
Abstract
The tumor suppressor gene TP53 is the most frequently mutated gene in human cancers and has been a popular area of research in the field of oncology. The p53 protein, encoded by the TP53 gene, not only binds to many targeted genes but also regulates apoptosis, autophagy, cell cycle arrest, metabolism, senescence and the tumor immune microenvironment to suppress tumorigenesis. In recent years, an increasing number of new functions of p53 have been discovered, and p53-mediated tumor suppressor functions have been greatly expanded. Mutations in TP53 not only abolish its ability to suppress tumorigenesis but also confer carcinogenic properties to p53-mutant cells. Because of the prevalence of p53 dysfunction in various disease types, p53 has long been considered an attractive target for new anticancer drugs. However, drugs targeting p53 are still under investigation in early clinical trials and have not been approved for clinical use. This finding is consistent with the speculation that p53 is widely regarded as “undruggable.” Surprisingly, several novel therapeutic approaches targeting p53, including MDM2/4 antagonists, compounds that target specific p53 mutants or restore the wild-type function of the mutated p53 protein, p53-based genetic therapies and p53-based tumor immunotherapy, have been developed in recent years. Here, we present a review of the structure, inactivation, and roles of p53 in diseases. In addition, this review discusses the efforts to target diseases associated with p53 dysfunction and the challenges encountered in the clinical development of these approaches.
Similar content being viewed by others

Introduction
The p53 protein, encoded by the TP53 gene, is expressed in almost all human tissues and plays a pivotal role in cancer suppression as a tumor suppressor. The transcription factor activity of p53 modulates the expression of specific genes involved in the following processes: apoptosis, cell cycle arrest, metabolism, senescence, ferroptosis, and autophagy.1,2 Generally, TP53 is regarded as the “guardian of the genome” and is widely considered one of the most critical genes that protects humans from tumorigenesis.3 However, mutations in TP53 occur in most human cancers,4 which can abrogate the anticancer activity of TP53 and confer cancerogenic properties to mutant p53 proteins.5 Mutations in TP53 are thought to occur in more than 50% of all human malignancies.6,7 The TP53 mutation rate exceeds 70% in certain subtypes of cancers, including squamous esophageal cancer, small-cell lung cancer, squamous cell lung cancer, and complex karyotype acute myeloid leukemia (AML), and is lower in partial tumors such as lymphoma, melanoma, and overall AML.8,9,10,11
Because of its tumor suppressor function and universal mutation in human cancer, p53 is a highly appealing target for new anticancer therapies. Therapeutic strategies targeting TP53 mutations require reactivation of proteins. Nevertheless, major small-molecule drugs used for cancer therapy act by suppressing protein activity. Therefore, p53 is widely regarded as “undruggable”.12 Although a series of therapeutic approaches targeting p53 mutations began developing decades ago, little progress has been made. A small number of drugs have been tested in clinical trials, and none of these compounds have been approved for use by the Food and Drug Administration (FDA) or the European Medicines Agency (EMA). In recent years, a growing number of methods for p53-based therapy, such as the use of small molecules that can inhibit the interaction between p53 and its negative regulators, mouse double minute 2/4 (MDM2/4), small molecules that can reactivate the function of mutant p53, and drugs that target specific types of p53 mutants, have emerged. Compared with previous reviews published in recent years,1,2,11,13,14 this review has the following innovations. First, this review not only discusses the structure and systems biology of p53 but also introduces the application of designed p53 therapy approaches based on machine learning and deep learning (DL) algorithms. Second, this review provides a comprehensive and detailed analysis of the relationship between p53 and nontumour diseases and the treatment of nontumour diseases. More importantly, this review still provides a more critical assessment of conflicting findings, such as the different mechanisms of CP31398 and APR-246. Thus, the purpose of this review is to discuss the structure, inactivation, and roles of p53 in diseases and therapeutic strategies targeting p53 in cancer and noncancer diseases.
Structure, inactivation, and roles of p53 in diseases
p53 structural domains and their functional implications
p53 structural domains
The p53 protein, encoded by the TP53 gene located on chromosome 17p13.1, is composed of 393 amino acids (AAs) and contains a wide range of functional domains.15 Specifically, the p53 protein in humans is composed of five distinct domains: the transactivation domain (TAD), the proline-rich domain (PRD), the central DNA-binding domain (DBD), the tetramerization domain (TD), and the C-terminal regulatory domain (CTD) (Fig. 1a). The N-terminal domain (NTD) comprises a disordered TAD and a PRD.16 The TADs include TAD1 (AA residues 1–40) and TAD2 (AA residues 41–61), which regulate the transcriptional activities of several genes.17 The TAD is a disordered binding region for a larger cohort of interacting proteins, including molecules involved in the transcriptional machinery, the negative regulators MDM2/4, and the transcriptional coactivators p300/CREB-binding protein (CBP).15,18,19 Short fragments of approximately 20 residues undergo disordered-to-ordered changes upon binding to proteins.20,21 The MDM2 binding area of the p53 TAD overlaps with some of the binding regions for p300, which is critical for the transcriptional function of p53. Thus, the interaction with p300 prevents the MDM2-mediated proteasomal degradation of p53. In contrast, binding to negative regulators (MDM2/4) inhibits the interaction of p300 with components of the transcription machinery. Under stress conditions, p53 activation involves the phosphorylation of many N-terminal serine and threonine residues by distinct protein kinases.22,23 In general, TADs not only facilitate transcription initiation by combining with transcriptional coactivators and transcription machinery elements but also inhibit transcriptional activity by binding to negative regulators. The TADs are followed by a PRD (AA residues 62–94) that includes five PXXP (P: proline; X: any AA) motifs.15 This region plays a critical role in promoting cell cycle arrest and p53-associated death.24,25 Furthermore, this region combines with the Src homology 3 domain to perform its signal transduction function.26 In fact, the specific structure and functionality of the PRD are largely unclear.

The main domain structure of the full-length p53 protein and the structures of p53 and MDM2/4. a The p53 protein in humans is composed of five distinct domains: the transactivation domain (TAD, AA 1–61), the proline-rich domain (PRD, AA 62–94), the central DNA-binding domain (DBD, AA 95–292), the tetramerization domain (TD, AA 325–356), and the C-terminal regulatory domain (CTD, AA 357–393). A linker region (LR) links the DBD to the TD. AA: amino acid. b p53 monomer structure and 6 hotpots (PDB: 8F2I). c Structure of the human MDMX protein bound to the p53 tumor suppressor transactivation domain (PDB: 3DAB). d MDM2 bound to the transactivation domain of p53 (PDB: 1YCR). e MDMX is shown as a surface (PDB: 3DAB). f MDM2 is shown as a surface (PDB: 1YCR). This figure was created by BioRender.com
The domain following the PRD is the DBD (AA residues 95–292), which is the core domain of p53. The basic scaffold of the DBD contains an immunoglobulin-like β-sandwich and two large loops (L2 and L3).27 The DBD surface provided by the immunoglobulin-like β-sandwich can be divided into two structural motifs that interact with the minor groove and major groove of the target DNA. Zinc ions can stabilize L2 and L3. Thus, zinc deficiency leads to the loss of DNA-binding specificity by decreasing the thermodynamic stability and increasing aggregation tendencies.28 The stability of the human DBD is relatively poor, and it can dissolve at temperatures slightly above body temperature, which affects the stability of the full-length p53 protein.28,29 The DBD is capable of combining specific sequences of targeted DNA to promote transcriptional initiation and adjust the transcriptional processes of a series of genes functioning in different pathways.30,31 p53 regulates gene transcription through binding the response element (RE), which consists of two decameric half-site palindromes of the general form 5’-RRRCWWGYYY-3’ (R = A, G; W = A, T; Y = C, T) and is divided by 0–13 base pairs.32,33 Partial REs are clustered regions with more half-sites. Structural analysis has elucidated the molecular mechanism by which p53 recognizes and binds DNA. The Arg-248 residue in the L3 loop binds the DNA minor groove. Residues Arg-273 and Asp-281 are involved in DNA-backbone contacts. The Arg-280 residue stably interacts with guanine bases in the major groove region of the target DNA, whereas the interactions involving Lys-120, Cys-277, and Ala-276 are sequence specific.34 Upon DNA binding, the structures of the DBD and the DNA helix change. The interaction of the p53-DBD with the Bax RE results in DNA deformation, and its partial disordering around the spacer regions allows p53-DBD to unwind and compress this region to promote protein‒protein interactions.35
In addition, the DBD regulates the interactions of various proteins that can affect the activity of p53.11 53BP1 and 53BP2 were first identified to bind to p53 and interact with the p53-DBD via the L2 and L3 loops.36,37 Several viral oncoproteins, including the human papillomavirus (HPV) oncoprotein E6 large T antigen (LTag) of simian virus 40 (SV40), also interact with the p53-DBD.14,38 For example, the binding sites for HPV E6 and iASPP in p53 basically coincide; thus, iASPP may suppress HPV E6 binding to promote p53 degradation.39 Notably, the DBD is the most common location of p53 mutations.17
A linker region (LR) links the DBD to the TD, which in turn is followed by the CTD. The structure of the TD is considered a dimer of primary dimers. The monomeric TD is composed of a short β-strand and an α-helix. A primary dimer consists of two monomers. Two primary dimers form a four-helix bundle tetramer with D2 symmetry through their helices. The surface of the tetramer is stabilized mainly through hydrophobic interactions. Notably, although tetramers are generated at high protein concentrations, the truncation of either of the two key hydrophobic residues (Leu-344 and Leu-348) can shift the oligomerization state toward the formation of a stable dimer.15 A p53 biogenesis study suggested that dimer formation occurs simultaneously on polysomes, whereas tetramers are synthesized in solution after dimerization.40 R337C/H/P and L344P influence the synthesis and transcriptional activity of p53 tetramers and are involved in the occurrence of Li-Fraumeni syndrome and Li-Fraumeni-like syndrome.41 The G334V mutation promotes amyloid formation by inducing beta-dominated structural changes.42 In unperturbed cells, the p53 protein forms monomers, dimers, and tetramers, with dimers prevailing.43 Upon exposure to a series of stress conditions, such as DNA damage, oncogene activation, ribosomal stress, nutrient deprivation, and hypoxia, TD (AA residues 325–356) is responsible for the tetramerization of full-length p53 (p53 functions as a tetramer in the process of DNA binding). Moreover, TD contains nuclear export signal sequences that are responsible for the subcellular location of p53.44 Like the NTD, the CTD (AA residues 357–393) is an unstructured region that plays critical roles in several processes, including cofactor recruitment, nonspecific DNA binding, protein stability, and the regulation of subcellular location.45 Notably, the CTD possibly undergoes local disordered-to-ordered changes upon binding to some proteins or nonspecific DNA.46,47 In addition, the CTD is necessary for contact with negative regulators (MDM2/4) and to induce many posttranslational modifications, such as acetylation, phosphorylation, methylation, and ubiquitination, which adjust its functionality and stability.48 Currently, the function of the CTD and the roles of individual modifications have not been fully elucidated, and controversial results associated with specific mechanisms have been reported. For example, the acetylation of CTD lysines regulates p53 transcriptional activity by increasing the binding of sequence-specific DNA to short oligonucleotide probes and long DNA fragments both in vitro and in vivo.49 However, the extent or whether this elevated effect can be observed seems to rely on the length and structural background of the DNA.50,51
Biological function of p53
Under physiological conditions, the expression of p53 remains low because of the interaction between p53 and MDM2, which promotes p53 proteasome degradation through its E3 ubiquitin ligase activity.52 Cellular stress, such as DNA damage, hypoxia, and oncogene activation, disturbs the interaction between p53 and MDM2 and inhibits p53 degradation, resulting in p53 accumulation. Accumulated p53 is stabilized and activated through posttranslational modifications, including methylation, phosphorylation and acetylation,53 which regulate gene transcription and affect a series of signaling pathways involved in apoptosis, cell cycle arrest, senescence, autophagy, ferroptosis, cellular metabolism and the tumor immune microenvironment (TME)54,55,56 (Fig. 2). Notably, DNA damage is one of the most potent triggers of p53 activation. Upon DNA damage, p53 is activated to induce cell cycle arrest, thereby providing the cell with sufficient time, resources, and energy to repair damaged DNA. If the damage is severe, p53 can also be activated to induce apoptosis and senescence to eliminate damaged cells. Ultimately, whether p53 induces cell cycle arrest, apoptosis, or senescence depends on the type of cell and the nature of the DNA damage.

A simplified scheme of p53 signaling pathways. Under normal circumstances, the p53 protein is tightly regulated by the negative regulator MDM2/4 to maintain low levels of p53 protein expression. The interaction of MDM2/4 promotes the proteasomal degradation of p53 through ubiquitination. Under stress conditions such as DNA damage, hypoxia, and the expression of oncogenes, p53 is activated and stabilized through posttranslational modifications. p53 subsequently accumulates into tetramers in the nucleus and binds to many target genes to regulate a series of processes, including cell cycle arrest, apoptosis, autophagy, senescence, DNA repair, and metabolism. This figure was created by BioRender.com
Cell cycle arrest
At low doses of cell stress, p53 can activate the transcriptional target gene cyclin-dependent kinase inhibitor 1A (CDKN1A) to induce cell cycle arrest. The p21 protein encoded by CDKN1A is a cyclin-dependent kinase (CDK) inhibitor that binds to CDKs to inhibit Rb phosphorylation. Hypophosphorylated Rb combines with E2F (a transcription factor), which inhibits E2F transcriptional activity and results in cell cycle arrest.57 p53 transcriptionally regulates PCNA and 14-3-3d expression to suppress DNA replication and induce G2/M phase arrest, respectively.58,59 Furthermore, as a p53 target gene and member of the growth arrest gene family, GADD45 expression is regulated by p53 to induce G2 phase arrest.60
Senescence
Senescence is the permanent arrest of the cell cycle, and it is well accepted as one of the major barriers to inhibiting tumorigenesis. Senescence is usually triggered by various stress signals, which elicit abnormal cells. The most well-studied pathways associated with cellular senescence are the p53/p21CIP1 and/or p16INK4A/Rb tumor suppressor pathways.61,62 The acetylation of p53 at certain sites hinders the phosphorylation of several serines located in the p53 NTD, which activates genes with high p53 affinity, including CDKN1A. p21CIP1 is encoded by CDKN1A and is a critical component of the senescence pathway.63 Once activated, p21CIP1 promotes senescence by regulating the expression of many genes.64 CDK1 and CDK2 are activated by ATM and ATR in response to UV exposure, and p53 and p21CIP1 are then activated to induce senescence.65 The p16INK4A/Rb pathway also plays a critical role in senescence. CDK4/6 are inactivated by p16INK4A, promoting the accumulation of phosphorylated Rb and the inactivation of E2F transcription, which induces senescence.66 In addition, plasminogen activator inhibitor-1 (PAI-1), a p53 target gene, is a marker of senescence and is necessary for replicative senescence. The suppression of PAI-1 induces replicative senescence.67 Notably, the p53/p21CIP1 and p16INK4a/Rb pathways are not independent but interact with each other through many crosstalk mechanisms. Prior to the upregulation of p16, p53 inactivation can inhibit the induction of senescence. However, the downregulation of p53 cannot prevent cell cycle arrest or the induction of senescence after the overexpression of p16.68
DNA repair
Cellular stress activates cell cycle checkpoints, leading to p53 stabilization. With the help of cell cycle arrest, DNA repair pathways are subsequently activated to inhibit cell cycle progression, providing a window of repair for the cell.69 Proteins, including PCNA, XPC, DDB2, RRM2B, PIG3 and POLH, are activated to promote DNA repair.70,71 Radiosensitive tissues such as the thymus, spleen, and lymphoid organs exhibit prolonged p53 signaling and transcription of p53 target genes after radiation. However, in epithelial cells such as those in the small and large intestines, more transitory p53 activation is associated with transient expression of its target genes.72 DNA damage induces a robust DNA checkpoint response in the germline cells of nematodes.73 When DNA double-strand breaks (DSBs) are induced by ionizing radiation (IR), mitotically dividing germline cells respond by modulating DNA checkpoint activity to induce cell cycle arrest. After DNA damage repair is complete, germline stem cells resume proliferation. Notably, only cells in the late stages of meiosis undergo apoptosis in response to DNA damage. Under DNA damage conditions, p53 in mammalian cells is still activated via the DNA damage checkpoint signaling pathway.74 Currently, p53 can regulate a variety of genes to induce cell cycle arrest. To a large extent, p53 maintains genomic stability by causing death or growth arrest in damaged cells. When cells undergo DNA damage before entering the S phase, p53 induces cell cycle arrest in the G1 phase through the activation of CDKN1A.75 Under the influence of DNA damage, p53 not only promotes DNA repair by inducing cell cycle arrest but also directly regulates multiple DNA repair pathways. Nucleotide excision repair (NER) eliminates a series of helix-distorting lesions that are mostly induced by UV irradiation,76,77 whereas base excision repair (BER) primarily acts on the modification of oxidative bases,78,79 and mismatch repair (MMR) scans for incorrectly inserted nucleotides during replication.80 DSBs that are typically caused by IR are repaired by nonhomologous end joining (NHEJ) or by homologous recombination (HR).81,82 RECQ helicases play important roles in maintaining genomic stability during recombination repair and replication.
Glycometabolism
In addition to regulating cell cycle arrest and DNA repair, p53 regulates energy metabolism, mainly glycometabolism, to inhibit the proliferation of damaged cells with DNA breaks. The phenomenon by which glucose uptake and lactate generation in cancer cells increase is called the Warburg effect.83 p53 regulates glucose transporters (GLUTs) that promote glucose transport across the cellular plasma membrane to inhibit glucose uptake.84,85 p53 also regulates several critical enzymes to regulate glucose metabolism.86,87 For example, p53 decreases fructose-2,6-bisphosphate levels by inhibiting PFKFB3 and PFKFB4 expression, resulting in glycolysis suppression.88 p53 also regulates several signaling pathways correlated with glycolysis suppression to inhibit glycolysis. p53 negatively affects the expression of PTEN and Parkin, which are regulators of the PI3K/AKT signaling pathway and suppress glycolysis.89,90
Lipid metabolism
p53 also regulates lipid metabolism through different pathways to inhibit stressed cell proliferation and subsequently tumor progression. p53 combines with the promoter region of SREBP-1, which is a transcription factor that regulates various genes involved in lipid metabolism, leading to the inhibition of SREBP-1 expression.91 p53 suppresses cholesterol esterification by decreasing the expression of SOAT1 and USP19.92 p53 also activates malonyl-CoA decarboxylase (MCD), PANK1, and CPT1C to accelerate fatty acid oxidation.93,94,95 The growth of tumor cells requires abundant ammonia generated via amino acid metabolism. p53 inhibits three critical enzymes of the urea cycle (ARG1, CPS1, and OTC) to regulate the ammonia content and further suppress tumor growth.96
Apoptosis
When cells are under severe stress conditions, such as DNA damage, which cannot be repaired through cell cycle arrest, p53 induces apoptosis to eliminate damaged cells (Table 1). Apoptosis is programmed cell death and is precisely regulated at the gene level, leading to the efficient elimination of damaged cells.97 The mechanisms of apoptosis are highly complex and involve a cascade of signaling pathways. Apoptosis can be triggered through the caspase-mediated extrinsic pathway or the intrinsic pathway. Both pathways ultimately lead to the activation of caspases, which in turn cause morphological and biochemical changes in the cell.98 In extrinsic-mediated pathways, p53 regulates the expression of Fas and KILLER/DR5 on the cell membrane, and then, caspase-8 is activated, resulting in apoptosis.99 In intrinsically mediated pathways, Bcl-2 family members expressed in the mitochondria mainly induce apoptosis. Proapoptotic proteins, including Bak, Bax, Bid, Puma, and Noxa, are transactivated by p53, which induces apoptosis.35,100,101 As a transcription factor, p53 can transcriptionally activate a series of proapoptotic genes, such as Puma, Noxa, and Bax, to induce apoptosis.35,101 p53 can induce apoptosis through direct protein‒protein interactions. For example, after cell stress, p53 interacts with the proapoptotic protein Bak, which promotes the oligomerization of Bak and the release of cytochrome c from mitochondria to induce apoptosis.102 p53 also binds to several antiapoptotic proteins, including Bcl-2, Mcl-1, and Bcl-xL, indirectly inducing apoptosis.27 In addition, p53 still promotes the activation of several genes that are not part of the extrinsic or intrinsic apoptotic pathways, and these genes may regulate the response of cells to death-inducing factors. For example, p53 can regulate the expression of various microRNA species, such as miR34a, which targets Bcl-2.103,104 Thus, p53 can reduce the level of Bcl-2 to increase the sensitivity of cells to apoptotic stimuli. As another transcriptional target of p53, the specific function of Zmat3 remains unclear, but it has been shown to influence the sensitivity of cells to apoptotic stimuli.105,106 In addition, p53 also induces mitochondrial apoptosis in a transcription-independent manner.54 The observation that deficiency of Puma (or combined deficiency of Puma and Noxa or Puma and p21) promoted the development of C-myc-driven lymphoma,107,108 which suggested that p53-mediated apoptosis could exert obvious tumor-suppressive effects.
Autophagy
Autophagy is an intracellular process by which damaged proteins and cytoplasmic organelles are degraded through lysosomal hydrolases, thus maintaining biological activities and cell viability in the context of a variety of stressors, such as nutritional deficiency (Table 1)109. Once the autophagy process is initiated, cytoplasmic components are embedded into an isolation membrane and form autophagosomes with enclosed double-membrane structures. Subsequently, the autophagosomes fuse with lysosomes to produce autolysosomes, where the contents are degraded to provide nutrients.110,111 Many links exist between p53 and autophagy. Chromatin immunoprecipitation sequencing and RNA sequencing identified a variety of autophagy genes (Atgs), including Atg2, Atg4, Atg7, and Atg10, as direct p53 target genes.112 Interestingly, p53 regulates autophagy in a dual manner, where the pool of nuclear p53 protein promotes autophagy through a transcription-dependent process. p53 is activated upon genotoxic and oxidative stress, which activates AMP-responsive protein kinase (AMPK) through the tuberous sclerosis (TSC) 1/TSC2 complex and inhibits mTOR activity to promote autophagy.113 The protein kinase ULK1, a crucial regulator of autophagy initiation, is also phosphorylated and activated by AMPK, which induces autophagy by regulating pivotal downstream effectors such as Beclin-1 through phosphorylation.114,115 Sestrin-1 and Sestrin-2, which are transcriptional target genes, activate AMPK and suppress mTOR activity to induce autophagy by phosphorylating TSC2 and stimulating its GAP activity.116 As a p53 target gene, damage-regulated autophagy modulator (DRAM) is an effector of p53-mediated death and encodes a lysosomal protein that promotes autophagy.117 However, the cytoplasmic p53 protein inhibits autophagy in a transcription-independent manner.118 One study showed that p53 regulates autophagy through LC3.119
Autophagy and apoptosis play critical roles in maintaining cellular and organismal homeostasis. Ideally, both autophagy and apoptosis can inhibit the development of tumors, as autophagy eliminates tumor cells, and apoptosis suppresses the survival of tumor cells. Autophagy exerts cytoprotective effects through the inhibition of DNA damage-induced mutations, which can promote carcinogenesis. However, as cancer progresses, this stress-relieving property of autophagy is utilized by tumor cells to meet their extensive metabolic and nutritional requirements necessary for proliferation and survival. The interaction between apoptosis and autophagy is a complicated process and is a main determinant of cell fate under stress conditions, which is dependent on the type of cell and duration of stress. The link between the apoptosis and autophagy pathways can be attributed to the fact that autophagy decreases the levels of proapoptotic proteins in cells, increasing the threshold of stress required for the pressure threshold required for apoptosis to induce cell death. The survival mechanism of autophagy is that cells adapt to stress stimulation and continue to survive, avoiding cell death through apoptosis.120 Conversely, the cascade of caspase activation correlated with apoptosis inhibits the autophagic machinery.121,122 However, in the presence of sublethal levels, stress can cause autophagy to exceed a critical threshold for apoptosis, inducing an apoptotic response. Once apoptosis is initiated, autophagy ceases.121
Ferroptosis
In recent years, a series of studies have shown that p53-associated metabolic activity is also important for its antitumor activity.123,124 p53 expression modulates multiple metabolic pathways to respond to cellular stress and maintain metabolic homeostasis.125 Furthermore, gain-of-function (GOF) mutant p53 proteins in tumor cells induce tumor progression through metabolic reprogramming.126 Ferroptosis is a novel form of cell death that differs from previously elucidated methods of cell death. p53 expression suppresses the expression of SLC7A11, which is a critical element of the cystine–glutamate antiporter that represses cystine uptake.124 As an acetylation-defective mutant, p533KR cannot induce apoptosis, cell cycle arrest, or senescence, but it can regulate the expression of SLC7A11 and induce ferroptosis mediated by reactive oxygen species (ROS).124 SAT1, a rate-limiting enzyme in polyamine catabolism, has been identified as a transcription target of p53.87 SAT1 expression induces lipid peroxidation and ferroptosis through ROS-mediated stress. Furthermore, the expression of SAT1 is associated with arachidonate 15-lipoxygenase (ALOX15) levels, and PD146176, an ALOX15 inhibitor, suppresses ferroptosis induced by SAT1.127 H2Bub1 expression has been reported to activate SLC7A11. p53 expression negatively affects the expression level of H2Bub1 through acceleration of the nuclear transcription of the deubiquitinase USP7 in an independent manner, which further promotes ferroptosis.123
Tumor immune microenvironment
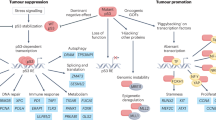
Recently, several studies have shown a correlation between p53 expression and the efficacy of tumor immunotherapy, which also indicates that the status of p53 affects the TME128,129,130 (Fig. 3). In other words, functional wild-type p53 promotes a cancer-suppressing TME in tumor cells, whereas a decrease in or loss of wild-type p53 promotes a cancer-promoting TME in tumor cells. Moreover, partial mutant p53 proteins that acquire GOF function further limit the ability of the immune system to kill tumor cells. Specifically, wild-type p53 expression can decrease the expression of PDL1 by increasing the expression of miR34a and promoting the expression of the natural killer (NK) cell-activating ligands UL16-binding protein 1 (ULBP1) and ULBP2.131,132 Subsequently, p53 exerts transcriptional effects through the binding of targeted genes to increase the killing ability of cytotoxic T cells and NK cells.131 In addition, wild-type p53 expression can enhance antitumor effects through regulating the secretion of cytokines and accelerating macrophage polarization toward the M1 phenotype.133 Notably, M1 macrophages increase the lethality of the TME against tumor cells. In contrast, M2 macrophages enhance the ability of the TME to protect tumor cells. One study suggested that p53 activation altered the TME by activating endogenous retroviruses to increase the sensitivity of tumor cells to immune checkpoint inhibitors.133 In addition, p53 status can affect innate immune signaling pathways (Fig. 4). Specifically, cytoplasmic DNA is recognized by the DNA-binding protein cyclic GMP–AMP synthase (cGAS), which facilitates cGAS homodimerization and the production of the second messenger cyclic GMP–AMP (cGAMP).134,135 The endoplasmic reticulum-resident stimulator of interferon genes (STING) is activated by the cGAMP molecule and then moves to the Golgi to form a complex with IFN regulatory factor 3 (IRF3) and TANK-binding kinase 1 (TBK1). Subsequently, IRF3 is phosphorylated by TBK1 in the complex, which promotes IRF3 homodimerization and translocation to the nucleus to further regulate the expression of a series of genes.136 The activation of the cGAS/STING pathway results in the production of type I interferons (IFNs) and innate immune signaling.134 Innate immune signaling is crucial for tumor suppression because it acts through cell-autonomous and noncell-autonomous mechanisms.137,138 Mutant p53 interacts with TBK1, disrupts the downstream cGAS/STING signaling pathway, and prevents the phosphorylation of IRF3. More importantly, mutant p53 induces immune evasion by inhibiting the activity of IRF3 in vivo.128 Since mutant p53 changes immune cell infiltration and phenotypes, we can overexpress TBK1 to reactivate IRF3, which disrupts mutant p53 binding to TBK1 and activates the immune system to suppress tumor growth.128 These findings provide a new therapeutic idea for mutant p53-expressing tumors whose cGAS/STING signaling pathways are altered. However, other (TBK1-independent) activities of mutant p53 may also affect the tumor microenvironment.139,140 Compared with either drug monotherapy, combination therapy with PDL1 and APG-115 resulted in increased antitumor effects, whereas this effect was not observed in Trp53-knockout mice, which further demonstrated that p53 activation is essential for the TME.141

p53 can regulate the tumor immune microenvironment to suppress tumorigenesis. a The tumor immune microenvironment associated with wild-type p53 plays an antitumor role. Wild-type p53 can increase the expression levels of miR34a and UL16-binding protein 1/2 (ULBP1/2). miR34a reduces PDL1 expression levels through inhibiting PDL1 mRNA translation and promoting PDL1 degradation, which enhances CD8+ T-cell-mediated killing. ULBP1/2, as activating ligands of natural killer (NK) cells, can enhance the cytotoxicity of NK cells. Conversely, wild-type p53 expression can promote the secretion of antitumor cytokines to inhibit tumorigenesis. b In TP53-mutated cells, downregulation of miR34a and ULBP1/2 expression decreases the killing ability of CD8+ T cells and NK cells. Mutated p53 proteins promote the secretion of exosomes that transform M1 macrophages (antitumor cells) into M2 macrophages (protumor cells) by delivering microRNAs such as miR-1246. In addition, cytokines secreted by protumor cells can attenuate the antitumor response. This figure was created by BioRender.com

The activity status of the cGAS/STING pathway under the regulation of wild-type versus mutant TP53. a Normally, the activation of the cGAS/STING pathway induces type I interferons (IFNs) and initiates immune signaling pathways to suppress tumorigenesis. b Mutated p53 can disrupt the downstream cGAS/STING signaling pathway to promote tumorigenesis and immune evasion. This figure was created by BioRender.com
Inactivation of p53
Mechanisms of mutation and aggregation
TP53 somatic mutations
Among somatically mutated genes, TP53 mutations are the most common.6 Mutations in several tumor suppressor genes, including APC, VHL, and RB1, are largely nonsense mutations or deletions, which reduce the expression of proteins. Nevertheless, missense mutations constitute the primary type of TP53 mutation11 (Fig. 5a Data are derived from the TP53 database (http://p53.fr/tp53-database/mutation-database)). Most mutations in TP53 are consequential, such as a single amino acid substitution or the emergence of an altered protein or faulty protein. Although TP53 mutations are widespread in many tumor types, the TP53 mutation rates differ across different organs (Fig. 5b). In terms of the overall overview of mutant p53 in cancer, the ovary is the most common organ for demonstrating TP53 mutations, and the kidney has the lowest mutation rate. Exons 5, 7, and 8 are the most common sites for mutations in TP53 (Fig. 5c). Although missense mutations can occur throughout the molecule, they most frequently occur in the DBD.142,143 The most frequent mutation locations of the DBD are known as “hotspots,” which contain codons G245, R249, R248, R282, R273, Y220, and R175144 (Fig. 1b).

TP53 mutation status in cancer. The data were derived from the TP53 database. (http://p53.fr/tp53-database/mutation-database) a Variant type and proportion of TP53. b TP53 mutation rates in different organs. c Distribution of exons and introns with TP53 mutations. This figure was created by BioRender.com
Somatic mutations in TP53 can be loosely divided into two types: DNA contact mutations and structural mutations.17 DNA contact mutations such as R248W, R273H, and R273C affect the interaction between p53 and DNA by affecting crucial DNA interaction sites. Mutations in important arginine residues located on the p53-DNA-binding surface facilitate the loss of transcriptional activity and sequence-specific DNA binding.28,145 Structural mutations such as R175H, R249S, and G245S decrease the thermodynamic and kinetic stability of the DBD by disturbing its structure, resulting in a decrease in the protein melting temperature and the emergence of unfolded proteins at body temperature.48 In addition, several structural mutations not only destabilize proteins but also impair the interactions between DNA and proteins. p53 mutations affect protein dynamics, further affecting drug binding. For example, the Y220C structural mutation significantly reduced p53 stability, leading to massive unfolding of the protein at physiological temperatures,3 exposing new hydrophobic pockets and providing potential binding sites for drug design. Thus, drugs targeting the Y220C mutation could stabilize p53 in its proper folded state and restore its wild-type function by binding to these pockets.
Notably, mutations in TP53 not only change its conformation but also cause p53 aggregation, and p53 aggregation in cells is more consequential than conformational changes.146,147 Mutations reduce the structural stability of p53, resulting in the exposure of adhesion sequences located in the hydrophobic core and p53 aggregation.148 Because of various experimental circumstances, the DBD can aggregate in a variety of morphologies, such as prion-like, amorphous, and fibrillar morphologies, after it unfolds.149 The dynamics of aggregation are unique. The aggregates participate in initiating processes in which molecules of p53 widely unfold and then aggregate, resulting in the emergence of amyloid-containing structures.150,151 p53 aggregation not only causes the loss of DNA recognition capacity and loss of the proapoptotic function of p53 but also promotes GOF effects.152,153 The classification of p53 as an amyloid protein is relatively novel, and amyloid proteins can increase the invasiveness of cancers.154 Therefore, for cancers with p53 aggregates, the development and exploration of molecules that can stabilize the folded structure, suppress aggregation, or disrupt the formation of aggregates are of considerable therapeutic interest.
In general, the stabilization of mutant p53 proteins is considered a tumor-specific event. A previous study simulated the process and functions of mutant p53 proteins in vivo via mutant p53 reporters and traced the precancerous clones in nontumor tissue.155 Mutant p53 cannot inhibit tumorigenesis because various mutant p53 proteins cannot activate genes targeted by wild-type p53. In addition, several p53 mutations induce the expression of oncogenic proteins, resulting in a dominant-negative (DN) effect on wild-type p53.156,157 The DN effect is exerted via the formation of mixed tetramers in heterozygous cells, which decreases the function of wild-type p53.157 The DN effect may play a critical role in the phenotypic diversity of tumors. In addition to loss-of-function (LOF), several core domain mutants also result in GOF. GOF has been reported to be correlated with processes such as uncontrolled cell proliferation, tumor metastasis, antitumor drug resistance, cancer invasion, and apoptosis inhibition.1,158 Notably, only some p53 mutations produce GOF. The probable factors by which mutant p53 attains new abilities are the upregulation of the expression of chromatin-regulating genes, including MLL1, MOZ, MLL2, acetyltransferase, and methyltransferases, and the regulation of other transcription factors, such as SP1, SREPB, p63, and p73.156,159 As mentioned above, TP53 mutations not only lose the ability to suppress tumorigenesis but also promote cancer development.
Since autophagy and apoptosis are the two primary cell death pathways with profound implications for cancer cell fate, we illustrate here how p53 mutations affect autophagy and apoptosis. Overall, mutant p53 promotes cancer cell survival under tumor- and therapy-associated stress conditions by suppressing autophagic and apoptotic responses. p73 plays an important role in the sensitivity of cells to anticancer drugs. Mutated p53 promoted chemotherapy resistance in head and neck cancer (HNSC) tumors by inhibiting the p73-mediated proapoptotic program.160 Mutated p53 inhibited the apoptosis of tumor cells with high p53 levels by inhibiting caspase-9 activity.161 Mutated p53 still binds to caspase-3 and inhibits its activity to further inhibit apoptosis.162 Mutant p53 has been proven to inhibit autophagy via different mechanisms.163,164,165,166 Mutant p53 inhibits AMPK activity in two different ways. The first process involves protein‒protein interactions between mutant p53 and the AMPK complex. The DBD of mutant p53 interacts with the AMPKα1 and AMPKα2 subunits, whereas the NTD disturbs the interaction between AMPKα and its upstream kinase LKB1 to inhibit autophagy by preventing AMPK phosphorylation and activation.163 The second pathway involves the transcriptional inhibition of AMPK activators such as Sestrin-1/Sestrin-2 by mutant p53. Sestrin-1 and Sestrin-2 can interact with the AMPK complex and promote its phosphorylation.164 In other words, Sestrin-1 and Sestrin-2 are conformational AMPK activators. Mutant p53 inhibits autophagy by decreasing the mRNA or protein levels of Sestrins, preventing the activation of the AMPK signaling pathway and the formation of autophagic vesicles.165 Mutant p53 also inhibits autophagy by stimulating mTOR signaling.114,165 The main causes of autophagy inhibition by mTOR are Ser757 phosphorylation and ULK1 suppression, which perturb the interaction between ULK1 and AMPK.114,166 Mutant p53 affects the mTOR signaling pathway by reducing the level of Beclin-1 phosphorylation to suppress autophagy.165 In addition, the machinery that inhibits autophagy involves Atgs. In regulatory regions of the Atg12 promoter, mutant p53 can recruit the nuclear factor-kappa B (NF-κB) p50‒p50 homodimer to form a mutant p53‒p50 complex that inhibits Atg12 and blocks the autophagic process.165 Autophagy also has an important influence on the stability of mutant p53. The overexpression of critical autophagic genes exhausts the levels of the mutant p53 protein, and treating cells with autophagy inhibitors increases the stability of mutant p53.167 The finding that autophagy decreases the stability of mutant p53 suggests that we may be able to treat tumors with mutant p53 by developing novel drugs or natural compounds that are able to trigger autophagy initiation.
Posttranslational modifications (PTMs), including phosphorylation, acetylation, methylation, and ubiquitination, play key roles in the regulation of p53.168,169 In the case of mutant p53, these modifications may further alter its functional and kinetic properties, thereby affecting its behavior in tumorigenesis and progression.170 For example, ubiquitination regulates p53 degradation. The ubiquitination level of mutant p53 may affect its accumulation and stability in cells, thereby affecting its tumor-promoting function. However, the lack of a thorough understanding of the role of PTMs in shaping the behavior of p53 mutants limits the development of more precise therapeutic strategies. We can change the conformation and function of mutant p53 via pharmacological intervention in specific posttranslational modification pathways. For example, inhibiting the activity of certain kinases can reduce the phosphorylation of mut-p53, thereby reducing its stability.
TP53 germline mutations
Germline mutation of TP53 in the heterozygous LOF form usually presents with a rare autosomally dominant genetic disease with a significantly increased risk of cancer development known as Li-Fraumeni syndrome (LFS). LFS was first discovered in 1969 and is characterized by the early onset of breast cancer as well as various sarcomas, adrenocortical carcinomas, gliomas, leukemias, and other cancers, especially in children and early adulthood.171 Although germline TP53 tumor suppressor gene mutations are linked to LFS, it is believed that 20–30% of people with this disorder do not have this mutation.172 TP53 LOF variants are linked to more than LFS variants on the basis of the affected domain and the severity of the mutation. Defective p53 activity is increasingly implicated in the etiology of several additional uncommon genetic disorders, such as Diamond–Blackfan anemia (DBA), CHARGE syndrome, and ATR-Seckel syndrome.173,174,175 DBA is an uncommon type of congenital intrinsic erythroid hypoplasia and was identified as the first human ribosomopathy in 2005.176 The hallmark of DBA is defective ribosomal RNA (rRNA) maturation caused by ribosomal protein (RP) gene mutations.177,178 CHARGE syndrome (coloboma, atresia of the choanae, heart defects, growth and mental retardation, ear anomalies, and genital anomalies) is an autosomal dominant genetic disease with a low incidence and is usually diagnosed in the neonatal or prenatal period because many congenital abnormalities and malformations have been identified.179,180 Deficiency or point mutation of chromodomain helicase DNA-binding protein 7 (CHD7), a chromatin remodeler with adenosine triphosphatase (ATPase) activity, is the causative factor of CHARGE syndrome.181 ATR plays a critical role in the response to DNA damage and the development of somatic cells.182 ATR-Seckel syndrome is an autosomal recessive disease that is characterized by developmental delay and remarkable microcephaly. The prerequisites for the diagnosis of ATR-Seckel syndrome are clinical characteristics, including obvious microcephaly, a proportionate short stature, distinctive facial features, and mental retardation.183 They can also lead to myelodysplastic syndrome (MDS) with multiple hits (multiple hits), which is consistent with the biallelic targeting of TP53 inactivation. This phenomenon is associated with increased blast counts, immunodeficiency, and increased risk of leukemic transformation (myeloid neoplasms) with poor prognosis.184 Additionally, there is evidence that germline p53 dysfunction can induce inflammation in a way that is dependent on the NF-kB pathway.185,186 Importantly, this immune defect and immune dysregulation demonstrated that tumorigenesis could not be caused only by p53 deletion. In many precancerous or inflammatory situations, elevated inflammation, oxidative stress, and p53 are correlated. Additionally, there is evidence that, in contrast to a matched group of family members without TP53 mutations, cancer-free LFS patients present clinical indicators of elevated oxidative stress and autoimmunity.187 Toll-like receptor (TLR) expression is regulated by p53, which also affects the immunological response in germline TP53-mutated patients.188 It is now known that TLRs, which are pattern recognition receptors involved in the innate immune response and may react to both endogenous and exogenous ligands, play a part in adaptive immunity. Moreover, TLRs have complicated functions in cancer, exhibiting both pro- and anticancer activity. There is some evidence that TLR signaling actively contributes to carcinogenesis during chronic inflammation, especially in relation to tumor angiogenesis, proinflammatory cytokine and chemokine production, and immune surveillance evasion.188
Germline GOF-heterozygous variants have also been reported in the TP53 gene. In 2018, a study reported two unrelated individuals who exhibited bone marrow failure, anemia, progressive thrombocytopenia, and hypogammaglobulinemia at 2 months (individual 1) and 15 days (individual 2) after birth.189 An analysis of the bone marrow revealed selective erythroid and slight B-cell hypoplasia. Other syndromic traits were observed in both patients, including seizures, developmental delay, cognitive impairment, microcephalism, hypogonadism, dental abnormalities, and reticular skin pigmentation. The anemia of individual 1 spontaneously resolved when he was 13 years old, indicating a clonal genetic reversion event. However, the bone marrow of individual 1 had minor trilineage hypoplasia, and his platelet levels steadily decreased. Genetic analysis of these patients revealed identical protein truncation with a loss of 32 residues from the C-terminal end, and surprisingly, more p53 exhibited transcriptional and functional p53 activity than controls did. This resulted from the loss of the domain required for the binding of negative transcriptional regulators (MDM2).189
In conclusion, TP53 germline mutations lead to a wide range of diseases by influencing various genes and signaling pathways. Understanding the functional effects of different mutations is critical for personalized treatment and genetic counseling, but understanding these effects is challenging because of the rarity of mutations in many individuals.
Interactions with negative regulators (MDM2/MDM4)
Wild-type p53 interacts with its negative regulators, which mainly include MDM2/4 (Fig. 1c-f), to maintain its low expression.190,191 The C-terminus of MDM2 is crucial for export of wild-type p53 from the nucleus to the cytoplasm, where it is degraded via E3 ubiquitin ligases.192,193 Furthermore, the N-terminus of MDM2 combines with the DBD of p53, which suppresses the transcriptional activity of wild-type p53.194 The interaction by which p53 activates MDM2 and regulates the expression of negative p53 regulators occurs in the p53-MDM2 negative feedback loop. The structure of MDM4 is similar to that of MDM2; therefore, MDM4 is considered the structural homolog of MDM2. MDM4 maintains p53 at low levels through several mechanisms. First, MDM4 binds directly to the N-terminus of p53 and inhibits its transcriptional activity.195 Second, MDM4 promotes p53 degradation mediated by MDM2; unlike that of MDM2, the E3 ubiquitin ligase activity of MDM4 is deficient. However, MDM4 can potentiate the ubiquitin activity of MDM2 by interacting with MDM2 and forming dimers.196,197 Third, the overexpression of MDM2/4 results in the inactivation of p53, which has been observed in various human tumors.198,199
p53 in other pathological settings
p53 not only plays an important role in the occurrence and development of tumors but is also closely related to many nontumor diseases, such as neurodegenerative diseases (NDDs) and metabolic diseases (Fig. 6).

p53-related diseases, including tumors and nontumor diseases. Nontumor diseases include neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease, and Huntington's disease, and metabolic diseases, such as obesity, diabetes, nonalcoholic fatty liver disease (NAFLD), myocardial infarction, and diabetic cardiomyopathy. This figure was created by BioRender.com
Neurodegenerative diseases
NDDs are a group of heterogeneous diseases characterized by progressive neuronal loss. We have previously suggested that p53 can regulate apoptosis, but extensive apoptotic events can damage the normal functions of many tissues, resulting in severe outcomes for tissues with limited regenerative potential. In fact, disordered apoptosis has become a fundamental pathogenic mechanism in degenerative diseases. Because of the intimate connection between p53 and apoptosis, p53 is considered a pathogenic factor in NDDs.200 p53 is also a predominant regulator of ferroptosis; thus, a rational speculation is that p53 promotes the progression of NDDs by regulating ferroptosis.201 A mutual regulatory effect between the p53 level/activity and proteins associated with NDDs has been identified.
Alzheimer’s disease
Alzheimer’s disease (AD) is the most common age-related NDD.202 Approximately 5% of AD cases are familial and are characterized by early onset before 65 years of age. Familial AD is usually correlated with mutations in 3 genes encoding amyloid β precursor protein (APP) and the enzymatic components of γ-secretase complexes, including presenilin 1 (PS1) and presenilin 2 (PS2).203,204 The pathological characteristics of AD are mainly neurofibrillary tangles and senile plaques.204 The accumulation of the hyperphosphorylated tau protein, a type of microtubule-associated protein (MAP), induces the formation of neurofibrillary tangles, and the aggregation of the amyloid-beta 1-42 (Aβ42) fragment produced from APP results in the formation of senile plaques by disturbing the activity of PS1 and PS2.205,206 Apoptosis is one of the mechanisms of cell death and has been a major topic of discussion. A previous study investigated the expression of various genes correlated with cell death cascades and suggested that the expression of the proapoptotic protein Bax-α was increased in the hippocampus of AD patients, whereas the expression of the antiapoptotic protein Bcl-2 was not altered.207 In the entorhinal cortex and hippocampus of AD patients, high-molecular-weight DNA fragmentation and altered nuclear morphology suggest increased apoptosis.208,209 Similarly, apoptotic cell death in brain tissues from AD patients and patients with familial AD carrying PA-1 mutations is usually correlated with the intracellular accumulation of Aβ42.210 In addition, apoptosis has been observed not only in neuronal cells but also in astrocytes.211 Compared with the brains of healthy individuals, the brains of AD patients have elevated p53 levels.212,213 Similarly, neurons in a transgenic mouse model accumulate p53 and Aβ42, which also indicates a correlation between the intracellular Aβ42/p53 pathway and neuronal loss.214 p53 can regulate the expression of various proteins associated with AD pathology to influence the process of this disease. p53 can transcriptionally inhibit the activity of PS1.215,216 Interestingly, PS1 overexpression reduces p53 activity, which forms a negative feedback mechanism to regulate p53 activity.217 Notably, mutant PS1 disturbs this mechanism because mutant PS1 significantly increases the transcriptional activity of p53.217 p53 can also increase tau protein phosphorylation in human cells to promote neuroblastoma cell death and the formation of neurotoxic neurofibrillary tangles.218,219 p53 may promote the phosphorylation of the tau protein by glycogen synthase kinase 3β (GSK3β) in individuals with AD.220 GSK3β can also regulate p53 levels via MDM2 phosphorylation, which mediates p53 degradation.221 Upon exposure to cellular stress, decreased MDM2 levels and increased p53 levels induce a consistent interaction between p53 and GSK3β, possibly promoting tau hyperphosphorylation.221 Proteins associated with AD can also regulate p53 levels and activity. Conditional knockdown of both PS1 and PS2 in the animal brain decreases p53 levels and activity. Fibroblasts deficient in PS1/PS2 and cells treated with PS1/PS2 inhibitors also exhibit reduced p53 expression and activity.217 Similarly, PS2 overexpression increases p53 levels and activity to promote p53-dependent cell death, which indicates that APP and its proteolytic products play important roles in the regulation of p53 levels and activity.222 In addition, the overexpression of other components of the γ-secretase complex, including nicastrin, anterior pharynx-defective 1 (Aph-1), and presenilin enhancer 2 (Pen 2), also suppresses p53 expression in AD animal models.223,224 Overall, p53 is considered to have complicated interactions with proteins associated with NDDs.225
Parkinson’s disease
Parkinson’s disease (PD) is the second most common NDD in terms of prevalence worldwide. The major brain locus affected by PD is a small region in the ventral midbrain known as the pars compacta of the substantia nigra and consists of dopaminergic neurons.226 The characteristic of PD is the formation of intracellular inclusions named Lewy bodies, composed of α-synuclein proteins.227,228 Like patients with AD, most PD patients are diagnosed with the sporadic form. Familial PD is usually associated with mutations in genes such as parkin, leucine-rich repeat kinase 2 (LRRK2), and α-synuclein.229 Although the pathogenesis of PD remains unclear, apoptosis may lead to the degeneration of dopaminergic neurons in PD patients. The activities and levels of multiple cellular effectors involved in the regulation of cell death, including caspase-3, caspase-9, and cytochrome C, indicate that cells die in individuals with PD,230,231 which has also been proven by the number of TUNEL-positive cells and in situ DNA fragmentation.232 p53 levels and activity are influenced in PD-affected patients and PD experimental models. Protein aggregation in individuals with NDDs usually damages the function of the proteasome, which is accompanied by cell death.233 Compared with that in healthy controls, the activity of the 20S proteasome is decreased in PD patients, and 20S proteasome activity is negatively correlated with PD duration and severity.230 Therefore, a reduction in proteasomal activity leads to p53 accumulation, as p53 is degraded by MDM2 through the proteasome.234 The p53, interferon-gamma, and NF-κB protein levels in the caudate nucleus are significantly higher in PD patients than in controls, whereas p53 levels are not altered in the substantia nigra, putamen, or cerebral cortex.235 Upon exposure to nitric oxide stress, the Ser15 residue of p53 is phosphorylated to increase p53 protein stability, which activates the nitric oxide-mediated apoptosis-like cell death pathway.236 α-Synuclein can decrease p53 expression and transcriptional activity to protect neuronal cells from proapoptotic effectors, in contrast to the A53T mutant form of the protein, which is correlated with PD.222 As a Lewy body constituent and a partner of alpha-synuclein, synphilin-1 interacts with α-synuclein to diminish the degeneration of neurons in the brain and motor abnormalities in mice with the A53T mutation.237,238 Synphilin-1 also significantly decreases p53 expression and transcriptional activity and reduces p53 mRNA levels and p53 promoter transactivation, which are caused mainly by the production of C-terminal products through caspase-3-mediated cleavage.239 Parkin is considered an E3 ubiquitin ligase and a transcription factor, and mutations in parkin lead to most autosomal recessive forms in juveniles.240,241 Parkin can transcriptionally repress p53. Specifically, parkin suppresses p53 promoter transactivation and reduces p53 mRNA levels through its Ring1 domain.240 Overexpressed parkin interacts with the p53 promoter, which disturbs the transactivation of the p53 promoter and promotes DNA binding.240 As a protein, DJ-1 acts as a chaperone protein and oxidative stress sensor, and DJ-1 mutations are associated with early-onset juvenile PD.242,243 In mammalian and zebrafish PD animal models, DJ-1 protects cells from apoptosis by inhibiting p53 transcriptional activity and decreasing Bax protein levels.244,245 DJ-1 also binds to the DNA-binding domain of p53, diminishing the DNA-binding affinity of p53 and inhibiting the expression of p53 target genes in a manner that relies on the oxidation of cysteine 106.246 Like in AD, p53 also binds to genes correlated with PD and regulates their expression. The upregulation of p53 remarkably increases the promoter activity, mRNA levels, and protein expression of α-synuclein through the DNA-binding characteristics of p53.247 Parkin is considered a target gene of p53, as it is involved in regulating the functions of p53 energy metabolism, including antioxidant defenses and the Warburg effect.248 Interestingly, reciprocal transcriptional control may exist because α-synuclein and parkin inhibit p53 activity and expression.222,240 Overall, p53 activation increases the expression of α-synuclein or parkin, which subsequently decreases p53 levels and parkin and α-synuclein transcription to reach equilibrium. In addition, p53 controls DJ-1 gene expression indirectly in a parkin-dependent manner. X-box binding protein 1 (XBP-1) is a transcription factor that promotes XBP-1 expression.249 In the brains of transgenic parkin-deficient mice, the inhibition of p53 caused by parkin deficiency increases XBP-1 expression, which in turn promotes DJ-1 expression by binding and transactivating the DJ-1 gene promoter.249 The interaction between p53 and DJ-1 increases the complexity of reciprocal interactions.
Other diseases
Huntington’s disease (HD), amyotrophic lateral sclerosis (ALS), and multiple sclerosis (MS) are also NDDs. HD is a genetic disease resulting from the propagation of a glutamine (CAG) stretch within the huntingtin protein (Htt). HD affects mainly the large pyramidal neurons of the cerebral cortex and the medium spiny neurons of the striatum.250 The pathological characteristics of HD are nuclear and cytosolic inclusions filled with aggregates of mutated huntingtin (mHtt) or its truncated N-terminal fragment.251,252 ALS is a disorder caused by the degeneration of groups of upper and lower motor neurons, including those in the cortex, spinal cord, and brainstem, damaging the function of voluntary muscles.253 The etiology of ALS is unclear, and ALS can be fatal within five years. Similar to AD and PD patients, approximately 90% of individuals with ALS are diagnosed with the sporadic form with no known gene mutations, and the remaining 10% of patients carry mutated genes. Notably, more than 20% of inherited cases are caused by mutations in the gene encoding SOD1.253 MS is an inflammatory demyelinating neurodegenerative disease of the central nervous system and is also an autoimmune disorder mediated by T cells.254 Although few genetic factors are known for ALS, multiple susceptibility variants exist in the major histocompatibility complex (MHC), especially in the human leukocyte antigen (HLA).254 p53 is also closely related to the aforementioned diseases. Affected brain regions in HD patients, HD animal models, and cells overexpressing mHtt present elevated p53 levels.251,255 p53 loss alleviates changes in mHtt expression and mitochondrial dysfunction and prevents neuronal degeneration to relieve some of the neurobehavioral defects associated with HD.250 Although p53 deficiency cannot fully inhibit the formation of inclusions containing mHtt, mice lacking p53 presented a milder disease phenotype through decreased mHtt levels.250 Similarly, mHtt containing expanded polyglutamine (polyQ) tracts increases p53 levels and transcriptional activity in neuronal cells by binding to p53.251 The results of immunoblotting and immunocytochemistry of central nervous system tissues suggested elevated p53 protein levels and the abnormal localization of p53 in ALS patients.256 Specifically, in normal cells of the central nervous system, p53 is usually expressed in nonneuronal cells. However, in ALS patients, p53 is overexpressed in motor neurons in the motor cortex and spinal cord.256 Another study revealed elevated levels of p53 and proapoptotic proteins, including caspase-3, caspase-8, Bax, and Fas, in spinal motor neurons from individuals with ALS, whereas p53 levels were not altered in neurons of the motor cortex.257 In mouse models with a mutated superoxide dismutase (SOD1) gene (G86R), p53 transcriptional activation and alterations in Bcl-x/Bax ratio were observed.258 Moreover, the ectopic expression of mutant SOD1 (G86R) promoted p53 phosphorylation and increased p53 levels in PC12 cells.258 Mutant optineurin (OPTN) also activated p53 in the neurons of ALS patients.259 Wild-type OPTN inhibits the activity of NF-κB; thus, the mutant OPTN detected in individuals with ALS is unable to suppress NF-κB activity. Mutant OPTN promotes the expression of downstream target genes of NF-κB, such as p53 and proapoptotic proteins, to induce cell death by triggering p53-dependent apoptosis.259
These results indicate that p53 activation is a critical event in NDDs. Increased p53 protein levels have been observed in the neuronal cells of brain tissues from NDD patients and animal models. Moreover, proteins correlated with NDDs can regulate p53 activity and expression. Interestingly, the relationships between p53 and proteins correlated with NDDs are complex and reciprocal. Activated p53 impacts the symptoms of NDDs in two ways. For example, p53 activation regulates the expression of a variety of genes to protect NDD-associated neurons from DNA damage.260 Conversely, activated p53 also increases NDD symptoms when neuronal homeostasis is disrupted.
Metabolic diseases
p53 regulates a series of metabolic diseases, including obesity, diabetes, and fatty liver diseases, through its complicated functions.125,261,262 Interestingly, different results concerning the role of p53 in metabolic diseases are often reported. Therefore, the specific role of p53 in metabolic diseases must be correlated with a specific context.
Obesity
The adipose tissue of mammals is divided into brown adipose tissue (BAT) and white adipose tissue (WAT). The predominant function of BAT is heat generation by fat metabolism.263 The principal role of the WAT is energy storage and the modulation of the energy balance.264 A negative relationship has been observed between obesity and BAT activity.265,266 Disorders of metabolic homeostasis in obese individuals promote several pathological processes, such as type 2 diabetes, insulin resistance, and an increased incidence of various cancers.267,268 p53 inhibits the differentiation of WAT in several in vitro models and protects against diet-induced obesity in vivo. Conversely, p53 positively regulates the differentiation of BAT. Moreover, p53 deficiency in committed skeletal muscle cells decreases their ability to differentiate into BAT.269 Therefore, p53 regulates adipogenic differentiation in a dual manner that relies on a specific adipogenic differentiation program. p53 deficiency in AgRP neurons significantly induces an obese phenotype characterized by mild hyperphagia, elevated adiposity, and decreased energy expenditure under conditions of excess nutrition.270 More importantly, p53 is regulated only in AgRP neurons and not in major neuronal populations of the arcuate nucleus of the hypothalamus (ARC), which suggests that p53 alterations associated with energy balance are nucleus-specific.270 Endothelial cell-specific p53 deficiency can improve insulin sensitivity and decrease fat accumulation to relieve the occurrence and development of obesity.271 The above studies revealed the role of p53 in preventing obesity, but some reports indicate that p53 can promote the occurrence of obesity. The majority of genetic polymorphisms are considered innocuous, whereas several single-nucleotide polymorphisms (SNPs) can exert remarkable biological effects.25,272 The most common site of SNPs in the p53 gene is amino acid codon 72, in which proline (P72) or arginine (R72) is encoded. Alterations in the p53 codon 72 polymorphism significantly influence the function of p53.273 In a humanized mouse model with p53 codon 72 variants fed a high-fat diet (HFD), mice with the p53 R72 variant exhibited more severe glucose intolerance and obesity than did mice with the p53 P72 variant. Specifically, mice with the p53 R72 variant developed islet hypertrophy, insulin resistance, and fatty liver disease.274
Diabetes
Although no clear clinical evidence is available that disordered p53 directly promotes type I diabetes, dysfunctional p53 can induce critical biological processes in diabetes, including disturbances in glucose homeostasis at the organismal level, pancreatic β-cell deficiency, and insulin resistance in peripheral tissues.261 Many studies have suggested that activated p53 induces the death of pancreatic β-cells in response to increased free fatty acid levels and oxidative stress.275,276,277 The insulin-like growth factor 1 (IGF-1)-AKT-mTOR pathway responds to nutrient availability and mitogens through signaling related to cell division and death. AMPK, TSC2, PTEN, and IGF-BP3, which are regulated by p53, negatively regulate the activity of the IGF-1-AKT-mTOR pathway upon exposure to stress.278 p53 regulates the expression of these genes in a cell type- and tissue-specific manner, coordinately regulating TSC2 and PTEN protein levels in various tissues, including the heart, skeletal muscle, white fat, kidney, and liver, via insulin-dependent energy metabolism.278 More importantly, p53 can adjust gluconeogenesis, glycolysis, and glucose uptake to affect glucose metabolism in peripheral tissues. Disruptions in glucose homeostasis caused by p53 increase circulating glucose levels, which is a characteristic of type 2 diabetes (T2D). p53 also impacts insulin sensitivity via autocrine loops and the autonomous regulation of cells. Various stimuli, including telomere dysfunction and oxidative stress, induce cellular senescence through the p53 and Rb proteins, and senescent cells can promote disorders associated with aging or age-related diseases. When T2D-like disease model mice are fed a high-calorie diet, the accumulation of oxidative stress in adipose tissue and senescence-associated alterations, including increases in p53 levels, proinflammatory cytokine levels, and senescence-associated beta-galactosidase activity, are observed.279 Suppressing p53 activity significantly improves senescence-like changes and insulin resistance in mice with T2D-like disease. In contrast, p53 upregulation in adipose tissue promotes insulin resistance by aggravating the inflammatory response.279 The finding that p53 expression in adipose tissue could regulate insulin sensitivity provides a new insight that cellular aging signals in adipose tissue could be a new therapeutic target for diabetes. Another study suggested that DNA damage in adipocytes can disturb secretory function and adipocyte metabolism and activate the p53 signaling pathway, inducing adipose tissue inflammation and insulin resistance.280 In addition, as an inhibitor of the interaction between MDM2 and p53, Nutlin-3 directly influences the immune system by increasing circulating interleukin-12 (IL-12) p40 levels, alleviating the severity of streptozotocin-induced diabetes.281 Interestingly, the mechanisms by which p53 regulates glucose homeostasis involve skeletal muscles and angiogenic cells. Endothelial nitric oxide synthase can increase the levels of peroxisome proliferator-activated receptor-γ coactivator-1α in skeletal muscle. The inhibition of endothelial p53 activation facilitates oxygen consumption and mitochondrial biogenesis by inhibiting the inactivation of endothelial nitric oxide synthase.271 Feeding endothelial cell-specific inactivated mice a high-calorie diet improved insulin resistance and decreased fat accumulation,271 suggesting that the suppression of endothelial p53 could be a new target for the treatment of obese patients with metabolic abnormalities. As a stomach-derived peptide, ghrelin can activate hypothalamic AMP-activated protein kinase to promote food intake. Finally, p53 regulates food intake by modulating ghrelin in the central nervous system to induce obesity and associated metabolic diseases.282
Fatty liver disease
Fatty liver disease can be classified into alcoholic and nonalcoholic fatty liver disease (NAFLD). NAFLD is a common metabolic disorder associated with obesity and has a wide range of pathological alterations, such as fatty liver and nonalcoholic steatohepatitis (NASH), which contribute to hepatocellular carcinomas (HCCs).261 Various stresses, including iron accumulation, high levels of saturated fatty acids, and disturbed metabolism, induce not only chronic liver injury but also p53 activity. In contrast, abnormal p53 activity can also lead to liver diseases. p53 deficiency decreases hepatic lipid peroxidation, inhibits the apoptosis of hepatocytes, and suppresses the development of nutritional steatohepatitis by suppressing the p66Shc signaling pathway.283 Compared with those in normal liver samples, the protein levels of p53, p21, and p66Shc are markedly increased in liver samples from NAFLD patients.283 p53 protein levels are associated with the severity of steatosis in NAFLD patients, which is consistent with the role of p53 in liver disorders. Increased p53 expression in hepatocytes has been observed in some mouse models of NAFLD.284,285 Notably, one of the critical questions in this field is how to modulate p53 activity in patients with NAFLD and diabetes to exploit new therapeutic approaches for these metabolic diseases. In a human hepatocellular carcinoma cell line, p53 inhibited miR-17-92 expression to upregulate the levels of connective tissue growth factor (CTGF), which is the hepatic fibrogenic master switch that promotes liver fibrosis.286 Compared with those in the control group, the hepatic CTGF levels and severity of liver fibrosis were lower in the hepatocyte-specific p53-deficient group, whereas the extent of hepatocyte apoptosis was similar. A previous study286 revealed not only that p53 can modulate liver fibrosis by altering CTGF expression but also that the p53/CTGF pathway may be a new target in the treatment of liver fibrosis. Although the above findings show potential for clinical application, p53 inhibition is not a long-term option because of the significant side effects of p53 inhibition. Notably, animal models treated with pifithrin-α p-nitro (PFT) (a p53 inhibitor) present decreased steatosis, oxidative stress, apoptosis, and insulin resistance.287 Paradoxically, animal models of NAFLD and NASH treated with low-dose doxorubicin exhibited moderate p53 activation, decreased inflammation and lipogenesis, and alleviated liver damage, which improved steatohepatitis and nonalcoholic steatosis caused by the diet.288
In conclusion, the above studies demonstrated that p53 plays a critical role in metabolic diseases. However, these studies do not indicate effective and potential clinical therapeutic approaches for metabolic diseases. A deeper understanding and further studies of the correlation between p53 and metabolic diseases are needed to elucidate the underlying mechanisms for the development of potential clinical strategies.
Cardiovascular diseases
Cardiovascular diseases (CVDs) are currently the predominant cause of death worldwide. The 2020 update of data from the American Heart Association Heart Disease and Stroke Statistics revealed that the number of CVD-related deaths in 2017 was more than 18 million and that the cost of treating CVDs exceeded that of other chronic medical diseases.289 Therefore, further investigations of the underlying mechanisms of CVDs and the identification of potential targets for CVD prevention are important. In recent years, p53 has been proven to regulate the heart under both physiological and pathological conditions. Under physiological conditions, p53 acts to sustain the cardiac architecture and adjust the levels of transcripts correlated with metabolism, the cardiac architecture, and excitation‒contraction coupling. A lack of wild-type p53 promotes heart hypertrophy and decreases heart function.290 In addition, p53 plays a critical role in regulating CVDs, including myocardial ischemia/reperfusion injury, heart failure, pressure overload- and diabetes-induced maladaptive remodeling and atherosclerosis.291,292,293
Myocardial infarction (MI) is the leading cause of CVD-related death. MI is characterized by significant metabolic and ionic disturbances caused by ischemia, impaired cardiac myocytes, and postinfarction cardiac malfunction and remodeling.294 During periods of ischemia, reperfusion injury, and postinfarction cardiac remodeling, p53 modulates cardiomyocyte death, including autophagy, apoptosis, and necrosis.293,295,296 Under conditions of DNA damage, Rho-associated coiled-coil containing protein kinase 1 (ROCK1)-mediated apoptosis is increased in the MI model. ROCK1 increases p53 protein levels by phosphorylating p53 at Ser15.297 p53 is significantly acetylated at the Lys (118) residue in a rat model of ischemia/reperfusion injury, which regulates p53 binding and accumulation at the Bax promoter.296 Notably, its acetylation can be reversed by oxygenation, and TIP60 acetylase can be regulated in MI hearts. In oxygenated MI hearts, deacetylated p53 promotes survival by binding to the promoter of the corresponding endothelial nitric oxide synthase gene.296
The persistence of diabetes maintains the heart in an environment characterized by hyperglycemia and elevated cytokine and fatty acid levels. Diabetic cardiomyopathy is characterized by disordered cardiac structure and function in the absence of other cardiac risk factors.298 Although diabetic cardiomyopathy was described many years ago, the incidence rates of this disease remain underappreciated.299 Although the number of studies on diabetic cardiomyopathy has increased significantly in recent years, the specific pathogenesis of this disease remains unclear. The early manifestation of diabetic cardiomyopathy is isolated diastolic dysfunction, which transitions to systolic dysfunction as the disease progresses.300 Alterations in p53-mediated processes, such as apoptosis, senescence, and mitochondrial respiration, are involved in the development of diabetic cardiomyopathy.291 Acetylation is a posttranslational modification of p53. Increased p53 acetylation promotes cardiomyocyte apoptosis in models of diabetic cardiomyopathy and heart failure. SIRT1, an NAD (+)-dependent protein deacetylase, plays an important role in the modulation of vascular function. The inhibition of SIRT1 activity can induce cardiomyocyte apoptosis by increasing p53 acetylation and upregulating Bax expression.301 Apoptosis mediated by p53 can be reversed by targeting sirtuin-1.302
Atherosclerosis is an arterial disease caused by various factors. The pathological characteristic of atherosclerosis is the formation of pathological plaques containing endothelial cells (ECs), macrophages, and vascular smooth muscle cells (VSMCs).303 Upon exposure to multiple stresses, dysfunctional p53 can prevent the occurrence of atherosclerosis or promote the development of atherosclerosis in distinct cell types through p53-dependent apoptosis and the growth arrest response.304 p53 colocalizes with apoptotic or nonproliferating macrophages at atherosclerotic sites. Apoptotic macrophages expressing p53 have decreased MDM2 levels.304 Compared with control mice, p53-deficient mice develop atherosclerosis more quickly in the presence of comparable serum cholesterol levels.305 More importantly, compared with those in control mice, the atherosclerotic sites in p53-deficient mice presented a remarkable increase in the cell proliferation rate and an insignificant increase in apoptosis.305 APOE*3-Leiden transgenic mice constitute an animal model of human-like atherosclerosis. A study intensely irradiated APOE*3-Leiden mice and reconstituted the bone marrow with bone marrow from either p53-deficient (p53(-/-)) or control (p53(+/+)) donor mice.306 Compared with mice reconstituted with p53(+/+) bone marrow, mice reconstituted with p53(-/-) bone marrow presented more severe atherosclerotic lesions and more lesion necrosis.306 These results indicate that p53 expressed in macrophages plays a critical role in the inhibition of atherosclerosis progression and identify a novel target for regulating plaque stability. The above studies suggest a correlation between p53 deficiency and increased atherosclerotic lesion size. However, the proatherogenic mechanisms of p53 deficiency cannot yet be explained by a decrease in apoptosis or cellular proliferation.
Under physiological conditions, the p53 protein is maintained at low levels through the ubiquitin‒proteasome system (UPS).307 The UPS contributes to increased p53 expression in individuals with hypertrophic and dilated cardiomyopathy.308 Elevated p53 expression was observed in heart samples from patients with dilated cardiomyopathy.308 Compared with those in normal tissues, the levels of MDM2 and herpesvirus-associated ubiquitin-specific protease (HAUSP), which is an enzyme that targets p53 for deubiquitination, are significantly increased. Moreover, increased levels of ubiquitination and proteasomal activity are observed in tissues from individuals with dilated cardiomyopathy.308 These results suggest that the cardiac UPS cannot degrade excess ubiquitinated proteins because of overwork. Adult mice with a deletion of MDM4, an MDM2 homolog and a negative regulator of p53, exhibited dilated cardiomyopathy.309 The mechanism of dilated cardiomyopathy may involve increased apoptosis in cardiomyocytes regulated by the p53 pathway.309
In conclusion, p53 expression is increased in individuals with various CVDs. Elevated p53 expression facilitates the development of CVDs by promoting programmed cell death, preventing angiogenesis, and regulating metabolism and cell cycle arrest. On the basis of these mechanisms, potential preclinical treatments targeting p53 have been explored. For example, a small-molecule inhibitor of p53, pifithrin-α, was isolated by Komarov et al. to decrease the side effects caused by radio- and chemotherapy.310 In an in vivo diabetic cardiomyopathy model, pifithrin-α impaired angiogenesis, restored cellular senescence, and decreased glycolysis by suppressing the transcription of p53-dependent target genes, including HIF-1α, p16, p21, p16, glucose transporter 1 (GLUT-1), and glucose transporter 4 (GLUT-4).311 In a rat ischemia/reperfusion model, pifithrin-α ameliorated cardiac dysfunction by decreasing the ratio of Bax to Bcl-2 mediated by p53 and alleviating cardiomyocyte apoptosis.312 Further studies are needed to confirm whether this option is appropriate for clinical use.
Therapeutic strategies targeting p53 in cancer
The pursuit of therapeutic approaches targeting p53 began several decades ago and has been constantly evolving since then. In recent years, with the increased understanding of p53's biological function, therapies targeting p53, including small-molecule drugs, cancer immunotherapies targeting p53, and gene therapies targeting p53, have gradually improved.
Small-molecule drugs
Small-molecule drugs targeting mutant TP53 and its negative regulators are different (Fig. 7). Strategies targeting mutant p53 restore the wild-type conformation and activity of mutant proteins and promote mutant p53 degradation by disrupting the stability of mutant p53. For cancers expressing wild-type p53, the strategies most generally involve inhibiting wild-type p53 degradation.

Small-molecule drugs targeting p53 in cancer. a Small-molecule drugs that target tumors with missense mutant p53 include those that act broadly on multiple p53 mutants and those that act specifically on the p53 mutant. The former includes CP31398, APR-246, etc., and the latter includes PC14586, which targets the p53 Y220C mutant, etc.). b Small-molecule drugs that target wild-type p53 tumors mainly include MDM2 inhibitors, MDM4 inhibitors, and MDM2 and MDM4 inhibitors. c Small-molecule drugs that deplete mutant p53 include HDAC inhibitors, HSP90 inhibitors, and statins. This figure was created by BioRender.com
Targeting missense mutations in p53
Mutant p53 expression is considered a promising target for antitumor treatment. The main type of TP53 mutation is a missense mutation, which results in conformational changes in mutant p53 and the generation of unfolded proteins.126 Attempts to restore the wild-type conformation of mutant p53 are speculated to restore a wild-type-like form and wild-type function. Thus, thousands of compounds targeting mutant p53 were developed several decades ago. Compounds that target mutant p53 are loosely divided into broad-spectrum compounds that target diverse p53 mutants and allele-specific compounds that target a single mutant or a different group of similar mutants (Table 2).
Broad-spectrum compounds
CP31398 was the first compound discovered that can reactivate mutant p53. Pfizer obtained CP31398 by screening a synthetic compound library.313 CP31398 administration can recover the transcriptional activity of partial mutant p53 and inhibit the thermal denaturation of wild-type p53 because of its Michael acceptor characteristics.313 Michael acceptors first interact with soft nucleophiles, which are mainly thiol groups of various molecules, such as cysteines and reduced glutathione, in cellular proteins. Ten cysteines located in the p53-DNA-binding core domain are likely adorned by Michael addition.314 CP31398 can possibly target new synthetic mutant p53 and change its conformation in living cells.315 It has been suggested that CP31398 has anticancer activities against several cancers, including colon cancer and melanoma.313 In addition, CP31398 administration can promote DNA damage and suppress p53 ubiquitylation to stabilize the conformation of wild-type p53.316,317 Subsequent studies have revealed more complicated mechanisms, such as a p53-independent increase in Bax expression, slight upregulation of MDM2 expression, and changes in the DNA‒p53 core domain complex.315 However, CP31398 has not undergone clinical development. Notably, CP31398 is a molecule that reactivates mutant p53; however, some studies suggest that its activity may also involve p53-independent pathways.318,319 One study revealed that CP31398 could facilitate the binding of mutant p53 to p53 response elements in vivo and restore the DNA-binding activity of mutant p53 in vitro. Moreover, CP31398 has no influence on the DNA-binding activity of p53 homologs (p63 and p73).318 Another study evaluated the efficacy of CP31398 in a transgenic UPII-SV40T mouse model of human transitional cell carcinoma (TCC), and the results showed that mice that received CP31398 suppressed invasive TCC. Bladder tumors exhibit increased levels of apoptosis markers (p53, p21, Bax, and Annexin).319 Overall, the different mechanisms of action of CP31398 provide more possibilities for its application in cancer therapy. CP31398 can treat tumors expressing mutant p53 by reactivating mutant p53 and can treat cancers with p53 deficiency by binding to other molecules.
PRIMA-1, obtained by screening the U.S. National Cancer Institute (NCI) library of chemical compounds, is a small molecule that restores the wild-type conformation and function when combined with mutant p53. PRIMA-1 has been reported to inhibit carcinogenesis and promote the expression of target genes such as Puma, Bax, and CDKN1A.320,321,322 Mechanistically, PRIMA-1 functions through its degradation product, the methylene quinuclidinone (MQ). As a potential Michael acceptor, MQ combines with p53 cysteine thiols to recover the wild-type conformation.320 Although all cystines located on the surface of p53 can be modified by the Michael addition reaction, cystine 124 is considered the most important site.323,324 APR-246, also known as eprenetapopt or PRIMA-1Met, is a methylated derivative of PRIMA-1.130 Like PRIMA-1, APR-246 is also a prodrug of MQ. However, APR-246 has greater lipophilicity and better cell permeability than PRIMA-1 does, which endows APR-246 with stronger antitumor effects.321 APR-246 suppresses the growth of tumors expressing mutant p53 through various pathways, such as degrading cellular glutathione levels, promoting endoplasmic reticulum (ER) responses, and adjusting oxidation‒reduction responses to induce ferroptosis.325,326,327
APR-246 is the first small molecule that has reached clinical trials. Multiple studies have demonstrated the antitumor activities of APR-246 in vivo and in vitro in preclinical tumor systems.314,328 APR-246 has been reported to induce the apoptosis of acute myeloid leukemia (AML) cell lines and primary AML patient cells.329 Paradoxically, a study showed that AML cell death after APR-246 treatment is inhibited by iron chelators, inhibitors of lipid peroxidation, and lipophilic antioxidants and is characterized by the accumulation of lipid peroxidation, thus meeting the definition of ferroptosis.326 These results showed that APR-246 induces ferroptosis in AML cell lines. Using small-cell lung cancer (SCLC) cell lines, Roza et al. demonstrated that APR-246 administration promoted apoptosis, suppressed tumor growth in vitro, and inhibited tumor growth in mice injected with SCLC cells in vivo.330 Studies have suggested that combined therapies using APR-246 and chemotherapies enhance antitumor activities. The combination of APR-246 with various compounds, including cisplatin, doxorubicin, and carboplatin, has been reported to induce similar synergistic effects in ovarian and lung mouse models.331,332
Currently, a wide range of clinical trials are ongoing in which APR-246 administration has finished or has not yet been completed12,333,334,335 (Table 3). A phase Ib/II clinical trial (NCT03072043) revealed abundant effects when APR-246 was combined with azacitidine (AZA) in MDS patients and AML patients carrying mutant p53.333 This combination has been shown to have similar safety to AZA monotherapy. The FDA subsequently granted the APR-246 orphan drug name and approved APR-246 for MDS treatment. Recently, a phase II trial utilizing a combination of APR-246 and AZA as post-transplant maintenance treatments evaluated their efficacy in MDS and AML patients with mutant p53. The relapse-free survival (RFS) 1 year after transplantation and median overall survival (OS) in this study were 58% and 19.3 months, respectively. The RFS and median OS of patients in the previous study were 30% and 5–8 months, respectively, which were lower than those reported in this study.12 A phase 1, dose-finding and expansion study (NCT04214860) was conducted to evaluate combined therapy using APR-246 and venetoclax with or without AZA in AML patients with p53 mutation.336 No dose-limiting toxicities (DLTs) were observed, and the recommended phase 2 dose for the APR-246 combination was 4.5 g/day on days 1–4. More than 20% of patients experienced ≥grade 3 adverse events (AEs), including febrile neutropenia (47%, 23/49), thrombocytopenia (37%, 18/49), leukopenia (25%, 12/49), and anemia (22%, 11/49). The overall response rate for APR-246 and venetoclax with AZA was 64% (25/39), and the complete response rate (CRR) was 38% (15/39). These results suggest that the combination of APR-246 and venetoclax with AZA has surprising activity and acceptable safety.336 Several underlying factors, such as differences in the experimental design, model systems, or patient heterogeneity, may account for the differences in the mechanisms of action of CP31398 and APR-246 and cause different therapeutic responses. Specifically, the concentration and timing of drug administration significantly impact therapeutic responses. For example, in vitro studies with CP31398 have shown that different concentrations and exposure times can lead to varying degrees of apoptosis and cell cycle arrest in cancer cells.337 Similarly, APR-246 has dose-dependent effects on the viability and apoptosis of various cell lines.338 Proper control groups are essential for evaluating drug efficacy. In a study of the combination of APR-246 and AZA for treating TP53-mutant MDS and AML, a control group receiving only AZA was included to assess the synergistic effects of the combination.339 In terms of the model system, different cell lines may respond differently to the same drug due to their genetic backgrounds. For example, CP31398 induced apoptosis in A431 cells expressing mutant p53 but not in normal human epidermal keratinocytes (NHEKs) expressing wild-type p53.337 Similarly, APR-246 has shown varying responses in cell lines with different p53 mutation statuses.339 In terms of patient heterogeneity, the stage and pathological characteristics of the disease can impact treatment outcomes. For example, in studies of APR-246 for multiple myeloma (MM), varying responses were observed in cell lines and primary cell samples with different p53 mutation statuses. In addition, the genetic background of patients and the statuses of other cellular pathways can influence the drug response. For example, APR-246 works by reducing intracellular glutathione levels and increasing reactive oxygen species (ROS) levels, but this mechanism may be affected by other metabolic pathways within the cell.326,338
Arsenic trioxide (ATO) is a conventional drug that has been approved for acute promyelocytic leukemia (APL) treatment by the FDA and has achieved superior effects.340 Most APL patients express the special fusion protein PML-RARα, which comprises PML zinc finger protein sequences and retinoic acid receptor α sequences. A study showed that ATO bound to cysteine residues located in the zinc finger regions of PML and PML-RARα, resulting in PML oligomerization and degradation.341 A wide range of studies have suggested that ATO promotes apoptosis in cells harboring mutant p53.342,343 Recently, ATO was reported to restore the thermostability and transcriptional activity of structural p53 (R175H) mutants. ATO has also been shown to inhibit tumor growth in vitro, decrease tumor volume, and improve survival in mouse xenograft models.344 Notably, ATO administration can rescue a wide range of structural p53 mutants, including R175H, R175L, G245S, and R249S, but it cannot exert the same function in DNA contact mutants, such as R248Q and R273H.344 Another study evaluated the ability of ATO to promote the recovery of 800 p53 mutants by inhibiting tumor cell growth in vitro and suppressing tumor activity in vivo, and 390 mutants were recovered.345 According to their different rescue abilities, 390 p53 mutants were classified as type 1, type 2a, or type 2b mutations. Type 1 mutations can be rescued to a level comparable to that of wild-type p53. In other words, their function can be restored to a significant extent, similar to that of normal p53. Type 2a mutations can be partially rescued but not to the same extent as type 1 mutations. Their rescue level is intermediate, indicating some restoration of function but not as effectively as type 1. Type 2b mutations result in the least degree of rescue. Compared with those of type 1 and type 2a mutants, their function is minimally restored, indicating that they are less responsive to the rescue compound. Thirty-three type 1 mutations were rescued to the same extent as the wild type, and ATO preferentially suppressed the growth of type 1 and type 2a mutant tumors.345 Currently, clinical trials are being performed for the ATO treatment of dysfunctional p53 (Table 3). The fact that ATO administration reactivates mutant p53 suggests that some FDA-approved drugs may be mutant p53 reactivators.
ReACp53, designed by Soragni et al., is a peptide that suppresses mutant p53 aggregation, restores the wild-type conformation of mutant p53, and promotes its nuclear localization to increase its transcriptional activity.153 Owing to these functions, ReACp53 induces apoptosis and cell cycle arrest in vitro and inhibits tumor growth in vivo.153 Notably, ReACp53 also affects p53-silenced tumor cells by adding this peptide to cancer cells.346
As a cationic tripyridylamide, ADH-6 was identified by screening an oligopyridylamide library. Drugs in the library are speculated to suppress amyloid formation in Alzheimer’s disease.347 ADH-6 has been tested on several p53 mutants, including the p53 R248W mutant, p53 R175H mutant and other aggregation-prone p53 mutants.347 ADH-6 stabilizes the p53 R248W mutant by interacting with the p53 core domain, preventing the formation of these aggregates.347 In addition to the p53 R248W mutant, ADH-6 was also effective against the p53 R175H mutant. Like p53 R248W, the p53 R175H mutant is prone to aggregation and contributes to tumor progression. ADH-6 showed significant efficacy in dissociating preformed aggregates and stabilizing the mutant protein, thereby reducing tumor growth in in vivo models. Studies have reported that ADH-6 administration simulates the structure of proteins and inhibits protein aggregation.348 ADH-6 has been reported to degrade mutant p53 aggregates and selectively induce mutant p53-expressing tumor cell apoptosis.347 Furthermore, ADH-6 administration also decreases the unfolded status of mutant p53 and promotes the expression of targeted genes such as p21, Bax, and Noxa.347
On the basis of compounds that bind to amyloid proteins, L1 and LH have been designed.349 L1 and LH are bifunctional ligands and have similar structures. L1 and LH adjust mutant p53 aggregation by increasing zinc ion levels in cells and promoting interactions between zinc ions and mutant p53. L1 and LH also facilitate the expression of p21 and Noxa.349
Targeting allele-specific compounds
Allele-specific compounds that target a single mutant or a group of similar mutants were proposed several decades ago. The Y220C (Fig. 5) mutation is a hotspot in p53 and is a relatively important mutation. Tyrosine is converted to cystine in p53 (Y220C) mutant proteins, resulting in an accessible crevice.350 Owing to the accessible crevice and the long distance between Y220 and the site at which p53 binds to DNA, Y220C is a perfect treatment target. PhiKan083 has been reported to bind crevices and restore the wild-type conformation.351 Like PhiKan083, PK7088, identified by screening synthesized fragment libraries, also binds unique crevices and restores transcriptional activity. PK7088 promotes apoptosis by increasing the expression levels of apoptotic proteins such as p21 and Noxa.352 In addition, PK7088 has a synergistic effect with Nutlin-3 in promoting apoptosis.352 Subsequently, PC14586 was identified through the same approach. PC14586 is an orally bioavailable small molecule that binds to crevices to restore the wild-type conformation and inhibit tumor growth.11 An ongoing phase I/II clinical trial (NCT 04585750) has assessed the safety, tolerability, pharmacokinetics, pharmacodynamics, and efficacy of PC14586 administration in patients with advanced solid tumors harboring the p53 Y220C mutation. The novel results of this study were shown at the 2022 ASCO meeting,353 and there were no DLTs. Among the 21 evaluable patients, 5 patients achieved partial response (PR), and 7 patients experienced stable disease (SD). Most adverse effects (AEs) were grade 1/2.353 However, the number of compounds that target the p53 Y220C mutant is limited.
Zn2+ plays an important role in p53-DNA recognition and enhances the interaction between p53 and DNA.354 Therefore, a lack of or decrease in Zn2+ promotes incorrect folding of proteins and influences the recognition of p53-DNA. R175H is a hotspot mutation of p53 that induces structural or conformational changes by destroying the conformation of the DBD.12 Specifically, the p53 R175H mutation decreases the binding affinity of the DBD and Zn2+ and promotes incorrect folding and inactivation of the p53 protein.12 Thus, compounds that target the p53 R175H mutant have been developed. NSC319726, also known as ZMC1, was identified through screening NCI60 antitumor drugs. ZMC1 functions because of its high affinity for Zn2+ as a metal ion chelator.355 ZMC1 induces apoptosis in a p53-dependent manner in vitro and inhibits tumor growth in vivo. Although ZMC1 targets the p53 R175H mutant, it also affects several p53 R273H and p53 R248Q mutants that lack binding affinity between p53 and DNA.355 Furthermore, ZMC1 exerts its anticancer function through increasing intracellular ROS levels and reducing glutathione levels.355,356 Similarly, ZMC2 and ZMC3 administration also restored the wild-type conformation of mutant p53.357
COTI-2 is a type of thiosemicarbazone and is the only compound that has been tested in clinical trials for drugs targeting the p53 (R175H) mutant. COTI-2 has been shown to suppress tumor growth by inducing apoptosis and cell cycle arrest in both p53-dependent and p53-independent manners.356,358 COTI-2 also recovers the DNA-binding properties of the p53-mutant protein.359,360 Notably, COTI-2 not only preferentially affects tumor cells expressing mutant p53 but also affects tumor cells with wild-type p53.324 A previous study evaluated the antitumor activity of COTI-2 in head and neck squamous cell carcinoma (HNSCC) and demonstrated the tumor inhibition function of COTI-2 in HNSCC cells and a mouse model146. The results of this study also suggest that COTI-2 promotes apoptosis or senescence through DNA damage and replication stress responses.356
Targeting negative regulators of p53
The above drugs are not applicable to cancers expressing wild-type p53. Normally, wild-type p53 is expressed at low levels because of its interaction with its negative regulators, such as MDM2/4. MDM2/4 overexpression has been found in many tumors expressing wild-type p53. Thus, blocking the interaction between MDM2/4 and p53 is the preferred strategy for treating tumors expressing homozygous wild-type p53. Currently, a series of drugs that block the interaction between p53 and its negative regulators have been developed (Table 3).
MDM2 inhibitors
Nutlins, which were identified by screening a synthetic chemical library, were the first compounds reported to inhibit the interaction between MDM2 and p53.361 Preclinical studies have suggested that nutlins stabilize p53 and activate p53-targeted genes by inhibiting the interaction between p53 and MDM2. Reactivation of the p53 pathway induces cell cycle arrest and apoptosis.362 Although nutlins have promising antitumor activity in tumors expressing wild-type p53, they have not been used in clinical applications because of their toxicity and poor bioavailability. RG7112 is a nutlin-3a analog and the first MDM2 inhibitor used in clinical trials. In a proof-of-mechanism study, researchers administered RG7112 to MDM2-amplified liposarcoma patients and assessed the correlation of MDM2 inhibition markers with RG7112 administration. Fourteen of the 17 evaluable patients had MDM2 gene expression amplification. One patient achieved a PR, and 14 patients experienced SD. Compared with those before treatment, the p53 and p21 levels observed after treatment with RG7112 increased 4-fold and 3-fold, respectively. Eight patients had serious AEs, which were mainly neutropenia and thrombocytopenia.363 MDM2 inhibition and wild-type p53 activation, which involves the activation of p53-targeted genes, including CDKN1A, BBC3, and Bax, and the induction of apoptosis, have beneficial effects on refractory/relapsed (R/R) AML and chronic myeloid leukemia (CML) patients.364 In addition, antileukemia activity has been observed in some AML patients. Although RG7112 has better efficacy, the toxicities caused by RG7112 are varied and serious, which hinder its clinical use.
RG7388 (idasanutlin), a nutlin derivative, has greater selectivity and efficacy than RG7112, an MDM2 inhibitor. Currently, clinical trials are being conducted to evaluate the safety and efficacy of RG7388 in patients with various cancers365,366 (Table 3). A phase 3 clinical trial evaluated the safety and efficacy of RG7388 plus cytarabine in patients with R/R AML.365 The primary endpoint was not achieved. The OS between the RG7388 plus cytarabine group and the RG7388 plus placebo group was not significantly different (8.3 vs. 9.1 months). However, the CRR and overall response rate (ORR) of the RG7388 plus cytarabine group were higher than those of the RG7388 plus placebo group (CRR, 20.3% vs. 17.1%; ORR, 38.8% vs. 22.0%). The most common AEs were diarrhea, febrile neutropenia, and nausea.365 A phase I trial of RG7388 in high-risk polycythemia vera (PV) and essential thrombocythemia (ET) patients was subsequently conducted. The ORR was 58% (7/12) after 6 cycles of RG7388 monotherapy. Although DLTs did not occur in all patients, most patients had low-grade gastrointestinal disease. These results suggest that RG7388 has promising efficacy.366 However, its safety still needs to be further confirmed in clinical applications.
AMG-232, a piperidone analog, is currently being tested in partial clinical trials (Table 3). As a potent and orally bioavailable MDM2 inhibitor, AMG-232 has shown antitumor activity in SJSA-1 osteosarcoma cells.367 A study evaluated the efficacy of AMG-232 and RG7112 in glioblastoma cells, and the results suggested that the effects of AMG-232 were much better than those of RG7112 in partial glioblastoma cells expressing wild-type p53. Furthermore, AMG-232 has been shown to suppress stemness-related factors and has a greater affinity for wild-type p53 than RG7112 does.368 Compared with AMG-232 monotherapy or chemotherapy alone, therapies involving the combination of AMG-232 and chemotherapies have superior antitumor activity through the promotion of apoptosis and the restoration of p53 activity.369 In the phase 1b trial of AMG-232 monotherapy or trametinib in patients with R/R AML,370 1 patient had DLT, 31% (4/13) of patients expressing wild-type p53 had a response, and no patients expressing mutant p53 had a response. The main AEs were nausea, diarrhea, and vomiting. AMG-232 was approved by the FDA following this trial.
APG-115 is a potent and orally bioavailable MDM2 inhibitor. It has been or is being tested in several clinical trials, resulting in fast-track designation by the FDA (Table 3). In China, APG-115 is the first MDM2 inhibitor used in the clinic. According to a preclinical study of APG-115 in AML patients, APG-115 strongly inhibits proliferation and promotes apoptosis through activation of the p53/p21 pathway in wild-type p53-expressing AML cells and in vivo. APG-115 administration enhances radiosensitivity, induces apoptosis, and causes cell cycle arrest in gastric cancer cells expressing wild-type p53.371 Compared with single-agent treatment, the combination of APG-115 and radiation strongly inhibits tumor growth in vivo.
In addition to the above drugs, MDM2 inhibitors tested in clinical trials also include HDM20, MK-8242, BI-907828, and milademetan (DS-3032b). However, wild-type p53 is expressed in normal tissues, resulting in possible toxicity. In other words, MDM2 inhibitors are not drugs that target specific sites in cancer. Furthermore, MDM2 inhibitors have a lower affinity for MDM4 and have inferior efficacy in tumors with amplified MDM4.
MDM4 inhibitors
Currently, there are no MDM4 inhibitors in clinical trials. SAH-p53-8 is a cell-penetrating stapled peptide that disrupts the interaction between MDM4 and p53 and has a high affinity for MDM4.372 SAH-p53-8 inhibits the growth of melanoma cells expressing wild-type p53, which depends on the presence of wild-type p53.373 Furthermore, SAH-p53-8 has no effect on normal melanoma cells. A previous study suggested that SAH-p53-8-induced cell death is correlated with cytoplasmic leakage through disruption of the cell membrane.374 Cytotoxicity to normal cells is one of the reasons for the omission of SAH-p53-8 in clinical trials.
MDM2 and MDM4 inhibitors
ALRN-6924 is an MDM2 and MDM4 inhibitor that targets both MDM2 and MDM4. Currently, ALRN-6924 is being studied in several clinical trials (Table 3). ALRN-6924 is active in breast cancer cells expressing wild-type p53 and induces p21 expression.375 Notably, ALRN-6924 does not affect cancer cells expressing mutant p53. ALRN-6924 induces apoptosis and cell cycle arrest to inhibit tumor growth and activate transcription in AML cells.376 ALRN-6924 also prolonged the survival of the AML xenograft model mice. A phase I trial of ALRN-6924 in solid tumor and lymphoma patients expressing wild-type p53 was conducted.377 Ninety-six percent of patients had grade 1–2 AEs, and 5 patients had DLTs, including fatigue, hypotension, anemia, neutropenia, and alkaline phosphatase elevation. The disease control rate (DCR) was 45% (2CR, 2PR, 21SD) in 55 evaluable patients.377
Deleting mutant p53
The expression of p53 mutants induces several vulnerabilities, such as specific dependencies for survival. Some strategies have been proposed to treat p53-mutant cancers by overcoming these limitations. These strategies include the exhaustion of mutant p53 via the application of histone deacetylase (HDAC) inhibitors and heat shock protein 90 (HSP90) inhibitors to suppress tumor cell growth, blocking growth signals regulated by mutated p53.378 However, only a small number of therapeutic approaches for the indirect regulation of mutant p53-expressing cancers have reached clinical trials. HSPs can disturb the interaction between mutant p53 and CHIP E3 or MDM2 ubiquitin ligases, which blocks the degradation of mutant p53. HDAC6 is a positive regulator of HSP90 and increases HSP90 levels to inhibit mutant p53 degradation. Therefore, the application of HSP90 or HDAC inhibitors increases the sensitivity of tumor cells to chemotherapy drugs by promoting mutant p53 degradation.379,380 A phase I clinical trial in which pazopanib and vorinostat were combined to treat patients with p53-mutant tumors suggested that HDAC inhibitors can abolish the proangiogenic function of mutant p53. Wild-type p53 can inhibit cancer occurrence through inhibition of the mevalonate pathway. However, stabilized mutant p53 proteins facilitate the metastatic behavior of tumor cells by increasing the activity of the mevalonate pathway.381 Surprisingly, statins can decrease the levels of mevalonate-5-phosphate and prevent mutated p53 from binding to HSPs, thus promoting the degradation of structural p53 mutants by MDM2 or CHIP E3.382,383 These findings suggest that statins may be used to treat cancers with mutant p53. Currently, numerous clinical trials of statins, such as NCT04767984, NCT03358017, and NCT03560882, are detecting p53 mutation status or p53 protein levels. The safety and pharmacological characteristics of statins are well known because of their long-term use in clinical practice, which is beneficial for the use of statins to treat cancers expressing mutant p53.
Immunotherapies
Cancer immunotherapy targeting p53 is also a prospective therapeutic approach aimed at improving the ability of the immune system to recognize and kill tumor cells expressing abnormal p53. The primary objective of cancer immunotherapy targeting p53 is to increase the ability of the immune system to recognize and eliminate tumor cells expressing abnormal p53. Cancer cells with TP53 missense mutations usually overexpress the p53 protein, and their surface may contain more p53-derived peptides produced by MHCs, which is the basis for effective immunotherapy.
Vaccines
Vaccinations aimed at increasing the response of immune cells to tumor cells expressing large amounts of p53 began in the 1990s.5,384 Although vaccination attempts to eliminate tumor cells overexpressing mutant p53 began more than 20 years ago, currently, no vaccine has been approved for clinical use because various trials have shown no efficacy in patients.
A phase I/II clinical trial evaluated the immunogenicity and safety of a p53 synthetic long peptide (p53-SLP) vaccine in metastatic colorectal cancer patients.385 The adverse events were mainly grade 1/2 and occurred primarily at the vaccination site. p53-specific T-cell responses induced by p53-SLP administration occur in almost all patients.385 A phase II single-arm clinical trial was conducted to evaluate whether low doses of cyclophosphamide could increase the immunogenicity of p53-SLP in patients with recurrent ovarian cancer. The results showed that patients with recurrent ovarian cancer had elevated p53-SLP immunogenicity after low doses of cyclophosphamide were administered.386 Another clinical trial evaluated the long-term clinical and immunological effects of p53-SLP on patients with recurrent ovarian cancer, but significant differences in disease-specific survival or clinical response rates to secondary chemotherapy were not observed between immunized patients and previous controls (P = 0.925).387
Early clinical trials subsequently evaluated modified vaccinia Ankara (MVA), which encodes wild-type p53, in patients with resistant epithelial ovarian cancer and refractory gastrointestinal malignancies.388,389 Among the eleven patients, five patients induced CD4+ T-cell responses, and six patients induced CD8+ T-cell responses. Patients with a greater T-cell response had longer progression-free survival (PFS) than did those with a small T-cell response.388 A powerful CD8+ T-cell response was induced by p53-MVA after the first immunization (P = 0.03), and no AEs above grade 2 occurred.390 Other clinical trials combining p53-MVA and the anti-PD1 antibody pembrolizumab are ongoing (NCT02432963 and NCT03113487).
Dendritic cell (DC) vaccines promote p53 expression on MHC class I and II molecules by modifying autologous DCs.391 One study enrolled patients with small-cell lung cancer who were treated with a new DC vaccine and evaluated its immunologic and clinical effects.392 The results suggested that most patients had p53-specific T-cell responses to vaccination and a relatively high objective clinical response to chemotherapy (61.9%), which rapidly followed vaccination.392 In another study, 21 patients with SCLC were treated with chemotherapy followed by the p53 DC vaccine, 13 of whom achieved a clinical response.393 However, the results of a phase II randomized trial revealed that patients treated with paclitaxel following DC–p53 vaccination did not achieve objective clinical response rates compared with the control group.394 After treatment with autologous wild-type p53 mRNA-transfected DCs, 13 of 18 breast cancer patients presenting with increased p53 expression presented a p53-specific IFN-γ T-cell response, which was greater than that of healthy donors (10%; 1 of 10) and patients with low p53 expression (11%; 2 of 18).395
Antibodies
In addition, p53-specific antibody therapies have been developed in recent years.
TCR-like antibodies, also called T-cell receptor mimic (TCRm) antibodies, recognize peptides produced by intracellular proteins by recognizing the epitopes displayed by MHC class I molecules on the cell surface. Consequently, novel TCR-like antibodies T1-116C and P1C1TM were developed and evaluated.396,397 T1-116C recognizes the wild-type p53 T-cell epitope, combines with normal peripheral blood mononuclear cells, and kills tumor cells through the activation of immune effector functions.396 P1C1TM, a mature human antibody, induces antibody-dependent cellular cytotoxicity toward tumor cells expressing mutant p53.397 In addition, P1C1TM inhibits the growth of tumor cells expressing mutant p53 through the conjugation of antibodies and drugs.397
Bispecific antibodies are also promising strategies for p53 mutation-related tumor immunotherapy. H2-scDb, a bispecific single-chain antibody, is subsequently generated.398 Although the density of the complex composed of the p53 peptide and HLA-A is low on the cell surface, H2-scDb can still effectively kill tumor cells because of its high affinity for the p53 R175H mutant in the complex.398 This approach is likely to receive more attention in the future.
Notably, despite the growing number of immunotherapies targeting p53, these approaches are still in the early stages of development; thus, whether these immunotherapies will actually provide long-term benefits for patients with p53 mutations remains unclear. Another problem with clinical conversion is the toxicity of immunotherapies. We cannot predict and control the toxicity caused by immunotherapies because the understanding of the complex processes of immunotherapies is incomplete. Great efforts are still needed to overcome various difficulties and promote immunotherapy progress in the future.
Gene therapies
In recent years, interest in gene therapy has increased. Gene therapy can directly repair or replace the mutated p53 gene to restore its normal tumor suppressor function. Currently, three main categories of gene therapies targeting p53 have been developed: adenovirus-based therapies, nanoparticles, nonviral approaches for p53 gene delivery, and CRISPR–Cas9-based therapies.
Adenovirus-based therapies
Gendicine, the first p53 gene therapy, is a recombinant human p53 adenovirus that was approved in 2003 by the China Food and Drug Administration (CFDA) for the treatment of HNSCC.399 Many clinical studies have demonstrated that gendicine is safe and has better efficacy when combined with radiotherapy or chemotherapy than does gendicine alone.399 However, various concerns, including adverse events, immunogenicity, and inefficiency in the successful use of viral vectors, remain. These results provide a basis for the future of the combination of adenoviral p53 therapy and immune checkpoint inhibitors. With the emergence and development of more novel viral vectors, p53 gene therapy may become an effective part of combination therapy in the future. Other adenovirus-based therapies, including advexin and SCH-58500, have shown promising results in clinical trials.400,401,402 Although advexin and SCH-58500 have not yet entered clinical use, these therapies have received much attention.
Nanoparticles
Compared with viruses, nanoparticles have lower immunogenicity, longer circulation times, and fewer adverse events.403 Unlike gendicine, nanoparticles are usually injected intravenously, making them suitable for treating distant metastases. Moreover, heterologous delivery of gene products into tumor cells greatly increases the efficacy and specificity of nanoparticles, increasing their ability to restore p53 expression and enhance their antitumor effects.404 SGT-53, a nanoparticle that has WT TP53 cDNA packed in a plasmid, was shown to inhibit tumor growth, increase the induction of apoptosis, and significantly improve median survival in patients treated in combination with temozolomide.405 A CXCR4-targeted mRNA nanoparticle was subsequently developed to induce p53 expression in HCC.389 The combination of CXCR4-targeted p53 mRNA nanoparticles and PD1 reprogrammed the molecular and cell components of the TME in p53-null orthotopic and ectopic HCC models.406
Nonviral approaches for p53 gene delivery
Given the increased attention given to p53 cDNA and mRNA delivery, small interfering RNA (siRNA) oligonucleotides that target specific p53 mutants to reduce GOF and DN effects are also being developed. Recently, diverse siRNAs capable of targeting four hotspot mutations, which account for approximately 20% of all p53 mutations, have been generated.407 These siRNAs eliminate the DN effect and depend on mutant p53 to induce cell death and inhibit tumor growth in xenografts. More importantly, these siRNAs are highly specific for targeted p53 mutants and have no effects on wild-type p53 expression.407
CRISPR–Cas9-based therapies
Following the development of CRISPR–Cas9 technology, this technology has also been explored for p53-based antitumor therapy.408 For example, the researchers performing one study introduced CRISPR–Cas9 to transform a TP53 missense mutation into a wild-type sequence in breast cancer cell lines.409 Approximately 7.6% of the cells successfully achieved TP53 correction, whereas fewer than 1% of the cells showed proof of indel formation. However, a recent study suggested that the introduction of CRISPR–Cas9 induced the emergence and amplification of TP53 inactivation mutations. Thus, CRISPR–Cas9 therapy targeting p53 should be used with caution. Notably, CRISPR–Cas9 can induce DNA damage-triggered p53 activation in cells expressing wild-type p53, promoting cell cycle arrest or cell death.408,410,411 As a programmable endonuclease, the CRISPR‒Cas system splits particular genomic sites through the guidance of a single guide RNA (gRNA).412 The beauty of CRISPR is not limited to DNA cleavage. The version of Cas9 in which mutations were introduced into the catalytic pocket of the Cas9 protein capably bind to DNA with no DNA cleavage. A series of combinations of deaminase, reverse transcriptase (RT), or a transcriptional repressor/activator with Cas9 subsequently achieved unprecedented benefits, such as the expression/activation of specific or multiple genes, DNA base pair conversions, or transversions.413,414 However, Cas9 may induce abnormal events. For example, Cas9 can successfully introduce DSBs at specific sites to introduce small insertions or deletions (indels) into the genome, and the introduced DSBs cause the formation of gene byproducts that disturb intracellular genome stability.415 CRISPR–Cas9 may also introduce DSBs at other sites because of off-target effects. The random fusion of many broken sites can cause chromosomal rearrangement, inducing structural variations, including chromosome translocation, deletion, and inversion, and eliciting cellular lesions.416 Programmable base editing (PBE) with an adenine base editor (ABE) consists of the fusion of impaired Cas9 and deoxyadenosine deaminase and efficiently transforms target A-T base pairs to G-C base pairs. Moreover, this approach results in higher product purity and fewer unnecessary byproducts, such as indels, than does CRISPR/Cas9. Compared with the current Cas9 nuclease-based approach, adenine base editors introduce point mutations more precisely and efficiently because of decreased off-target genome modifications and the absence of DNA cleavage.417,418 Optimistically, the CRISPR adenine base-editing system was developed to correct gene mutations. In contrast to the CRISPR–Cas9 vector, the CRISPR adenine base-editing system not only possibly eliminates targeted cells but also effectively corrects tumor-associated TP53 mutations.419 More importantly, the CRISPR adenine base-editing system does not select for TP53 mutations because it functions independently.
Gene therapy has important theoretical potential in cancer treatment, but it still faces many challenges in clinical application, including technology maturity, safety, efficacy, and patient acceptance. Major clinical challenges include immature technology, limited efficacy, and uncertain toxicity. Although some progress has been made in gene therapy research, the current technology is still not mature. For example, wild-type p53 replacement therapy, although widely studied and used, is unable to repair or eliminate aberrant genes and is often considered insufficiently effective in clinical practice. In addition, some nanoparticles enter cells by interacting with the cell membrane or fusing, which may cause nonspecific cytolysis and thus have cytotoxicity limitations.
Therapeutics for nonneoplastic diseases
The exploration of therapeutics targeting p53 in nontumorous diseases is not as widespread as that in cancers. Regulating p53 activity ameliorates various noncancer diseases through the extensive functions of p53. For example, p53 inhibition may suppress the development of NDDs and ischemic organ injury. The specific mechanisms of p53 in metabolic dysfunction and aging-related conditions have not been identified. Although p53 inhibitors are reported to treat some noncancer diseases, the risk of tumorigenesis is increased. A series of drugs and clinical trials for the treatment of nontumor diseases, including NDDs and metabolic diseases, have been conducted (Table 4).
Neurodegenerative diseases
After incessant efforts in recent years, some medicines have been approved to manage NDDs, whereas multiple medicines alleviate only symptoms. NDDs are also the main clinical concern in older individuals.420 The restrictive functions of the blood–brain barrier (BBB) block “foreign substances” from entering the brain, which is the predominant reason for the lack of pathogenesis-targeting therapies. The occurrence of nanoparticles has resolved this problem to a certain extent.421 In some individuals, the therapeutic purposes include alleviating symptoms, restoring balance and mobility, and easing pain.
Alzheimer’s disease
Three classes of drugs, including antibodies targeting amyloid-beta (Aβ) plaques, cholinesterase inhibitors, and glutamate regulators, have been approved for AD treatment. Aducanumab (Aduhelm) is the first disease-modifying drug approved for AD treatment and was approved in June 2021.422 As an IgG1 monoclonal antibody, aducanumab binds to extracellular Aβ plaques to eliminate them.422,423 Clinical data have shown that aducanumab can decrease the load of Aβ plaques but does not improve cognitive function in AD patients.424
Currently, cholinesterase inhibitors are the first-line drugs used for AD treatment. The most commonly used cholinesterase inhibitors in the clinic are donepezil, galantamine, and rivastigmine. Cholinesterase inhibitors can increase cholinergic activity by inhibiting acetylcholine degradation, which attenuates the cognitive decline of patients with dementia.425 Tacrine is the first cholinesterase inhibitor approved by the FDA for AD treatment. Tacrine was later discontinued because of liver toxicity and limited effectiveness.426 Donepezil was approved for patients with mild and moderate AD in 1996 and was approved for patients with severe AD in 2006. Donepezil application significantly improved the daily activities, behavior, and cognitive function of AD patients.427 A randomized, double-blind study evaluated the effectiveness and tolerability of donepezil at concentrations ranging from 10 to 23 mg/d in patients with moderate-to-severe AD.428 A total of 1467 AD patients were randomly assigned to receive 23 mg/d or 10 mg/d donepezil, and the results suggested that 23 mg/d donepezil significantly improved cognitive function compared with 10 mg/d donepezil. Notably, the incidence of adverse events in the 23 mg/day donepezil group was higher than that in the 10 mg/day donepezil group (73.7% vs. 63.7%), especially gastrointestinal toxicities, including nausea (11.8% vs. 3.4%), vomiting (9.2% vs. 2.5%), and diarrhea (8.3% vs. 5.3%). Moreover, the results of this study led to the approval of 23 mg/d donepezil for patients with moderate-to-severe AD in 2012. A post hoc analysis suggested that the improvement in cognition caused by 23 mg/d donepezil over 10 mg/d donepezil was not correlated with the age, weight, or sex of AD patients or the duration of treatment with previous low doses of donepezil.429 Rivastigmine was first approved by the FDA for the treatment of mild-to-moderate AD in 2000. The Investigation of Transdermal Exelon in Alzheimer’s Disease (IDEAL) was a 24-week, double-dummy and double-blind study430 that evaluated the efficacy and safety of rivastigmine patches with capsules and the placebo. A total of 1195 AD patients were enrolled. The results showed that rivastigmine had significant efficacy in the treatment group compared with the placebo group and that the efficacy of the 10 cm2 patch was similar to that of the capsules. Importantly, the incidences of nausea (7.2% vs. 23.1%) and vomiting (6.2% vs. 17.0%) were lower than those in the placebo group. Compared with capsules, 20 cm2 patches markedly increased cognitive scores and had tolerability similar to that of capsules.430 The activities of daily living and cognition (ACTION) trial compared the efficacy and safety of low-dose versus high-dose rivastigmine transdermal patches in patients with severe AD,431 which supported the approval of high-dose rivastigmine (13.3 mg/24 h) patches for patients with severe AD. Galantamine, which was approved in 2001 by the FDA, is an allosteric modulator of nicotinic and acetylcholine receptors and a competitive inhibitor of acetylcholinesterase.432 A multicenter, double-blind study randomly assigned patients to receive galantamine or a placebo to increase the safety of galantamine.433 Compared with 1021 patients treated with the placebo, 1024 patients treated with galantamine had a lower mortality rate (hazard ratio = 0.58, P = 0.011) and improved cognition (mean change in Mini-Mental State Examination scores at the endpoint of −2.14 for patients receiving galantamine compared with −1.41 for patients receiving the placebo). Glutamate impairs neurons and induces neurodegeneration through the activation of NMDA receptors and glutamate receptors in postsynaptic neurons. Memantine is an uncompetitive NMDA receptor antagonist and prevents damage to AD patients caused by NMDA receptor overactivation.434 Memantine was first approved for patients with moderate-to-severe AD in 2003. Monotherapy with memantine obviously improved the cognition of AD patients. The efficacy of a cholinesterase inhibitor combined with memantine was superior to that of memantine alone.435
Parkinson’s disease
The predominant principle of treating PD is to increase dopamine levels in the substantia nigra region of the brain.436 Levodopa preparations, monoamine oxidase-B (MAO-B) inhibitors, and dopamine agonists are helpful medicines for PD treatment.437 For young patients with obvious tremors, anticholinergic drugs need to be used cautiously.437 A pragmatic, open-label trial revealed that PD patients randomly assigned to initial treatment with levodopa experienced small but permanent benefits after 7 years compared with patients treated with MAO-B inhibitors or dopamine agonists, which was specifically shown by a 1.8-point improvement (95% CI, 0.5–3.0; P = 0.005) in the average score on the Parkinson Disease Questionnaire-39 mobility subscale (10-items; 0- to 40-point range).438 Compared with patients treated with levodopa, more patients treated with MAO-B inhibitors and dopamine agonists terminate treatment because of adverse events.438 Oral dopamine agonists such as ropinirole and pramipexole promote impulse control disorders, including abnormal sexual and eating behaviors, compulsive spending, gambling, and compulsive medication use, in more than 40% of patients.439 However, approximately 15%-20% of patients who discontinue the use of dopamine agonists experience withdrawal symptoms such as panic attacks, anxiety, diaphoresis, irritability, and pain.440 Therefore, although individuals experience severe adverse events, the use of dopamine agonists cannot be terminated instantly. Advanced therapies, including deep-brain stimulation, levodopa-carbidopa enteral suspension, and MRI-guided focused ultrasound, which can be used to treat the motor symptoms of PD patients, have been developed. Strict center assessments are needed before treatment, and the complications caused by these approaches should be noted. The Unified Parkinson Disease Rating Scale ranges from 0 to 108 points, usually with a minimal clinically critical discrepancy of 2.3–2.7 points. A meta-analysis indicated that deep-brain stimulation improved on-medication motor scores on the Unified Parkinson Disease Rating Scale by 4.56 points (95% CI, 3.11–6.00) compared with the best drug therapy and off-medication scores by 15.50 points (95% CI, 12.60–18.39).441 Online screening tools can seek and select proper candidates for deep-brain stimulation.442,443 The most critical screening approach is evaluation by experienced teams and a deep understanding of potential risk.
In addition, nonmotor symptoms (MNMs), such as constipation, dementia, and psychosis, can also be treated with drugs in PD patients.444 Rivastigmine plays a critical role in improving Parkinson’s disease dementia and is designated by the International Parkinson and Movement Disorder Society.445 In a double-blind clinical trial in which individuals were randomly assigned to receive rivastigmine (362 individuals) or the placebo (172 individuals), participants treated with rivastigmine had increased scores (mean improvement of 2.1 points) on the 70-point Alzheimer’s Disease Assessment Scale, whereas those in the placebo group had decreased scores (decrease of 0.7 points) (P < 0.001).446 Selective serotonin norepinephrine reuptake inhibitors, selective serotonin reuptake inhibitors, and tricyclic antidepressants may be effective treatments for depression in patients with PD, and nonpharmacologic approaches, including repetitive transcranial magnetic stimulation and cognitive‒behavioral therapy, may also be useful.445 Notably, optimal treatment approaches are still unavailable for some nonmotor symptoms. For example, insomnia and fatigue are common in patients with PD, but no proper medical treatments can be used for these symptoms.445,447 Overall, the selection of proper drug treatments for nonmotor symptoms should rely on the efficacy and extent of adverse events.
Other diseases
ALS patients die of respiratory failure after symptoms appear.448 Currently, two drugs, riluzole and edaravone, are approved for the treatment of ALS. Riluzole is a glutamate-receptor antagonist and was approved in 1995 as an oral tablet with a 100 mg/day dose. Two randomized controlled trials (RCTs) reported the efficacy of riluzole.449,450 The results of these RCTs revealed that, compared with placebo, riluzole administration prolongs the survival of ALS patients by 3–4 months. Edaravone is a free-radical scavenger and was approved in 2017 at a dosage of 60 mg/day. Edaravone can prevent the development of the disease.451
The above approaches and restrictions caused by the BBB cannot meet the demand for new treatments for NDDs.452 Nanotechnology has become a safe and optimal approach and can target drug/gene delivery to the central nervous system because of its characteristics, including a high drug loading capacity, low systemic toxicity, and good physical and chemical stability.453 Nanoparticles are divided into inorganic nanoparticles and organic nanoparticles. Metal nanoparticles easily cross the BBB and accumulate in the brain, which has attracted much attention.454 Organic nanoparticles are usually naturally occurring molecules, such as lipids and other organic molecules, which can be carriers for delivering nanomedicine because of their superior biocompatibility.455 Currently, several clinical trials of nanoparticle formulations for the treatment of NDDs exist.424 Only one clinical trial has been completed in which lipid nanoparticle-based formulations have been used for transthyretin-mediated amyloidosis. An exciting clinical trial started in 2003 in which nanoparticle-mediated delivery of APH-1105 was used to treat mild-to-moderate AD.
Metabolic diseases
The incidence of all metabolic diseases increased from 2000 to 2019, according to data from the World Health Organization (WHO).456 Additionally, metabolic diseases are often correlated with a series of complications, increasing mortality, and medical costs.457,458 Biomacromolecular approaches, including those involving proteins, oligonucleotides, and antibodies, play pivotal roles in the treatment of metabolic diseases.459,460 Various metabolic diseases are closely related and interact with each other. For example, NAFLD progression is strongly correlated with insulin resistance and type 2 diabetes mellitus (T2DM).461 NAFLD is closely related to obesity, and diet and exercise have proven to be effective in treating NAFLD.462 Therefore, targeting these interactions and adopting a comprehensive attitude toward the treatment of metabolic diseases can prove to be a very useful method.
Obesity
The effective treatment of obesity requires the combination of multidisciplinary methods, including dietary management, behavioral alterations, and physical activity. Currently, the 7 antiobesity medications (AOMs) approved by the FDA to treat obesity are phentermine, orlistat, phentermine/topiramate extended release (ER), naltrexone sustained release (SR)/bupropion SR, liraglutide (3.0 mg), semaglutide (2.4 mg), and tirzepatide.463 A retrospective study revealed that obese individuals (BMI = 27 kg/m2) treated with phentermine constantly for more than 12 months had lower weights than those treated with the medicine for less than 3 months.464 Orlistat can decrease fat absorption in the gastrointestinal tract by suppressing the activities of pancreatic and gastric lipases.465 The double-blind Xenical in the Prevention of Diabetes in Obese Subjects (XENDOS) trial evaluated the efficacy of orlistat.466 A total of 3305 patients were randomly assigned to receive 120 mg of orlistat or a placebo 3 times daily; individuals treated with orlistat lost more weight after 4 years, and the results were similar in those with or without prediabetes. Currently, the era of obesity pharmacotherapy is exciting. Compared with previous AOMs, novel agents, such as semaglutide and tirzepatide, promote more weight loss. More clinical trials associated with these medicines are ongoing.
Diabetes, fatty liver disease, and cardiovascular diseases
Currently, effective treatment approaches for NAFLD are still lacking. The predominant therapeutic strategies for NAFLD include the optimization of comorbidities and lifestyle alterations. However, lifestyle interventions are difficult to maintain, which influences the overall efficacy of treatment.467 Therefore, medical therapies are needed to improve liver-related outcomes effectively. Resmetirom, a thyroid hormone receptor-β (THR-β) agonist, was the first and only treatment approved by the FDA for NAFLD.468 Some drugs have been explored to improve therapeutic approaches for NAFLD. In an interim analysis of clinical trials, patients were assigned to receive resmetirom (80 mg or 100 mg) or a placebo.469 Fibrosis was attenuated in 24% and 26% of patients in the 80 mg/kg and 100 mg/kg groups, respectively, compared with 14% in the placebo group (P < 0.001). A remarkable decrease in low-density lipoprotein cholesterol levels (−14% and −16%, respectively) was observed in the 80 mg and 100 mg groups compared with a mild reduction in the placebo group. The most common adverse events in the resmetirom group were slight nausea, diarrhea, and vomiting, suggesting acceptable safety.469 Several clinical trials are ongoing to evaluate the long-term benefits of resmetirom. Approval may be retracted if the trials fail to meet their primary and secondary endpoints.
NAFLD includes a spectrum of disease severities ranging from simple steatosis to more severe disorders characterized by hepatocyte death, inflammation, and the development of NASH. Simple steatosis is correlated with an increased risk of other metabolic diseases, such as insulin resistance, dyslipidemia, T2DM, and hypertension.470 T2DM and obesity increase the risk of developing NAFLD, and improvements in insulin sensitivity promote histological improvements in fibrosis regression and NASH.470,471 CVDs are the main cause of death in patients with T2DM, accounting for up to two-thirds of all deaths.472 Elevated HbA1c levels are significantly associated with increased risks of heart disease and overall mortality.473 A meta- analysis revealed that strategies that decrease glucose levels, especially those that decrease body weight, reduce the risk of the main adverse cardiovascular events, which proves the efficacy of diabetic therapies in ameliorating cardiovascular events.474 Currently, a series of GLP1R agonists (liraglutide, exenatide, semaglutide, and dulaglutide) are approved for the treatment of T2DM.474 T2DM patients treated with either exenatide, semaglutide, or liraglutide experienced obvious benefits in decreasing hepatic lipid levels and the levels of inflammatory markers, which improved HbA1c levels and decreased body weight.475,476 Some of the beneficial effects of GLP1R agonism on NAFLD endpoints are tightly correlated with decreased weight and improved metabolic indices. Patients with NAFLD, obesity and T2DM have elevated DPP4 levels, which promote hepatocyte apoptosis and fibrosis.477 The suppression of DPP4 can also be used to treat metabolic diseases. Currently, four DPP4 inhibitors, alogliptin, saxagliptin, sitagliptin, and linagliptin, are approved for the treatment of T2DM. SGLT2 inhibitors, including canagliflozin, ertugliflozin, empagliflozin, and dapagliflozin, can increase the urinary excretion of glucose and are approved for the treatment of T2DM. Moreover, SGLT2 inhibitors promote weight loss in many individuals and decrease the risk of major cardiovascular events.478 Several trials have elevated the efficacy of SGLT2 inhibitors in patients with NAFLD and T2DM. Patients treated with dapagliflozin, canagliflozin, or empagliflozin presented decreased hyperglycemia and improvements in hepatic lipid content, liver fibrosis, and liver enzymes.479,480 Thiazolidinediones probably ameliorate peripheral insulin sensitivity by triggering the release of adipokines, facilitating triglyceride storage in adipose tissue and increasing the inhibitory action of insulin on lipolysis, leading to decreased plasma levels of free fatty acids and decreased hepatic lipid accretion.481 Rosiglitazone is the most effective drug among the two approved thiazolidinediones.482 Several studies have suggested that NASH patients who receive rosiglitazone have increased insulin sensitivity and decreased hepatic steatosis. However, considerable edema and weight gain were observed.483 As a nuclear receptor, the farnesoid X receptor (FXR) is overexpressed in the liver and intestine and modulates the expression of genes involved in the synthesis of cholesterol and bile acids.484 Obeticholic acid is a bile acid derivative that activates FXR, which can ameliorate insulin sensitivity and decrease the expression of markers of liver fibrosis and inflammation in NAFLD and T2DM patients.485 Recently, several studies have shown improvements in histological markers of NASH and fibrosis with obeticholic acid treatment.486
Overall, the treatment of metabolic diseases needs to take a holistic perspective because of the correlations and interactions between metabolic diseases. The influence of medicines on multiple metabolic diseases needs to be considered when these drugs are used. To date, effective and precise treatment approaches for several diseases, such as NAFLD, are lacking. Although many drugs have been investigated in the preclinical stage, they have not yet entered the clinic because of their toxicity or obstacles to their clinical translation. Further research and exploration are needed in the future.
Discussion of key questions regarding p53
Recently, systems biology and computational approaches have advanced our understanding of p53 regulation and helped predict treatment response. In one study, machine learning and directional networks were used to optimize the regulatory manner of p53.487 Machine learning was used to predict the mutation status of TP53 from transcriptomic data, and the directional network was used to reconstruct the effect of TP53 mutation on the transcription level of its target. Moreover, this study effectively demonstrated the inferential capacity of the model framework under irradiation and hypoxia conditions. For example, the study examined changes in p53 downstream pathways in the wild-type TP53 cell line H460 (a lung cancer cell line) both before and after radiation exposure. The results indicated that the downstream signaling pathways in the irradiated samples were significantly altered compared with those in the untreated cells. Furthermore, network analysis based on random comparisons revealed that the probabilities of achieving similarity scores at the 25%, 50%, 75%, and 90% cutoff levels were 100%, 67%, 67%, and 36%, respectively. Notably, when comparing the irradiated and nonirradiated cell lines, the similarity of the networks at the four critical value thresholds (25%, 50%, 75%, and 90%) was 100%, 0%, 0%, and 0%, respectively. These findings collectively underscore the significant impact of irradiation on the regulatory properties of p53 in lung cancer cell lines. This is the beginning of the use of regulatory networks to predict the impact of mutations and is also applicable to other functional abnormalities, with important implications for p53-targeting drug design and precision medicine. A proof-of-concept study employed a deep learning (DL) algorithm to predict the TP53 mutational status.488 This study used a convolutional neural network model to identify tumor regions and predict TP53 mutations in breast cancer, which was validated by immunohistochemistry (IHC) and next-generation sequencing (NGS). The results showed that 92% of the tumor regions were predicted to have TP53 mutations by the DL algorithm, which was consistent with the 90% predicted by IHC. Compared with traditional methods, DL-based algorithms provide promising prospects for enhanced biomarker detection and precision oncology, but further validation is needed for better integration with real-world clinical work. DL-based histopathological analysis plays an important role in improving patient management and treatment planning on the basis of molecular biomarkers. An integrated approach can reveal new drug combinations or guide personalized treatment strategies, ultimately bridging the gap between laboratory research and clinical application. Specifically, machine learning methods can efficiently predict the synergistic effects of drug combinations by integrating various data sources, such as gene expression data, drug chemical structures, and protein interaction networks. For example, the DeepSynergy method uses deep learning to integrate chemical and genomic information to predict the synergy of drug combinations.489 Additionally, DrugComboRanker integrates drug and cancer genomic features to predict drug combinations that target different cancer signaling modules.489 In addition, machine learning models can predict patients’ responses to specific drug combinations on the basis of their individual characteristics, such as mutation profiles and protein expression profiles, thereby providing a basis for personalized treatment. For example, the DCPT platform uses a random forest model to predict patients’ specific responses to drug combinations by integrating exome sequencing, RNA sequencing, and drug target information.490 On the basis of the above findings, we can also predict patients’ specific responses to p53-targeted therapies and design more effective combination therapies via systems biology methods. For example, multiomics data, including genomics, proteomics, and transcriptomics data, can be integrated to construct a comprehensive network model of the p53 signaling pathway. By analyzing these data with machine learning algorithms, such as support vector machines (SVMs) or random forests (RFs), we can predict patients’ responses to p53-targeted therapies.490 We can identify key targets and pathways that interact with the p53 signaling pathway through systems biology methods, such as network analysis and pathway enrichment analysis. For example, the multiple target optimal intervention (MTOI) method can identify potential drug targets and suggest optimal target intervention combinations.490 Furthermore, we can evaluate the impact of different drug combinations on the p53 signaling pathway via machine learning models, such as stochastic gradient boosting (SGB) or deep learning, to optimize combination therapies. By simulating the effects of drug combinations on cellular networks, we can select the combinations that can restore the network to its normal state to the greatest extent.490
In recent years, an increasing number of studies have explored the complex mechanisms of p53 under different physiological conditions and the influences and mechanisms of p53 mutations. The current understanding of p53 dysfunction has significantly promoted the development of therapeutic strategies targeting p53. In addition to its lack of activity, the mutant p53 protein tends to unfold and aggregate, inducing the formation of amyloid aggregates. Therefore, researchers are dedicated to maintaining the activity of mutant p53 and developing new molecules that directly inhibit the aggregation of p53.491 New molecules usually include small molecules and peptides, and the design principle is based on the latest molecular mechanism of the disease, namely, amyloid formation. For example, protein aggregation and amyloid formation occur in NDDs such as AD, PD, and ALS. Further research is needed to explore p53-targeted therapies that inhibit p53 aggregation.
Although we have described the structure, biological function, mutation, and therapeutic potential of p53 in detail, there are still some questions that need to be further discussed. First, p53 mutations contribute to the complexity and evolution of tumors through several mechanisms, enhancing tumor heterogeneity, invasiveness, and treatment resistance. p53 mutations can increase heterogeneity in gene expression, the cell cycle status, and metabolic features within the tumor cell population. This heterogeneity means that different subsets of tumor cells may respond differently to treatment, complicating therapeutic efforts. Mutant p53 can influence the remodeling of the extracellular matrix (ECM) and the expression of cell adhesion molecules, thereby increasing the invasiveness of tumor cells. For example, mutant p53 can activate specific signaling pathways (such as the PI3K/AKT pathway) to promote tumor cell migration and invasion.492 In some cases, mutant p53 can also modulate the tumor microenvironment by interacting with immune cells, further enhancing tumor invasiveness.129 More importantly, mutant p53 can promote resistance to chemotherapy and targeted therapies through various mechanisms. On the one hand, mutant p53 can suppress the expression of apoptosis-related genes (such as Puma), helping tumor cells evade the apoptosis induced by chemotherapeutic drugs.492 On the other hand, mutant p53 can upregulate the expression of multidrug resistance proteins (such as MDR1), leading to increased drug efflux and increased chemotherapy resistance.492 The diversity and complexity of p53 mutants pose significant challenges for targeted therapies. Different types of p53 mutants may promote tumor progression through distinct mechanisms, necessitating the development of specific therapeutic strategies for specific types. Likewise, tumor heterogeneity means that even within the same tumor, different cell populations may respond differently to the same treatment, increasing the complexity and difficulty of therapy. In summary, mutant p53 promotes the complexity and evolution of cancers through multiple mechanisms, complicating the use of targeted therapies. However, by gaining a deeper understanding of the mechanisms underlying p53 mutations, targeted therapeutic strategies can be developed to overcome these challenges and improve the effectiveness of cancer treatment.
Second, in a heterogeneous tumor environment, can therapies be designed to selectively eliminate cells with GOF p53 mutations without harming normal cells that rely on wild-type p53? Within the tumor ecosystem, targeting cells with mutant p53 could have profound effects on the TME. Mutant p53 may not only lose the tumor suppressor function of wild-type p53 but also acquire new tumor-promoting functions, such as promoting tumor invasion, metastasis, and drug resistance. Thus, therapies targeting mutant p53 may impact the tumor ecosystem by inhibiting protumor signaling, restoring immune surveillance, and reducing tumor heterogeneity. In heterogeneous tumor environments, achieving selective targeting of mutant p53 cells without damaging wild-type p53-dependent normal cells is a major challenge, mainly for the following reasons: some drugs targeting mutant p53 may also be toxic to wild-type p53 cells, and the complex tumor microenvironment may affect the efficacy of drugs. To overcome the above challenges, combination therapy is a promising strategy. By combining multiple drugs, it is possible to restore wild-type p53 function and block the tumor-promoting signal of mutant p53 simultaneously. This combination strategy not only inhibits tumor growth but also antagonizes adaptive and metastatic behaviors driven by mutant p53.
Third, the paradoxical behavior of p53 mutants deserves our attention. Traditionally, p53 has been regarded as a quintessential tumor suppressor that safeguards against malignancy by maintaining genomic stability and regulating apoptosis. However, mutations in p53 not only disrupt this vital function but also confer unexpected oncogenic properties to the protein. This duality challenges the conventional binary classification of genes as either tumor suppressors or oncogenes. Wild-type p53 plays crucial roles in maintaining genomic stability and regulating apoptosis. When the p53 gene is mutated, its tumor-suppressive function can be entirely lost, leading to increased genomic instability. In addition to their loss of function, certain mutants actively promote cancer cell survival, drive tumor progression, and enhance adaptability under stress. A deep exploration of the paradoxical behavior of p53 mutants is crucial for developing effective cancer treatment strategies. For example, reactivating the tumor-suppressive functions of wild-type p53 through small molecules or other approaches may help restore genomic stability and induce apoptosis in cancer cells. For GOF mutations, developing targeted therapies that specifically inhibit the oncogenic functions of GOF mutant p53 may help suppress the survival and progression of tumors. Combining strategies to restore wild-type p53 function with those targeting GOF mutations may represent a more comprehensive therapeutic approach.
Finally, mutant p53 GOF activities, including the facilitation of cell proliferation, invasion, survival, cancer-promoting metabolism, protumorigenic inflammation, and drug resistance, have been observed in a series of experimental models, and multiple GOF mechanisms have been proposed.140,493,494 Mutant p53 can interact with transcription factors and cofactors to regulate multiple gene expression programs that promote cell proliferation, survival, invasion, and drug resistance.493 Mutant p53 can activate downstream signaling pathways that contribute to tumor progression. For example, mutant p53 can induce noncanonical NF‒κB (NC‒NF‒κB) signaling through the cGAS‒STING pathway, which promotes tumor cell invasion, metastasis, and immunosuppression. This pathway activation is a result of CIN-induced cytosolic DNA release, which activates the cGAS‒STING pathway and downstream NC‒NF‒κB signaling.495 The GOF properties of mutant p53 involve more than just destabilizing the p53 protein. They also promote the reprogramming of cellular pathways to confer a survival advantage to cancer cells. This distinction has significant implications for therapeutic strategies. Effective cancer therapies should therefore focus on both restoring wild-type p53 function and targeting the oncogenic signaling pathways activated by mutant p53. This dual approach may represent a more robust strategy for treating cancers driven by p53 mutations.
Conclusions
As mentioned above, an increasing number of studies in recent years have led to a better understanding of the complex functions of p53 in different contexts, including the fundamental regulation, function, and therapeutic potential of p53. However, several problems still need to be solved. For example, p53-induced classical activities or other activities are more critical to tumor suppressor function. In addition, how to target p53 to treat tumors is still a great challenge. The existing therapies targeting p53 all have some degree of limitations. For mutant p53, most studies aim to restore the wild-type conformations and functions of p53 mutants. However, p53 mutants vary owing to the heterogeneity of p53 mutations, and one compound is effective against only one or a few p53 mutants. Furthermore, the specific mechanisms of these compounds are not clear. For tumors expressing wild-type p53, therapeutic approaches aim to inhibit wild-type p53 degradation by disrupting the interaction between p53 and MDM2/4. However, pure MDM4 inhibitors and MDM2/4 bispecific inhibitors are lacking, and further exploration of new effective drugs is still needed. In addition, drugs that inhibit the interaction between p53 and MDM2/4 induce wild-type p53 accumulation, most likely resulting in toxicity in normal cells and tissues. Although most of the above drugs have been studied in clinical trials, most clinical trials are phase I/II trials. Partial clinical trials have not achieved their primary endpoints and have reported DLTs; these results do not support the clinical use of these drugs. Notably, traditional compounds used for treating other diseases should be reevaluated to determine whether they work in tumors expressing mutant p53. For example, ATO has long been used to treat APL and has been shown to stabilize the p53 (R175H) mutant and restore p53 activity.
In recent years, novel therapies targeting p53, such as immunotherapy and gene therapy, have been developed, including vaccines, p53-specific antibodies, nanoparticles, siRNA delivery, and CRISPR–Cas9. Although the above methods have made great progress, many problems remain to be solved. For example, DNA and RNA need to be modified to ensure the delivery of nucleic acids and prevent their degradation. In addition, the clearance role of the liver is a major obstacle to gene delivery therapy. Great efforts are still needed in the future to develop delivery particles that are low in immunogenicity and can act directly on target tissues.
p53 has long been considered “undruggable,” but the emergence of an increasing number of drugs and novel technologies targeting p53 seems to have changed this view. Like p53, KRAS was also considered “undruggable” several decades ago. Currently, the KRAS inhibitor G12C has been approved by the FDA. We can gain confidence from the successful approval of KRAS inhibitors, and there are reasons to believe that drugs targeting p53 will eventually be successful in the clinic. The successful application of drugs targeting p53 will certainly be a breakthrough owing to the widespread occurrence of p53 mutations.
References
Hafner, A., Bulyk, M. L., Jambhekar, A. & Lahav, G. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell. Biol. 20, 199–210 (2019).
Liu, Y., Su, Z., Tavana, O. & Gu, W. Understanding the complexity of p53 in a new era of tumor suppression. Cancer Cell 42, 946–967 (2024).
Hu, J. et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J. Hematol. Oncol. 14, 157 (2021).
Robles, A. I., Jen, J. & Harris, C. C. Clinical outcomes of TP53 mutations in cancers. Cold Spring Harb. Perspect. Med. 6, a026294 (2016).
Sabapathy, K. & Lane, D. P. Therapeutic targeting of p53: all mutants are equal, but some mutants are more equal than others. Nat. Rev. Clin. Oncol. 15, 13–30 (2018).
Kandoth, C. et al. Mutational landscape and significance across 12 major cancer types. Nature 502, 333–339 (2013).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Song, Y. et al. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 509, 91–95 (2014).
Peifer, M. et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat. Genet. 44, 1104–1110 (2012).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Wang, H., Guo, M., Wei, H. & Chen, Y. Targeting p53 pathways: mechanisms, structures, and advances in therapy. Signal Transduct. Target. Ther. 8, 92 (2023).
Hassin, O. & Oren, M. Drugging p53 in cancer: one protein, many targets. Nat. Rev. Drug Discov. 22, 127–144 (2023).
Peuget, S., Zhou, X. & Selivanova, G. Translating p53-based therapies for cancer into the clinic. Nat. Rev. Cancer 24, 192–215 (2024).
Oren, M. & Prives, C. p53: a tale of complexity and context. Cell 187, 1569–1573 (2024).
Joerger, A. C. & Fersht, A. R. Structural biology of the tumor suppressor p53. Annu. Rev. Biochem. 77, 557–582 (2008).
Krois, A. S., Dyson, H. J. & Wright, P. E. Long-range regulation of p53 DNA binding by its intrinsically disordered N-terminal transactivation domain. Proc. Natl. Acad. Sci. USA 115, E11302–e11310 (2018).
Duffy, M. J., Synnott, N. C., O’Grady, S. & Crown, J. Targeting p53 for the treatment of cancer. Semin. Cancer Biol. 79, 58–67 (2022).
Teufel, D. P., Freund, S. M., Bycroft, M. & Fersht, A. R. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl. Acad. Sci. USA 104, 7009–7014 (2007).
Di Lello, P. et al. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 22, 731–740 (2006).
Mohan, A. et al. Analysis of molecular recognition features (MoRFs). J. Mol. Biol. 362, 1043–1059 (2006).
Vacic, V. et al. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 6, 2351–2366 (2007).
Bode, A. M. & Dong, Z. Post-translational modification of p53 in tumorigenesis. Nat. Rev. Cancer 4, 793–805 (2004).
Toledo, F. & Wahl, G. M. Regulating the p53 pathway: in vitro hypotheses, in vivo veritas. Nat. Rev. Cancer 6, 909–923 (2006).
Hoyos, D., Greenbaum, B. & Levine, A. J. The genotypes and phenotypes of missense mutations in the proline domain of the p53 protein. Cell Death Differ. 29, 938–945 (2022).
Dumont, P., Leu, J. I., Della Pietra, A. C. 3rd, George, D. L. & Murphy, M. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat. Genet. 33, 357–365 (2003).
Jiang, M. et al. p53 binds the nuclear matrix in normal cells: binding involves the proline-rich domain of p53 and increases following genotoxic stress. Oncogene 20, 5449–5458 (2001).
Wei, H. et al. Structural insight into the molecular mechanism of p53-mediated mitochondrial apoptosis. Nat. Commun. 12, 2280 (2021).
Bullock, A. N., Henckel, J. & Fersht, A. R. Quantitative analysis of residual folding and DNA binding in mutant p53 core domain: definition of mutant states for rescue in cancer therapy. Oncogene 19, 1245–1256 (2000).
Ang, H. C., Joerger, A. C., Mayer, S. & Fersht, A. R. Effects of common cancer mutations on stability and DNA binding of full-length p53 compared with isolated core domains. J. Biol. Chem. 281, 21934–21941 (2006).
Karakostis, K. et al. p53 mRNA and p53 protein structures have evolved independently to interact with MDM2. Mol. Biol. Evol. 33, 1280–1292 (2016).
Vaughan, C. A. et al. The oncogenicity of tumor-derived mutant p53 is enhanced by the recruitment of PLK3. Nat. Commun. 12, 704 (2021).
Friedman, P. N., Chen, X., Bargonetti, J. & Prives, C. The p53 protein is an unusually shaped tetramer that binds directly to DNA. Proc. Natl. Acad. Sci. USA 90, 3319–3323 (1993).
Riley, T., Sontag, E., Chen, P. & Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell. Biol. 9, 402–412 (2008).
Kitayner, M. et al. Structural basis of DNA recognition by p53 tetramers. Mol. Cell 22, 741–753 (2006).
Chen, Y. et al. Structure of p53 binding to the BAX response element reveals DNA unwinding and compression to accommodate base-pair insertion. Nucleic Acids Res. 41, 8368–8376 (2013).
Baldock, R. A. et al. ATM Localization and Heterochromatin Repair Depend on Direct Interaction of the 53BP1-BRCT2 Domain with γH2AX. Cell Rep. 13, 2081–2089 (2015).
Derbyshire, D. J. et al. Crystal structure of human 53BP1 BRCT domains bound to p53 tumour suppressor. EMBO J. 21, 3863–3872 (2002).
Martinez-Zapien, D. et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature 529, 541–545 (2016).
Chen, S. et al. iASPP mediates p53 selectivity through a modular mechanism fine-tuning DNA recognition. Proc. Natl. Acad. Sci. USA 116, 17470–17479 (2019).
Nicholls, C. D., McLure, K. G., Shields, M. A. & Lee, P. W. Biogenesis of p53 involves cotranslational dimerization of monomers and posttranslational dimerization of dimers. Implications on the dominant negative effect. J. Biol. Chem. 277, 12937–12945 (2002).
Kamada, R., Toguchi, Y., Nomura, T., Imagawa, T. & Sakaguchi, K. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 106, 598–612 (2016).
Higashimoto, Y. et al. Unfolding, aggregation, and amyloid formation by the tetramerization domain from mutant p53 associated with lung cancer. Biochemistry 45, 1608–1619 (2006).
Gaglia, G., Guan, Y., Shah, J. V. & Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. USA 110, 15497–15501 (2013).
Gencel-Augusto, J. et al. Dimeric p53 mutant elicits unique tumor-suppressive activities through an altered metabolic program. Cancer Discov. 13, 1230–1249 (2023).
Laptenko, O., Tong, D. R., Manfredi, J. & Prives, C. The tail that wags the dog: how the disordered C-terminal domain controls the transcriptional activities of the p53 tumor-suppressor protein. Trends Biochem. Sci. 41, 1022–1034 (2016).
Mujtaba, S. et al. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 13, 251–263 (2004).
Friedler, A., Veprintsev, D. B., Freund, S. M., von Glos, K. I. & Fersht, A. R. Modulation of binding of DNA to the C-terminal domain of p53 by acetylation. Structure 13, 629–636 (2005).
Joerger, A. C. & Fersht, A. R. The p53 pathway: origins, inactivation in cancer, and emerging therapeutic approaches. Annu. Rev. Biochem. 85, 375–404 (2016).
Luo, J. et al. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Natl. Acad. Sci. USA 101, 2259–2264 (2004).
Espinosa, J. M. & Emerson, B. M. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell 8, 57–69 (2001).
McKinney, K., Mattia, M., Gottifredi, V. & Prives, C. p53 linear diffusion along DNA requires its C terminus. Mol. Cell 16, 413–424 (2004).
Bieging, K. T., Mello, S. S. & Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 14, 359–370 (2014).
Chen, L., Liu, S. & Tao, Y. Regulating tumor suppressor genes: post-translational modifications. Signal Transduct. Target. Ther. 5, 90 (2020).
Aubrey, B. J., Kelly, G. L., Janic, A., Herold, M. J. & Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 25, 104–113 (2018).
Mihara, M. et al. p53 has a direct apoptogenic role at the mitochondria. Mol. Cell 11, 577–590 (2003).
Wang, X., Simpson, E. R. & Brown, K. A. p53: protection against tumor growth beyond effects on cell cycle and apoptosis. Cancer Res. 75, 5001–5007 (2015).
Engeland, K. Cell cycle regulation: p53-p21-RB signaling. Cell Death Differ. 29, 946–960 (2022).
Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 25, 114–132 (2018).
Boutelle, A. M. & Attardi, L. D. p53 and tumor suppression: it takes a network. Trends Cell. Biol. 31, 298–310 (2021).
Taylor, W. R. & Stark, G. R. Regulation of the G2/M transition by p53. Oncogene 20, 1803–1815 (2001).
Sharpless, N. E. & Sherr, C. J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 15, 397–408 (2015).
Chen, J. et al. Contribution of p16INK4a and p21CIP1 pathways to induction of premature senescence of human endothelial cells: permissive role of p53. Am. J. Physiol. Heart Circ. Physiol. 290, H1575–H1586 (2006).
Knights, C. D. et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J. Cell. Biol. 173, 533–544 (2006).
Benson, E. K. et al. p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 33, 3959–3969 (2014).
Abuetabh, Y. et al. DNA damage response revisited: the p53 family and its regulators provide endless cancer therapy opportunities. Exp. Mol. Med. 54, 1658–1669 (2022).
Mijit, M., Caracciolo, V., Melillo, A., Amicarelli, F. & Giordano, A. Role of p53 in the regulation of cellular senescence. Biomolecules 10, 420 (2020).
Kortlever, R. M., Higgins, P. J. & Bernards, R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat. Cell. Biol. 8, 877–884 (2006).
Campisi, J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 120, 513–522 (2005).
Heltberg, M. S. et al. Enhanced DNA repair through droplet formation and p53 oscillations. Cell 185, 4394–4408.e4310 (2022).
Williams, A. B. & Schumacher, B. p53 in the DNA-damage-repair process. Cold Spring Harb. Perspect. Med. 6, a026070 (2016).
Kotsinas, A., Aggarwal, V., Tan, E. J., Levy, B. & Gorgoulis, V. G. PIG3: a novel link between oxidative stress and DNA damage response in cancer. Cancer Lett 327, 97–102 (2012).
Stewart-Ornstein, J. et al. p53 dynamics vary between tissues and are linked with radiation sensitivity. Nat. Commun. 12, 898 (2021).
Gartner, A., Milstein, S., Ahmed, S., Hodgkin, J. & Hengartner, M. O. A conserved checkpoint pathway mediates DNA damage-induced apoptosis and cell cycle arrest in C. elegans. Mol. Cell 5, 435–443 (2000).
Shiloh, Y. & Ziv, Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell. Biol. 14, 197–210 (2013).
el-Deiry, W. S. et al. WAF1, a potential mediator of p53 tumor suppression. Cell 75, 817–825 (1993).
Sengupta, S. et al. Tumor suppressor p53 represses transcription of RECQ4 helicase. Oncogene 24, 1738–1748 (2005).
Hastak, K., Adimoolam, S., Trinklein, N. D., Myers, R. M. & Ford, J. M. Identification of a functional in vivo p53 response element in the coding sequence of the xeroderma pigmentosum group C gene. Genes Cancer 3, 131–140 (2012).
Hamann, I., König, C., Richter, C., Jahnke, G. & Hartwig, A. Impact of cadmium on hOGG1 and APE1 as a function of the cellular p53 status. Mutat. Res. 736, 56–63 (2012).
Mulder, J. E., Bondy, G. S., Mehta, R. & Massey, T. E. Up-regulation of nucleotide excision repair in mouse lung and liver following chronic exposure to aflatoxin B₁ and its dependence on p53 genotype. Toxicol. Appl. Pharmacol. 275, 96–103 (2014).
van Oers, J. M. et al. The MutSβ complex is a modulator of p53-driven tumorigenesis through its functions in both DNA double-strand break repair and mismatch repair. Oncogene 33, 3939–3946 (2014).
Deriano, L. & Roth, D. B. Modernizing the nonhomologous end-joining repertoire: alternative and classical NHEJ share the stage. Annu. Rev. Genet. 47, 433–455 (2013).
Jasin, M. & Rothstein, R. Repair of strand breaks by homologous recombination. Cold Spring Harb. Perspect. Biol. 5, a012740 (2013).
Wang, Y. & Patti, G. J. The Warburg effect: a signature of mitochondrial overload. Trends Cell. Biol. 33, 1014–1020 (2023).
Schwartzenberg-Bar-Yoseph, F., Armoni, M. & Karnieli, E. The tumor suppressor p53 down-regulates glucose transporters GLUT1 and GLUT4 gene expression. Cancer Res 64, 2627–2633 (2004).
Kawauchi, K., Araki, K., Tobiume, K. & Tanaka, N. p53 regulates glucose metabolism through an IKK-NF-kappaB pathway and inhibits cell transformation. Nat. Cell. Biol. 10, 611–618 (2008).
Liu, J. et al. Parkin targets HIF-1α for ubiquitination and degradation to inhibit breast tumor progression. Nat. Commun. 8, 1823 (2017).
Kondoh, H. et al. Glycolytic enzymes can modulate cellular life span. Cancer Res. 65, 177–185 (2005).
Ros, S. et al. 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 4 is essential for p53-null cancer cells. Oncogene 36, 3287–3299 (2017).
Liu, J., Zhang, C., Hu, W. & Feng, Z. Parkinson’s disease-associated protein Parkin: an unusual player in cancer. Cancer Commun. 38, 40 (2018).
Viotti, J. et al. Glioma tumor grade correlates with parkin depletion in mutant p53-linked tumors and results from loss of function of p53 transcriptional activity. Oncogene 33, 1764–1775 (2014).
Xu, X., Wang, J., Xu, L., Li, P. & Jiang, P. p53 suppresses lipid droplet-fueled tumorigenesis through phosphatidylcholine. J. Clin. Invest. 134, e171788 (2024).
Zhu, Y. et al. P53 deficiency affects cholesterol esterification to exacerbate hepatocarcinogenesis. Hepatology 77, 1499–1511 (2023).
Liu, Y. et al. Ribosomal protein-Mdm2-p53 pathway coordinates nutrient stress with lipid metabolism by regulating MCD and promoting fatty acid oxidation. Proc. Natl. Acad. Sci. USA 111, E2414–E2422 (2014).
Sanchez-Macedo, N. et al. Depletion of the novel p53-target gene carnitine palmitoyltransferase 1C delays tumor growth in the neurofibromatosis type I tumor model. Cell Death Differ. 20, 659–668 (2013).
Assaily, W. et al. ROS-mediated p53 induction of Lpin1 regulates fatty acid oxidation in response to nutritional stress. Mol. Cell 44, 491–501 (2011).
Li, L. et al. p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 567, 253–256 (2019).
Fuchs, Y. & Steller, H. Programmed cell death in animal development and disease. Cell 147, 742–758 (2011).
Wong, R. S. Apoptosis in cancer: from pathogenesis to treatment. J. Exp. Clin. Cancer Res. 30, 87 (2011).
Maecker, H. L., Koumenis, C. & Giaccia, A. J. p53 promotes selection for Fas-mediated apoptotic resistance. Cancer Res. 60, 4638–4644 (2000).
Oda, E. et al. Noxa, a BH3-only member of the Bcl-2 family and candidate mediator of p53-induced apoptosis. Science 288, 1053–1058 (2000).
Nakano, K. & Vousden, K. H. PUMA, a novel proapoptotic gene, is induced by p53. Mol. Cell 7, 683–694 (2001).
Leu, J. I., Dumont, P., Hafey, M., Murphy, M. E. & George, D. L. Mitochondrial p53 activates Bak and causes disruption of a Bak-Mcl1 complex. Nat. Cell. Biol. 6, 443–450 (2004).
He, L. et al. A microRNA component of the p53 tumour suppressor network. Nature 447, 1130–1134 (2007).
Bommer, G. T. et al. p53-mediated activation of miRNA34 candidate tumor-suppressor genes. Curr. Biol. 17, 1298–1307 (2007).
Allen, M. A. et al. Global analysis of p53-regulated transcription identifies its direct targets and unexpected regulatory mechanisms. eLife 3, e02200 (2014).
Bersani, C., Xu, L. D., Vilborg, A., Lui, W. O. & Wiman, K. G. Wig-1 regulates cell cycle arrest and cell death through the p53 targets FAS and 14-3-3σ. Oncogene 33, 4407–4417 (2014).
Valente, L. J., Grabow, S., Vandenberg, C. J., Strasser, A. & Janic, A. Combined loss of PUMA and p21 accelerates c-MYC-driven lymphoma development considerably less than loss of one allele of p53. Oncogene 35, 3866–3871 (2016).
Michalak, E. M. et al. Puma and to a lesser extent Noxa are suppressors of Myc-induced lymphomagenesis. Cell Death Differ. 16, 684–696 (2009).
Viry, E. et al. Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity. Biochem. Pharmacol. 92, 31–42 (2014).
Mizushima, N. Autophagy: process and function. Genes Dev. 21, 2861–2873 (2007).
Yang, Z. & Klionsky, D. J. An overview of the molecular mechanism of autophagy. Curr. Top. Microbiol. Immunol. 335, 1–32 (2009).
Kenzelmann Broz, D. et al. Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev. 27, 1016–1031 (2013).
Feng, Z., Zhang, H., Levine, A. J. & Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 102, 8204–8209 (2005).
Kim, J., Kundu, M., Viollet, B. & Guan, K. L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell. Biol. 13, 132–141 (2011).
Russell, R. C. et al. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell. Biol. 15, 741–750 (2013).
Budanov, A. V. & Karin, M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 134, 451–460 (2008).
Crighton, D. et al. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell 126, 121–134 (2006).
Tasdemir, E. et al. Regulation of autophagy by cytoplasmic p53. Nat. Cell. Biol. 10, 676–687 (2008).
Scherz-Shouval, R. et al. p53-dependent regulation of autophagy protein LC3 supports cancer cell survival under prolonged starvation. Proc. Natl. Acad. Sci. USA 107, 18511–18516 (2010).
González-Polo, R. A. et al. The apoptosis/autophagy paradox: autophagic vacuolization before apoptotic death. J. Cell Sci. 118, 3091–3102 (2005).
Mariño, G., Niso-Santano, M., Baehrecke, E. H. & Kroemer, G. Self-consumption: the interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell. Biol. 15, 81–94 (2014).
Youle, R. J. & Narendra, D. P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 12, 9–14 (2011).
Wang, Y. et al. Epigenetic regulation of ferroptosis by H2B monoubiquitination and p53. EMBO Rep. 20, e47563 (2019).
Jiang, L. et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62 (2015).
Liu, Y. & Gu, W. The complexity of p53-mediated metabolic regulation in tumor suppression. Semin. Cancer Biol. 85, 4–32 (2022).
Kennedy, M. C. & Lowe, S. W. Mutant p53: it’s not all one and the same. Cell Death Differ. 29, 983–987 (2022).
Ou, Y., Wang, S. J., Li, D., Chu, B. & Gu, W. Activation of SAT1 engages polyamine metabolism with p53-mediated ferroptotic responses. Proc. Natl. Acad. Sci. USA 113, E6806–e6812 (2016).
Ghosh, M. et al. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell 39, 494–508.e495 (2021).
Zhu, M. et al. Loss of p53 and mutational heterogeneity drives immune resistance in an autochthonous mouse lung cancer model with high tumor mutational burden. Cancer Cell 41, 1731–1748.e1738 (2023).
Zhang, T. et al. Comprehensive analysis of TP53 mutation characteristics and identification of patients with inferior prognosis and enhanced immune escape in diffuse large B-cell lymphoma. Am. J. Hematol. 97, E14–e17 (2022).
Cortez, M. A. et al. PDL1 regulation by p53 via miR-34. J. Natl. Cancer Inst. 108, djv303 (2016).
Textor, S. et al. Human NK cells are alerted to induction of p53 in cancer cells by upregulation of the NKG2D ligands ULBP1 and ULBP2. Cancer Res. 71, 5998–6009 (2011).
Zhou, X. et al. Pharmacologic activation of p53 triggers viral mimicry response thereby abolishing tumor immune evasion and promoting antitumor immunity. Cancer Discov. 11, 3090–3105 (2021).
Sun, L., Wu, J., Du, F., Chen, X. & Chen, Z. J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013).
Wu, J. et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830 (2013).
Ishikawa, H., Ma, Z. & Barber, G. N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792 (2009).
Gulen, M. F. et al. Signalling strength determines proapoptotic functions of STING. Nat. Commun. 8, 427 (2017).
Woo, S. R. et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 (2014).
Cooks, T. et al. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 9, 771 (2018).
Kim, M. P. & Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 25, 161–168 (2018).
Fang, D. D. et al. MDM2 inhibitor APG-115 synergizes with PD-1 blockade through enhancing antitumor immunity in the tumor microenvironment. J. Immunother. Cancer 7, 327 (2019).
Freed-Pastor, W. A. & Prives, C. Mutant p53: one name, many proteins. Genes Dev. 26, 1268–1286 (2012).
de Andrade, K. C. et al. Cancer incidence, patterns, and genotype-phenotype associations in individuals with pathogenic or likely pathogenic germline TP53 variants: an observational cohort study. Lancet Oncol. 22, 1787–1798 (2021).
Baugh, E. H., Ke, H., Levine, A. J., Bonneau, R. A. & Chan, C. S. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differ. 25, 154–160 (2018).
Dearth, L. R. et al. Inactive full-length p53 mutants lacking dominant wild-type p53 inhibition highlight loss of heterozygosity as an important aspect of p53 status in human cancers. Carcinogenesis 28, 289–298 (2007).
de Oliveira, G. A. P. et al. The status of p53 oligomeric and aggregation states in cancer. Biomolecules 10, 548 (2020).
Li, J. et al. p53 amyloid aggregation in cancer: function, mechanism, and therapy. Exp. Hematol. Oncol. 11, 66 (2022).
Wang, G. & Fersht, A. R. First-order rate-determining aggregation mechanism of p53 and its implications. Proc. Natl. Acad. Sci. USA 109, 13590–13595 (2012).
Krummel, K. A., Lee, C. J., Toledo, F. & Wahl, G. M. The C-terminal lysines fine-tune P53 stress responses in a mouse model but are not required for stability control or transactivation. Proc. Natl. Acad. Sci. USA 102, 10188–10193 (2005).
Silva, J. L. et al. Targeting biomolecular condensation and protein aggregation against cancer. Chem. Rev. 123, 9094–9138 (2023).
Okorokov, A. L. et al. The structure of p53 tumour suppressor protein reveals the basis for its functional plasticity. EMBO J. 25, 5191–5200 (2006).
Wang, G. & Fersht, A. R. Propagation of aggregated p53: cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. USA 112, 2443–2448 (2015).
Soragni, A. et al. A designed inhibitor of p53 aggregation rescues p53 tumor suppression in ovarian carcinomas. Cancer Cell 29, 90–103 (2016).
Ferretti, G. D. S., Quarti, J., Dos Santos, G., Rangel, L. P. & Silva, J. L. Anticancer therapeutic strategies targeting p53 aggregation. Int. J. Mol. Sci. 23, 11023 (2022).
Yao, P. et al. Protein-level mutant p53 reporters identify druggable rare precancerous clones in noncancerous tissues. Nat. Cancer 4, 1176–1192 (2023).
Boettcher, S. et al. A dominant-negative effect drives selection of TP53 missense mutations in myeloid malignancies. Science 365, 599–604 (2019).
Flowers, B. M. et al. Cell of origin influences pancreatic cancer subtype. Cancer Discov. 11, 660–677 (2021).
Ding, D. et al. Gain-of-function mutant p53 together with ERG proto-oncogene drive prostate cancer by beta-catenin activation and pyrimidine synthesis. Nat. Commun. 14, 4671 (2023).
Zhu, J. et al. Gain-of-function p53 mutants co-opt chromatin pathways to drive cancer growth. Nature 525, 206–211 (2015).
Bergamaschi, D. et al. p53 polymorphism influences response in cancer chemotherapy via modulation of p73-dependent apoptosis. Cancer Cell 3, 387–402 (2003).
Chee, J. L. et al. Wild-type and mutant p53 mediate cisplatin resistance through interaction and inhibition of active caspase-9. Cell Cycle 12, 278–288 (2013).
Frank, A. K., Pietsch, E. C., Dumont, P., Tao, J. & Murphy, M. E. Wild-type and mutant p53 proteins interact with mitochondrial caspase-3. Cancer Biol. Ther. 11, 740–745 (2011).
Zhou, G. et al. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell 54, 960–974 (2014).
Morrison, A. et al. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 29, 408–417 (2015).
Cordani, M. et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 10, 1008–1029 (2016).
Egan, D., Kim, J., Shaw, R. J. & Guan, K. L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 7, 643–644 (2011).
Choudhury, S., Kolukula, V. K., Preet, A., Albanese, C. & Avantaggiati, M. L. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy? Cell Cycle 12, 1022–1029 (2013).
Liu, J. et al. UFMylation maintains tumour suppressor p53 stability by antagonizing its ubiquitination. Nat. Cell. Biol. 22, 1056–1063 (2020).
Xu, P. et al. Inhibition of p53 sulfoconjugation prevents oxidative hepatotoxicity and acute liver failure. Gastroenterology 162, 1226–1241 (2022).
Nguyen, T. A., Menendez, D., Resnick, M. A. & Anderson, C. W. Mutant TP53 posttranslational modifications: challenges and opportunities. Hum. Mutat. 35, 738–755 (2014).
Malkin, D. Li-fraumeni syndrome. Genes Cancer 2, 475–484 (2011).
Kratz, C. P. et al. Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin. Cancer Res. 23, e38–e45 (2017).
Farooqui, A. A., Liss, L. & Horrocks, L. A. Neurochemical aspects of Alzheimer’s disease: involvement of membrane phospholipids. Metab. Brain Dis. 3, 19–35 (1988).
Lee, Y. et al. ATR maintains select progenitors during nervous system development. EMBO J. 31, 1177–1189 (2012).
Van Nostrand, J. L. et al. Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature 514, 228–232 (2014).
Liu, Y. & Karlsson, S. Perspectives of current understanding and therapeutics of Diamond-Blackfan anemia. Leukemia 38, 1–9 (2024).
Choesmel, V. et al. Impaired ribosome biogenesis in Diamond-Blackfan anemia. Blood 109, 1275–1283 (2007).
Wlodarski, M. W. et al. Diagnosis, treatment, and surveillance of Diamond-Blackfan anaemia syndrome: international consensus statement. Lancet Haematol. 11, e368–e382 (2024).
van Ravenswaaij-Arts, C. & Martin, D. M. New insights and advances in CHARGE syndrome: Diagnosis, etiologies, treatments, and research discoveries. Am. J. Med. Genet. C Semin. Med. Genet. 175, 397–406 (2017).
Thomas, A. T. et al. Phenotypic characteristics and variability in CHARGE syndrome: a PRISMA compliant systematic review and meta-analysis. J. Neurodev. Disord. 14, 49 (2022).
Stathopoulou, A. et al. CHARGE syndrome-associated CHD7 acts at ISL1-regulated enhancers to modulate second heart field gene expression. Cardiovasc. Res. 119, 2089–2105 (2023).
Kirtay, M. et al. ATR regulates neuronal activity by modulating presynaptic firing. Nat. Commun. 12, 4067 (2021).
O’Driscoll, M., Gennery, A. R., Seidel, J., Concannon, P. & Jeggo, P. A. An overview of three new disorders associated with genetic instability: LIG4 syndrome, RS-SCID and ATR-Seckel syndrome. DNA Repair 3, 1227–1235 (2004).
Bernard, E. et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat. Med. 26, 1549–1556 (2020).
Zhang, S. et al. Trp53 negatively regulates autoimmunity via the STAT3-Th17 axis. FASEB J. 25, 2387–2398 (2011).
Fischer, N. W., Ma, Y. V. & Gariépy, J. Emerging insights into ethnic-specific TP53 germline variants. J. Natl. Cancer Inst. 115, 1145–1156 (2023).
Schwitalla, S. et al. Loss of p53 in enterocytes generates an inflammatory microenvironment enabling invasion and lymph node metastasis of carcinogen-induced colorectal tumors. Cancer Cell 23, 93–106 (2013).
Shatz, M., Menendez, D. & Resnick, M. A. The human TLR innate immune gene family is differentially influenced by DNA stress and p53 status in cancer cells. Cancer Res. 72, 3948–3957 (2012).
Toki, T. et al. De Novo mutations activating germline TP53 in an inherited bone-marrow-failure syndrome. Am. J. Hum. Genet. 103, 440–447 (2018).
Li, X. et al. Design of ultrahigh-affinity and dual-specificity peptide antagonists of MDM2 and MDMX for P53 activation and tumor suppression. Acta Pharm. Sin. B 11, 2655–2669 (2021).
Wasylishen, A. R. & Lozano, G. Attenuating the p53 pathway in human cancers: many means to the same end. Cold Spring Harb. Perspect. Med. 6, a026211 (2016).
Huang, Y., Che, X., Wang, P. W. & Qu, X. p53/MDM2 signaling pathway in aging, senescence and tumorigenesis. Semin. Cancer Biol. 101, 44–57 (2024).
Deng, L., Meng, T., Chen, L., Wei, W. & Wang, P. The role of ubiquitination in tumorigenesis and targeted drug discovery. Signal Transduct. Target. Ther. 5, 11 (2020).
Kussie, P. H. et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 274, 948–953 (1996).
Francoz, S. et al. Mdm4 and Mdm2 cooperate to inhibit p53 activity in proliferating and quiescent cells in vivo. Proc. Natl. Acad. Sci. USA 103, 3232–3237 (2006).
Brummer, T. & Zeiser, R. The role of the MDM2/p53 axis in antitumor immune responses. Blood 143, 2701–2709 (2024).
Linke, K. et al. Structure of the MDM2/MDMX RING domain heterodimer reveals dimerization is required for their ubiquitylation in trans. Cell Death Differ. 15, 841–848 (2008).
Manfredi, J. J. Mdm2 and MdmX: partners in p53 destruction. Cancer Res. 81, 1633–1634 (2021).
Bhattacharya, S., Chakraborty, D., Basu, M. & Ghosh, M. K. Emerging insights into HAUSP (USP7) in physiology, cancer and other diseases. Signal Transduct. Target. Ther. 3, 17 (2018).
Chang, J. R. et al. Role of p53 in neurodegenerative diseases. Neurodegener. Dis. 9, 68–80 (2012).
Reichert, C. O. et al. Ferroptosis mechanisms involved in neurodegenerative diseases. Int. J. Mol. Sci. 21, 8765 (2020).
Hebert, L. E., Weuve, J., Scherr, P. A. & Evans, D. A. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783 (2013).
Selkoe, D. J. The molecular pathology of Alzheimer’s disease. Neuron 6, 487–498 (1991).
Suh, Y. H. & Checler, F. Amyloid precursor protein, presenilins, and alpha-synuclein: molecular pathogenesis and pharmacological applications in Alzheimer’s disease. Pharmacol. Rev. 54, 469–525 (2002).
Buée, L. et al. From tau phosphorylation to tau aggregation: what about neuronal death? iochem. Soc. Trans. 38, 967–972 (2010).
Masters, C. L. et al. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc. Natl. Acad. Sci. USA 82, 4245–4249 (1985).
Sajan, F. D. et al. Apoptotic gene expression in Alzheimer’s disease hippocampal tissue. Am. J. Alzheimers Dis. 22, 319–328 (2007).
Anderson, A. J., Stoltzner, S., Lai, F., Su, J. & Nixon, R. A. Morphological and biochemical assessment of DNA damage and apoptosis in Down syndrome and Alzheimer disease, and effect of postmortem tissue archival on TUNEL. Neurobiol. Aging 21, 511–524 (2000).
Su, J. H., Anderson, A. J., Cummings, B. J. & Cotman, C. W. Immunohistochemical evidence for apoptosis in Alzheimer’s disease. Neuroreport 5, 2529–2533 (1994).
Chui, D. H. et al. Apoptotic neurons in Alzheimer’s disease frequently show intracellular Abeta42 labeling. J. Alzheimers Dis. 3, 231–239 (2001).
Kobayashi, K. et al. Correlation between astrocyte apoptosis and Alzheimer changes in gray matter lesions in Alzheimer’s disease. J. Alzheimers Dis. 6, 623–632 (2004).
Cenini, G., Sultana, R., Memo, M. & Butterfield, D. A. Elevated levels of pro-apoptotic p53 and its oxidative modification by the lipid peroxidation product, HNE, in brain from subjects with amnestic mild cognitive impairment and Alzheimer’s disease. J. Cell. Mol. Med. 12, 987–994 (2008).
Kitamura, Y. et al. Changes of p53 in the brains of patients with Alzheimer’s disease. Biochem. Biophys. Res. Commun. 232, 418–421 (1997).
Ohyagi, Y. et al. Intracellular Abeta42 activates p53 promoter: a pathway to neurodegeneration in Alzheimer’s disease. FASEB J. 19, 255–257 (2005).
Pastorcic, M. & Das, H. K. Regulation of transcription of the human presenilin-1 gene by ets transcription factors and the p53 protooncogene. J. Biol. Chem. 275, 34938–34945 (2000).
Roperch, J. P. et al. Inhibition of presenilin 1 expression is promoted by p53 and p21WAF-1 and results in apoptosis and tumor suppression. Nat. Med. 4, 835–838 (1998).
Alves da Costa, C. et al. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J. Neurosci. 26, 6377–6385 (2006).
Hooper, C. et al. p53 is upregulated in Alzheimer’s disease and induces tau phosphorylation in HEK293a cells. Neurosci. Lett. 418, 34–37 (2007).
Mookherjee, P. & Johnson, G. V. Tau phosphorylation during apoptosis of human SH-SY5Y neuroblastoma cells. Brain Res. 921, 31–43 (2001).
Billingsley, M. L. & Kincaid, R. L. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem. J. 323, 577–591 (1997).
Proctor, C. J. & Gray, D. A. GSK3 and p53 - is there a link in Alzheimer’s disease? Mol. Neurodegener. 5, 7 (2010).
Alves Da Costa, C., Paitel, E., Vincent, B. & Checler, F. Alpha-synuclein lowers p53-dependent apoptotic response of neuronal cells. Abolishment by 6-hydroxydopamine and implication for Parkinson’s disease. J. Biol. Chem. 277, 50980–50984 (2002).
Pardossi-Piquard, R. et al. p53-dependent control of cell death by nicastrin: lack of requirement for presenilin-dependent gamma-secretase complex. J. Neurochem. 109, 225–237 (2009).
Campbell, W. A. et al. Zebrafish lacking Alzheimer presenilin enhancer 2 (Pen-2) demonstrate excessive p53-dependent apoptosis and neuronal loss. J. Neurochem. 96, 1423–1440 (2006).
Dunys, J. et al. p53-Dependent Aph-1 and Pen-2 anti-apoptotic phenotype requires the integrity of the gamma-secretase complex but is independent of its activity. J. Biol. Chem. 282, 10516–10525 (2007).
Barzilai, A. & Melamed, E. Molecular mechanisms of selective dopaminergic neuronal death in Parkinson’s disease. Trends Mol. Med. 9, 126–132 (2003).
Goedert, M., Spillantini, M. G., Del Tredici, K. & Braak, H. 100 years of Lewy pathology. Nat. Rev. Neurol. 9, 13–24 (2013).
Gwinn-Hardy, K. & Singleton, A. A. Familial Lewy body diseases. J. Geriatr. Psychiatry Neurol. 15, 217–223 (2002).
Chartier-Harlin, M. C. et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364, 1167–1169 (2004).
Blandini, F. et al. Peripheral proteasome and caspase activity in Parkinson disease and Alzheimer disease. Neurology 66, 529–534 (2006).
Junn, E. & Mouradian, M. M. Apoptotic signaling in dopamine-induced cell death: the role of oxidative stress, p38 mitogen-activated protein kinase, cytochrome c and caspases. J. Neurochem. 78, 374–383 (2001).
Tatton, N. A. & Kish, S. J. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience 77, 1037–1048 (1997).
Zhou, H. Y. et al. Proteasome inhibitor lactacystin induces cholinergic degeneration. Can. J. Neurol. Sci. 37, 229–234 (2010).
Haupt, Y., Maya, R., Kazaz, A. & Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 387, 296–299 (1997).
Mogi, M., Kondo, T., Mizuno, Y. & Nagatsu, T. p53 protein, interferon-gamma, and NF-kappaB levels are elevated in the parkinsonian brain. Neurosci. Lett. 414, 94–97 (2007).
Lee, S. J., Kim, D. C., Choi, B. H., Ha, H. & Kim, K. T. Regulation of p53 by activated protein kinase C-delta during nitric oxide-induced dopaminergic cell death. J. Biol. Chem. 281, 2215–2224 (2006).
Engelender, S. et al. Synphilin-1 associates with alpha-synuclein and promotes the formation of cytosolic inclusions. Nat. Genet. 22, 110–114 (1999).
Smith, W. W. et al. Synphilin-1 attenuates neuronal degeneration in the A53T alpha-synuclein transgenic mouse model. Hum. Mol. Genet. 19, 2087–2098 (2010).
Giaime, E. et al. Caspase-3-derived C-terminal product of synphilin-1 displays antiapoptotic function via modulation of the p53-dependent cell death pathway. J. Biol. Chem. 281, 11515–11522 (2006).
da Costa, C. A. et al. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat. Cell. Biol. 11, 1370–1375 (2009).
Shimura, H. et al. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat. Genet. 25, 302–305 (2000).
Olzmann, J. A. et al. Familial Parkinson’s disease-associated L166P mutation disrupts DJ-1 protein folding and function. J. Biol. Chem. 279, 8506–8515 (2004).
Bonifati, V. et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 (2003).
Fan, J. et al. DJ-1 decreases Bax expression through repressing p53 transcriptional activity. J. Biol. Chem. 283, 4022–4030 (2008).
Bretaud, S., Allen, C., Ingham, P. W. & Bandmann, O. p53-dependent neuronal cell death in a DJ-1-deficient zebrafish model of Parkinson’s disease. J. Neurochem. 100, 1626–1635 (2007).
Kato, I. et al. Oxidized DJ-1 inhibits p53 by sequestering p53 from promoters in a DNA-binding affinity-dependent manner. Mol. Cell. Biol. 33, 340–359 (2013).
Duplan, E., Giordano, C., Checler, F. & Alves da Costa, C. Direct α-synuclein promoter transactivation by the tumor suppressor p53. Mol. Neurodegener. 11, 13 (2016).
Zhang, C. et al. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc. Natl. Acad. Sci. USA 108, 16259–16264 (2011).
Duplan, E. et al. ER-stress-associated functional link between Parkin and DJ-1 via a transcriptional cascade involving the tumor suppressor p53 and the spliced X-box binding protein XBP-1. J. Cell Sci. 126, 2124–2133 (2013).
Ryan, A. B., Zeitlin, S. O. & Scrable, H. Genetic interaction between expanded murine Hdh alleles and p53 reveal deleterious effects of p53 on Huntington’s disease pathogenesis. Neurobiol. Dis. 24, 419–427 (2006).
Bae, B. I. et al. p53 mediates cellular dysfunction and behavioral abnormalities in Huntington’s disease. Neuron 47, 29–41 (2005).
DiFiglia, M. et al. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 (1997).
Rosen, D. R. et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 362, 59–62 (1993).
McCarthy, C. & Thorpe, J. Some recent advances in multiple sclerosis. J. Neurol. 263, 1880–1886 (2016).
Grison, A. et al. Ser46 phosphorylation and prolyl-isomerase Pin1-mediated isomerization of p53 are key events in p53-dependent apoptosis induced by mutant huntingtin. Proc. Natl. Acad. Sci. USA 108, 17979–17984 (2011).
Martin, L. J. p53 is abnormally elevated and active in the CNS of patients with amyotrophic lateral sclerosis. Neurobiol. Dis. 7, 613–622 (2000).
Ranganathan, S. & Bowser, R. p53 and cell cycle proteins participate in spinal motor neuron cell death in ALS. Open Pathol. J. 4, 11–22 (2010).
González de Aguilar, J. L. et al. Alteration of the Bcl-x/Bax ratio in a transgenic mouse model of amyotrophic lateral sclerosis: evidence for the implication of the p53 signaling pathway. Neurobiol. Dis. 7, 406–415 (2000).
Akizuki, M. et al. Optineurin suppression causes neuronal cell death via NF-κB pathway. J. Neurochem. 126, 699–704 (2013).
Merlo, P. et al. p53 prevents neurodegeneration by regulating synaptic genes. Proc. Natl. Acad. Sci. USA 111, 18055–18060 (2014).
Lacroix, M., Riscal, R., Arena, G., Linares, L. K. & Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol. Metab. 33, 2–22 (2020).
Men, H. et al. The regulatory roles of p53 in cardiovascular health and disease. Cell. Mol. Life Sci. 78, 2001–2018 (2021).
Lidell, M. E. & Enerbäck, S. Brown adipose tissue-a new role in humans? Nat. Rev. Endocrinol. 6, 319–325 (2010).
Cristancho, A. G. & Lazar, M. A. Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell. Biol. 12, 722–734 (2011).
Vegiopoulos, A. et al. Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 328, 1158–1161 (2010).
Cypess, A. M. et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 360, 1509–1517 (2009).
Khandekar, M. J., Cohen, P. & Spiegelman, B. M. Molecular mechanisms of cancer development in obesity. Nat. Rev. Cancer 11, 886–895 (2011).
Gesta, S., Tseng, Y. H. & Kahn, C. R. Developmental origin of fat: tracking obesity to its source. Cell 131, 242–256 (2007).
Molchadsky, A. et al. p53 is required for brown adipogenic differentiation and has a protective role against diet-induced obesity. Cell Death Differ. 20, 774–783 (2013).
Quiñones, M. et al. p53 in AgRP neurons is required for protection against diet-induced obesity via JNK1. Nat. Commun. 9, 3432 (2018).
Yokoyama, M. et al. Inhibition of endothelial p53 improves metabolic abnormalities related to dietary obesity. Cell Rep. 7, 1691–1703 (2014).
De Iuliis, F., Salerno, G., Taglieri, L. & Scarpa, S. Are pharmacogenomic biomarkers an effective tool to predict taxane toxicity and outcome in breast cancer patients? Literature review. Cancer Chemother. Pharmacol. 76, 679–690 (2015).
Bergamaschi, D. et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet. 38, 1133–1141 (2006).
Kung, C. P. et al. The P72R polymorphism of p53 predisposes to obesity and metabolic dysfunction. Cell Rep. 14, 2413–2425 (2016).
Zhang, Y., Zeng, S. X., Hao, Q. & Lu, H. Monitoring p53 by MDM2 and MDMX is required for endocrine pancreas development and function in a spatio-temporal manner. Dev. Biol. 423, 34–45 (2017).
Tornovsky-Babeay, S. et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in β cells. Cell Metab. 19, 109–121 (2014).
Hoshino, A. et al. Inhibition of p53 preserves Parkin-mediated mitophagy and pancreatic β-cell function in diabetes. Proc. Natl. Acad. Sci. USA 111, 3116–3121 (2014).
Feng, Z. et al. The regulation of AMPK beta1, TSC2, and PTEN expression by p53: stress, cell and tissue specificity, and the role of these gene products in modulating the IGF-1-AKT-mTOR pathways. Cancer Res. 67, 3043–3053 (2007).
Minamino, T. et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 15, 1082–1087 (2009).
Vergoni, B. et al. DNA damage and the activation of the p53 pathway mediate alterations in metabolic and secretory functions of adipocytes. Diabetes 65, 3062–3074 (2016).
Secchiero, P. et al. The MDM2 inhibitor Nutlin-3 attenuates streptozotocin-induced diabetes mellitus and increases serum level of IL-12p40. Acta Diabetol. 50, 899–906 (2013).
Velásquez, D. A. et al. The central Sirtuin 1/p53 pathway is essential for the orexigenic action of ghrelin. Diabetes 60, 1177–1185 (2011).
Tomita, K. et al. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. J. Hepatol. 57, 837–843 (2012).
Farrell, G. C. et al. Apoptosis in experimental NASH is associated with p53 activation and TRAIL receptor expression. J. Gastroenterol. Hepatol. 24, 443–452 (2009).
Panasiuk, A., Dzieciol, J., Panasiuk, B. & Prokopowicz, D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World J. Gastroenterol. 12, 6198–6202 (2006).
Kodama, T. et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Invest. 121, 3343–3356 (2011).
Derdak, Z. et al. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. J. Hepatol. 58, 785–791 (2013).
Porteiro, B. et al. Pharmacological stimulation of p53 with low-dose doxorubicin ameliorates diet-induced nonalcoholic steatosis and steatohepatitis. Mol. Metab. 8, 132–143 (2018).
Virani, S. S. et al. Heart disease and stroke statistics-2020 update: a report from the American Heart Association. Circulation 141, e139–e596 (2020).
Mak, T. W., Hauck, L., Grothe, D. & Billia, F. p53 regulates the cardiac transcriptome. Proc. Natl. Acad. Sci. USA 114, 2331–2336 (2017).
Nakamura, H. et al. p53 promotes cardiac dysfunction in diabetic mellitus caused by excessive mitochondrial respiration-mediated reactive oxygen species generation and lipid accumulation. Circ. Heart Fail. 5, 106–115 (2012).
Mercer, J., Figg, N., Stoneman, V., Braganza, D. & Bennett, M. R. Endogenous p53 protects vascular smooth muscle cells from apoptosis and reduces atherosclerosis in ApoE knockout mice. Circ. Res. 96, 667–674 (2005).
Xu, T. et al. ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening. Redox Biol. 20, 414–426 (2019).
Frangogiannis, N. G. Pathophysiology of myocardial infarction. Compr. Physiol. 5, 1841–1875 (2015).
Liu, C. Y. et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat. Commun. 9, 29 (2018).
Gogna, R., Madan, E., Khan, M., Pati, U. & Kuppusamy, P. p53’s choice of myocardial death or survival: oxygen protects infarct myocardium by recruiting p53 on NOS3 promoter through regulation of p53-Lys(118) acetylation. EMBO Mol. Med. 5, 1662–1683 (2013).
Su, D. et al. ROCK1/p53/NOXA signaling mediates cardiomyocyte apoptosis in response to high glucose in vitro and vivo. Biochim. Biophys. Acta Mol. Basis Dis. 1863, 936–946 (2017).
Tan, Y. et al. Mechanisms of diabetic cardiomyopathy and potential therapeutic strategies: preclinical and clinical evidence. Nat. Rev. Cardiol. 17, 585–607 (2020).
Bouthoorn, S. et al. The prevalence of left ventricular diastolic dysfunction and heart failure with preserved ejection fraction in men and women with type 2 diabetes: a systematic review and meta-analysis. Diabetes Vasc. Dis. Res. 15, 477–493 (2018).
Seferović, P. M. & Paulus, W. J. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur. Heart J. 36, 1718–1727 (2015). 1727a-1727c.
Mu, W. et al. Overexpression of a dominant-negative mutant of SIRT1 in mouse heart causes cardiomyocyte apoptosis and early-onset heart failure. Sci. China Life Sci. 57, 915–924 (2014).
Vahtola, E. et al. Sirtuin1-p53, forkhead box O3a, p38 and post-infarct cardiac remodeling in the spontaneously diabetic Goto-Kakizaki rat. Cardiovasc. Diabetol. 9, 5 (2010).
Tabas, I. p53 and atherosclerosis. Circ. Res. 88, 747–749 (2001).
Ihling, C. et al. Co-expression of p53 and MDM2 in human atherosclerosis: implications for the regulation of cellularity of atherosclerotic lesions. J. Pathol. 185, 303–312 (1998).
Guevara, N. V., Kim, H. S., Antonova, E. I. & Chan, L. The absence of p53 accelerates atherosclerosis by increasing cell proliferation in vivo. Nat. Med. 5, 335–339 (1999).
van Vlijmen, B. J. et al. Macrophage p53 deficiency leads to enhanced atherosclerosis in APOE*3-Leiden transgenic mice. Circ. Res. 88, 780–786 (2001).
Manfredi, J. J. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev 24, 1580–1589 (2010).
Birks, E. J. et al. Elevated p53 expression is associated with dysregulation of the ubiquitin-proteasome system in dilated cardiomyopathy. Cardiovasc. Res. 79, 472–480 (2008).
Xiong, S., Van Pelt, C. S., Elizondo-Fraire, A. C., Fernandez-Garcia, B. & Lozano, G. Loss of Mdm4 results in p53-dependent dilated cardiomyopathy. Circulation 115, 2925–2930 (2007).
Komarov, P. G. et al. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 285, 1733–1737 (1999).
Gu, J. et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 9, 82 (2018).
Liu, P., Xu, B., Cavalieri, T. A. & Hock, C. E. Inhibition of p53 by pifithrin-alpha reduces myocyte apoptosis and leukocyte transmigration in aged rat hearts following 24 h of reperfusion. Shock 30, 545–551 (2008).
Foster, B. A., Coffey, H. A., Morin, M. J. & Rastinejad, F. Pharmacological rescue of mutant p53 conformation and function. Science 286, 2507–2510 (1999).
Bykov, V. J. N., Eriksson, S. E., Bianchi, J. & Wiman, K. G. Targeting mutant p53 for efficient cancer therapy. Nat. Rev. Cancer 18, 89–102 (2018).
Rippin, T. M. et al. Characterization of the p53-rescue drug CP-31398 in vitro and in living cells. Oncogene 21, 2119–2129 (2002).
Rao, C. V., Swamy, M. V., Patlolla, J. M. & Kopelovich, L. Suppression of familial adenomatous polyposis by CP-31398, a TP53 modulator, in APCmin/+ mice. Cancer Res. 68, 7670–7675 (2008).
Xu, J. et al. Targeting wild-type and mutant p53 with small molecule CP-31398 blocks the growth of rhabdomyosarcoma by inducing reactive oxygen species-dependent apoptosis. Cancer Res. 70, 6566–6576 (2010).
Demma, M. J., Wong, S., Maxwell, E. & Dasmahapatra, B. CP-31398 restores DNA-binding activity to mutant p53 in vitro but does not affect p53 homologs p63 and p73. J. Biol. Chem. 279, 45887–45896 (2004).
Madka, V. et al. p53-stabilizing agent CP-31398 prevents growth and invasion of urothelial cancer of the bladder in transgenic UPII-SV40T mice. Neoplasia 15, 966–974 (2013).
Lambert, J. M. et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 15, 376–388 (2009).
Bykov, V. J. et al. PRIMA-1(MET) synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 24, 3484–3491 (2005).
Bykov, V. J. et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat. Med. 8, 282–288 (2002).
Augustyn, K. E., Merino, E. J. & Barton, J. K. A role for DNA-mediated charge transport in regulating p53: Oxidation of the DNA-bound protein from a distance. Proc. Natl. Acad. Sci. USA 104, 18907–18912 (2007).
Wassman, C. D. et al. Computational identification of a transiently open L1/S3 pocket for reactivation of mutant p53. Nat. Commun. 4, 1407 (2013).
Hong, Y. et al. APR-246 triggers ferritinophagy and ferroptosis of diffuse large B-cell lymphoma cells with distinct TP53 mutations. Leukemia 36, 2269–2280 (2022).
Birsen, R. et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica 107, 403–416 (2022).
Mlakar, V. et al. PRIMA-1(MET)-induced neuroblastoma cell death is modulated by p53 and mycn through glutathione level. J. Exp. Clin. Cancer Res. 38, 69 (2019).
Duffy, M. J., Synnott, N. C. & Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 83, 258–265 (2017).
Ali, D. et al. APR-246 exhibits anti-leukemic activity and synergism with conventional chemotherapeutic drugs in acute myeloid leukemia cells. Eur. J. Haematol. 86, 206–215 (2011).
Zandi, R. et al. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 17, 2830–2841 (2011).
Wang, Z. et al. Correction to: The anti-cancer agent APR-246 can activate several programmed cell death processes to kill malignant cells. Cell Death Differ. 30, 1096 (2023).
Mohell, N. et al. APR-246 overcomes resistance to cisplatin and doxorubicin in ovarian cancer cells. Cell Death Dis. 6, e1794 (2015).
Sallman, D. A. et al. Eprenetapopt (APR-246) and azacitidine in TP53-mutant myelodysplastic syndromes. J. Clin. Oncol. 39, 1584–1594 (2021).
Cluzeau, T. et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: a phase II study by the Groupe Francophone des Myélodysplasies (GFM). J. Clin. Oncol. 39, 1575–1583 (2021).
Lehmann, S. et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J. Clin. Oncol. 30, 3633–3639 (2012).
Garcia-Manero, G. et al. Eprenetapopt combined with venetoclax and azacitidine in TP53-mutated acute myeloid leukaemia: a phase 1, dose-finding and expansion study. Lancet Haematol 10, e272–e283 (2023).
Tang, X. et al. CP-31398 restores mutant p53 tumor suppressor function and inhibits UVB-induced skin carcinogenesis in mice. J. Clin. Invest. 117, 3753–3764 (2007).
Menichini, P. et al. Antitumor effects of PRIMA-1 and PRIMA-1(Met) (APR246) in hematological malignancies: still a mutant P53-dependent affair? Cells 10, 98 (2021).
Sallman, D. A. To target the untargetable: elucidation of synergy of APR-246 and azacitidine in TP53 mutant myelodysplastic syndromes and acute myeloid leukemia. Haematologica 105, 1470–1472 (2020).
Soignet, S. L. et al. Complete remission after treatment of acute promyelocytic leukemia with arsenic trioxide. N. Engl. J. Med. 339, 1341–1348 (1998).
Zhang, X. W. et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science 328, 240–243 (2010).
Zheng, T. et al. Nutlin-3 overcomes arsenic trioxide resistance and tumor metastasis mediated by mutant p53 in Hepatocellular Carcinoma. Mol. Cancer 13, 133 (2014).
Liu, Q., Hilsenbeck, S. & Gazitt, Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL. Blood 101, 4078–4087 (2003).
Chen, S. et al. Arsenic trioxide rescues structural p53 mutations through a cryptic allosteric site. Cancer Cell 39, 225–239.e228 (2021).
Song, H. et al. Diverse rescue potencies of p53 mutations to ATO are predetermined by intrinsic mutational properties. Sci. Transl. Med. 15, eabn9155 (2023).
Wang, G. & Fersht, A. R. Multisite aggregation of p53 and implications for drug rescue. Proc. Natl. Acad. Sci. USA 114, E2634–e2643 (2017).
Palanikumar, L. et al. Protein mimetic amyloid inhibitor potently abrogates cancer-associated mutant p53 aggregation and restores tumor suppressor function. Nat. Commun. 12, 3962 (2021).
Kumar, S. & Hamilton, A. D. α-Helix mimetics as modulators of Aβ self-assembly. J. Am. Chem. Soc. 139, 5744–5755 (2017).
Miller, J. J. et al. Bifunctional ligand design for modulating mutant p53 aggregation in cancer. Chem. Sci. 10, 10802–10814 (2019).
Guiley, K. Z. & Shokat, K. M. A small molecule reacts with the p53 somatic mutant Y220C to rescue wild-type thermal stability. Cancer Discov. 13, 56–69 (2023).
Boeckler, F. M. et al. Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc. Natl. Acad. Sci. USA 105, 10360–10365 (2008).
Liu, X. et al. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 41, 6034–6044 (2013).
Dumbrava, E. E. et al. First-in-human study of PC14586, a small molecule structural corrector of Y220C mutant p53, in patients with advanced solid tumors harboring a TP53 Y220C mutation. J. Clin. Oncol. 40, 3003–3003 (2022).
Jin, Q. et al. Zinc-doped Prussian blue nanoparticles for mutp53-carrying tumor ion interference and photothermal therapy. Asian J. Pharm. Sci. 17, 767–777 (2022).
Yu, X., Vazquez, A., Levine, A. J. & Carpizo, D. R. Allele-specific p53 mutant reactivation. Cancer Cell 21, 614–625 (2012).
Lindemann, A. et al. COTI-2, a novel thiosemicarbazone derivative, exhibits antitumor activity in HNSCC through p53-dependent and -independent mechanisms. Clin. Cancer Res. 25, 5650–5662 (2019).
Yu, X. et al. Thiosemicarbazones functioning as zinc metallochaperones to reactivate mutant p53. Mol. Pharmacol. 91, 567–575 (2017).
Bormio Nunes, J. H. et al. Cancer cell resistance against the clinically investigated thiosemicarbazone COTI-2 is based on formation of intracellular copper complex glutathione adducts and ABCC1-mediated efflux. J. Med. Chem. 63, 13719–13732 (2020).
Fallatah, M. M. J., Law, F. V., Chow, W. A. & Kaiser, P. Small-molecule correctors and stabilizers to target p53. Trends Pharmacol. Sci. 44, 274–289 (2023).
Green, J. A., Von Euler, M. & Abrahmsen, L. B. Restoration of conformation of mutant p53. Ann. Oncol. 29, 1325–1328 (2018).
Vassilev, L. T. et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 (2004).
Klein, C. & Vassilev, L. T. Targeting the p53-MDM2 interaction to treat cancer. Br. J. Cancer 91, 1415–1419 (2004).
Ray-Coquard, I. et al. Effect of the MDM2 antagonist RG7112 on the P53 pathway in patients with MDM2-amplified, well-differentiated or dedifferentiated liposarcoma: an exploratory proof-of-mechanism study. Lancet Oncol 13, 1133–1140 (2012).
Andreeff, M. et al. Results of the phase I trial of RG7112, a small-molecule MDM2 antagonist in leukemia. Clin. Cancer Res. 22, 868–876 (2016).
Konopleva, M. Y. et al. Idasanutlin plus cytarabine in relapsed or refractory acute myeloid leukemia: results of the MIRROS trial. Blood Adv. 6, 4147–4156 (2022).
Mascarenhas, J. et al. Oral idasanutlin in patients with polycythemia vera. Blood 134, 525–533 (2019).
Sun, D. et al. Discovery of AMG 232, a potent, selective, and orally bioavailable MDM2-p53 inhibitor in clinical development. J. Med. Chem. 57, 1454–1472 (2014).
Her, N. G. et al. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 9, 792 (2018).
Canon, J. et al. The MDM2 inhibitor AMG 232 demonstrates robust antitumor efficacy and potentiates the activity of p53-inducing cytotoxic agents. Mol. Cancer Ther. 14, 649–658 (2015).
Erba, H. P. et al. Phase 1b study of the MDM2 inhibitor AMG 232 with or without trametinib in relapsed/refractory acute myeloid leukemia. Blood Adv. 3, 1939–1949 (2019).
Yi, H. et al. A novel small molecule inhibitor of MDM2-p53 (APG-115) enhances radiosensitivity of gastric adenocarcinoma. J. Exp. Clin. Cancer Res. 37, 97 (2018).
Bernal, F., Tyler, A. F., Korsmeyer, S. J., Walensky, L. D. & Verdine, G. L. Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide. J. Am. Chem. Soc. 129, 2456–2457 (2007).
Gembarska, A. et al. MDM4 is a key therapeutic target in cutaneous melanoma. Nat. Med. 18, 1239–1247 (2012).
Li, Y. C. et al. A versatile platform to analyze low-affinity and transient protein-protein interactions in living cells in real time. Cell Rep. 9, 1946–1958 (2014).
Pairawan, S. et al. First in class dual MDM2/MDMX inhibitor ALRN-6924 enhances antitumor efficacy of chemotherapy in TP53 wild-type hormone receptor-positive breast cancer models. Breast Cancer Res. 23, 29 (2021).
Carvajal, L. A. et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 10, eaao3003 (2018).
Meric-Bernstam, F. et al. Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas. J. Clin. Oncol. 35, 2505–2505 (2017).
Meng, X. et al. Strategies for Molecularly Enhanced Chemotherapy To Achieve Synthetic Lethality In Endometrial Tumors With Mutant p53. Obstet. Gynecol. Int. 2013, 828165 (2013).
Alexandrova, E. M. et al. Improving survival by exploiting tumour dependence on stabilized mutant p53 for treatment. Nature 523, 352–356 (2015).
Li, D., Marchenko, N. D. & Moll, U. M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 18, 1904–1913 (2011).
Freed-Pastor, W. A. et al. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell 148, 244–258 (2012).
Parrales, A. et al. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell. Biol. 18, 1233–1243 (2016).
Tutuska, K., Parrilla-Monge, L., Di Cesare, E., Nemajerova, A. & Moll, U. M. Statin as anti-cancer therapy in autochthonous T-lymphomas expressing stabilized gain-of-function mutant p53 proteins. Cell Death Dis. 11, 274 (2020).
Brosh, R. & Rotter, V. When mutants gain new powers: news from the mutant p53 field. Nat. Rev. Cancer 9, 701–713 (2009).
Speetjens, F. M. et al. Induction of p53-specific immunity by a p53 synthetic long peptide vaccine in patients treated for metastatic colorectal cancer. Clin. Cancer Res. 15, 1086–1095 (2009).
Vermeij, R. et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: a single-arm phase II study. Int. J. Cancer 131, E670–E680 (2012).
Leffers, N. et al. Long-term clinical and immunological effects of p53-SLP® vaccine in patients with ovarian cancer. Int. J. Cancer 130, 105–112 (2012).
Hardwick, N. R. et al. p53-reactive T cells are associated with clinical benefit in patients with platinum-resistant epithelial ovarian cancer after treatment with a p53 vaccine and gemcitabine chemotherapy. Clin. Cancer Res. 24, 1315–1325 (2018).
Hardwick, N. R. et al. Correction: p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin. Cancer Res. 23, 322 (2017).
Hardwick, N. R. et al. p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin. Cancer Res. 20, 4459–4470 (2014).
Barfoed, A. M. et al. Cytotoxic T-lymphocyte clones, established by stimulation with the HLA-A2 binding p5365-73 wild type peptide loaded on dendritic cells In vitro, specifically recognize and lyse HLA-A2 tumour cells overexpressing the p53 protein. Scand. J. Immunol. 51, 128–133 (2000).
Antonia, S. J. et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin. Cancer Res. 12, 878–887 (2006).
Chiappori, A. A., Soliman, H., Janssen, W. E., Antonia, S. J. & Gabrilovich, D. I. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin. Biol. Ther. 10, 983–991 (2010).
Chiappori, A. A. et al. Randomized-controlled phase II trial of salvage chemotherapy after immunization with a TP53-transfected dendritic cell-based vaccine (Ad.p53-DC) in patients with recurrent small cell lung cancer. Cancer Immunol. Immunother. 68, 517–527 (2019).
Met, O., Balslev, E., Flyger, H. & Svane, I. M. High immunogenic potential of p53 mRNA-transfected dendritic cells in patients with primary breast cancer. Breast Cancer Res. Treat. 125, 395–406 (2011).
Li, D. et al. Development of a T-cell receptor mimic antibody against wild-type p53 for cancer immunotherapy. Cancer Res. 77, 2699–2711 (2017).
Low, L., Goh, A., Koh, J., Lim, S. & Wang, C. I. Targeting mutant p53-expressing tumours with a T cell receptor-like antibody specific for a wild-type antigen. Nat. Commun. 10, 5382 (2019).
Hsiue, E. H. et al. Targeting a neoantigen derived from a common TP53 mutation. Science 371, eabc8697 (2021).
Zhang, W. W. et al. The first approved gene therapy product for cancer Ad-p53 (Gendicine): 12 years in the clinic. Hum. Gene Ther. 29, 160–179 (2018).
Gabrilovich, D. I. INGN 201 (Advexin): adenoviral p53 gene therapy for cancer. Expert Opin. Biol. Ther. 6, 823–832 (2006).
Atencio, I. A. et al. Biological activities of a recombinant adenovirus p53 (SCH 58500) administered by hepatic arterial infusion in a Phase 1 colorectal cancer trial. Cancer Gene Ther. 13, 169–181 (2006).
Buller, R. E. et al. A phase I/II trial of rAd/p53 (SCH 58500) gene replacement in recurrent ovarian cancer. Cancer Gene Ther. 9, 553–566 (2002).
Lewinski, N., Colvin, V. & Drezek, R. Cytotoxicity of nanoparticles. Small 4, 26–49 (2008).
Chen, C. et al. Polyethyleneimine-modified calcium carbonate nanoparticles for p53 gene delivery. Regen. Biomater. 3, 57–63 (2016).
Senzer, N. et al. Phase I study of a systemically delivered p53 nanoparticle in advanced solid tumors. Mol. Ther. 21, 1096–1103 (2013).
Xiao, Y. et al. Combining p53 mRNA nanotherapy with immune checkpoint blockade reprograms the immune microenvironment for effective cancer therapy. Nat. Commun. 13, 758 (2022).
Ubby, I. et al. Cancer therapeutic targeting using mutant-p53-specific siRNAs. Oncogene 38, 3415–3427 (2019).
Funk, J. S. et al. Deep CRISPR mutagenesis characterizes the functional diversity of TP53 mutations. Nat. Genet. 57, 140–153 (2025).
Komor, A. C., Kim, Y. B., Packer, M. S., Zuris, J. A. & Liu, D. R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016).
Enache, O. M. et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 52, 662–668 (2020).
Ihry, R. J. et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nat. Med. 24, 939–946 (2018).
Jinek, M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012).
Moradpour, M. & Abdulah, S. N. A. CRISPR/dCas9 platforms in plants: strategies and applications beyond genome editing. Plant Biotechnol. J. 18, 32–44 (2020).
Saber Sichani, A., Ranjbar, M., Baneshi, M., Torabi Zadeh, F. & Fallahi, J. A review on advanced CRISPR-based genome-editing tools: base editing and prime editing. Mol. Biotechnol. 65, 849–860 (2023).
Tsai, S. Q. et al. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods 14, 607–614 (2017).
Yin, J. & Hu, J. The origin of unwanted editing byproducts in gene editing. Acta Biochim. Biophys. Sin. 54, 767–781 (2022).
Gaudelli, N. M. et al. Programmable base editing of A•T to G•C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017).
Grünewald, J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol. 37, 1041–1048 (2019).
Sayed, S. et al. Efficient correction of oncogenic KRAS and TP53 mutations through CRISPR base editing. Cancer Res. 82, 3002–3015 (2022).
Choonara, Y. E. et al. Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int. J. Mol. Sci. 10, 2510–2557 (2009).
Harilal, S. et al. Advancements in nanotherapeutics for Alzheimer’s disease: current perspectives. J. Pharm. Pharmacol. 71, 1370–1383 (2019).
Dunn, B., Stein, P. & Cavazzoni, P. Approval of aducanumab for Alzheimer disease-the FDA’s perspective. JAMA Intern. Med. 181, 1276–1278 (2021).
Walsh, S., Merrick, R., Milne, R. & Brayne, C. Aducanumab for Alzheimer’s disease? BMJ 374, n1682 (2021).
Lamptey, R. N. L. et al. A review of the common neurodegenerative disorders: current therapeutic approaches and the potential role of nanotherapeutics. Int. J. Mol. Sci. 23, 1851 (2022).
Hampel, H. et al. Revisiting the cholinergic hypothesis in Alzheimer’s disease: emerging evidence from translational and clinical research. J. Prev. Alzheimers Dis. 6, 2–15 (2019).
Raina, P. et al. Effectiveness of cholinesterase inhibitors and memantine for treating dementia: evidence review for a clinical practice guideline. Ann. Intern. Med. 148, 379–397 (2008).
Birks, J. & Harvey, R. J. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst. Rev., Cd001190 (2006).
Farlow, M. R. et al. Effectiveness and tolerability of high-dose (23 mg/d) versus standard-dose (10 mg/d) donepezil in moderate to severe Alzheimer’s disease: A 24-week, randomized, double-blind study. Clin. Ther. 32, 1234–1251 (2010).
Sabbagh, M. et al. Evaluating the cognitive effects of donepezil 23 mg/d in moderate and severe Alzheimer’s disease: analysis of effects of baseline features on treatment response. BMC Geriatr. 13, 56 (2013).
Winblad, B. et al. IDEAL: a 6-month, double-blind, placebo-controlled study of the first skin patch for Alzheimer disease. Neurology 69, S14–S22 (2007).
Farlow, M. R., Grossberg, G. T., Sadowsky, C. H., Meng, X. & Somogyi, M. A 24-week, randomized, controlled trial of rivastigmine patch 13.3 mg/24 h versus 4.6 mg/24 h in severe Alzheimer’s dementia. CNS Neurosci. Ther. 19, 745–752 (2013).
Lilienfeld, S. Galantamine-a novel cholinergic drug with a unique dual mode of action for the treatment of patients with Alzheimer’s disease. CNS Drug Rev. 8, 159–176 (2002).
Hager, K. et al. Effects of galantamine in a 2-year, randomized, placebo-controlled study in Alzheimer’s disease. Neuropsychiatr. Dis. Treat. 10, 391–401 (2014).
Johnson, J. W. & Kotermanski, S. E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 6, 61–67 (2006).
Reisberg, B. et al. Memantine in moderate-to-severe Alzheimer’s disease. N. Engl. J. Med. 348, 1333–1341 (2003).
Alexander, G. C. et al. Revisiting FDA approval of aducanumab. N. Engl. J. Med. 385, 769–771 (2021).
Fox, S. H. et al. International Parkinson and movement disorder society evidence-based medicine review: update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 33, 1248–1266 (2018).
Gray, R. et al. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson’s disease (PD MED): a large, open-label, pragmatic randomised trial. Lancet 384, 1196–1205 (2014).
Garcia-Ruiz, P. J. et al. Impulse control disorder in patients with Parkinson’s disease under dopamine agonist therapy: a multicentre study. J. Neurol. Neurosurg. Psychiatry 85, 840–844 (2014).
Pondal, M. et al. Clinical features of dopamine agonist withdrawal syndrome in a movement disorders clinic. J. Neurol. Neurosurg. Psychiatry 84, 130–135 (2013).
Bratsos, S., Karponis, D. & Saleh, S. N. Efficacy and safety of deep brain stimulation in the treatment of Parkinson’s disease: a systematic review and meta-analysis of randomized controlled trials. Cureus 10, e3474 (2018).
Okun, M. S. et al. Development and initial validation of a screening tool for Parkinson disease surgical candidates. Neurology 63, 161–163 (2004).
Wächter, T., Mínguez-Castellanos, A., Valldeoriola, F., Herzog, J. & Stoevelaar, H. A tool to improve pre-selection for deep brain stimulation in patients with Parkinson’s disease. J. Neurol. 258, 641–646 (2011).
Armstrong, M. J. & Okun, M. S. Diagnosis and treatment of Parkinson disease: a review. JAMA 323, 548–560 (2020).
Seppi, K. et al. Update on treatments for nonmotor symptoms of Parkinson’s disease-an evidence-based medicine review. Mov. Disord. 34, 180–198 (2019).
Emre, M. et al. Rivastigmine for dementia associated with Parkinson’s disease. N. Engl. J. Med. 351, 2509–2518 (2004).
Elbers, R. G., Verhoef, J., van Wegen, E. E., Berendse, H. W. & Kwakkel, G. Interventions for fatigue in Parkinson’s disease. Cochrane Database Syst. Rev. 2015, Cd010925 (2015).
Bucchia, M. et al. Therapeutic development in amyotrophic lateral sclerosis. Clin. Ther. 37, 668–680 (2015).
Bensimon, G., Lacomblez, L. & Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. ALS/Riluzole Study Group. N. Engl. J. Med. 330, 585–591 (1994).
Lacomblez, L., Bensimon, G., Leigh, P. N., Guillet, P. & Meininger, V. Dose-ranging study of riluzole in amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis/Riluzole Study Group II. Lancet 347, 1425–1431 (1996).
Jaiswal, M. K. Riluzole and edaravone: a tale of two amyotrophic lateral sclerosis drugs. Med. Res. Rev. 39, 733–748 (2019).
Teleanu, D. M., Negut, I., Grumezescu, V., Grumezescu, A. M. & Teleanu, R. I. Nanomaterials for drug delivery to the central nervous system. Nanomaterials 9, 371 (2019).
Chauhan, P. S., Yadav, D., Koul, B., Mohanta, Y. K. & Jin, J. O. Recent advances in nanotechnology: a novel therapeutic system for the treatment of Alzheimer’s disease. Curr. Drug Metab. 21, 1144–1151 (2020).
Fatima, S. et al. Nanomedicinal strategies as emerging therapeutic avenues to treat and manage cerebral ischemia. CNS Neurol. Disord. Drug Targets 20, 125–144 (2021).
Pinzón-Daza, M. L. et al. Nanoparticle- and liposome-carried drugs: new strategies for active targeting and drug delivery across blood-brain barrier. Curr. Drug Metab. 14, 625–640 (2013).
Chew, N. W. S. et al. The global burden of metabolic disease: Data from 2000 to 2019. Cell Metab. 35, 414–428.e413 (2023).
Tomic, D., Shaw, J. E. & Magliano, D. J. The burden and risks of emerging complications of diabetes mellitus. Nat. Rev. Endocrinol. 18, 525–539 (2022).
Xu, Z. et al. Association between the oxidative balance score and all-cause and cardiovascular mortality in patients with diabetes and prediabetes. Redox. Biol. 76, 103327 (2024).
de Medeiros, A. F., de Queiroz, J. L. C., Maciel, B. L. L. & de Araújo Morais, A. H. Hydrolyzed proteins and vegetable peptides: anti-inflammatory mechanisms in obesity and potential therapeutic targets. Nutrients 14, 690 (2022).
Asahara, N. et al. A monoclonal antibody activating AdipoR for type 2 diabetes and nonalcoholic steatohepatitis. Sci. Adv. 9, eadg4216 (2023).
Cotter, T. G. & Rinella, M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology 158, 1851–1864 (2020).
Vilar-Gomez, E. et al. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology 149, 367–378.e365 (2015).
Schmitz, S. H., Saunders, K. H. & Aronne, L. J. Cutting-edge approaches to obesity management: the latest pharmacological options. Endocrinol. Metab. Clin. North Am. 54, 85–102 (2025).
Lewis, K. H. et al. Safety and effectiveness of longer-term phentermine use: clinical outcomes from an electronic health record cohort. Obesity (Silver Spring) 27, 591–602 (2019).
Zhi, J. et al. Retrospective population-based analysis of the dose-response (fecal fat excretion) relationship of orlistat in normal and obese volunteers. Clin. Pharmacol. Ther. 56, 82–85 (1994).
Torgerson, J. S., Hauptman, J., Boldrin, M. N. & Sjöström, L. XENical in the prevention of diabetes in obese subjects (XENDOS) study: a randomized study of orlistat as an adjunct to lifestyle changes for the prevention of type 2 diabetes in obese patients. Diabetes Care 27, 155–161 (2004).
European Association for the Study of the Liver (EASL), European Association for the Study of Diabetes (EASD), European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines on the management of metabolic dysfunction-associated steatotic liver disease (MASLD). J. Hepatol. 81, 492–542 (2024).
Kim, H. Y. & Rinella, M. E. Emerging therapies and real-world application of metabolic dysfunction-associated steatotic liver disease treatment. Clin. Mol. Hepatol. (2025).
Harrison, S. A. et al. A phase 3, randomized, controlled trial of resmetirom in NASH with liver fibrosis. N. Engl. J. Med. 390, 497–509 (2024).
Younossi, Z. M. et al. Epidemiology of chronic liver diseases in the USA in the past three decades. Gut 69, 564–568 (2020).
Kleiner, D. E. et al. Association of histologic disease activity with progression of nonalcoholic fatty liver disease. JAMA Netw. Open 2, e1912565 (2019).
Low Wang, C. C., Hess, C. N., Hiatt, W. R. & Goldfine, A. B. Clinical update: cardiovascular disease in diabetes mellitus: atherosclerotic cardiovascular disease and heart failure in type 2 diabetes mellitus - mechanisms, management, and clinical considerations. Circulation 133, 2459–2502 (2016).
Selvin, E. et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N. Engl. J. Med. 362, 800–811 (2010).
Gimeno, R. E., Briere, D. A. & Seeley, R. J. Leveraging the gut to treat metabolic disease. Cell Metab. 31, 679–698 (2020).
Newsome, P. et al. Effect of semaglutide on liver enzymes and markers of inflammation in subjects with type 2 diabetes and/or obesity. Aliment. Pharmacol. Ther. 50, 193–203 (2019).
Armstrong, M. J. et al. Glucagon-like peptide 1 decreases lipotoxicity in non-alcoholic steatohepatitis. J. Hepatol. 64, 399–408 (2016).
Watt, M. J., Miotto, P. M., De Nardo, W. & Montgomery, M. K. The liver as an endocrine organ-linking NAFLD and insulin resistance. Endocr. Rev. 40, 1367–1393 (2019).
Heerspink, H. J., Perkins, B. A., Fitchett, D. H., Husain, M. & Cherney, D. Z. Sodium glucose cotransporter 2 inhibitors in the treatment of diabetes mellitus: cardiovascular and kidney effects, potential mechanisms, and clinical applications. Circulation 134, 752–772 (2016).
Kahl, S. et al. Empagliflozin effectively lowers liver fat content in well-controlled type 2 diabetes: a randomized, double-blind, phase 4, placebo-controlled trial. Diabetes Care 43, 298–305 (2020).
Shimizu, M. et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes. Metab. 21, 285–292 (2019).
Mayerson, A. B. et al. The effects of rosiglitazone on insulin sensitivity, lipolysis, and hepatic and skeletal muscle triglyceride content in patients with type 2 diabetes. Diabetes 51, 797–802 (2002).
Lehmann, J. M. et al. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J. Biol. Chem. 270, 12953–12956 (1995).
Ratziu, V. et al. Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 135, 100–110 (2008).
Ahmad, T. R. & Haeusler, R. A. Bile acids in glucose metabolism and insulin signalling - mechanisms and research needs. Nat. Rev. Endocrinol. 15, 701–712 (2019).
Mudaliar, S. et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 145, 574–582.e571 (2013).
Younossi, Z. M. et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 394, 2184–2196 (2019).
Triantafyllidis, C. P. et al. A machine learning and directed network optimization approach to uncover TP53 regulatory patterns. iScience 26, 108291 (2023).
Frascarelli, C. et al. Deep learning algorithm on H&E whole slide images to characterize TP53 alterations frequency and spatial distribution in breast cancer. Comput. Struct. Biotechnol. J. 23, 4252–4259 (2024).
Güvenç Paltun, B., Kaski, S. & Mamitsuka, H. Machine learning approaches for drug combination therapies. Brief. Bioinform. 22, bbab293 (2021).
Wu, L. et al. Machine learning methods, databases and tools for drug combination prediction. Brief. Bioinform. 23, bbab355 (2022).
Kwan, K., Castro-Sandoval, O., Gaiddon, C. & Storr, T. Inhibition of p53 protein aggregation as a cancer treatment strategy. Curr. Opin. Chem. Biol. 72, 102230 (2023).
Chen, X. et al. Mutant p53 in cancer: from molecular mechanism to therapeutic modulation. Cell Death Dis. 13, 974 (2022).
Pilley, S., Rodriguez, T. A. & Vousden, K. H. Mutant p53 in cell-cell interactions. Genes Dev. 35, 433–448 (2021).
Mantovani, F., Collavin, L. & Del Sal, G. Mutant p53 as a guardian of the cancer cell. Cell Death Differ. 26, 199–212 (2019).
Zhao, M. et al. Mutant p53 gains oncogenic functions through a chromosomal instability-induced cytosolic DNA response. Nat. Commun. 15, 180 (2024).
Acknowledgements
This work has been supported by National Natural Science Foundation of China grants (W2412122, 82470195, 82200208), Natural Science Foundation of Tianjin grants (24JCYBJC00780, 24ZXZSSS00050), State Key Laboratory of Druggability Evaluation and Systematic Translational Medicine grants (QZ 23-6, 23-4), Jin Men Medical Talents of Tianjin (TJSJMYXYC-D2-039), Clinical Oncology Research Fund of CSCO grant (Y-2024AZ(BTK)MS-0040) and Tianjin Key Medical Discipline (Specialty) Construction Project grant (TJYXZDXK-009A).
Author information
Authors and Affiliations
Contributions
Wenhua Wang, Xia Liu, and Hengqi Liu selected of literature, drafted the paper, and prepared the figures. Hassan Abolhassani and Han Yan collected the related references and organized the tables. Huilai Zhang and Xianhuo Wang conceived, designed, and edited the manuscript. All the authors have read and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, W., Liu, X., Liu, H. et al. p53: from understanding its structure to advances in therapeutic targeting. Sig Transduct Target Ther 11, 121 (2026). https://doi.org/10.1038/s41392-025-02549-5
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-025-02549-5
