phase 2, randomized, placebo-controlled, double-blind trials. Lancet Oncol. 16, 108–116 (2015).
Garcia, J. M., Friend, J. & Allen, S. Therapeutic potential of anamorelin, a novel, oral ghrelin mimetic, in patients with cancer-related cachexia: a multicenter, randomized, double-blind, crossover, pilot study. Support. Care Cancer 21, 129–137 (2013).
Golan, T. et al. LY2495655, an antimyostatin antibody, in pancreatic cancer: a randomized, phase 2 trial. J. Cachexia Sarcopenia Muscle 9, 871–879 (2018).
Jatoi, A. et al. A placebo-controlled, double-blind trial of infliximab for cancer-associated weight loss in elderly and/or poor performance non-small cell lung cancer patients (N01C9). Lung Cancer 68, 234–239 (2010).
Del Fabbro, E. et al. Effects of melatonin on appetite and other symptoms in patients with advanced cancer and cachexia: a double-blind placebo-controlled trial. J. Clin. Oncol. 31, 1271–1276 (2013).
Beijer, S. et al. Effect of adenosine 5’-triphosphate infusions on the nutritional status and survival of preterminal cancer patients. Anticancer Drugs 20, 625–633 (2009).
Cuvelier, G. D. et al. A randomized, double-blind, placebo-controlled clinical trial of megestrol acetate as an appetite stimulant in children with weight loss due to cancer and/or cancer therapy. Pediatr. Blood Cancer 61, 672–679 (2014).
Couluris, M. et al. The effect of cyproheptadine hydrochloride (periactin) and megestrol acetate (megace) on weight in children with cancer/treatment-related cachexia. J. Pediatr. Hematol. Oncol. 30, 791–797 (2008).
Laviano, A., Muscaritoli, M. & Rossi-Fanelli, F. Phase II study of high-dose fish oil capsules for patients with cancer-related cachexia: a Cancer and Leukemia Group B study. Cancer 103, 651–652 (2005).
Abe, K. et al. Effects of ω-3 fatty acid supplementation in patients with bile duct or pancreatic cancer undergoing chemotherapy. Anticancer Res. 38, 2369–2375 (2018).
Cheon, C. et al. Efficacy and safety of Sipjeondaebo-Tang for Anorexia in Patients with cancer: a pilot, randomized, double-blind, placebo-controlled trial. Evid. Based Complement. Altern. Med. 2017, 8780325 (2017).
de Clercq, N. C. et al. Fecal microbiota transplantation from overweight or obese donors in cachectic patients with advanced gastroesophageal cancer: a randomized, double-blind, placebo-controlled, phase II study. Clin. Cancer Res. 27, 3784–3792 (2021).
Acknowledgements
We thank all the lab members for critical reading and comments on the manuscript. We are grateful to Dr. Qin Liu and Ms. Ziyun Qin, who made substantial contributions to the preparation of this comprehensive review. We would like to thank the anonymous reviewers and the editorial team for their constructive comments and expertise, which have been very beneficial for improving the quality of our paper. This work was sponsored by grants from the National Key Research and Development Program of China (No. 2023YFC3402100 Bin Wang), the National Natural Science Foundation of China (NSFC Nos. 82192890 and 82192891 to Xiu-wu Bian, 82203318 to Zongsheng He), the China Postdoctoral Science Foundation (No. 2024M763883 to Xuan Zhang), the Science and Technology Innovation Key R&D Program of Chongqing (CSTB2023TIAD-STX0002 to Bin Wang), the Natural Science Foundation of Chongqing (Nos. CSTB2023NSCQ-LZX0156 to Bin Wang, CSTB2022NSCQ-MSX0880 to Zongsheng He, CSTB2024NSCQ-BSX0017 to Ke Li), and funding from the JinFeng laboratory to Bin Wang.
Author information
Authors and Affiliations
Contributions
Yuting Tan, Rui Xue, Yuwei Pan, and Zongsheng He collected and analyzed references and data and wrote the manuscript. Yuting Tan, Rui Xue, Yuwei Pan, Zongsheng He, and Xiao Hu designed and organized the figures and tables. Xiao Hu, Yaping L,i and Ke Li provided technical support. Xuan Zhang, Xiu-wu Bian, and Bin Wang proposed the review topic and supervised the study. Zongsheng He, Xuan Zhang, Xiu-wu Bian, and Bin Wang edited the manuscript and approved its submission. All the authors have read and approved the review article.
Corresponding authors
Ethics declarations
Competing interests
Xiu-wu Bian is the editorial board member of Signal Transduction and Targeted Therapy, but he has not been involved in the process of manuscript handling. The other authors declare no potential conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, Y., Xue, R., Pan, Y. et al. Cancer cachexia: molecular basis and therapeutic advances. Sig Transduct Target Ther 11, 16 (2026). https://doi.org/10.1038/s41392-025-02331-7
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-025-02331-7
