
Abstract
Acute kidney injury (AKI) is a serious condition characterized by decreased urine output, often accompanied by psychiatric symptoms like depression. However, there are limited pharmacological treatments available for AKI and its associated depressive symptoms. In this study, we investigated whether cisplatin-induced AKI in mice leads to depression-like behaviors and whether ketamine could alleviate both the kidney injury and these behaviors. Mice with cisplatin-induced AKI exhibited elevated levels of creatinine and urea, kidney damage, increased kidney injury molecule-1 protein, and pathological changes in the liver, colon, and spleen. They also showed depression-like behaviors and reduced expression of synaptic proteins in the prefrontal cortex. Remarkably, a single dose of ketamine significantly reduced these symptoms and pathological changes. Interestingly, the beneficial effects of ketamine on the kidneys, other organs, and depression-like behaviors, were reversed by the tropomyosin receptor kinase B (TrkB) inhibitor ANA-12. Western blot analysis revealed the involvement of the TrkB and ERK (extracellular signal-regulated kinase)-CREB (cAMP response element binding protein) signaling pathway. Additionally, metabolomics analysis indicated that blood metabolites, such as C16-ceramide, may contribute to the effects of ketamine in this model. These findings suggest that cisplatin-induced nephrotoxicity in AKI mice contributes to depression-like behaviors, and ketamine can alleviate both kidney damage and depression-like symptoms by modulating the TrkB and ERK-CREB signaling pathways, as well as altering blood metabolites. However, the role of the kidney-brain axis in these depression-like behaviors remains unclear. Furthermore, ketamine may have therapeutic potential for treating kidney diseases such as AKI, along with associated depressive symptoms.
Similar content being viewed by others

Introduction
Acute kidney injury (AKI) is characterized by a rapid deterioration of renal function within hours or days, marked by significantly elevated serum levels of creatinine and blood urea nitrogen (BUN), and often presenting as oliguria or anuria in patients. AKI is prevalent, affecting an estimated one-fifth of hospitalized patients at some point [1, 2]. It is also a common complication in intensive care units (ICU), impacting 25 to 57% of ICU patients globally, with mortality rates ranging from 15 to 60% [3, 4]. AKI is often caused by renal hypoperfusion due to severe trauma, disease, or surgery. However, the detailed mechanisms remain poorly understood, and current clinical treatments are limited, necessitating the discovery of new therapeutic targets and drugs.
Kidney function impairment frequently coexists with neuropathy and psychiatric symptoms. Among patients with chronic kidney disease (CKD), numerous studies have shown that the prevalence of depressive symptoms can range from 25 to 100%, depending on diagnostic criteria and population demographics [5]. Additionally, the decline in kidney function is linked to cognitive impairments, such as Alzheimer’s disease [6]. A cohort study investigating the long-term prognosis of AKI patients revealed that 22% experienced depression [7]. However, there are limited reports on the correlations and mechanisms underlying the “kidney-brain axis,” particularly concerning the relationship between AKI and depression.
Ketamine is an anesthetic with anti-inflammatory, and antioxidant effects [8,9,10,11]. Multiple clinical studies have shown that ketamine can provide rapid and sustained antidepressant effect in patients with treatment-resistant patients with major depressive disorder (MDD) [12, 13]. Furthermore, ketamine’s therapeutic effects have been confirmed in several animal models, including Parkinson’s disease [14], multiple sclerosis [15, 16], and stroke [17]. Increasing preclinical studies suggest that signaling pathways such as brain-derived neurotrophic factor (BDNF)-tropomyosin receptor kinase B (TrkB) and ERK (extracellular signal-regulated kinase)-CREB (cAMP response element binding protein) signaling pathways play a role in the beneficial effects of ketamine [18,19,20,21,22,23,24,25,26,27,28]. However, the therapeutic effects of ketamine on AKI and AKI-induced psychiatric symptoms are not yet fully understood.
The current study has four main objectives. First, we examined whether cisplatin-induced AKI mice exhibit depression-like behaviors. Second, we investigated whether ketamine can alleviate both cisplatin-induced AKI and the associated depression-like behaviors. Third, we used the TrkB inhibitor ANA-12 to explore the role of TrkB signaling in ketamine’s mechanism of action. Additionally, we performed Western blot analysis of the TrkB and ERK–CREB signaling pathways in the prefrontal cortex (PFC) and kidneys. Finally, we conducted metabolomics analysis of serum samples, as the gut–brain axis, including metabolites, plays a role in depression-like behaviors [29, 30].
Materials and methods
Animals
Male C57BL/6 mice (8 weeks old, 20–25 g) were purchased from HuaFuKang Co., Ltd. (Beijing, China). The mice were housed in a specific-pathogen-free (SPF) animal facility, maintaining a controlled environment with temperatures set at 23 ± 1°C and a 12-h light/dark cycle (lights on from 07:00 to 19:00), and provided ad libitum access to SPF mice feed (Beijing Keao Xieli Feed Co., Ltd., Beijing, China) and water. The experimental protocol of this study was approved by the Institutional Animal Care and Use Committee of the West China Hospital, Sichuan University (Permission Number 20230807003). The animals were deeply anesthetized with isoflurane and rapidly killed by cervical dislocation. All efforts were made to minimize mice suffering.
Drugs
Cisplatin (20 mg/kg, Tokyo Chemical Industry Co., Ltd., Tokyo, Japan) was dissolved in phosphate-buffered saline (PBS). Ketamine, obtained from Shanghai Yuansi Standard Science and Technology Co., Ltd., was used with approval from the public security department of Shanghai and Chengdu, China. Ketamine (10 mg/kg) was dissolved in saline as previously reported [24, 25, 31]. ANA-12 (N-[2-[[(hexahydro-2-oxo-lH-azepin-3-yl)amino]carbonyl]phenyl]benzo[b]thiophene-2-carboxamide: 0.5 mg/kg) (MedChem Express, NJ, USA) was dissolved in PBS containing 17% dimethylsulfoxide (DMSO) in line with previous research [20, 21, 31, 32]. All other reagents were commercially sourced.
Schedule of treatment and sample collection
The cisplatin-induced acute kidney injury (AKI) model was conducted following the methodology previously described [33]. Briefly, mice were randomly divided into the following four groups: PBS + saline, cisplatin + saline, PBS + ketamine, and cisplatin + ketamine. Following a 1-week habituation period, mice received a single intraperitoneal injection of either cisplatin (20 mg/kg) or PBS (10 ml/kg). Ketamine (10 mg/kg) was administered intraperitoneally 24 h after the initial injection. The locomotion tests (LMT) were conducted 2 h post-ketamine injection on day 8. Subsequently, the forced swimming tests (FST) were performed 2 h after the LMT. The sucrose preference tests (SPT) were conducted on day 9, and mice were euthanized for collection of serum, kidney, colon and prefrontal cortex (PFC) on day 10 (Fig. 1A).

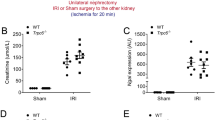
A Schedule of treatment and sample collection. B Survival curves (Log-rank test: F3,36 = 12.62, P = 0.006) and body weight (repeated-measures one-way ANOVA: F3,36 = 6.581, P = 0.002) changes of the four groups. C Levels of blood urea nitrogen (BUN) (two-way ANOVA: cisplatin: F1,36 = 87.39, P < 0.001, ketamine: F1,36 = 31.62, P < 0.001, interaction: F1,36 = 30.29, P < 0.001) and creatinine (CREA) (two-way ANOVA: cisplatin: F1,36 = 82.17, P < 0.001, ketamine: F1,36 = 29.35, P < 0.001, interaction: F1,36 = 29.12, P < 0.001) among the four groups. D Representative HE staining images of renal tissue. E Representative immunohistochemical staining of KIM-1 on renal tissue. Data represent the mean ± SEM (n = 10). *P < 0.05, **P < 0.001. KT, ketamine; LMT, locomotion test; FST, forced swimming test; SPT, sucrose preference test.
In the ANA-12 treatment experiment, cisplatin (20 mg/kg) was administered on day 7 as described above. Saline, ketamine (10 mg/kg), ANA-12 (0.5 mg/kg), or a mixture of ketamine and ANA-12, was injected intraperitoneally 24 h after the initial cisplatin injection. The groups were as follows: PBS + saline group, cisplatin + saline group, cisplatin + ketamine group, cisplatin + ketamine/ANA-12, cisplatin + ANA-12, PBS + ANA-12 group. The SPT were performed on day 9, and mice were sacrificed for collections of serum, PFC, kidneys, liver, colon, and spleen on day 10 (Fig. 3A).
Behavioral tests
Behavioral tests, including the LMT, FST, and 1% SPT were performed as previously reported [34,35,36].
Locomotion test (LMT)
The mouse was placed within experimental cages (500 mm (length) × 500 mm (width) × 350 mm (height)), and locomotor activities were recorded for 60 min. The locomotor activity was measured by an animal movement analysis system EthoVision XT 12 (Noldus Information Technology, VA, USA). The cages were cleaned between test sessions to minimize any potential confounding factors.
Forced swimming test (FST)
The mouse was placed in a cylindrical container (diameter: 15 cm; height: 25 cm) filled with water to a depth of 15 cm, maintained at a temperature of 23 ± 1 °C. Its behavior was observed and recorded for a period of 6 min. Immobility duration was quantified using the EthoVision XT 12 system (Noldus Information Technology, VA, USA) automatically.
Sucrose preference test (SPT)
Mice were exposed to both water and a 1% sucrose solution for 48 h, followed by a 4 h period of water and food deprivation. Subsequently, the animals were given unrestricted access to water and the 1% sucrose solution for 1 h. The intake of both water and the 1% sucrose solution during this hour was measured, and the proportion of sucrose solution consumed relative to the total fluid intake was calculated.
Measurement of blood urea nitrogen (BUN) and creatinine (CREA)
The serum levels of BUN and CREA were quantified utilizing an automatic biochemical analyzer (TOSHIBA, Tokyo, Japan), following the manufacturer’s instructions.
Hematoxylin-Eosin (HE) staining
HE staining of the kidneys, liver, colon, and spleen was performed as previously reported [35, 36]. Briefly, the sections were dewaxed and hydrated, rinsed in water, and then stained with hematoxylin for 5 min, followed by a 10 min rinse. The sections were fixed with a bluing reagent, rinsed again, and stained with eosin for 1-2 min. Finally, the sections were dehydrated through a series of ethanol washes, cleared in xylene, and mounted.
Immunohistochemistry
Following high-temperature and high-pressure antigenic retrieval (EDTA, pH 9.0, 95°C, 30 min), the prepared tissue sections were blocked with 3% H2O2 and washed with PBS. Next, the sections were blocked with 3% BSA for 30 min and then incubated overnight at 4 °C with primary antibody targeting KIM-1 (1:900, Cat Number: # PA5-98302, Invitrogen). The following day, after triple washing with 0.05 M Tris–HCl saline (TBS) with 0.1% Tween-20 (TBST), the sections were incubated with HRP-conjugated goat anti-rabbit IgG (1:200, Cat Number: # AS014, ABclonal) at 37 °C for 45 min, followed by another round of triple washing with TBST. Subsequent to DAB staining (Cat#DAB4033, MXB® Biotechnologies) and hematoxylin staining, the slides were washed with PBS. Dehydration and clearing of the sections were accomplished using absolute alcohol. Neutral gum was applied to cover the slices. The sections were then automatically scanned for images using a Vectra Polaris Automated Quantitative Pathology Imaging System (PerkinElmer, Waltham, MA, USA).
Western blot analysis
Western blot analysis was performed as previously reported [34,35,36,37]. Briefly, kidney and prefrontal cortex (PFC) were homogenized in Laemmli lysis buffer. Protein quantification was conducted using a BCA protein assay kit (Beyotime, Shanghai, China), and 50 micrograms of protein were heated at 95 °C for 5 min with a quarter volume of SDS–PAGE sample loading buffer. Subsequently, the proteins were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis, using 10% SurePAGE-gels (Cat Number: M00665, GenScript, Piscataway, NJ, USA). Proteins were transferred to polyvinylidene difluoride (PVDF) membranes using a Trans-Blot Mini Cell (Bio-Rad). For immunodetection, the blots were blocked with 2% BSA in TBST for 1 h at room temperature (RT) and incubated overnight at 4 °C with primary antibodies against PSD-95 (1:1000, Cat Number: 51-6900, Invitrogen, Camarillo, CA, USA), GluA1 (1:1000, Cat Number: ab31232, Abcam, Cambridge, MA, USA), TrkB (1:500, Cat Number: #AF6461, Affinity Bioscience), phospho-TrkB (1:2000, Cat Number: #AF3461, Affinity Bioscience), ERK (1:1000, Cat Number: #AF0155, Affinity Bioscience), p-ERK (1:2000, Cat Number: #AF1015, Affinity Bioscience), CREB (1:500, Cat Number: #AF6188, Affinity Bioscience), and p-CREB (1:2000, Cat Number: #AF3189, Affinity Bioscience), and β-actin (1:20000, Cat Number: #AF7018, Affinity Bioscience). The following day, the blots were washed three times in TBST and incubated with goat anti-rabbit IgG (H + L) (1:10000, Cat Number: #S0001, Affinity Bioscience) for 1 h at RT. After the final three washes, band detection was performed using a Western Blotting Detection System (GE Healthcare Bio-science), and images were captured using a ChemiDoc™ Touch Imaging System (Bio–Rad Laboratories, Hercules, CA). Image analysis was conducted using Image LabTM 3.0 software (Bio-Rad Laboratories).
Gene expression analysis by quantitative real-time PCR
Total RNA was isolated from kidney tissue using an FastPure Cell/Tissue Total RNA Isolation Kit (Vazyme, Nanjing, China). After quantifying the levels/quality of each isolated RNA sample using a Nanodrop spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA), cDNA templates were synthesized from each sample (1 μg total RNA/mouse) utilizing the HiScript III RT SuperMix (Vazyme, Nanjing, China). Quantitative real-time PCR was then performed using SYBR Green master mix (Vazyme, Nanjing, China), with primer sequences detailed in Supplementary Table 1. Each sample was analyzed in triplicate, and the relative RNA expression levels were normalized to GAPDH. All data were analyzed using the 2−ΔΔCt method.
Serum metabolome analysis and data preprocessing
Metabolome analysis was performed as previously reported [37]. Briefly, the serum metabolites were extracted and transferred to sample vials for LC–MS analysis. An equal aliquot of each sample (10 μl) was mixed as the quality control (QC) sample. Raw data were processed using Progenesis QI 2.3 (Nonlinear Dynamics, Waters, USA) for peak detection, extraction, alignment, and integration. In total, 1588 and 1933 peaks were detected in positive and negative ionization modes, respectively. Metabolic features that were detected in < 80% of any sample set and exhibited a relative standard deviation (RSD) of QC > 30% were excluded from further analysis. Subsequently, 1378 (positive mode) and 1693 (negative mode) metabolite features remained, with any remaining missing values imputed with the minimum value. The features from both positive and negative ionization modes were combined for subsequent statistical analyses.
Statistical analysis
All statistical analyses were conducted using R version 4.3.1. Sample size was selected based on our previous reports [34,35,36,37]. Experimental data were presented as the mean ± standard error of the mean (SEM). Body weight data were analyzed using repeated one-way analysis of variance (ANOVA), followed by post-hoc Tukey’s multiple comparison tests. Behavioral tests and Western blot data in the ketamine experiment were analyzed using two-way ANOVA, followed by post-hoc Tukey’s multiple comparison tests. Additionally, behavioral tests and western blot data from the ketamine and ANA-12 experiments were separately analyzed using one-way ANOVA. P-values < 0.05 were considered statistically significant.
Metabolomic data underwent log10 transformation and were scaled to unit variance for multivariate analysis. Subsequently, the data were analyzed using the non-parametric Kruskal–Wallis rank-sum test, followed by Dunn’s test. P-values < 0.05 were considered statistically significant. Metabolites with (1) variable importance in the projection of > 1, (2) fold change of > 1.2 or < −0.83, and (3) P < 0.05 (Kruskal–Wallis rank-sum test) were considered as differentially abundant. Volcano plots were generated utilizing the Greipel and ggplot2 packages, while heatmaps were constructed using the pheatmap package.
Results
Effects of ketamine on cisplatin-induced acute kidney injury
We initially investigated the nephroprotective effects of ketamine in cisplatin-induced AKI mice (Fig. 1A). A single injection of ketamine significantly reduced the mortality and body weight loss caused by cisplatin injection (Fig. 1B). Moreover, ketamine treatment resulted in a decrease in the levels of BUN and creatinine in AKI mice (Fig. 1C). Additionally, the mRNA levels of two known tubule damage biomarkers, neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule-1 (KIM-1), were increased by cisplatin injection but attenuated by ketamine treatment (Fig. S1). H&E staining also revealed that ketamine significantly reduced cisplatin-induced kidney damage, infiltration of inflammatory cells, tubular degeneration, and vacuolization (Fig. 1D). Furthermore, immunohistochemical staining demonstrated higher expression of KIM-1 in the kidneys of cisplatin-treated mice, which was reduced by ketamine treatment (Fig. 1E). These data suggest that ketamine has a therapeutic effect on cisplatin-induced AKI in mice.
Depression-like behaviors in AKI mice and antidepressant effects of ketamine
Next, we conducted behavioral tests to investigate whether cisplatin-induced AKI mice exhibit depression-like phenotypes. Cisplatin significantly reduced mice’s activity in the locomotion test and increased their immobility time in the FST. Although ketamine had no effect on the activity level of mice, it reduced the immobility time in FST and enhanced their preference for sucrose in the SPT (Fig. 2A-C). Furthermore, Western blot analysis revealed that ketamine attenuated the decreased expression of synaptic proteins (e.g., PSD-95 and GluA1) in the PFC in AKI mice (Fig. 2D, E). These data indicate that cisplatin-induced AKI mice exhibit depression-like phenotypes, which can be improved by ketamine.

A Locomotion test (LMT) (two-way ANOVA: cisplatin: F1,36 = 47.95, P < 0.001, ketamine: F1,36 = 0.004, P = 0.949, interaction: F1,36 = 0.021, P = 0.886). B Forced swimming test (FST) (two-way ANOVA: cisplatin: F1,36 = 12.27, P = 0.001, ketamine: F1,36 = 5.092, P = 0.030, interaction: F1,36 = 3.055, P = 0.089). C: Sucrose preference test (SPT) (two-way ANOVA: cisplatin: F1,36 = 6.954, P = 0.012, ketamine: F1,36 = 4.674, P = 0.037, interaction: F1,36 = 10.12, P = 0.003). D PSD-95 (two-way ANOVA: cisplatin: F1,36 = 4.341, P = 0.044, ketamine: F1,36 = 4.159, P = 0.048, interaction: F1,36 = 5.512, P = 0.245). E GluA1 (two-way ANOVA: cisplatin: F1,36 = 6.462, P = 0.016, ketamine: F1,36 = 4.276, P = 0.046, interaction: F1,36 = 5.406, P = 0.258). Data represent the mean ± SEM (n = 10). *P < 0.05, **P < 0.001, ***P < 0.001. KT, ketamine.
Based on the crucial role of the gut-brain axis in depression and to investigate the impact of cisplatin and ketamine on gut microbiota, we attempted to collect fecal samples from these groups, particularly the cisplatin + saline and cisplatin + ketamine groups. However, cisplatin-induced severe intestinal damage, resulting in no feces being collected from the mice (Fig. S2).
Inhibitory effects of TrkB inhibitor ANA-12 on the beneficial effects of ketamine
It has been reported that ketamine exerts antidepressant effects via the BDNF-TrkB signaling pathway [20, 21, 23]. Therefore, we used the TrkB inhibitor, ANA-12, to further elucidate the mechanism of ketamine (Fig. 3A). Treatment with ANA-12 blocked the improvement in survival rate and body weight loss in cisplatin-induced AKI mice treated with ketamine (Fig. 3B). Additionally, treatment with ANA-12 significantly blocked the effects of ketamine on cisplatin-induced increases in the mRNA levels of NGAL and KIM-1 (Fig. S3). Moreover, ANA-12 alone did not alter these mRNA levels in either cisplatin-treated mice or control mice (Fig. S3). H&E staining results showed that kidney damage in the cisplatin + ketamine + ANA-12 group of mice was more severe compared to the cisplatin + ketamine group (Fig. 3C). Furthermore, ANA-12 blocked the effects of ketamine on reduced sucrose preference and reduced expression of synaptic proteins in the PFC of AKI mice (Fig. 3D). Additionally, ANA-12 alone did not alter these changes, such as sucrose preference and synaptic proteins in the PFC, in either cisplatin-treated or control mice (Fig. 3D).

A Schedule of treatment and sample collection. B Survival curves (Log-rank test: F5,54 = 16.56, P = 0.005) and body weight (repeated-measures one-way ANOVA: F5,54 = 30.493, P < 0.001) changes of the four groups. C Representative HE staining images of renal tissue. D From left to right: SPT (one-way ANOVA: F5,54 = 10.81, P < 0.001), western blot analysis of PSD-95 (one-way ANOVA: F5,54 = 34.83, P < 0.001) and GluA1 (one-way ANOVA: F5,54 = 69.24, P < 0.001). Data represent the mean ± SEM (n = 10). **P < 0.001, ***P < 0.001. SPT, sucrose preference tests; KT, ketamine; ANA, ANA-12.
To further validate the involvement of the BDNF-TrkB pathway in the therapeutic action of ketamine, we performed Western blot analysis of TrkB and phosphorylated TrkB (p-TrkB) in the PFC and kidney. Previous studies have shown that the ERK-CREB signaling pathway is downstream of BDNF-TrkB signaling [21, 23]. In this study, we examined the effects of ketamine and ANA-12 on the ERK and CREB signaling pathway. We assessed the expression levels of ERK, p-ERK, CREB, and p-CREB in the PFC and kidney. Ketamine activated the TrkB signaling pathway and its downstream molecules in both the kidney and PFC, notably inducing phosphorylation of TrkB, ERK, and CREB (Fig. 4A-B). Interestingly, treatment with ANA-12 significantly blocked the beneficial effects of ketamine on these changes in the PFC (Fig. 4A-B). In contrast, ANA-12 alone did not alter these expressions in either cisplatin-treated or control mice in the PFC (Fig. 4A-B). Similar results were observed in the kidney (Fig. S4).

A Representative western blot images in the PFC tissue. B p-TrkB/TrkB ratio (one-way ANOVA: F5,54 = 30.58, P < 0.001), p-ERK/ERK ratio (one-way ANOVA: F5,54 = 110.7, P < 0.001), p-CREB/CREB ratio (one-way ANOVA: F5,54 = 20.2, P < 0.001) in the PFC tissue. Data represent the mean ± SEM (n = 10). *P < 0.05, **P < 0.001, ***P < 0.001. PFC, prefrontal cortex; KT, ketamine; ANA, ANA-12.
In addition to the kidneys, we conducted H&E staining to examine the effects of ketamine on other organs, such as the liver, colon, and spleen. Cisplatin caused liver damage, including a large number of vacuolated cell deaths, structural disorders in the colon, and the disappearance of red and white pulp in the spleen (Fig. S5). Interestingly, ketamine ameliorated these cisplatin-induced pathological changes in these organs, while ANA-12 antagonized the beneficial effects of ketamine (Fig. S5).
These data suggest that ketamine can improve both kidney damage and depression-like behaviors in AKI mice through the activation of the TrkB and ERK-CREB signaling pathways. Additionally, ketamine may improve cisplatin-induced pathological changes in other organs, such as the liver, colon, and spleen via the TrkB signaling pathway.
Serum metabolomic changes in AKI mice after administration of ketamine
Untargeted metabolomics analysis of serum samples was performed to further elucidate the impact of ketamine on metabolism. After alignment with the database, a total of 3,051 metabolites were accurately identified. Significant differences were observed among the cisplatin + saline, cisplatin + ketamine group and cisplatin + ketamine + ANA-12 group through OPLS-DA analysis, indicating dramatic changes in serum metabolites after ketamine and ANA-12 treatments (Fig. 5A-C).

A OPLS-DA between the PBS + saline group and the cisplatin + saline group (R2Y = 0.991, Q2 = 0.839, P < 0.05). B OPLS-DA between the cisplatin + saline group and the cisplatin + ketamine group (R2Y = 0.975, Q2 = 0.516, P < 0.05). C OPLS-DA between the cisplatin + ketamine group and the cisplatin + ketamine + ANA-12 group (R2Y = 0.993, Q2 = 0.671, P < 0.05). D Volcano plot demonstrated metabolites changes in the PBS + saline group vs the cisplatin + saline group. E Volcano plot demonstrated metabolites changes in the cisplatin + saline group vs the cisplatin + ketamine group. F Volcano plot demonstrated metabolites changes in the cisplatin + ketamine group vs the cisplatin + ketamine + NA-12 group. The X axis indicates log2-transformed fold change of metabolite abundances, and Y axis indicates the negative value of log10-transformed P-value using the Kruskal–Wallis rank-sum test. The horizontal lines represent P < 0.05 and vertical lines indicate fold change of > 1.2 or < −0.83. Metabolites elevated or decreased are highlighted in red and blue, respectively, and metabolites with the highest VIP and lowest P are labeled with text. OPLS-DA, orthogonal partial least squares discriminant analysis; KT, ketamine; ANA, ANA-12.
Subsequently, we analyzed various metabolites among the four groups. Compared to the control group, 182 metabolites were significantly increased and 10 metabolites were significantly decreased in cisplatin-induced AKI mice (Fig. 5D, Table S2). In comparison to the cisplatin + saline group, 31 metabolites demonstrated a significant increase and 3 metabolites exhibited a significant decrease in the cisplatin + ketamine group (Fig. 5E, Table S3). Furthermore, following the administration of ANA-12 in AKI mice treated with ketamine, 11 metabolites were increased and 3 metabolites were decreased (Fig. 5F, Table S4). PubChem CID numbers were used instead of names when the structures of the compounds were complex (https://pubchem.ncbi.nlm.nih.gov/). Refer to Table S5 for the corresponding CID numbers. Additionally, metabolites with the highest VIP (variable importance in projection) scores and lowest P-values were depicted (Fig. S6A and S6B). Interestingly, we found that C16-ceramide levels increased after cisplatin injection, while ketamine inhibited this increase, and ANA-12 blocked the effect of ketamine.
These data suggest that C16-ceramide may contribute to cisplatin-induced kidney damage and depression-like behaviors, as well as ketamine’s beneficial effects.
Associations between altered metabolites, behavioral tests and proteins in the PFC
Subsequently, we performed a correlation analysis to investigate the associations between serum metabolites, sucrose preference test results, and protein levels (Fig. 6A). Most metabolites, such as C16-ceramide, hexyl benzoate, and rotundifoline, were negatively correlated with SPT results, synaptic proteins, BDNF, and the ratio of p-TrkB/TrkB in the kidney and brain. Additionally, metabolites such as dehydronorketamine (ketamine metabolite) and cabergoline were positively correlated with the sucrose preference index and protein expression in the brain and kidney. Notably, aside from BDNF in the kidney, C16-ceramide levels were negatively correlated with sucrose preference results and the expression of all other proteins (Fig. 6B).

A The heatmap depicted the relationships between the sucrose preference, proteins and metabolites. B Associations of C16-Ceramide with sucrose preference, PSD-95, GluA1 and p-TrkB/TrkB in the PFC and p-TrkB/TrkB in the kidney. PFC, prefrontal cortex.
Discussion
The major findings of this study are as follows. First, ketamine attenuated cisplatin-induced kidney injury, including kidney destruction, increased BUN and creatinine levels in the blood, and elevated KIM-1 expression in the kidney. Additionally, ketamine attenuated cisplatin-induced pathological changes in the liver, colon, and spleen. Second, we found that cisplatin also led to the development of depression-like phenotypes in mice, including decreased sucrose preference and reduced expression of PSD-95 and GluA1 in the PFC, which can be reversed by ketamine. Third, the TrkB inhibitor ANA-12 reduced the beneficial effects of ketamine on cisplatin-induced kidney injury and depression-like behaviors by suppressing the TrkB and ERK-CREB signaling, especially by inhibiting the phosphorylation of TrkB, ERK, and CREB in the kidney and PFC. Additionally, ANA-12 blocked the beneficial effects of ketamine on cisplatin-induced pathological changes in the liver, colon, and spleen. Finally, metabolomics analysis revealed that various metabolites (e.g., C16-ceramide) increased after cisplatin injection but decreased after ketamine treatment, suggesting their potential roles in the effects of cisplatin and ketamine treatment. Taken together, ketamine can improve cisplatin-induced kidney injury and depression-like behaviors, possibly by activating the TrkB and ERK-CREB signaling pathways and altering blood metabolites.
Cisplatin is known to cause toxicity in several organs, including the kidneys, liver, colon, and spleen [38, 39], and this is consistent with our current data. Additionally, TrkB plays a significant role in these peripheral organs. In the kidneys, BDNF-TrkB signaling has been implicated in protecting against AKI and maintaining renal function [40,41,42]. Research has shown that BDNF-TrkB signaling regulates nephrotoxicity in a rat model of chronic cyclosporin A nephropathy [42]. Furthermore, TrkB signaling in the liver can influence hepatic regeneration and responses to injury [43, 44]. In the colon, TrkB is associated with the regulation of cellular proliferation and differentiation, which is crucial for maintaining the integrity of the mucosal lining [45]. In the immune system, TrkB in the spleen is involved in regulating immune responses, including the activation and survival of lymphocytes [46]. In this study, we found that ketamine attenuated cisplatin-induced pathological changes in these organs, which were reversed by the TrkB inhibitor ANA-12. These findings suggest that ketamine could ameliorate cisplatin-induced pathological changes in these organs through the activation of the TrkB signaling pathway.
In our study, we observed that ketamine administration improved both systemic health markers, such as creatinine and urea levels, and behavioral outcomes. While these systemic improvements could contribute to the reduction in depression-like behaviors, our additional analyses of specific signaling pathways, such as TrkB and ERK-CREB pathway in the PFC, suggest that ketamine also has direct effects on the brain that likely contribute to these behavioral changes. Furthermore, the reversal of ketamine’s beneficial effects on both systemic and behavioral measures by ANA-12 indicates a significant role of TrkB signaling in these outcomes. However, future studies are needed to further explore the relationship between ketamine-induced systemic health improvements and antidepressant-like effects.
In addition to its robust antidepressant effects, ketamine also possesses potent anti-inflammatory and antioxidant properties [8,9,10,11]. Our study revealed that ketamine has therapeutic effects on cisplatin-induced AKI and depression-like behaviors, as well as pathological changes in the liver, colon, and spleen. These findings suggest that ketamine provides protective effects against cisplatin-induced damage in various organs. However, the full extent of its benefits and the underlying mechanisms remain largely unexplored, necessitating further investigation.
Preclinical studies suggest that arketamine, the (R)-enantiomer of ketamine, exhibits stronger and more prolonged antidepressant-like effects than esketamine, (S)-enantiomer of ketamine, through the BDNF-TrkB and ERK-CREB signaling pathway [8,9,10,11, 20, 21, 23]. Clinical trials of arketamine are currently being conducted by several pharmaceutical companies worldwide [11, 47]. BDNF-TrkB is widely expressed in the mammalian brain and peripheral organs. Conversely, ANA-12 inhibited TrkB phosphorylation, thereby suppressing the BDNF-TrkB signaling pathway. In this study, we observed that ANA-12 inhibits the beneficial effects of ketamine on depression-like behaviors and pathological damages in the kidney, as well as in other organs such as the liver, colon, and spleen. Western blot analysis revealed that ANA-12 reduced the ratios of p-TrkB/TrkB, p-ERK/ERK, p-CREB/CREB in both the kidney and PFC. This suggests that the therapeutic effects of ketamine on the PFC and kidney may also involve the TrkB and ERK-CREB signaling pathway. However, further research using ketamine and its potent enantiomer arketamine is needed to ascertain the role of BDNF-TrkB and ERK-CREB signaling in the kidney and other organs.
The present study demonstrated that ketamine mitigated the increase in blood metabolites, such as C16-ceramide and N-acetyl-DL-leucine, induced by cisplatin injection. C16-ceramide, a ceramide with a fatty acid chain length of 16, is associated with neuronal cell death, oxidative stress responses, and the expression of pro-inflammatory genes [48, 49]. Elevated ceramide levels are recognized as a common feature of neurodegenerative diseases, including Alzheimer’s disease, Parkinson’s dementia, and frontotemporal lobar degeneration [50]. Collectively, it seems that elevated C16-ceramide may contribute to kidney damage and depression-like behaviors in AKI mice. While C16-ceramide was found at elevated levels, this observation alone does not prove it as the causative compound in the cisplatin-induced AKI and associated depression-like behaviors. Nonetheless, the precise effects and mechanisms of C16-ceramide in cisplatin-induced toxicity require further validation. N-acetyl-DL-leucine, a derivative of leucine, is known for its ability to enhance blood-brain barrier permeability. Studies have shown that leucine can significantly improve cerebellar ataxia in humans [51]. However, a double-blind, randomized, placebo-controlled clinical crossover trial found that NADLL was not superior to a placebo in the treatment of cerebellar ataxia [52]. Further research is needed to investigate the role of leucine and its derivative NADLL in AKI mice.
Additionally, we observed that cisplatin led to a decrease in dopamine 4-sulfate levels in the blood, while ketamine increased these levels. Dopamine 4-sulfate, a product of dopamine sulfation, is primarily secreted by nerve cells. It is also suggested that the majority of dopamine 4-sulfate in the blood originates in the upper gastrointestinal tract [53]. The reduction in dopamine 4-sulfate suggests that cisplatin may damage the central or peripheral nervous system, which may partially explain the development of depression associated with cisplatin. Overall, these metabolites may contribute to the effects of cisplatin-induced neurotoxicity and kidney toxicity. However, further research is necessary to elucidate their roles in cisplatin-induced kidney toxicity and psychological symptoms.
Our study highlights the potential of ketamine as a therapeutic agent for treating AKI and its associated depressive symptoms. The observed improvements in both systemic health markers and depression-like behaviors suggest that ketamine could be beneficial in managing the complex interplay between renal and mental health. However, using ketamine in clinical settings, especially for patients with compromised renal function, requires careful consideration of dosing, administration, and monitoring to minimize potential side effects. While our mouse model provides valuable insights, there are inherent limitations when extrapolating these findings to humans. Differences in physiology, metabolism, and drug responses between mice and humans can affect the efficacy and safety profiles of treatments. Additionally, the controlled experimental conditions in animal studies may not fully replicate the complexities of human AKI and its comorbidities. Therefore, further research, including clinical trials, is essential to determine the safety, optimal dosage, and therapeutic efficacy of ketamine (or arketamine) in human populations with AKI and associated psychiatric symptoms.
In this study, we found that cisplatin-treated mice exhibit depression-like behaviors and toxicity in peripheral organs, such as the kidney. These findings suggest that the effects on the kidney and brain may be independent, although a kidney-brain connection cannot be ruled out [54]. Indirect pathways or systemic factors, such as inflammatory mediators, could contribute to both depression-like behaviors and organ toxicity in AKI mice. Future studies employing targeted mechanistic approaches, such as direct manipulation of kidney-brain signaling pathways or the analysis of specific biomarkers, are necessary to clarify the interaction between kidney damage and brain dysfunction in cisplatin-treated mice.
In conclusion, this study demonstrated that AKI mice exhibit depression-like behaviors and abnormal blood metabolite composition, accompanied by kidney damage and pathological changes in other organs such as the liver, colon, and spleen. Ketamine was shown to alleviate cisplatin-induced pathological damages in the kidney and other organs, as well as depression-like behaviors, through the TrkB and ERK-CREB signaling pathways and changes in blood metabolites. Further research is needed to explore the specific role of the two enantiomers of ketamine in this model.
Data availability
The data generated in this study are available from the corresponding author YP upon reasonable request.
References
Yang L, Xing G, Wang L, Wu Y, Li S, Xu G, et al. Acute kidney injury in China: a cross-sectional survey. Lancet (London, England). 2015;386:1465–71.
Huang J, Xu D, Yang L. Acute kidney injury in Asia: disease burden. Sem Nephrology. 2020;40:443–55.
Singbartl K, Kellum JA. AKI in the ICU: definition, epidemiology, risk stratification, and outcomes. Kidney Int. 2012;81:819–25.
Hoste EA, Bagshaw SM, Bellomo R, Cely CM, Colman R, Cruz DN, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41:1411–23.
Palmer S, Vecchio M, Craig JC, Tonelli M, Johnson DW, Nicolucci A, et al. Prevalence of depression in chronic kidney disease: systematic review and meta-analysis of observational studies. Kidney Int. 2013;84:179–91.
Tang X, Han YP, Chai YH, Gong HJ, Xu H, Patel I, et al. Association of kidney function and brain health: a systematic review and meta-analysis of cohort studies. Ageing Res Rev. 2022;82:101762.
Balogun RA, Omotoso BA, Xin W, Ma JZ, Scully KW, Arogundade FA, et al. Major depression and long-term outcomes of acute kidney injury. Nephron. 2017;135:23–30.
Hashimoto K. Rapid-acting antidepressant ketamine, its metabolites and other candidates: a historical overview and future perspective. Psychiatry Clin Neurosci. 2019;73:613–27.
Hashimoto K. Molecular mechanisms of the rapid-acting and long-lasting antidepressant actions of (R)-ketamine. Biochem Pharmacol. 2020;177:113935.
Wei Y, Chang L, Hashimoto K. Molecular mechanisms underlying the antidepressant actions of arketamine: beyond the NMDA receptor. Mol Psychiatry. 2022;27:559–73.
Hashimoto K, Zhao M, Zhu T, Wang X, Yang J Ketamine and its two enantiomers in anesthesiology and psychiatry: a historical review and future directions. J Anes Transl Med. 2024:3:65–132.
Zarate CA Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64.
Fava M, Freeman MP, Flynn M, Judge H, Hoeppner BB, Cusin C, et al. Double-blind, placebo-controlled, dose-ranging trial of intravenous ketamine as adjunctive therapy in treatment-resistant depression (TRD). Mol Psychiatry. 2020;25:1592–603.
Fujita A, Fujita Y, Pu Y, Chang L, Hashimoto K. MPTP-induced dopaminergic neurotoxicity in mouse brain is attenuated after subsequent intranasal administration of (R)-ketamine: a role of TrkB signaling. Psychopharmacol. 2020;237:83–92.
Wang X, Chang L, Tan Y, Qu Y, Shan J, Hashimoto K. (R)-ketamine ameliorates the progression of experimental autoimmune encephalomyelitis in mice. Brain Res Bull. 2021;177:316–23.
Wang X, Chang L, Wan X, Tan Y, Qu Y, Shan J, et al. (R)-ketamine ameliorates demyelination and facilitates remyelination in cuprizone-treated mice: a role of gut-microbiota-brain axis. Neurobiol Dis. 2022;165:105635.
Xiong Z, Chang L, Qu Y, Pu Y, Wang S, Fujita Y, et al. Neuronal brain injury after cerebral ischemic stroke is ameliorated after subsequent administration of (R)-ketamine, but not (S)-ketamine. Pharmacol Biochem Behav. 2020;191:172904.
Garcia LS, Comim CM, Valvassori SS, Réus GZ, Barbosa LM, Andreazza AC, et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog Neuropsychopharmacol Biol Psychiatry. 2008;32:140–4.
Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Cheng PF, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–5.
Yang C, Shirayama Y, Zhang JC, Ren Q, Yao W, Ma M, et al. R-ketamine: a rapid-onset and sustained antidepressant without psychotomimetic side effects. Transl Psychiatry. 2015;5:e632.
Yang C, Ren Q, Qu Y, Zhang JC, Ma M, Dong C, et al. Mechanistic target of rapamycin-independent antidepressant effects of (R)-ketamine in a social defeat stress model. Biol Psychiatry. 2018;83:18–28.
Kim JW, Autry AE, Na ES, Adachi M, Björkholm C, Kavalali ET, et al. Sustained effects of rapidly acting antidepressants require BDNF-dependent MeCP2 phosphorylation. Nat Neurosci. 2021;24:1100–9.
Yao W, Cao Q, Luo S, He L, Yang C, Chen J, et al. Microglial ERK-NRBP1-CREB-BDNF signaling in sustained antidepressant actions of (R)-ketamine. Mol Psychiatry. 2022;27:1618–29.
Liu WX, Wang J, Xie ZM, Xu N, Zhang GF, Jia M, et al. Regulation of glutamate transporter 1 via BDNF-TrkB signaling plays a role in the anti-apoptotic and antidepressant effects of ketamine in chronic unpredictable stress model of depression. Psychopharmacology (Berl). 2016;233:405–15.
Tang XH, Diao YG, Ren ZY, Zang YY, Zhang GF, Wang XM, et al. A role of GABAA receptor α1 subunit in the hippocampus for rapid-acting antidepressant-like effects of ketamine. Neuropharmacology. 2023;225:109383.
Zhang JC, Yao W, Hashimoto K. Arketamine, a new rapid-acting antidepressant: a historical review and future directions. Neuropharmacology. 2022;218:109219.
Zhang K, Yao Y, Hashimoto K. Ketamine and its metabolites: potential as novel treatments for depression. Neuropharmacology. 2023;222:109305.
Huang C, Wu Z, Wang D, Qu Y, Zhang J, Jiang R, et al. Myelin-associated oligodendrocytic basic protein-dependent myelin repair confers the long-lasting antidepressant effect of ketamine. Mol Psychiatry. 2024;29:1741–53.
Chang L, Wei Y, Hashimoto K. Brain-gut-microbiota axis in depression: a historical overview and future directions. Brain Res Bull. 2022;182:44–56.
Wei Y, Wang T, Liao L, Fan X, Chang L, Hashimoto K. Brain-spleen axis in health and diseases: a review and future perspective. Brain Res Bull. 2022;182:130–40.
Zhang JC, Yao W, Dong C, Yang C, Ren Q, Ma M, et al. Comparison of ketamine, 7,8-dihydroxyflavone, and ANA-12 antidepressant effects in the social defeat stress model of depression. Psychopharmacology. 2015;232:4325–35.
Ren Q, Ma M, Yang C, Zhang JC, Yao W, Hashimoto K. BDNF-TrkB signaling in the nucleus accumbens shell of mice has key role in methamphetamine withdrawal symptoms. Transl Psychiatry. 2015;5:e666.
Pu Y, Huang R, Chai L, Yang H, Wang D, Wei Z. et al. Multimode evaluating the fluctuation of lipid droplets polarity in acute kidney injury and tumor models. Sensors Actuators B Chem. 2023:380:133343.
Pu Y, Tan Y, Qu Y, Chang L, Wang S, Wei Y, et al. A role of the subdiaphragmatic vagus nerve in depression-like phenotypes in mice after fecal microbiota transplantation from Chrna7 knock-out mice with depression-like phenotypes. Brain Behav Immun. 2021;94:318–26.
Pu Y, Zhang Q, Tang Z, Lu C, Wu L, Zhong Y, et al. Fecal microbiota transplantation from patients with rheumatoid arthritis causes depression-like behaviors in mice through abnormal T cells activation. Transl Psychiatry. 2022;12:223.
Pu Y, He Y, Zhao X, Zhang Q, Wen J, Hashimoto K, et al. Depression-like behaviors in mouse model of Sjögren’s syndrome: a role of gut-microbiota-brain axis. Pharmacol Biochem Behav. 2022;219:173448.
Pu Y, Wu Q, Zhang Q, Huang T, Wen J, Wei L, et al. Mesenchymal stem-cell-derived microvesicles ameliorate MPTP-induced neurotoxicity in mice: a role of the gut–microbiota–brain axis. Psychopharmacology. 2023;240:1103–18.
Manohar S, Leung N. Cisplatin nephrotoxicity: a review of the literature. J Nephrol. 2018;31:15–25.
Dasari S, Njiki S, Mbemi A, Yedjou CG, Tchounwou PB. Pharmacological effects of cisplatin combination with natural products in cancer chemotherapy. Int J Mol Sci. 2022;23:1532.
Endlich N, Lange T, Kuhn J, Klemm P, Kotb AM, Siegerist F, et al. BDNF: mRNA expression in urine cells of patients with chronic kidney disease and its role in kidney function. J Cell Mol Med. 2018;22:5265–77.
Hsu CY, Sheu WH, Lee IT. Brain-derived neurotrophic factor associated with kidney function. Diabetol Metab Syndr. 2023;15:16.
Tao YS, Piao SG, Jin YS, Jin JZ, Zheng HL, Zhao HY, et al. Expression of brain-derived neurotrophic factor in kidneys from normal and cyclosporine-treated rats. BMC Nephrol. 2018;19:63.
García-Suárez O, González-Martínez T, Perez-Perez M, Germana A, Blanco-Gélaz MA, Monjil DF, et al. Expression of the neurotrophin receptor TrkB in the mouse liver. Anat Embryol (Berl). 2006;211:465–73.
Grzelak N, Kaczmarek D, Mrówczyński W. Comparison of the effects of BDNF/TRKB signalling on metabolic biomarkers in the liver of sedentary and trained rats with normal and knockout BDNF genotypes. Front Physiol. 2023;14:1268648.
Quan X, Chen W, Liang C, Jia Y, Wang Y, Luo H, et al. Downregulation of BDNF-TrkB signaling may contribute to the colonic motility disorders in mice with streptozocin-induced diabetes. Neurogastroenterol Motil. 2023;35:e14647.
Pérez-Pérez M, García-Suárez O, Blanco-Gelaz MA, Esteban I, Ciriaco E, Laurà R, et al. TrkB mRNA and protein in mouse spleen: structure of the spleen of functionally deficient TrkB mice. Cell Tissue Res. 2004;316:179–87.
Hashimoto K Are “mystical experiences” essential for antidepressant actions of ketamine and the classic psychedelics? Eur Arch Psychiatry Clin Neurosci. 2024. https://doi.org/10.1007/s00406-024-01770-7.
Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, et al. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–31.
El Alwani M, Wu BX, Obeid LM, Hannun YA. Bioactive sphingolipids in the modulation of the inflammatory response. Pharmacol Ther. 2006;112:171–83.
de Wit NM, den Hoedt S, Martinez-Martinez P, Rozemuller AJ, Mulder MT, de Vries HE. Astrocytic ceramide as possible indicator of neuroinflammation. J Neuroinflammation. 2019;16:48.
Strupp M, Teufel J, Habs M, Feuerecker R, Muth C, van de Warrenburg BP, et al. Effects of acetyl-DL-leucine in patients with cerebellar ataxia: a case series. J Neurol. 2013;260:2556–61.
Feil K, Adrion C, Boesch S, Doss S, Giordano I, Hengel H, et al. Safety and efficacy of acetyl-DL-leucine in certain types of cerebellar ataxia: The ALCAT randomized clinical crossover trial. JAMA Netw Open. 2021;4:e2135841.
Eisenhofer G, Aneman A, Friberg P, Hooper D, Fåndriks L, Lonroth H, et al. Substantial production of dopamine in the human gastrointestinal tract. J Clin Endocrinol Metab. 1997;82:3864–71.
Yan Q, Liu M, Xie Y, Lin Y, Fu P, Pu Y, et al. Kidney-brain axis in the pathogenesis of cognitive impairment. Neurobiol Dis. 2024;200:106626.
Acknowledgements
This study was supported by National Natural Science Foundation of China (to YP, NO. 82101616), Natural Science Foundation of Sichuan Province (to YP, NO. 2023NSFSC1703), 1·3·5 project for Outstanding interdisciplinary project of West China Hospital, Sichuan University (to YL, NO. ZYGD18003).
Author information
Authors and Affiliations
Contributions
KH, YL and YP conceived and designed the study. TH, YH, RC, QZ, XZ and YP performed the behavioral and biochemical experiments, and analyzed the data. All authors contributed to the interpretation of the results. TH, YH, and YP drafted the first version of the manuscript. KH and YL reviewed and edited the manuscript. All authors provided critical comments and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
KH is the inventor of filed patent applications on “The use of R-Ketamine in the treatment of psychiatric diseases”, “(S)-norketamine and salt thereof as pharmaceutical”, “R-Ketamine and derivative thereof as prophylactic or therapeutic agent for neurodegeneration disease or recognition function disorder”, “Preventive or therapeutic agent and pharmaceutical composition for inflammatory diseases or bone diseases”, and “R-Ketamine and its derivatives as a preventive or therapeutic agent for a neurodevelopmental disorder” by the Chiba University. KH has also received speakers’ honoraria, consultant fee, or research support from Otsuka. The other authors declare no competing interest.
Ethical declarations
The experimental protocol of this study was approved by the Institutional Animal Care and Use Committee of the West China Hospital, Sichuan University (Permission Number 20230807003).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, T., He, Y., Cheng, R. et al. Ketamine attenuates kidney damage and depression-like behaviors in mice with cisplatin-induced acute kidney injury. Transl Psychiatry 14, 468 (2024). https://doi.org/10.1038/s41398-024-03176-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-024-03176-4
