
Abstract
Pyroptosis, which rapidly releases cellular contents through pyroptotic pores, is an ideal method for inducing in situ cancer vaccines, evoking systemic antitumor immunity, and suppressing primary and metastatic tumors. However, the clinical translation of pyroptosis-based therapy is hindered by the inability to spatially control the activation of inert precursors and the inefficient catalytic activity of nanozymes, which often fail to generate sufficient reactive oxygen species for effective treatment. To address this, we designed a conductive coordination nanozyme prodrug, Cu–DHN. Its π-conjugated polyphenol backbone functions as an intrinsic “electron highway,” enabling rapid electron shuttling to utilize the entire nanoparticle volume for catalysis, thereby achieving exceptional peroxidase-like activity. Upon systemic administration, Cu–DHN remains inert in circulation but is precisely activated within the tumor microenvironment by a tandem GSH-depletion and H2O2-responsive logic gate. This triggers a self-cascade reaction that locally transforms the coordinated prodrug into juglone, which concurrently reverses gasdermin D epigenetic silencing and activates the NLRP3 inflammasome for caspase-1-mediated cleavage. This single-agent, tumor-specific initiation of pyroptosis, augmented by concomitant cuproptosis, elicits potent immunogenic cell death and robust systemic antitumor immunity, effectively suppressing primary and metastatic tumors while exhibiting a pristine safety profile. Our work establishes electron-shuttling coordination polymers as a versatile platform for developing safe and potent catalytic immunotherapies.
Similar content being viewed by others

Introduction
Cancer remains one of the most formidable threats to human health, ranking as the second leading cause of global mortality, where over 90% of cancer-related fatalities stem from metastatic progression.1 While personalized vaccines with whole tumor components have emerged as a transformative modality demonstrating superior efficacy against both primary and metastatic tumors by eliciting systemic antitumor immunity,2 the cumbersome preparation process, along with the high costs of storage and transportation, impedes their clinical implementation.3 Recent breakthroughs have identified pyroptosis as a strategic immunomodulatory pathway. This caspase-mediated process is initiated through proteolytic cleavage of gasdermin (GSDM) proteins, generating N-terminal fragments that oligomerize into plasma membrane pores.4,5 Characterized by GSDM pore-driven bubble-like protrusions (pyroptotic bodies), tumor pyroptosis exhibits unique therapeutic advantages: (i) rapid release of tumor-associated antigens (TAAs) and damage-associated molecular patterns (DAMPs, e.g., calreticulin (CRT), high mobility group box-1 protein (HMGB-1), adenosine triphosphate (ATP)) creates in situ cancer vaccines;6 (ii) enhanced dendritic cell (DC) maturation facilitates efficient cross-presentation of TAAs and DAMPs to CD8+ T cells; and (iii) systemic activation of tumor-specific cytotoxic T lymphocytes establishes durable antimetastatic immunity.7 These attributes position pyroptosis induction as a paradigm-shifting strategy for triggering endogenous vaccination effects, amplifying systemic antitumor immune surveillance, and ultimately suppressing posttreatment recurrence and metastasis. Recent advancements in nanoparticle-enabled pyroptosis induction have unveiled novel therapeutic avenues for cancer immunotherapy, yet critical challenges persist in achieving tumor-specific efficacy while mitigating systemic toxicity. Zhang et al. demonstrated the potential of a biohybrid nanozyme that triggers caspase-1-mediated pyroptosis via cascade metabolic interference;8 however, this strategy is inherently limited by its dependence on abundance in GSDM protein—a condition rarely met in clinical malignancies due to DNA methyltransferase-driven epigenetic silencing of GSDM genes in most human cancers versus their constitutive expression in normal tissues.9 This expression dichotomy renders nonspecific caspase activation strategies not only therapeutically ineffective against GSDM-deficient tumors but also prone to severe off-target toxicity, including neurotoxicity, thrombocytopenia, and nephrotoxicity.10,11,12,13
To overcome the limitation of pyroptosis induction in GSDM-deficient tumors, current strategies focus on elevating intratumoral GSDM levels through DNA methyltransferase inhibitors (e.g., gemcitabine) or direct delivery of GSDM proteins/expression plasmids.14,15,16 Nevertheless, these methods necessitate additional caspase-activating agents to cleave GSDM proteins for pyroptosis initiation. For instance, Wang et al. co-delivered cisplatin with GSDME plasmids to induce pyroptosis,15 yet the asynchronous drug release kinetics of these agents significantly compromised synergistic efficacy. Moreover, persistent off-target toxicity remains a critical limitation. To improve biosafety, Chen et al. developed a pH-ultrasensitive “off-on” nanosystem coencapsulating photosensitizer Ce6 and quencher QSY21 that demonstrated tumor-selective pyroptosis through acidic microenvironment-triggered reactive oxygen species (ROS) generation and subsequent caspase-3/GSDME activation.17 Nevertheless, two fundamental constraints endure: (i) lysosomal preleakage of Ce6 in normal cells induces phototoxic damage and off-target pyroptosis, and (ii) therapeutic efficacy only flourishes in a minority of GSDME-expressing tumors. Therefore, developing pyroptosis inducers that simultaneously achieve tumor specificity, biosafety, and therapeutic efficacy remains a critical unmet challenge in precision oncology paradigms. Prodrug strategies have garnered significant attention in precision oncology due to their inherent capacity to remain pharmacologically inert in normal tissues while achieving targeted activation at disease sites. Pioneering work by Liang et al. demonstrated an acid-responsive PEG-DOX prodrug conjugated via hydrazone bonds, which selectively releases doxorubicin in tumor microenvironments to activate caspase-3 and induce pyroptosis in GSDME-high malignancies.18 Wang et al. also reported a ROS-activated nano-platform delivering 5-azacytidine (a DNA methyltransferase inhibitor) to epigenetically upregulate GSDME expression for tumor-specific pyroptosis.19 However, these systems also face some constraints: (i) a discrepancy in pharmacokinetic profiles of caspase-activating compounds and GSDM-elevating therapeutics compromises pyroptosis induction efficiency; and (ii) complex manufacturing processes hinder clinical translation. This highlights an unmet need for single-agent prodrug systems capable of tumor-specific activation to simultaneously trigger caspase cascades and elevate GSDM expression—a critical breakthrough for optimizing the therapeutic index in pyroptosis-based therapies.
Intriguingly, our investigation of juglone, a natural naphthoquinone from traditional Chinese medicine, revealed novel dual mechanisms: (i) K+ efflux-mediated NLRP3 inflammasome activation leading to caspase-1 cleavage,20 and (ii) DNA methyltransferase inhibition (RNA-seq validated) that reverses GSDMD epigenetic silencing (Supplementary Fig. 1). This dual-action profile enables complete pyroptosis execution through coordinated caspase activation (caspase-1), GSDMD upregulation, and subsequent N-terminal cleavage to form lytic pores. Preliminary data demonstrate concentration- and time-dependent pyroptosis induction (Supplementary Fig. 2), while systemic toxicity concerns are mitigated by recruiting its prodrug form 1,5-dihydroxynaphthalene (DHN), a commercially available compound exhibiting negligible normal tissue toxicity until ROS-mediated oxidation converts it to active juglone specifically in tumors.21 This innovative single-agent strategy resolves inherent pharmacokinetic mismatches in combination therapies and eliminates intricate synthesis requirements. By leveraging tumor-specific ROS generation to activate DHNs, our strategy uniquely addresses the precision medicine challenges of current pyroptosis induction paradigms, establishing a novel therapeutic axis with superior tumor-targeting specificity, enhanced biosafety profile, and potentiated therapeutic efficacy. The endogenous stimulus-responsive generation of ROS has emerged as a research hotspot due to its unique spatiotemporal-unrestricted characteristics.22 Recent advances demonstrate that copper-based nanozymes can catalytically produce ROS through logic-gated responses to the tumor microenvironment’s high glutathione (GSH) “AND” hydrogen peroxide (H2O2) concentrations, effectively inducing tumor ferroptosis while minimizing normal tissue toxicity.23 Notably, the generated ROS can also oxidize DHN into juglone, subsequently triggering tumor-specific pyroptosis. However, ROS generation efficiency confronts several barriers, including (i) tumor microenvironment-specific limitations involving CAT-driven H2O2 scavenging24 and (ii) nanozyme-intrinsic structural constraints characterized by insufficient surface accessibility of Cu catalytic sites and underutilization of embedded Cu reservoirs due to inefficient electron transfer.25 Additionally, Golub et al. revealed that copper ions can induce tumor cuproptosis, whose therapeutic efficacy critically depends on intracellular copper transport efficiency.26
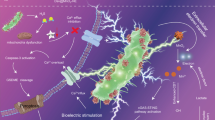
Based on these findings, this work proposes a conductive coordination nanozyme prodrug system (Cu–DHN) through concise coordination coassembly of Cu2+, DHN, and cysteine (L-Cys) with high drug-loading capacity (Supplementary Table 1). The electron-conducting conjugated architecture of DHNs facilitates electron transfer between the surface and internal Cu2+,27,28 while Cys-mediated particle size customization optimizes the specific surface area to expose more catalytic sites, synergistically enhancing peroxidase (POD) activity. This design enables (i) efficient H2O2 conversion into hydroxyl radicals (•OH) with concurrent self-cascade oxidation of DHN to juglone; (ii) juglone-mediated CAT inhibition that amplifies intracellular H2O2 accumulation, establishing a spatiotemporally enhanced ROS-juglone positive feedback loop for precise pyroptosis/ferroptosis; and (iii) functioning as an efficient copper ion carrier, Cu–DHN enhances tumor-specific cellular uptake of Cu2+, thereby effectively inducing cuproptosis and stimulating the release of ATP that augments tumor immunogenicity. Following intratumoral administration, the nontoxic prodrug state of Cu–DHN undergoes tumor-specific activation by tumor microenvironmental GSH and H2O2, synergistically triggering pyroptosis with inherent cuproptosis/ferroptosis effects, thereby promoting in situ tumor vaccination and robust systemic antitumor immunity. As a tumor-specific conductive coordination nanozyme prodrug system that precisely induces pyroptosis, this breakthrough addresses the long-standing challenges of inefficient pyroptosis induction and nonspecific systemic inflammation, establishing a novel paradigm for safe and potent primary and metastatic tumor therapy through synergistic pyroptosis/cuproptosis/ferroptosis-mediated systemic antitumor immune activation (Fig. 1).

A catalytic-immunotherapy cycle driven by conductive nanozyme Cu–DHN. The electron-conducting Cu–DHN nanozyme achieves tumor-specific activation through a biochemical “AND” gate (GSH “AND” H2O2), leading to on-site •OH generation and prodrug conversion to juglone. This process initiates a powerful, integrated cell death program (pyroptosis/cuproptosis/ferroptosis). The subsequent immunogenic cell death releases TAAs and DAMPs to fuel a systemic antitumor immune response, ultimately inhibiting primary and metastatic tumor growth. This figure was created with BioRender.com
Results
Synthesis and characterization of the conductive coordination nanozyme prodrug Cu–DHN
The conductive coordination nanozyme prodrug Cu–DHN was synthesized according to the protocol illustrated in Fig. 2a. Given that the enzymatic activity is critically dependent on the exposure ratio of active catalytic sites, smaller nanoparticles with higher specific surface areas generally exhibit enhanced POD through increased exposure of active sites.29 Inspired by the critical role of modulators in governing the growth of metal-organic frameworks (MOFs),30,31 we employed a modulator strategy using L-cysteine (Cys) to govern the coordination assembly process.

Engineering the architecture and coordination environment of the Cu–DHN nanozyme via L-Cysteine (Cys) modulation. a Synthesis scheme of Cu–DHN, illustrating the role of L-Cys as a coordination terminator and size regulator. The zoomed-in region illustrates the intermolecular hydrogen bonding between L-Cys molecules. This scheme was created with BioRender.com. b Hydrodynamic size distribution profiles (n = 3), and c TEM images of Cu–DHN with varying Cu:DHN:Cys feed molar ratios, revealing a non-monotonic size trend. d Variation profiles of hydrodynamic size for different M-DHN (M = Fe, Mn, Co) nanoparticles (n = 3) demonstrating the generalizability of Cys-mediated size control. e Fourier transform infrared (FT-IR) spectra confirming the involvement of phenolic –OH and thiol –SH groups; f normalized Cu K-edge XANES spectra, g Fourier transforms of k3-weighted Cu K-edge EXAFS spectra, and h high-resolution Cu 2p XPS spectra of Cu–DHN with varying Cu:DHN:Cys molar ratios, collectively evidencing the evolution of a mixed (O, S)-coordination shell and a rising Cu⁺/Cu²⁺ ratio with increasing Cys content. i HR-TEM image and corresponding SAED pattern, and j elemental distribution mapping and corresponding line-scan profile of Cu–DHN at a Cu:DHN:Cys molar ratio of 0.6:1:0.2. k XRD patterns, and l Brunauer–Emmett–Teller (BET) N2 adsorption–desorption isotherms demonstrating the pore structure evolution of Cu–DHN with varying Cu:DHN:Cys molar ratios, confirming the progressive surface passivation that defines the core–shell pore structure. Data are represented as mean ± SD
As shown in Fig. 2b, the hydrodynamic diameter of Cu–DHN decreased dramatically from 160 nm (Cu:DHN:Cys = 0.5:1:0, without Cys) to 70 nm when the molar ratio reached 0.6:1:0.2. However, further increasing the Cys content to craft Cu–DHN (Cu:DHN:Cys = 0.75:1:0.5) and Cu–DHN (Cu:DHN:Cys = 1:1:1) enlarged the particles to 110 and 140 nm, respectively. Thus, we demonstrated that L-Cys served as a potent molecular modulator to precisely control the size of Cu–DHN, a critical parameter for optimizing its peroxidase (POD)-mimetic activity. This size control was achieved by tuning the competition between infinite coordination polymerization and Cys-mediated termination, with excessive Cys inducing a reversal of size reduction guided by crystallization due to intermolecular hydrogen bonding. The efficacy of this modulation is directly evidenced by the nonmonotonic variation in nanoparticle dimensions.
The parent polyphenol ligand 1,5-DHN, featuring two phenolic hydroxyl groups, promotes uncontrolled polymer growth by coordinating with Cu2+ ions. The initial coordination at the 1-position creates steric hindrance, forcing the 5-position hydroxyl to coordinate with another Cu2+, leading to uncontrolled growth of infinite coordination polymers and larger nanoparticles. The introduction of Cys, possessing a single thiol group, effectively terminated this infinite coordination assembly, thereby restricting particle growth and yielding smaller nanoparticles.30 However, excessive Cys triggered hydrogen bond-mediated self-assembly between Cys molecules,32 counteracting the size-reduction effect. At higher concentrations, the excess free Cys in the synthesis solution (at pH ≈ 8) predominantly existed in a zwitterionic form. This facilitates the formation of robust intermolecular hydrogen bonds between the NH3+ and COO− groups and may further be stabilized by disulfide-linked L-cystine formation. This hydrogen bond-mediated self-assembly and crystallization of Cys acts as a secondary aggregation force, counteracting the primary size-reduction effect and leading to the observed increase in hydrodynamic diameter. Multifaceted characterization robustly confirmed the precision, universality, and stability of Cys-mediated size control. This strategy not only yielded optimally sized nanoparticles for our Cu-based metal-phenolic network but also proved applicable across a spectrum of catalytic metals, underscoring its potential as a general platform for nanozyme design.
Complementary TEM and SEM characterization (Fig. 2c, Supplementary Fig. 3) confirmed these spherical-like morphologies of all nanostructures and validated the size trends observed by DLS. The pristine Cu–DHN (0.5:1:0) exhibited a ~150 nm diameter, while Cys-optimized samples (0.6:1:0.2) showed a dramatic size reduction to ~50 nm. Subsequent Cys overloading (0.75:1:0.5 and 1:1:1) progressively increased particle sizes to 100 and 120 nm, respectively, demonstrating precise size modulation through stoichiometric control. The optimal Cu:DHN:Cys = 0.6:1:0.2 achieves a critical balance between leashing infinite coordination polymerization and preventing hydrogen bond-driven agglomeration. The slight discrepancy between the smaller dry-state (TEM/SEM) and larger hydrodynamic (DLS) diameters is expected, attributable to the thin hydration layer surrounding the nanoparticles in solution.
Furthermore, we extended the synthesis to three additional POD-active metal ions (Fe3+, Mn2+, and Co2+) for coordination coassembly with DHN and Cys (Fig. 2d). Dynamic light scattering (DLS) analysis revealed that while the absolute particle sizes of M-DHN (M = Fe, Mn, Co) varied depending on the metal ion species, the nonmonotonic size variation pattern in response to changing Cys composition remained consistent across all systems. Moreover, Mg–DHN (Mg:DHN:Cys = 0.6:1:0.2), Cu–Cys (Cu:DHN:Cys = 0.5:0:1, without DHN), and the parent drug group Cu–juglone (Cu:juglone:Cys = 0.6:1:0.2) were synthesized as control materials for further evaluation (Supplementary Fig. 4). Multifaceted characterization robustly confirmed the precision, universality, and stability of Cys-mediated size control. This strategy not only yielded optimally sized nanoparticles for our Cu-based metal-phenolic network but also proved applicable across a spectrum of catalytic metals, underscoring its potential as a general platform for nanozyme design. Dispersing the prodrug nanoparticle Cu–DHN in various media (Supplementary Fig. 5) demonstrated that it largely maintained its integrity and uniformity, with only a slight size reduction observed in acidic environments (pH < 5), indicating good colloidal stability for subsequent biological applications.
Subsequently, we investigated the driving forces behind the coordination coassembly of Cu–DHN. Initial UV‒vis absorption spectral analysis revealed characteristic peaks of DHN in Cu–DHN (Supplementary Fig. 6). Notably, broad-band absorption in the infrared region was observed, attributed to enhanced conjugation resulting from J-aggregation of the DHN assembly,33 suggesting successful coordination between DHN and Cu2+ ions. Fourier transform infrared spectroscopy (FT-IR) analysis demonstrated significant attenuation of the phenolic hydroxyl group’s in-plane bending vibration at 1421 cm−1 in DHN following Cu2+ coordination (Fig. 2e). Concurrently, the characteristic S–H stretching vibration of Cys at 2646 cm−1 disappeared after Cu2+ interaction, indicating restricted vibrational modes of both phenolic hydroxyl and thiol groups upon metal coordination. These observations preliminarily confirm the coassembly of Cu2+ with phenolic hydroxyl groups of DHN and thiol groups of Cys, leading to the formation of a Cu–DHN conductive coordination nanozyme prodrug. Our comprehensive spectroscopic analysis also revealed that L-Cys orchestrated a multifunctional coordination network for optimized nanozyme performance. Apart from being a mere comonomer, it critically governs the coordination geometry, modulates the copper valence state, and ultimately tailors the prodrug’s catalytic potency and therapeutic potential. This integrated role is fundamental to achieving both controlled nanoparticle size and high catalytic activity.
To further elucidate the coordination mechanism, we employed XPS and synchrotron radiation characterization. Beyond simple coordination, L-Cys actively engineered a hybrid Cu⁺/Cu²⁺ interface, which is a kernel for catalytic activation. XPS and synchrotron radiation analyses provided atomic-level insights into this sophisticated mechanism. EXAFS data unambiguously confirmed the coexistence of Cu–O (from DHN) and Cu–S (from Cys) coordination shells (Fig. 2f, g). X-ray spectroscopy (XPS) analysis revealed a 1.33 eV negative shift in the O 1s binding energy for DHN after Cu2+ coordination, while a 0.27 eV positive shift occurred in the S 2p binding energy for Cys (Supplementary Fig. 7), suggesting a distinct electron cloud distribution in the Cu–O and Cu–S bonds. Fourier transforms of the k3-weighted Cu extended X-ray absorption fine structure (XAFS) provided direct evidence of coordination environments (Fig. 2g): Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited a dominant peak at 1.56 Å corresponding to Cu-–O coordination shells, consistent with the CuO reference. Conversely, Cu–Cys showed a characteristic Cu–S coordination peak at 1.80 Å, confirming a heterogeneous coordination environment with both oxygen and sulfur atoms in the first shell (Supplementary Table 2).
In addition, progressive increases in Cys content (Cu:DHN:Cys from 0.6:1:0.2 to 1:1:1) resulted in intermediate first-shell radii between the Cu–O and Cu–S configurations, confirming the coexistence of both coordination modes (Supplementary Fig. 8, Supplementary Table 2). Cu K-edge X-ray absorption near-edge structure (XANES) spectra revealed significant valence state variations: an incremental shift occurred from Cu2+ toward Cu+ as the Cys content increased (Fig. 2f). The Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited Cu K-edge characteristics matching CuO, indicating the exclusive presence of Cu2+. Increasing the Cys content progressively shifted the absorption edge toward the Cu2S reference, with Cu–Cys displaying an intermediate edge position between CuS and Cu2S, suggesting a hybrid of Cu+/Cu2+ valence states in Cys-containing Cu–DHN. XPS deconvolution of Cu 2p peaks quantified this valence transition: Cu+ content increased from 14% to 70% as the Cys ratio increased from 0.2 to 1 equivalent (Fig. 2h, Supplementary Fig. 9). This was a direct consequence of the reducing capacity of Cys, which reduced a portion of the Cu2+ precursors to Cu+ during synthesis. This dual valency was proven to be a cornerstone of our design: it ensured storage stability as a Cu2+-based coordination polymer, while preloading the nanoparticle with a significant surface reservoir of the more bioactive Cu+. The redox-mediated valence modulation is rooted in the reducing capacity of Cys, which reduces Cu2+ to Cu+ while terminating nanoparticle growth, resulting in surface-enriched Cu+ species. The surface-preloaded Cu+ species endow Cu–DHN with dual therapeutic advantages: (i) facile catalytic activation for rapid •OH generation from H2O2 upon cellular internalization34 and (ii) enhanced cuproptosis induction through elevated Cu+ levels.26
In this process, Cys plays three indispensable roles. As a size regulator, it terminated the infinite coordination polymerization by surface-limited coordination; as a coordination partner, it created a mixed (O, S)-ligand field; as a redox modulator, it generated a stable yet Fenton reaction-ready Cu+/Cu2+ hybrid state. This sophisticated coordination design yields a prodrug that is stable in storage yet primed for rapid catalytic activation and cuproptosis induction in the tumor microenvironment, perfectly aligning the synthesis strategy with the therapeutic objective.
To elucidate the mechanism underlying Cys-regulated particle size control in Cu–DHN, we conducted systematic structural and compositional analyses. Our structural analyses established that L-Cys governed the particle size by terminating the uncontrolled coordination growth of the copper–polyphenol framework Cu–DHN, leading to the formation of a unique crystalline core–amorphous shell structure. High-resolution TEM (HR-TEM) imaging combined with elemental mapping of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) revealed distinct structural features. Elemental line scanning and mapping of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) provided critical evidence for this mechanism. Elemental mapping and line scanning revealed that sulfur (S) signals were predominantly localized at the particle surface, whereas Cu and oxygen (O) were distributed uniformly throughout the core–shell architecture (Fig. 2i). This visual evidence echoed the mechanism where initial, unrestricted coordination between the bidentate phenolic hydroxyl groups of DHN and Cu2+ forms the crystalline core, while subsequent surface termination by monodentate Cys creates the disordered shell (Fig. 2j). This spatial segregation confirmed that Cys-derived sulfur participates in surface coordination without penetrating the crystalline core.35 The intact crystalline core region (evidenced by clear lattice fringes and diffraction spots) was encapsulated by a loose, amorphous outer shell. This spatial segregation confirms that Cys does not incorporate into the growing crystal lattice but rather coordinates with Cu2+ at the particle interface, effectively passivating the surface and arresting further growth.
XRD and BET analyses unraveled this core–shell model and excluded a simple physical mixture. XRD patterns further corroborated the core–shell configuration (Fig. 2k). Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited characteristic diffraction peaks at 7.07°, 12.94°, 15.38°, 20.33°, 23.84°, 25.21°, 26.20°, and 36.36°, consistent with the HR-TEM observations. Progressive Cys incorporation (0–1 equivalent) diminished the diffraction peaks of the crystalline Cu–DHN core, recognized by gradual attenuation of Cu–DHN diffraction intensities and emergence of Cu–Cys signatures at 18.90°, 28.49°, 33.15°, and 34.49°. Complete suppression of Cu–DHN peaks occurred at the highest Cys ratio, where Cu:DHN:Cys = 1:1:1. However, physical mixtures of Cu–DHN (Cu:DHN:Cys = 0.5:1:0) and Cu–Cys maintained distinct and distinguishable diffraction patterns (Supplementary Fig. 10). This critical comparison confirmed that rather than being physically blended, the Cys-derived shell was chemically bound, forming a conformal coating that shielded the core from X-ray detection.36
BET analysis revealed pore volume variations supporting the proposed model (Fig. 2l). Compared to Cu–Cys (0.085 cm³/g), Cu–DHN exhibited a significantly higher pore volume (0.238 cm³/g), reflecting its larger pore dimensions favorable for gas diffusion. This discrepancy confirmed that the surface Cu–Cys coating acted as a molecular sieve, physically restricting gas access to the porous Cu–DHN core. In this regard, increasing Cys content progressively reduced measured pore volumes below theoretical values calculated for physical mixtures, with the deviation amplifying at higher Cys ratios (Supplementary Table 3). This phenomenon suggests that conformal Cu–Cys surface coatings restrict gas access to the porous Cu–DHN core through size-exclusion effects.37
The superior catalytic performance of Cu–DHN arises from synergistic size control and intrinsic electron shuttling
Initial performance screening to decode the principles and outcomes of size optimization is necessary. Subsequently, we demonstrated that the superior peroxidase (POD)-like activity and glutathione peroxidase (GSH-Px)-like activity of our nanozyme emerged from the sophisticated interplay of a minimized particle size, an optimized Cu+/Cu2+ valence state, and a conductive polyphenol backbone that enables efficient electron shuttling. As shown in Fig. 3, this triad collaborated to maximize active-site exposure, facilitate the crucial Fenton redox cycle, and engage the entire nanoparticle volume in catalysis.

The electron-shuttling architecture of Cu–DHN underpins its superior and sustained catalytic activity. a GSH-Px-like activity evaluation of Cu–DHN with varying Cu:DHN:Cys feed molar ratios via DTNB assay. The column chart presents the peak values of UV–Vis absorbance at 412 nm. b and c POD-like property of Cu–DHN with varying Cu:DHN:Cys molar ratios via (b) TMB colorimetric assay (the column chart presents the peak values of UV–Vis absorbance at 662 nm), and c ESR analysis using DMPO as the spin trapping agent, confirming •OH as the primary reactive species. d Catalytic Hill plots of Cu–DHN with varying Cu:DHN:Cys molar ratios in the presence of GSH using H2O2 as substrate. e, f Direct evidence for the electron-shuttling mechanism illustrated by high-resolution Cu 2p XPS, presenting highly efficient Cu⁺/Cu²⁺ cycling in e the agile behavior of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) and the retarded behavior of f Cu–Cys. g Schematic representations of electron transport via a conductive DHN-promoted pathway. This scheme was created with BioRender.com. h Electrical conductivity of Cu–DHN with varying Cu:DHN:Cys molar ratios. Data are represented as mean ± SD (n = 3). i Time-dependent MB degradation process after incubation with Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) in the presence of GSH and H2O2. j Juglone yield from Cu–DHN with varying Cu:DHN:Cys molar ratios after GSH and H2O2 incubation over time (n = 3). Data are represented as mean ± SD
It has been reported that Cu2+-based nanozymes can oxidize GSH to generate glutathione disulfide (GSSG) while being reduced to Cu⁺ with POD activity, thereby disrupting redox homeostasis in tumor cells.23 Therefore, the GSH-Px activity of Cu–DHN was quantitatively assessed using Ellman’s colorimetric assay probed by DTNB.38 As shown in Supplementary Fig. 11, Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) exhibited a time-dependent GSH-depletion capacity. Subsequent comparative analysis of Cu–DHN with different Cu:DHN:Cys feed ratios revealed that Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) demonstrated optimal GSH-depletion efficiency (Fig. 3a). This enhancement is attributed to its minimized particle size (approximately one-third that of Cu–DHN with Cu:DHN:Cys = 0.5:1:0), collectively yielding a 27-fold increase in specific surface area compared to Cu DHN (Cu:DHN:Cys = 0.5:1:0). The expanded surface area facilitates the exposure of catalytically active sites, thereby promoting interfacial redox reactions with GSH (Supplementary Fig. 12).
The POD activity was further investigated through TMB chromogenic assays measuring •OH generation from the dismutation of H2O2 catalyzed by Cu–DHN and Cu–Cys with varying Cu:DHN:Cys feeding ratios.39 Kinetic analysis demonstrated a negative correlation between nanoparticle size and •OH production, with smaller Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) exhibiting superior catalytic performance due to increased active-site accessibility (Fig. 3b). The type of ROS was identified by electron spin resonance (ESR) spectroscopy. DMPO probing presented characteristic 1:2:2:1 quartet signals, confirming •OH as the predominant species contributing to catalytic activities (Fig. 3c, Supplementary Fig. 13). Kinetic analysis fitting the Hill equation revealed that Cu–DHN’s maximum reaction rate (Vmax) inversely correlated with particle size and consistently surpassed all controls (Fig. 3d), including previously reported nanozymes (Supplementary Table 4). The distinct kinetic phases can be rationalized by the initial scavenging of •OH by GSH and DHN moieties, followed by a rapid acceleration phase once •OH generation outstrips consumption. This efficient kinetics was a direct consequence of the high-density, readily accessible active sites enabled by the electron-conducting framework.
Notably, despite comparable particle sizes between Cu–Cys and Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2), the former exhibited significantly inferior GSH-depletion performance. Cu–DHN outperformed Cu–Cys by 3.2-fold in •OH yield despite comparable dimensions. As anticipated, minimizing the particle size to ~70 nm (Cu:DHN:Cys = 0.6:1:0.2) dramatically enhanced both GSH-depletion and •OH generation, owing to the maximized specific surface area. Nevertheless, the critical limitation of relying solely on surface atoms was challenged by the direct comparison of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) with Cu–Cys: despite comparable sizes, Cu–DHN outperformed Cu–Cys by a significant margin in both GSH consumption and •OH yield. This disparity suggested that additional factors beyond particle size optimization contribute to the enhanced catalytic activity. Thus, we hypothesized that the conjugated backbone of DHN was critical for unlocking a robust and sustainable catalytic function. This “electron highway” of the π-conjugated structure not only drove superior enzyme kinetics but also facilitated a complete prodrug activation cycle, setting Cu–DHN apart from its counterparts. Consequently, we quantitatively captured the properties of this “electron highway” using XPS and four-point probe conductivity measurements.
Upon GSH incubation, XPS showed that in Cu–DHN, the Cu⁺ proportion surged from 14% to 81% (Fig. 3e), indicating a highly efficient and profound reduction process that engaged the nanoparticle core. Subsequent H2O2 exposure triggered near-complete (90%) reoxidation to Cu²⁺ (Fig. 3f), demonstrating a robust and reversible redox cycle essential for sustained •OH generation. In stark contrast, Cu–Cys showed lethargic valence changes (<20% variation), confirming its confinement to superficial, limited reactions (Supplementary Fig. 14). Conductivity measurements provided further evidence of enhanced charge transfer in Cu–DHN, showing significantly higher conductivity than Cu–Cys (Fig. 3h). This phenomenon was a direct result of the synergistic effects between efficient Cu+/Cu2+ valence transitions and DHN-mediated electron transport through the nanoparticle’s core–shell architecture (Fig. 3g). Postreaction characterization via TEM, DLS, and XRD confirmed that structural integrity was maintained with preserved crystallinity and consistent size distribution trends (Supplementary Figs. 15, 16), indicating that the GSH/H2O2 treatment had a negligible influence on the particle size and structure of Cu–DHN. This compelling evidence substantiates the superior efficacy of our dual strategy combining particle size optimization and electron transfer engineering for POD performance enhancement.
Methylene blue (MB) bleaching assays confirmed persistent •OH generation over 180 min (Fig. 3i). Critically, this sustained radical flux efficiently oxidizes the coordinated DHN into its active anticancer form, juglone. Distinct DHN-to-juglone conversion efficiencies were mediated by Cu–DHN with varying Cu:DHN:Cys ratios. After 3 h of coincubation, Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) achieved ~55% DHN oxidation to juglone, whereas Cu–DHN (Cu:DHN:Cys = 0.5:1:0) exhibited a substantially lower conversion efficiency (~30%) under identical conditions (Fig. 3j). Notably, the juglone conversion rates across Cu–DHN at different Cu:DHN:Cys ratios showed a positive correlation with their respective POD enzymatic activities. This proportional relationship conclusively linked the superior catalytic properties of Cu–DHN directly to its therapeutic function. To confirm that the observed therapeutic effects were due to the Cu-dependent catalytic activity, we synthesized a control material by replacing the active center with redox-inert Mg2+ ions and forming Mg–DHN (Mg:DHN:Cys = 0.6:1:0.2). The complete catalytic inertia (Supplementary Fig. 17) of the redox-inert Mg–DHN control, which exhibited negligible activity across all assays, underscored that the observed therapeutic effects are specifically due to the Cu-dependent catalytic activity, providing a rigorous negative control.
In conclusion, the orchestrated interplay of size reduction, a Cu⁺/Cu²⁺ hybrid valence state, and a conjugated ligand backbone is paramount. Particularly, the electron highway within Cu–DHN has done more than just enhance kinetics; it ensured a durable catalytic performance that powers the continuous on-site production of both cytotoxic radicals and the activated chemotherapeutic agent, solidifying its design as an integrated and self-sufficient prodrug system.
Cu–DHN elicits potent antitumor immunity via a synergistic cascade of pyroptosis-dominant programmed cell death
Cu–DHN orchestrated a potent and tumor-specific therapeutic outcome by sequentially activating a cascade of pyroptosis, cuproptosis, and ferroptosis, with pyroptosis serving as the dominant pathway. This tripartite cell death synergy not only effectively killed cancer cells but also, crucially, initiated powerful immunogenic cell death (ICD) to stimulate an in situ vaccination effect, as shown in Fig. 4.

In vitro effects of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) on 4T1 cells. a CLSM images and b mean fluorescence intensity (MFI) of 4T1 cells stained by DCFH-DA (ROS probe) after co-incubation with Cu–DHN for 6 h (n = 3). c Juglone yield in 4T1 cells after co-incubation with Cu–DHN for 12 h (n = 3). d Cell viability curves of 4T1 cells after co-incubation with Cu–DHN for 24 h (n = 6). e Bright-field microscopic images of 4T1 cells after co-incubation with Cu–DHN for 18 h (yellow arrows highlight membrane-bound vesicular protrusions indicative of pyroptotic morphology). f Intracellular K+ content in 4T1 cells after co-incubation with Cu–DHN for 6 h (n = 3). g NLRP3 content, h Caspase-1 activity, i GSDMD-FL content, j GSDMD-N content in 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). k IL-1β secretion from 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). l Internalized amounts of Cu by 4T1 cells after co-incubation with Cu–DHN for 6 h (n = 3). m FDX1 content in 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). n GPX4 activity in 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). o CLSM images of 4T1 cells stained by Liperfluo (LPO probe) after co-incubation with Cu–DHN for 12 h (n = 3). p Flow cytometry analysis of 4T1 cells labeled by AF488-CRT antibody after co-incubation with Cu–DHN for 18 h. Release of q HMGB-1, and r ATP from 4T1 cells after co-incubation with Cu–DHN for 18 h (n = 3). s BMDC maturation after co-incubation with Cu–DHN treated 4T1 cells for 24 h (n = 3). Cu concentration: 0.020 mM, DHN or juglone concentration: 0.033 mM. Data are represented as mean ± SD
The cellular uptake and lysosomal escape ability of Cu–DHN were evaluated first. As shown in Supplementary Fig. 18a, intracellular Cu–DHN accumulation exhibited a significant time-378-dependent increase during 6 h of coincubation, reaching saturation thereafter. Moreover, Cu–DHN exploits macropinocytosis for entry and triggers a self-limiting cascade of lysosomal destabilization (Supplementary Fig. 18b, 19). Afterwards, general intracellular •OH generation was probed by DCFH-DA via confocal laser scanning microscopy (CLSM) (Fig. 4a, b). Both the free CuCl2 and its coadministration groups with juglone and DHN exhibited significantly weaker DCF green fluorescence intensity compared to the coordinated composites Cu–DHN and Cu–juglone. This disparity arose from enhanced POD-like activity in Cu–DHN through particle size optimization and conductive structure modulation. In contrast, Cu–Cys showed significantly reduced •OH generation compared to Cu–DHN, since the absence of “redox capillaries” in ligands hampered efficient electron transfer and impeded the swift revival of catalytic activity. This •OH not only directly caused damage but also oxidized the prodrug DHN into juglone.
The •OH generation mechanism requires both catalysts and substrates, and the superior POD activity of Cu–DHN enables efficient •OH generation under sufficient H2O2 conditions.24 In this established self-amplifying loop, juglone, oxidized from DHN by •OH, inhibits catalase (CAT) activity (Supplementary Fig. 20), thereby elevating intracellular H2O2 to boost further •OH generation and DHN oxidation. Based on the exceptional •OH-generating capacity of Cu–DHN, we quantified the intracellular DHN-to-juglone conversion. CuCl2 + DHN combinations oxidized merely 5% DHN, while Mg–DHN nanoparticles (lacking POD activity) showed negligible conversion. In contrast, Cu–DHN achieved >50% DHN oxidation after 12 h of incubation (Fig. 4c), attributable to its robust •OH production.
Then, we established the superior and selective cytotoxicity of Cu–DHN. Cu–DHN exhibited micromolar-level IC50 values (IC50 = 0.026 mM Cu and 0.042 mM DHN) against 4T1 tumor cells, outperforming control nanozymes such as Cu–Cys (IC50 = 0.067 mM Cu) and matching the potency of the active parent drug version Cu–juglone (IC50 = 0.016 mM Cu, 0.027 mM juglone) (Fig. 4d). Crucially, this potency was tumor-selective: unlike the indiscriminate toxicity of the parent drug Cu–juglone (Supplementary Table 5), Cu–DHN maintained high viability in normal cells, demonstrating its successful activation only within the tumor’s unique redox environment (Supplementary Fig. 21). Cu–DHN maintained >75% viability in HUVECs, NIH 3T3 cells, and HSF normal cells at 0.1 mM DHN compared with Cu–juglone (<35% viability at 0.08 mM juglone) (Supplementary Fig. 21a). This specificity was governed by a tumor-selective GSH “AND” H2O2 logic gate and a self-amplifying catalytic loop: the •OH-generated juglone inhibited catalase, elevating intracellular H₂O₂ to fuel further •OH production and DHN-to-juglone conversion (>50% in 12 h), a process inaccessible to less catalytic analogs (Supplementary Fig. 21b).
In 4T1 cells, which are inherently gasdermin deficient,40 Cu–DHN successfully initiated pyroptosis, as evidenced by the formation of characteristic pyroptotic bodies (Fig. 4e, Supplementary Figs. 2, 22). Mechanistically, it activated the canonical pathway: K⁺ efflux triggered NLRP3 inflammasome assembly and Caspase-1 cleavage (Fig. 4f-h). Most importantly, Cu–DHN epigenetically upregulated GSDMD transcription through DNA methyltransferase inhibition (Fig. 4i, Supplementary Figs. 23a, b, 24), thereby “priming” low-GSDMD tumor cells for pyroptosis. This ensured the cleavage of GSDMD and the mature release of IL-1β through the resulting pores (Fig. 4j, k). This dual-pathway activation (epigenetic directing and functional reactivation) overcomes intrinsic resistance in GSDMD-low tumors while avoiding off-target pyroptosis risks, ensuring robust pyroptosis even in challenging models.
Beyond pyroptosis, our platform ingeniously overcomes a central paradox in cuproptosis therapy. While copper ions are potent inducers of cuproptosis, their efficacy is severely impeded by the cell’s sophisticated homeostatic mechanisms that minimize free copper flux.26 However, our design resolves this by exploiting macropinocytosis—a rampant endocytic pathway in cancer cells—to deliver copper as a coordinated polymer nanoparticle. As quantitatively demonstrated by our earlier ICP-MS uptake assays (Fig. 4l), the encapsulation of copper within a coordination polymer framework shifts the primary cellular entry pathway from regulated, low-capacity transporter-mediated uptake to efficient, nonspecific macropinocytosis. This paradigm shift in delivery mechanics was evidenced by the dramatically enhanced cellular copper accumulation achieved by all our nanoformulations (regardless of Cu–DHN, Cu–juglone, and Cu–Cys) compared to unbound CuCl2. This strategy bypassed physiological copper regulators and ensured a massive, uncontrolled intracellular delivery of copper.
Since macropinocytosis is a gateway that is particularly active in nutrient-demanding cancer cells, the preactivated Cu⁺ species within the Cu–DHN particle readily engage the cuproptosis pathway, leading to the marked downregulation of FDX1 and LIAS (Fig. 4m, Supplementary Fig. S25c). Thus, the same cellular uptake mechanism that underlies its dominant pyroptosis effect also empowers a potent (Supplementary Figs. 23c, d, 24a, b), synergistic cuproptosis, making Cu–DHN a comprehensive cell death inducer. Rescue experiments demonstrated that the contribution of cuproptosis is secondary to that of pyroptosis. Copper sequestration by the copper chelator TTM illustrated the central role of Cu–DHN as a motor of cuproptosis (Supplementary Fig. 25). Apart from pyroptosis and cuproptosis, Cu–DHN triggered lipid peroxidation by depleting GSH and disabling GPX4, initiating ferroptosis (Fig. 4n, o). The MDA concentration differentiated the GSH-Px-dependent ROS storm from the general oxidative attack. However, rescue experiments confirmed that the contribution of ferroptosis was subdominant (Supplementary Fig. 26), as its inhibition failed to fully reverse cell death.
As comprehensively detailed in Fig. 4, Cu–DHN elicited potent immunogenic cell death in 4T1 cells by synchronously activating pyroptotic, cuproptotic, and ferroptotic pathways. Flow cytometry analysis demonstrated that Cu–DHN induced a superior immunogenic cell death (ICD) effect, with CRT exposure reaching ~90% (Fig. 4p, Supplementary Fig. 23). This effect was complemented by HMGB-1 and ATP secretion levels comparable to those of the parent drug Cu–juglone, all of which were significantly higher than the levels observed in other control groups. Through massive TAA and DAMP release (Fig. 4q, r), the endogenous cancer vaccine formation mechanism revealed Cu–DHN as a critical ICD inducer comparable to Cu–juglone, showing 2-fold higher DC maturation rates than other control groups (Fig. 4s, Supplementary Fig. 28) and effectively priming a tumor-specific T-cell response. Notably, the DC maturation induced by pyroptosis-primed cells was 2-fold higher than that from cells undergoing only cuproptosis/ferroptosis, underscoring pyroptosis as the principal driver of ICD.
In vivo antitumor efficacy and mechanism of Cu–DHN
To validate that our nanozyme prodrug Cu–DHN executes its designed therapeutic mechanisms within the complex tumor microenvironment, we first employed an orthotopic 4T1 breast cancer model with local administration (Fig. 5a). Cu–DHN treatment significantly suppressed orthotopic tumor growth over 14 days, demonstrating comparable inhibition to the Cu–juglone group and superior to all controls (Fig. 5b, Supplementary Fig. 29). This potent antitumor activity of Cu–DHN translated into a 75% tumor growth inhibition (TGI) (Fig. 5c,d), significantly reduced tumor weights, and a notable survival benefit. The potent antitumor activity translated into significant survival prolongation in Cu–DHN-treated tumor-bearing mice relative to control groups (Fig. 5e). Critically, the Mg–DHN control, lacking catalytic activity, was indistinguishable from the PBS group, underscoring that the therapeutic effect is contingent on metal-dependent catalysis. Furthermore, the mixture of free CuCl2 + DHN showed minimal efficacy, attributable to its inadequate cellular internalization and insufficient ROS production for prodrug activation, highlighting the indispensable role of nanocoordination in ensuring codelivery and activation.

In vivo effects of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2) on 4T1 tumor. a Schematic illustration of inhibiting tumor growth via inducing pyroptosis, cuproptosis, and ferroptosis by a single intratumoral injection of Cu–DHN. b Longitudinal monitoring of tumor growth following single-dose Cu–DHN intratumoral administration (3.78 μmol kg−1 Cu, 6.24 μmol kg−1 DHN or juglone) (n = 5). c Weights of dissected tumors, and d tumor inhibition rate at 14 days post-treatment (n = 5). e Survival rate analysis of 4T1 tumor-bearing BALB/c mice treated with Cu–DHN (n = 8). f CLSM images and MFI of tumor slices labeled with g DHE (1 day post-treatment), h GSDMD-N antibody (14 days post-treatment), i FDX1 antibody (14 days post-treatment), j Liperfluo (1 day post-treatment), and H&E (14 days post-treatment) (n = 3). Data are represented as mean ± SD
Histopathological analysis of excised tumors revealed the molecular basis for Cu–DHN’s efficacy (Fig. 5f–j): a concerted attack via multiple programmed cell death pathways. We observed (i) the execution of pyroptosis, evidenced by upregulated GSDMD-N; (ii) the induction of cuproptosis, marked by downregulated FDX1; and (iii) the triggering of ferroptosis, demonstrated by elevated lipid peroxidation (LPO) levels. This multimodal cell death signature, corroborated by the marked reduction in tumor cellularity in H&E-stained cells, confirms that Cu–DHN’s catalytic activity successfully translates into a powerful and complementary therapeutic outcome in vivo.
Thus far, local administration of Cu–DHN has conclusively validated our design principle: the nanozyme efficiently generates •OH in the tumor milieu, activates the DHN-to-juglone conversion, and subsequently unleashes a potent, synergistic combination of pyroptosis, cuproptosis, and ferroptosis. The fact that Cu–DHN, a stable prodrug, matches the efficacy of the preformed toxic agent (Cu–Juglone) is definitive proof of its successful in vivo activation and mechanistic fidelity.
This robust foundational efficacy and mechanism validation upon local delivery strongly supports its further investigation under more clinically relevant, systemic administration routes. We transitioned from local (intratumoral) administration to a clinically relevant systemic (intravenous) delivery model (Supplementary Fig. 30a). Our results demonstrate that low-dose Cu–DHN exhibited relatively weak tumor growth inhibition, whereas moderate- and high-dose Cu–DHN significantly suppressed tumor progression. Moreover, compared to the high-dose group, the moderate-dose regimen did not cause significant body weight loss in tumor-bearing mice. Thus, moderate-dose Cu–DHN demonstrates both potent antitumor efficacy and favorable biosafety. (Supplementary Fig. 30b, c).
Pharmacokinetic evaluation revealed that moderate-dose Cu–DHN achieves a serum half-life of 2.01 h (Supplementary Fig. 30d), a performance closely comparable to PEGylated nanomaterials as reported. Biodistribution analysis confirmed the success of this strategy, showing significant tumor enrichment of Cu–DHN, peaking at 18.37% ± 1.89% ID g−1 at 12 h postinjection (Supplementary Fig. 30e).
The cornerstone of clinical translation is systemic safety. Beyond direct cancer cell killing, Cu–DHN orchestrated a potent systemic immune response. ELISA analyses revealed marked elevations in key antitumor cytokines, including TNF-α and IFN-γ, which peaked between day 3 and day 7 posttreatment. The subsequent slight decline reflects a natural and desirable immune regulatory process, preventing potential immune-related adverse events (irAEs) and indicating well-controlled activation (Supplementary Fig. 30f, g). This transient cytokine profile underscores Cu–DHN’s ability to ignite a powerful yet self-limiting immune response. Moreover, no statistically significant differences were observed in hepatorenal function markers, platelet indices, or body weight compared to the PBS control group (Supplementary Fig. 30h). This superior safety was a direct result of our “chemical triggering” design.
Inhibition of distant tumor growth via the ICD effect elicited by Cu–DHN
Pyroptosis, characterized by rapid execution and substantial release of DAMPs and TAAs, prompted us to investigate systemic antitumor immunity in Cu–DHN-treated tumor-bearing mice. Primary orthotopic breast tumors were established through 4T1 cell inoculation in the left mammary fat pad of 6-week-old female BALB/c mice, followed by intratumoral Cu–DHN administration when tumors reached ~200 mm3. Distant orthotopic tumors were induced in the contralateral mammary fat pad 24 h posttreatment (Fig. 6a). CRT labeling of primary tumors revealed significantly increased membrane exposure in Cu–DHN-treated groups (Fig. 6b, Supplementary Fig. 31), indicating effective immunogenic cell death (ICD) and in situ vaccination. Subsequent monitoring demonstrated marked suppression of distant tumor growth in Cu–DHN-treated mice, with complete tumor prevention observed in one subject over 22 days (Fig. 6c, Supplementary Fig. 32). The abscopal antitumor efficacy of Cu–DHN paralleled that of Cu–juglone prototypes, confirming comparable activation of ICD-mediated TAA release and systemic antitumor immunity. In contrast, the Mg–DHN and free CuCl2 + DHN groups showed negligible distant tumor control due to insufficient ICD induction, as evidenced by terminal tumor weight measurements (Fig. 6d). Mechanistic investigations combining flow cytometry and ELISA revealed multimodal immunostimulatory effects of Cu–DHN. Enhanced maturation of DCs in primary tumor-draining lymph nodes (TDLNs) (Fig. 6e, Supplementary Fig. 33), elevated CD8+ T-cell infiltration (although not CD4+ T cells) in distant tumors (Fig. 6f,g, Supplementary Figs. 34, 35), and significantly increased intratumoral TNF-α and IFN-γ levels (Fig. 6h, i). Antibody-mediated T lymphocyte subset depletion experiments established CD8+ T lymphocytes as the principal effectors of systemic immunity (Fig. 6j), with anti-CD8α antibody treatment dramatically abrogating therapeutic efficacy, while CD4+ depletion showed a negligible significant impact (Fig. 6k, Supplementary Fig. 36). These findings collectively demonstrate that intratumoral Cu–DHN administration enables site-specific activation of juglone derivatives, initiates in situ vaccination through ICD-mediated TAA release, promotes DC maturation, and orchestrates CD8+ T-cell-dependent systemic antitumor immunity.

In vivo evaluation of the abscopal effect of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2). a Schematic illustration of inhibiting distant tumor growth via the abscopal effect of Cu–DHN. b CLSM images of tumor slices labeled with AF488-CRT antibody at 4 days post-treatment. c Longitudinal monitoring of distant tumor growth in BALB/c mice after different treatments (n = 5). d Weights of dissected tumors at 22 days post-treatment (n = 5). e Quantitative matured DCs in primary tumor-draining lymph nodes (TDLNs) (n = 4). The percentage of CD8+ T cells in f CD3+ T cells and g all cells harvested from distant tumors (n = 4). h TNF-α and i IFN-γ contents in distant tumors (n = 4). j Experimental design schematic for investigating CD4+ and CD8+ T cell roles in Cu–DHN-triggered systemic antitumor immunity. k Distant tumor growth curves of 4T1 tumor-bearing mice after Cu–DHN administration along with lymphocyte depletion (n = 5). Data are represented as mean ± SD
Inhibition of pulmonary metastasis via the systemic immune response evoked by Cu–DHN
The highly aggressive pulmonary metastatic propensity of 4T1 mammary carcinoma substantially compromises survival outcomes in tumor-bearing murine models.1 To evaluate the antimetastatic ability of Cu–DHN, orthotopic 4T1 breast cancer models were established in 6-week-old female BALB/c mice through left mammary fat pad inoculation. Upon reaching tumor volumes of ~200 mm3, intratumoral administration of Cu–DHN was performed, followed by intravenous challenge with 4T1 cells 24 h post-treatment to establish pulmonary metastasis (Fig. 7a). Following 14 days of therapeutic intervention, quantitative analysis revealed that Cu–DHN treatment significantly reduced pulmonary metastasis burden, as evidenced by an ~50% decrease in lung wet weight and a >90% reduction in metastatic nodules compared to PBS controls (Fig. 7b–d). This antimetastatic efficacy translated into significant survival prolongation, with Cu–DHN-treated cohorts exhibiting a prolonged survival rate at the experimental endpoint comparable to Cu–juglone prototypes (Fig. 7e).

In vivo evaluation of the anti-metastasis effect of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2). a Schematic illustration of the mechanism by which Cu–DHN inhibits lung metastasis through systemic antitumor immunity induction. b Lung wet weight and c number of pulmonary metastatic nodules quantified 14 days post-treatment (n = 5). d Representative digital images of lung tissues and histological sections obtained at 14 days post-treatment (the red circles indicate metastatic pulmonary nodules). e Survival rate analysis of 4T1 tumor-bearing mice with pulmonary metastasis following Cu–DHN treatment (n = 8). Quantitative CD8+ T cells in f CD3+ T cells and g all cells obtained from primary tumor-draining lymph nodes (TDLNs) from mice at 3 days post-treatment (n = 3). h TNF-α and (i) IFN-γ contents in serum from mice at 3 days post-treatment (n = 4). j Lung wet weight and metastatic nodule enumeration at 14 days post Cu–DHN administration with concomitant lymphocyte depletion (n = 5). k Survival rate analysis of mice with lung metastasis of 4T1 tumors after Cu–DHN treatment along with lymphocyte depletion (n = 8). Data are represented as mean ± SD
Mechanistic investigations demonstrated the unique capacity of Cu–DHN to convert primary tumors into in situ vaccines, as evidenced by a significant increase in primary tumor-draining lymph node CD8+ T-cell infiltration (Fig. 7f, g, Supplementary Fig. 37) and 5-fold and 3-fold elevation in plasma TNF-α and IFN-γ levels versus the PBS group (Fig. 7h, i). Depletion studies also confirmed CD8+ T cells as pivotal mediators (Fig. 7j, k), with anti-CD8α antibody administration nullifying therapeutic effects (12-fold increase in metastatic nodules), while CD4+ cell depletion showed no significant impact (Supplementary Fig. 38). Notably, Cu–DHN exhibited comparable antimetastatic potency and immunostimulatory effects to Cu–juglone prototypes, attributable to its superior POD activity enabling sustained ROS generation and efficient DHN-to-juglone conversion. This multimodal mechanism synergistically enhanced systemic antitumor immunity, effectively suppressing metastatic progression and improving host survival.
Biosafety of the Cu-DHN prodrug compared with Cu–juglone
The development of pyroptosis-based antitumor therapies faces significant challenges in clinical translation due to the ubiquitous expression of GSDM proteins in normal tissues, which may lead to off-target pyroptosis and subsequent systemic toxicity.4 Herein, we systematically investigated the biosafety profiles of the tumor-specific activatable prodrug Cu–DHN compared with its Cu–juglone prototypes in 4T1 tumor-bearing BALB/c mice (Fig. 8a). Hematological analysis, blood biochemistry, body weight monitoring, and histopathological examination of major organs revealed that Cu–juglone treatment induced severe hepatorenal dysfunction, manifested through abnormal biochemical markers (Fig. 8b–g), characteristic glomerular swelling (Fig. 8j), and thrombocytopenia with reduced plateletcrit (Fig. 8h, i), accompanied by significant body weight loss (Fig. 8k). In contrast, Cu–DHN maintained comparable antitumor efficacy while demonstrating superior biosafety, showing no statistically significant differences in hepatorenal parameters, platelet indices, or histopathological features compared to the PBS group. This safety advantage stems from the tumor-specific activation mechanism of Cu–DHN, where the prodrug remains biologically inert in normal tissues despite partial redistribution from tumor sites (~45% clearance within 4 h post-intratumoral injection, Supplementary Fig. 39). The temporal dissociation between tumor retention and activation kinetics ensures that redistributed Cu–DHN maintains its dormant state in healthy tissues, whereas the constitutive activity of Cu–juglone causes immediate off-target pyroptosis. These findings collectively establish Cu–DHN as a precision pyroptosis inducer that achieves potent local tumor eradication and systemic antitumor immunity activation while circumventing the dose-limiting toxicities associated with conventional pyroptosis-inducing agents, thereby providing a promising platform for developing safe and effective in situ tumor vaccines.

In vivo biocompatibility evaluation of Cu–DHN (Cu:DHN:Cys = 0.6:1:0.2). a Schematic illustration of investigating biosafety of Cu–DHN via biochemical assessment, hematologic analysis, histological sections,and body weight variation. b–g ALT, AST, ALP, BUN, CREA, and LDH contents in serum harvested from 4T1 tumor-bearing mice at 3 days post-treatment (n = 4). h, i Platelets content and thrombocytocrit in whole blood harvested from 4T1 tumor-bearing mice at 3 days post-treatment (n = 4). j H&E staining of histological sections of major tissues at 5 days post-treatment. k Body weight curves of 4T1 tumor-bearing mice during 14 days of Cu-DHN treatment (n = 5). Data are represented as mean ± SD
Discussion
In recent years, pyroptosis has garnered attention for its unique capacity to rapidly release TAAs and DAMPs, thereby initiating potent endogenous cancer vaccination and provoking systemic antitumor immune responses. Still, the pursuit of pyroptosis as a mechanism for in situ cancer vaccination is fundamentally challenged by a critical therapeutic dilemma: how to achieve robust and selective gasdermin activation within tumors while sparing healthy tissues. As introduced, most existing strategies are constrained by their dependency on high basal GSDM expression in tumors or by the systemic administration of caspase activators, which poses significant off-target toxicity risks due to the ubiquitous expression of gasdermin proteins. Furthermore, combinatorial approaches designed to epigenetically prime tumors for pyroptosis often grapple with asynchronous drug release and compounded systemic toxicities, impeding clinical translation. Our study directly confronts this multifaceted problem by engineering the Cu–DHN conductive coordination nanozyme prodrug system, which is designed from the ground up for tumor-specific activation and synchronous multimodal cell death induction, thereby aiming to decouple therapeutic efficacy from systemic toxicity.
Our results demonstrate that Cu–DHN successfully integrates catalytic activity, redox modulation, and prodrug activation into a single agent, representing a convergence of several advanced therapeutic concepts. The observed synergy between pyroptosis, cuproptosis, and ferroptosis aligns with the emerging paradigm that engaging multiple, complementary regulated cell death pathways can amplify immunogenic cell death (ICD) and overcome compensatory resistance mechanisms common in mono-modal therapies. Specifically, the ability of Cu–DHN to induce pyroptosis even in GSDMD-low settings via juglone-mediated epigenetic reprogramming and NLRP3 inflammasome activation addresses a key limitation of current GSDME-biased strategies. This finding expands the therapeutic scope of pyroptosis beyond tumors with high endogenous gasdermin expression. Moreover, the “AND-gate” logic requiring both elevated GSH and H2O2 for activation provides a tumor-selective rationale that is conceptually supported by prior prodrug designs targeting the tumor microenvironment (TME), yet it advances upon them by coupling this selectivity with built-in catalytic signal amplification.
The structural innovation of Cu–DHN—its electron-conductive framework—is central to its efficacy, as it maximizes reactive oxygen species (ROS) generation essential for both prodrug conversion and redox disruption. The in vitro and in vivo data robustly support this design: Cu–DHN-treated cells exhibited hallmark ICD markers (CRT, HMGB1, ATP) at levels surpassing monotherapeutic controls, confirming the hypothesized synergy. In bilateral and metastatic models, the potent abscopal effect and CD8⁺ T-cell-dependent tumor suppression validate the successful generation of systemic antitumor immunity, a gold-standard outcome for in situ vaccination. However, a critical analysis must also consider the current boundaries of our findings. The superior efficacy was demonstrated primarily via intratumoral injection, which, while validating the proof-of-concept and minimizing systemic exposure, does not fully replicate the challenges of systemic delivery required for treating disseminated disease. Additionally, the long-term fate and potential immunogenicity of the copper-containing nano-assembly in vivo remain to be fully characterized. The potent feedback loop of ROS generation, while therapeutic, also necessitates caution, as dysregulated oxidative stress could theoretically foster an adaptive pro-tumorigenic environment in certain contexts.
The Cu–DHN platform opens several promising avenues for targeted therapy and research. Its core mechanism suggests immediate potential for local-regional treatment of accessible tumors, acting as a potent immunoadjuvant. For broader application, future work must prioritize engineering systemic delivery versions. This could involve conferring active targeting (e.g., via ligand conjugation) or leveraging physiological targeting (e.g., EPR effect enhancement) to achieve adequate tumor accumulation. Furthermore, the multimodal cell death signature of Cu–DHN presents a compelling rationale for combination therapies. It could be synergistically paired with immune checkpoint inhibitors to reverse T-cell exhaustion, with radiotherapy to amplify local ICD, or with targeted therapies to tackle heterogeneous tumor cell populations. From a translational research perspective, identifying predictive biomarkers—such as tumoral H₂O₂ flux, GSH levels, or NLRP3 status—will be crucial for patient stratification. Finally, a detailed investigation into the pharmacokinetics, biodegradation, and long-term biosafety of the nanozyme components is an indispensable next step toward clinical development.
In conclusion, we have developed and validated Cu–DHN, a first-in-class conductive coordination nanozyme prodrug that directly addresses the major limitations of selectivity and safety in tumor pyroptosis induction. By ingeniously combining tumor microenvironment-responsive activation, an electron-conductive structure for catalytic amplification, and a single-agent mechanism that synchronously triggers pyroptosis, cuproptosis, and ferroptosis, Cu–DHN transforms localized tumor ablation into a potent systemic immune response. This study not only provides a novel and effective nano-platform for cancer immunotherapy but also establishes a new design paradigm for spatiotemporally controlled induction of synergistic cell death, offering a promising pathway toward safer and more effective precision oncology strategies.
Materials and methods
Chemicals and reagents
Catalase (CAT) activity assay kit (Beijing Boxbio Science & Technology Co., Ltd., Beijing, China), 3,3’,5,5’-tetramethylbenzidine (TMB, Shandong Sparkjade Biotechnology Co., Ltd., China), glutathione peroxidase (GPX) activity assay kit (Solarbio Science & Technology Co., Ltd., Beijing, China), cell counting kit (CCK, FC101, TransGen Biotech, China), fetal bovine serum (FBS-303, Inner Mongolia Jinyuankang Biotechnology Co., Ltd., China), mouse interleukin 1β (IL-1β, 98024ES48, Yeasen Biotechnology Co., Ltd., Shanghai, China), mouse gasdermin D (GSDMD) ELISA kit and mouse N-terminal fragment of GSDMD (GSDMD-N) ELISA kit (Jiangsu Meimian Industrial Co., Ltd., China), mouse interferon-gamma (IFN-γ) ELISA kit (Shanghai Jonlnbio Industrial Co., Ltd., China), mouse tumor necrosis factor-α (TNF-α) ELISA kit (abs520010-96T, Absin Bioscience Inc., Shanghai, China), one-step SDS‒PAGE gel kit (LABLEAD Inc., Beijing, China), antifade mounting medium (KGA1529-25, KeygenBioTECH, Nanjing, China). Caspase-1 activity assay kit (KTA3020, Abbkine Inc., Wuhan, China). NOD-like receptor family pyrin domain containing 3 (NLRP-3) ELISA kit (Bioroyee Biotechnology Inc., Beijing, China). Mouse ferredoxin1 (FDX1) ELISA kit and mouse high mobility group protein B1 (HMGB1) ELISA kit (Wellbio Biotechnology Co., Ltd., China). Nigericine (free acid, powder), the pancaspase inhibitor z-VAD-fmk, Caspase-1 specific inhibitor Ac-YVAD-cmk, and dynasore (Acmec Biochemical Technology Co. Ltd., Shanghai, China) were used. Pitstop 2 (Yuanye Bio Co., Ltd., Shanghai, China). MβCD (Innochem-Beijing Technology Co. Ltd., China).
Synthesis of the conductive coordination nanozyme prodrug Cu–DHN
A conductive Cu–DHN coordination nanozyme prodrug was prepared based on the coordination interaction between Cu(II) ions and phenolic hydroxyl groups of DHN. Briefly, DHN was dissolved in DMSO to prepare a 10 mg mL–1 stock solution. A total of 800 μL of the DHN stock solution was added to 17 mL of DI water and thoroughly mixed. The mixture was stirred at 750 rpm on a magnetic stirrer, and 0.1 M NaOH aqueous solution was added dropwise to adjust the pH to 8.0, creating a weakly alkaline environment to facilitate the ionization of phenolic hydroxyl groups of DHN, which is therefore favorable for the coordination reaction. Subsequently, 5 mg mL−1 aqueous CuCl2 solution was added dropwise at a rate of 20 μL s−1, and continuous stirring was performed at room temperature for 3 h to facilitate the coordination assembly of Cu(II) ions with DHN. To obtain Cu–DHN with a tunable particle size, nontoxic L-Cys was introduced as a modulator into the reaction system (since L-Cys has only one sulfhydryl group that can coordinate with Cu(II) ions,41 and the Cu–S bond formed is significantly stronger than the Cu–O bond formed between Cu(II) ions and DHN, its coordination with Cu(II) ions will terminate subsequent coordination reactions. Therefore, the particle size of Cu–DHN can be adjusted by regulating the feeding ratio of L-Cys).42,43 Briefly, the reaction was extended for an additional hour by adding different volumes of 5 mg mL−1 aqueous L-Cys solution at a rate of 20 μL s−1 under an argon atmosphere to tailor the particle size of Cu–DHN. Upon completion of the reaction, the raw Cu–DHN nanoparticles at different Cu:DHN:Cys ratios were fractured using a probe sonicator (Scientz instrument, 360 W, 2 s on/3 s off, 3 mm probe diameter) for 30 min to ensure uniform distribution and consistent particle size. The product was then purified by high-speed centrifugation (26,000 rpm, 30 min, repeated three times), redispersed in 2 mL of PBS buffer (pH 7.4) to maintain stability, and stored at 4 °C for subsequent experiments. LIAS antibody (HY-P81072, MedChemExpress MCE, Shanghai, China)
Characterization
The crystallinity of Cu–DHN with various compositions was characterized by a powder X-ray diffractometer (XRD, D-POWER, GKINST Co., Ltd.). X-ray photoelectron spectra of Cu–DHN were measured by X-ray photoelectron spectroscopy (XPS, Axis Ultra DLD Kratos AXIS SUPRA) supported by Beijing Zhongkebaice Technology Service Co., Ltd. (www.zkbaice.cn). The surface area and pore volume were measured via the BET method (Micromeritics Tristar II 3020) by Ms. Kexin Zheng from Scientific Compass (www.shiyanjia.com). The XAFS spectra of these standard samples (CuO, Cu2O, CuS, and Cu2S) were recorded in transmission mode. The spectra were processed and analyzed by the software codes Athena and Artemis. The EXAFS fitting was performed using the Artemis package of Demeter (0.9.26).
POD activity of Cu–DHN
Hydroxyl radicals (•OH) were detected using TMB as a chromogenic substrate.44 Briefly, Cu–DHN ([Cu] = 1.0 mM) at different Cu:DHN:Cys ratios was mixed with GSH (1.0 mM) in PBS (pH 6.4) and incubated for 10 min. H2O2 (10 mM) was added, followed by 1 mM 3,3’,5,5’-tetramethylbenzidine (TMB, Shandong Sparkjade Biotechnology Co., Ltd.) in a quartz cuvette. The absorption spectrum was measured from 500 to 700 nm, with the increase in absorbance at 640 nm indicating •OH production. The kinetics of •OH from Cu–DHN reacting with varying concentrations of H2O2 were quantified using a microplate reader. Parameters such as Vmax and Km were calculated from the Hill equation to compare the POD activity of Cu–DHN at different Cu:DHN:Cys ratios.
Conversion efficiency of DHN to juglone
The conversion efficiency of Cu–DHN in oxidizing DHN by •OH to juglone was investigated using UV‒Vis absorption spectroscopy.45 Free DHN (100 μg mL−1) was added to Cu–DHN and Cu–Cys (0.5 mM Cu ion concentration), along with GSH (0.5 mM) and H2O2 (5.0 mM). Samples were collected at different time points and centrifuged at 26,000 rpm for 30 min, and the absorbance of the supernatant at 425 nm was measured. The results were compared against a juglone standard curve to calculate juglone conversion.
Intracellular ROS generation in 4T1 cells
4T1 cells were seeded at 3 × 105 cells per dish in a glass-bottom confocal dish and cultured overnight at 37 °C in 5% CO2. After cell attachment, the medium was removed, and the cells were rinsed with precooled PBS once. Then, medium containing Cu–DHN and control materials ([Cu] = 0.020 mM, [DHN] = 0.033 mM) was added, and the cells were incubated for another 6 h. After incubation, the medium was aspirated, and the cells were washed three times with precooled PBS. Before detection, the cells were incubated with 0.5 mL of 10 μM DCFH-DA solution for 30 min at 37 °C. Cells were then fixed with 4% paraformaldehyde for 15 min. After washing with PBS, the cells were stained with 10 μg/mL DAPI at 37 °C for 10 min, followed by six washes with precooled PBS. Intracellular ROS production was measured using confocal laser scanning microscopy (CLSM), with nuclei visualized by DAPI staining.
Cytotoxicity of Cu–DHN
4T1 cells were seeded at a density of 1 × 104 cells per well in 96-well plates. The cells were cultured overnight at 37 °C to allow adherence. After 12 h, the medium was replaced with that containing Cu–DHN and control materials ([Cu] = 0.020 mM, [DHN] = 0.033 mM) and incubated for an additional 24 h. Then, the culture medium was discarded, and the cells were washed gently with PBS (equilibrated at room temperature) to remove residual Cu–DHN and control materials. Afterwards, 100 µL of culture medium containing 10% CCK-8 was added to each well. The plates were incubated at 37 °C for 1 h. Absorbance was measured at a wavelength of 490 nm using a microplate reader.
To verify membrane rupture, 4T1 cells were seeded in glass-bottom confocal dishes and incubated overnight for adhesion. Cu–DHN ([Cu] = 0.020 mM, [DHN] = 0.033 mM) was added and incubated for 5 h. The surface of treated cells was rinsed with 0.1 M PBS three times and stained with Calcein AM/PI solution for 15 min at 37 °C. At the end of incubation, the staining solution was aspirated, and the cells were rinsed three times with 0.1 M PBS. The medium was replaced with serum-free, phenol red-free RPMI 1640, and the cells were observed under a Leica Thunder Imager.
Intracellular caspase-1 activity in 4T1 cells after Cu–DHN treatment
Caspase-1 activity in 4T1 cells treated with Cu–DHN and control materials was assessed using the Ac-YVAD-pNA assay from a caspase-1 activity assay kit. 4T1 cells (1 × 107) were seeded in 15 cm Petri dishes and incubated overnight at 37 °C in 5% CO2. After 12 h, the medium was replaced with that containing Cu–DHN and control materials ([Cu] = 0.020 mM, [DHN] = 0.033 mM) and incubated for an additional 18 h. Following incubation, the cells were washed with precooled PBS and collected for analysis. A total of 4 × 106 cells were centrifuged (1500 rpm, 5 min) to isolate the pellet, which was resuspended in 50 µL of cell lysis buffer containing DTT and lysed on ice for 30 min. After lysis, the cells were centrifuged at 4 °C (12,000 rpm, 5 min), and 50 μL of the supernatant was mixed with 50 μL of reaction buffer and 5 μL of Ac-YVAD-pNA (4 mM). This mixture was incubated for 2 h at 37 °C. The absorbance was measured at 405 nm using a microplate reader. Caspase-1 activity was calculated using the formula provided by the assay kit. To support pyroptosis as the mechanism, rescue experiments were conducted with the caspase-1 inhibitor Ac-YVAD-cmk before incubation with Cu–DHN commenced. The extent of IL-1β secretion, GSDMD cleavage, and cell viability restoration was evaluated in each group, with or without the protection of Ac-YVAD-cmk.
Intracellular GSDMD-FL and GSDMD-N contents in 4T1 cells after Cu–DHN treatment
To assess the effect of Cu–DHN and its control material on GSDMD-FL and GSDMD-N protein levels in 4T1 cells, ELISA kits specific to these proteins were utilized. 4T1 cells (1 × 107) were seeded in 15 cm Petri dishes and incubated overnight at 37 °C in 5% CO2. After 12 h, the medium was replaced with that containing Cu–DHN and control materials ([Cu] = 0.020 mM, [DHN] = 0.033 mM) and incubated for an additional 18 h. Following incubation, the cells were washed with precooled PBS and collected for analysis. A total of 4 × 106 cells were centrifuged (1500 rpm, 5 min) and resuspended in 1 mL extract. Cells were disrupted by sonication on ice (300 W, 2 s on/5 s off, total 3 min) and centrifuged at 4 °C (12,000 rpm, 5 min). A 5-fold dilution was prepared by adding 10 μL of the supernatant to 40 μL of diluent. Finally, GSDMD-FL and GSDMD-N protein levels were measured using the respective ELISA kits following the manufacturer’s instructions. Moreover, the intracellular GSDMD-FL and GSDMD-N protein levels were also measured using Western blotting.
Expression of FDX1 and LIAS in 4T1 cells after Cu-DHN treatment
4T1 cells (1 × 107) were seeded in 15 cm Petri dishes and incubated overnight at 37 °C in 5% CO2. After 12 h, the medium was replaced with that containing Cu–DHN and control materials ([Cu] = 0.020 mM, [DHN] = 0.033 mM) and incubated for an additional 18 h. Following incubation, the cells were washed with precooled PBS and collected for analysis. A total of 4 × 106 cells were centrifuged (1500 rpm, 5 min) and resuspended in 1 mL extract. Cells were disrupted by sonication on ice (300 W, 2 s on/5 s off, total 3 min) and centrifuged at 4 °C (12,000 rpm, 5 min). The processed samples were tested according to the manufacturer’s instructions for the FDX1 ELISA kits. The expression level of LIAS was measured by Western blotting. Through these steps, the impact of Cu–DHN on copper uptake in tumor cells and the expression of FDX1 and LIAS was comprehensively evaluated, supporting the efficient induction of tumor cuproptosis.
LPO level analysis of 4T1 cells after Cu–DHN treatment
To evaluate ferroptosis in 4T1 cells induced by Cu–DHN, LPO was assessed using a Liperfluro probe. 4T1 cells were seeded at a density of 3 × 105 cells per glass-bottom confocal dish and cultured overnight at 37 °C in 5% CO2. Following cell attachment, the medium was removed, and the cells were rinsed with precooled PBS. Medium containing Cu–DHN and control materials ([Cu] = 0.020 mM, [DHN] = 0.033 mM) was added, and the cells were incubated for 12 h. By the end of incubation, the medium was aspirated, and the cells were washed three times with precooled PBS. Then, 0.5 mL of 1.0 μM Liperfluro solution was added and incubated for 30 min at 37 °C. Cells were fixed with 4% paraformaldehyde for 15 min. After washing three times with room temperature PBS, the cells were stained with 10 μg mL−1 DAPI for 10 minutes at 37 °C and washed six times with precooled PBS. LPO levels were assessed by detecting green fluorescence from the Liperfluro dye using CLSM, with nuclei visualized under the DAPI channel. This step enabled accurate assessment of the effects of Cu–DHN and control materials on LPO levels, providing insights into the level of ferroptosis in 4T1 cells. To elaborate on the exact source of lipid peroxidation, rescue experiments using ferroptosis-specific inhibitors ferrostatin-1 (Fer-1) or the iron chelator deferoxamine (DFO) prior to Cu–DHN exposure were carried out. Cell viability was determined by CCK-8 assay. BODIPY 581/591 C11 staining was employed to reveal the potential remedial outcomes of Fer-1 and DFO on Cu–DHN-induced lipid peroxidation and distinguish from a general oxidative stress.
In vitro detection of DC maturation
For DC maturation, bone marrow-derived DCs (BMDCs) were isolated and cultured to assess their maturation in response to DAMPs released by Cu–DHN. Mice were euthanized, and their tibias and femurs were sterilized with 75% ethanol. Bone marrow was flushed from the cavity using serum-free RPMI 1640 medium and filtered through a 200-mesh screen. After centrifugation (1800 rpm, 5 min), the supernatant was discarded, and the cell pellet was resuspended in 2 mL red blood cell lysis buffer for 2 min. The cells were then resuspended in X-vivo 15 medium supplemented with 10 ng mL−1 IL-4 and 10 ng mL−1 GM-CSF and seeded in 10 cm Petri dishes at 2 × 106 cells per dish. The medium was replaced every 3 days with fresh medium containing 20 ng mL−1 IL-4 and GM-CSF. The cells were cultured at 37 °C in 5% CO2 for 6 days to generate immature dendritic cells (DCs). Immature DCs were seeded at 6 × 105 cells per well in six-well plates and incubated with 3 × 105 4T1 cells treated with Cu–DHN or control materials for 24 h. After incubation, cells were harvested and stained with anti-CD11c, anti-CD80, and anti-CD86 antibodies for 30 min. Maturation was assessed by flow cytometry, identifying the mature DCs as those expressing all three markers (CD11c, CD80, and CD86).
Animal ethics approval
Six-week-old female BALB/c mice and male BALB/c mice were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. and housed in SPF animal rooms. Animal experiments were conducted in accordance with the guidelines formulated by the Animal Management Committee of the National Center for Nanoscience and Technology, University of Chinese Academy of Sciences (Approval number: NCNST-LX-2203-27).
In vivo antitumor efficacy of Cu–DHN on orthotopic 4T1 tumors
Six-week-old BALB/c female mice were inoculated with 1 × 106 4T1 cells in the left breast pad of the abdomen. When the tumor volume grew to approximately 200 mm,3 BALB/c mice were intratumorally injected with Cu–DHN and different control materials (injection dose: 3.78 μmol kg−1 Cu, 6.24 μmol kg−1 DHN, or juglone). The tumor volume and body weight of the mice were measured every other day. When the mice died naturally or the tumor volume reached 1500 mm,3 the mice were recorded as dead. On day 14 after administration, all mice were euthanized, and the tumors were isolated for weighing. Moreover, these tumor tissues will be processed into histological sections and labeled with GSDMD-N and FDX1 antibodies to evaluate the extent of pyroptosis/cuproptosis occurring in the tumors. The survival curve of tumor-bearing mice was plotted to further evaluate the therapeutic effect of Cu–DHN. To address pharmacokinetics, serum stability, and biodistribution, Cu–DHN was delivered via intravenous injection at three dosage levels (low: 3.00 μmol kg−1 Cu, 4.95 μmol kg−1 DHN; moderate: 6.00 μmol kg−1 Cu, 9.90 μmol kg−1 DHN; high: 9.00 μmol kg−1 Cu, 14.86 μmol kg−1 DHN). Routine blood tests and quantification of copper levels in the main organs by ICP-MS were conducted to determine the systemic fate of Cu–DHN.
In vivo antitumor abscopal effect and antimetastatic ability of Cu–DHN
A bilateral 4T1 tumor model was established to evaluate the antitumor abscopal effect induced by Cu–DHN. BALB/c female mice were inoculated with 1 × 106 4T1 cells in the left breast pad of the abdomen to establish primary tumors. When the tumor volume grew to ~200 mm,3 BALB/c mice were intratumorally injected with Cu–DHN and different control materials (injection dose: 3.78 μmol/kg Cu, 6.24 μmol/kg DHN, or juglone). After 24 h, distant tumors were established by inoculating 8 × 105 4T1 cells into the right breast pad of the abdomen. The tumor volume of the mice was measured every other day. At the end of the experiment, all mice were euthanized, and the tumors were isolated for weighing. Moreover, the primary tumors were excised and prepared into histological sections and labeled with AF488-CRT antibody to evaluate the in vivo ICD effect using CLSM, followed by 72 h post administration of Cu-DHN.
A pulmonary metastasis model was established to evaluate the antimetastatic ability of Cu–DHN. BALB/c female mice were inoculated with 1 × 106 4T1 cells in the left breast pad of the abdomen to establish primary tumors. When the tumor volume grew to approximately 200 mm, BALB/c mice were intratumorally injected with Cu-DHN and different control materials (injection dose: 3.78 μmol/kg Cu, 6.24 μmol/kg DHN, or juglone). After 24 h, the pulmonary metastasis model was established by intravenous (i.v.) injection of 2 × 105 4T1 cells. The time-dependent profiles of serum TNF-α and IFN-γ were monitored over the 14-day therapeutic period. At 14 days post-administration, the pulmonary tissues were harvested for weighing and counting the number of pulmonary metastasis nodules. Moreover, the pulmonary tissues were prepared into histological sections and stained with H&E for microscopic evaluation of pulmonary metastasis nodular lesions. The survival curve of pulmonary metastasis-bearing mice was plotted to further evaluate the therapeutic effect of Cu–DHN.
Statistical analysis
Quantitative data were calculated and processed as the mean ± SD. The experimental data were analyzed using OriginPro and GraphPad Prism 9. Significance was determined by an unpaired Student’s t-test for the comparison of two groups and one-way analysis of variance (ANOVA) for multiple groups.
Data availability
The SourceData underlying Figs. 2b, d–i, k, l, 3a–f, h–j, 4b–d, f–n, p–s, 5b–e, g–j, 6c–i, k, 7b, c, e, f–k, 8b–i, k, Supplementary Figs. 1a–h, 4, 5a, b, 6, 7a, b, 8–16, 17a, b, 18a, b, 19c, 20a, b, 21a, 23a–d, 24a, b, e, f, 25a–c, 26a–d, 27b, 29, 30b–h, 31, 32, 35, 36a, b, 38, 39, Supplementary Table 1 are provided as a Source Data file. The remaining data are available within the Article, Supplementary Information or Source Data file. Source data are provided with this paper.
References
Li, Y. X. et al. Invasion and metastasis in cancer: molecular insights and therapeutic targets. Signal Transduct. Target. Ther. 10, 57 (2025).
Zhao, H. et al. Tailoring aggregation extent of photosensitizers to boost phototherapy potency for eliciting systemic antitumor immunity. Adv. Mater. 34, 2106390 (2022).
Ma, L. et al. Immunotherapy and prevention of cancer by nanovaccines loaded with whole-cell components of tumor tissues or cells. Adv. Mater. 33, 2104849 (2021).
Liu, X., Xia, S. Y., Zhang, Z. B., Wu, H. & Lieberman, J. Channelling inflammation: gasdermins in physiology and disease. Nat. Rev. Drug Discov. 20, 384–405 (2021).
Liu, Y. F. et al. Pyroptosis in health and disease: mechanisms, regulation and clinical perspective. Signal Transduct. Target. Ther. 9, 245 (2024).
Gong, N. et al. Enhancing in situ cancer vaccines using delivery technologies. Nat. Rev. Drug Discov. 23, 607–625 (2024).
Gao, W. T., Wang, X. Y., Zhou, Y., Wang, X. Q. & Yu, Y. Autophagy, ferroptosis, pyroptosis, and necroptosis in tumor immunotherapy. Signal Transduct. Target. Ther. 7, 196 (2022).
Zhang, S. et al. Biomineralized two-enzyme nanoparticles regulate tumor glycometabolism inducing tumor cell pyroptosis and robust antitumor immunotherapy. Adv. Mater. 34, 2206851 (2022).
Wang, Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103 (2017).
Ai, Y. L. et al. Mannose antagonizes GSDME-mediated pyroptosis through AMPK activated by metabolite GlcNAc-6P. Cell Res. 33, 904–922 (2023).
Su, M. et al. Gasdermin D-dependent platelet pyroptosis exacerbates NET formation and inflammation in severe sepsis. Nat. Cardiovas. Res. 1, 732–747 (2022).
Wei, C. et al. Brain endothelial GSDMD activation mediates inflammatory BBB breakdown. Nature 629, 893–900 (2024).
Ma, F. et al. Gasdermin E dictates inflammatory responses by controlling the mode of neutrophil death. Nat. Commun. 15, 386 (2024).
Wang, Q. et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature 579, 421–426 (2020).
Wang, N. et al. A cooperative nano-CRISPR scaffold potentiates immunotherapy via activation of tumour-intrinsic pyroptosis. Nat. Commun. 14, 779 (2023).
Zhang, Q. et al. Radiofrequency-activated pyroptosis of bi-valent gold nanocluster for cancer immunotherapy. ACS Nano 17, 515–529 (2023).
Chen, B. et al. A pyroptosis nanotuner for cancer therapy. Nat. Nanotechnol. 17, 788–798 (2022).
Liang, M. Y. et al. Stepwise size shrinkage cascade-activated supramolecular prodrug boosts antitumor immunity by eliciting pyroptosis. Adv. Sci. 9, 2203353 (2022).
Wang, Y. Y. et al. Dual-responsive epigenetic inhibitor nanoprodrug combined with oncolytic virus synergistically boost cancer immunotherapy by igniting gasdermin E-mediated pyroptosis. ACS Nano 18, 20167–20180 (2024).
Munoz-Planillo, R. et al. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153 (2013).
Koo, J. et al. Gigantic porphyrinic cages. Chem 6, 3374–3384 (2020).
Sun, Q. et al. Recent advances on endogenous/exogenous stimuli-triggered nanoplatforms for enhanced chemodynamic therapy. Coord. Chem. Rev. 451, 214267 (2022).
Liu, J. et al. Copper-based metal-organic framework overcomes cancer chemoresistance through systemically disrupting dynamically balanced cellular redox homeostasis. J. Am. Chem. Soc. 144, 4799–4809 (2022).
Wang, J. et al. Biodegradable ferrous sulfide-based nanocomposites for tumor theranostics through specific intratumoral acidosis-induced metabolic symbiosis disruption. J. Am. Chem. Soc. 144, 19884–19895 (2022).
Xu, W. et al. Photoexcited Ru single-atomic sites for efficient biomimetic redox catalysis. Proc. Natl. Acad. Sci. USA 120, e2220315120 (2023).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375, 1254–1261 (2022).
Xie, L. S., Skorupskii, G. & Dinca, M. Electrically conductive metal-organic frameworks. Chem. Rev. 120, 8536–8580 (2020).
Lu, B., Zeng, L., Xu, J., Le, Z. & Rao, H. Electrosynthesis of highly conducting poly(1,5-dihydroxynaphthalene) in BF3•Et2O. Eur. Polym. J. 45, 2279–2287 (2009).
Li, T. et al. A pH-activatable copper-biomineralized proenzyme for synergistic chemodynamic/chemo-immunotherapy against aggressive cancers. Adv. Mater. 35, 2210201 (2023).
Zhang, X. et al. Creating enzyme-mimicking nanopockets in metal-organic frameworks for catalysis. Sci. Adv. 8, eadd5678 (2022).
Zhao, Y., Wang, J. & Pei, R. Micron-sized ultrathin metal-organic framework sheet. J. Am. Chem. Soc. 142, 10331–10336 (2020).
Ma, B. et al. Self-assembled copper amino acid nanoparticles for in situ glutathione “AND” H2O2 sequentially triggered chemodynamic therapy. J. Am. Chem. Soc. 141, 849–857 (2019).
Zhang, Q. et al. Bright and stable NIR-II J-aggregated AIE dibodipy-based fluorescent probe for dynamic in vivo bioimaging. Angew. Chem. Int. Ed. 60, 3967–3973 (2021).
He, T. et al. Different valence states of copper ion delivery against triple-negative breast cancer. ACS Nano 18, 5434–5445 (2024).
Mukherjee, D., Gamler, J. T. L., Skrabalak, S. E. & Unocic, R. R. Lattice strain measurement of core@shell electrocatalysts with 4D scanning transmission electron microscopy nanobeam electron diffraction. ACS Catal. 10, 5529–5541 (2020).
He, L. et al. Solvent-assisted self-assembly of a metal-organic framework based biocatalyst for cascade reaction driven photodynamic therapy. J. Am. Chem. Soc. 142, 6822–6832 (2020).
Xu, Y. et al. An enzyme-engineered nonporous copper(I) coordination polymer nanoplatform for cuproptosis-based synergistic cancer therapy. Adv. Mater. 34, 2204733 (2022).
Zhang, M. et al. Rapid glutathione analysis with SERS microneedles for deep glioblastoma tissue differentiation. Anal. Chem. 96, 10200–10209 (2024).
Wang, Y. et al. Bimetallic single-atom nanozyme-based electrochemical-photothermal dual-function portable immunoassay with smartphone imaging. Anal. Chem. 96, 13663–13671 (2024).
Li, Z. et al. Enhancing gasdermin-induced tumor pyroptosis through preventing ESCRT-dependent cell membrane repair augments antitumor immune response. Nat. Commun. 13, 6321 (2022).
Ma, B. J. et al. Self-assembled copper amino acid nanoparticles for in situ glutathione “AND” H2O2 sequentially triggered chemodynamic therapy. J. Am. Chem. Soc. 141, 849–857 (2019).
Zhang, X. F. et al. Creating enzyme-mimicking nanopockets in metal-organic frameworks for catalysis. Sci. Adv. 8, eadd5678 (2022).
Howarth, A. J. et al. Best practices for the synthesis, activation, and characterization of metal-organic frameworks. Chem. Mater. 29, 26–39 (2017).
Zhang, Y. et al. A novel CuO nanoparticle loaded DMSN@MnO2 nanozyme as an optical platform for 2-mercaptobenzothiazole assays and biotoxicity mitigation. Chem. Eng. J. 505, 159363 (2025).
Nugmanova, A. G. et al. Electric field-induced amplification of graphene oxide’s visible light photocatalytic activity. J. Mater. Chem. A 13, 200–204 (2024).
Acknowledgements
This work was supported by the National Key R&D Program of China (no. 2021YFA1200900 to Chunying Chen), New Cornerstone Science Foundation Basic Science Center Project of the National Natural Science Foundation of China (no. 22388101 to Chunying Chen), National Natural Science Foundation of China (no. 32201099 to Hao Zhao), China Postdoctoral Science Foundation (nos. 2022T150151 and 2022M720933 to Hao Zhao), Natural Science Foundation of Chongqing (no. CSTB2025NSCQ-GPX0363 to Hao Zhao), Chongqing Medical Youth Top-notch Talent (no. YXQN202575 to Hao Zhao), Chongqing Postdoctoral Retention Grant (no. 2024-llyzz01 to Hao Zhao), and National Natural Science Foundation of China (no. 12075273 to Wei Xu).
Author information
Authors and Affiliations
Contributions
Hao Zhao and Chunying Chen conceived, supported and supervised this project, and designed the experiments. Yuqiao Wang and Hao Zhao synthesized the Cu–DHN prodrug nanozymes and performed their characterization and evaluation, and conducted cell experiments. Yuqiao Wang, Hao Zhao, Ke Sun, Dan Deng, Guanyu Liu, Lan Li, Tao Liu and Lin Bao carried out in vivo antitumor experiments and related studies on immune-mediated antitumor effects. Wei Xu analyzed the synchrotron radiation data. Hao Zhao conceived and designed conceptual diagrams and guided Yuqiao Wang in using BioRender for their creation. Yuqiao Wang and Hao Zhao analyzed the data, prepared all figures based on experimental data, and wrote the manuscript. All authors have reviewed and approved the article.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Zhao, H., Sun, K. et al. Conductive coordination nanozyme prodrugs precisely trigger pyroptosis, cuproptosis and ferroptosis for in situ cancer vaccination. Sig Transduct Target Ther 11, 96 (2026). https://doi.org/10.1038/s41392-026-02607-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41392-026-02607-6
