
Abstract
Both schizophrenia (SCZ) and Alzheimer’s disease (AD) are highly heritable brain disorders. Despite of the observed comorbidity and shared psychosis and cognitive decline between the two disorders, the genetic risk architecture shared by SCZ and AD remains largely unknown. Based on summary statistics of the currently available largest genome-wide association studies for SCZ (n = 130,644) and AD (n = 455,258) in individuals of European ancestry, we conducted conditional/conjunctional false discovery rate (FDR) analysis to enhance the statistical power for discovering more genetic associations with SCZ or AD and to detect the common genetic variants shared by both disorders. We found shared genetic architecture in SCZ conditioned on AD and vice versa across different levels of significance, indicating polygenic overlap. We found 268 (78 novel) SCZ-only and 125 (55 novel) AD-only SNPs at conditional FDR < 0.01, and 16 lead SNPs shared by SCZ and AD at conjunctional FDR < 0.05. Only half of the shared SNPs showed concordant effect direction, which was consistent with the modest genetic correlation (r = 0.097; P = 0.026) between the two disorders. This study provides evidence for polygenic overlap between SCZ and AD, suggesting the existence of the shared molecular genetic mechanisms, which may inform therapeutic targets that are applicable for both disorders.
Similar content being viewed by others

Introduction
Alzheimer’s disease (AD), a putative neurodegenerative disease, is the most common type of dementia among elderly people, which affects more than 30 million people in the world and the number will be tripled by 2050 [1]. While AD is primarily recognized for cognitive decline, particularly amnesia [2], psychotic symptoms like hallucinations and delusions are observed in about 40–60% patients with AD [3, 4]. These symptoms may lead to more pronounced cognitive impairment, accelerated cognitive deterioration, and ultimately, a poorer prognosis [3,4,5,6,7]. Notably, psychotic and cognitive symptoms are also the common symptoms of schizophrenia (SCZ) [8], a well-known debilitating psychiatric disorder with a lifetime prevalence of approximately 1% [9], although AD typically shows more frequent non-bizarre delusions and visual hallucinations than SCZ [4, 10, 11]. The investigation of the mechanisms underlying the shared symptoms between AD and SCZ may inform the therapeutic targets shared by both disorders, and therefore accelerate the development of treatments that may benefit both AD and SCZ.
AD and SCZ are highly heritable with an estimated heritability of 60–80% for AD [2, 12] and 80% for SCZ [12, 13]. Several genome-wide association studies (GWASs) have been conducted on AD [14, 15] and SCZ [16,17,18], respectively. The largest GWAS for AD identified 38 risk loci, indicating the contribution of microglia, immune cells, and protein catabolism to AD pathology [14]. Meanwhile, the largest GWAS for SCZ reported 287 risk loci, suggesting the involvement of neuronal function (especially the synaptic biological processes) in SCZ pathology [17]. However, these genetic risk loci can only account for a small proportion of the single nucleotide polymorphism (SNP)-based heritability of each disorder. Moreover, these studies only reported one genome-wide significant SNP (rs6504163) shared by the two disorders, which cannot fully explain for their shared symptoms. Given that the current sample sizes are insufficient for identifying all genetic risk SNPs associated with both disorders, more informative methodologies should be employed to discover additional risk SNPs for AD and SCZ as well as those shared by both disorders. For example, the conditional (condFDR) and conjunctional (conjFDR) FDR method [19] have been used to discover the more genetic associations for phenotypes and the shared genetic SNPs between phenotypes [20,21,22,23,24]. The discovery of additional risk SNPs shared by AD and SCZ may provide new insights into the pathophysiology (biological processes) shared by these two disorders, potentially informing future therapeutic strategies, such as the repurposing of existing antipsychotic drugs that target the shared pathophysiology to treat AD-related psychosis. This is extremely important because few pharmacological agents have been approved to treat AD-related psychosis [25, 26].
Two prior linkage disequilibrium (LD) score regression (LDSC) analyses failed to find a significant genetic correlation between AD and SCZ [27, 28], which may be caused by factors, such as the insufficient sample sizes of GWASs. In this study, we aimed to detect the risk SNPs between AD and SCZ by conducting the cond/conjFDR analyses based on the summary statistics of the currently largest GWASs for AD (n = 455,258) [15] and SCZ (n = 130,644) [17] conducted in individuals of European (EUR) ancestry. In addition to find the risk SNPs shared by AD and SCZ, we also aimed to identify additional risk SNPs associated with AD and SCZ using condFDR by exploiting the joint statistical power of these two GWASs through an empirical Bayesian framework [19].
Materials and methods
GWAS summary statistics for AD and SCZ
The EUR-GWAS summary statistics of AD was SCZ were obtained from 455,258 individuals, including 71,880 patients with clinically diagnosed AD or AD-by-proxy and 383,378 healthy controls [15], while the EUR-GWAS summary statistics of SCZ obtained from 130,644 individuals, including 53,386 patients with SCZ and 77,258 healthy controls [17]. The details about the diagnostic criteria, sample sizes, and sources of the two datasets are provided in Supplementary Table S1.
Genetic correlation analysis
To avoid the bias due to the complex regional LD patterns, we cut out SNPs within the major histocompatibility complex (MHC, hg19 location chr6: 25119106-33854733) and chromosome 8p23.1(hg19 location chr8: 7242715-12483982). These regions are characterized by strong SNP associations and long-range LD, which can obscure the effects of individual loci [19, 29]. The high gene density, extreme polymorphism, and functional clustering of related genes within the MHC region, combined with strong LD, make it particularly challenging to disentangle the contributions of specific loci [30, 31]. By excluding these regions, we aimed to reduce confounding effects and improve the clarity and reliability of genetic loci identification in our study [31, 32]. Then, LDSC [32] was conducted to roughly calculate SNP-based genetic correlations between SCZ and AD based on the Python package (https://github.com/bulik/ldsc).
Cond/conjFDR analysis
By leveraging genetic pleiotropy of complex traits including disorders with polygenic architecture, the cond/conjFDR method (https://github.com/alexploner/cfdr.pleio) utilizes an empirical Bayes statistical framework to improve the discovery of SNPs associated with each trait and to identify SNPs shared by two traits based on the GWAS summary statistics of both traits [19]. The condFDR analysis uses the overlapping SNP-trait associations from GWAS for another trait to re-rank the test statistics for a primary trait, conditioned on the associations of the secondary trait [33, 34]. By inverting the role of the two traits, the condFDR analysis identifies SNPs associated with the other trait. The conjFDR analysis identifies SNPs associated with both traits [34, 35], by defining the conjFDR value for each SNP as the maximum of the two condFDR values for this SNP, providing a conservative estimate of FDR for SNPs jointly associated with both traits [36]. The cond/conjFDR method can be applied to the cross-trait analysis between AD and SCZ because it has been successfully used to identify the shared SNPs between two traits even without overall genetic correlation [37]. Before the cond/conjFDR analyses, the conditional quantile-quantile (Q-Q) plot [19] was utilized to visualize cross-trait enrichment by illustrating the distribution of P values for the primary trait while conditioning on the significance of its association with the secondary trait at thresholds of P ≤ 1, 0.01, 0.001, and 0.0001 [20, 21, 35, 38]. The significant enrichment with both traits was confirmed when SNPs with lower P values for the primary trait increased as a function of the significance of the association with the secondary trait. In the condFDR analyses, we initially defined AD as the primary trait and SCZ as the second trait, and then we shifted the role of the two traits. Therefore, we could discover SNPs associated with SCZ and AD at condFDR <0.01, respectively. We defined conjFDR < 0.05 as the significance threshold for SNPs shared by SCZ and AD and condFDR <0.01 as the significance threshold for the conditional analyses. The two significance thresholds were selected because they were consistently applied in almost all previous cond/conjFDR studies [20, 21, 35]. In addition, we also assessed the effective direction of the common SNPs by observing the beta values. We excluded the extended major histocompatibility complex (hg19 location chr6: 25119106–33854733) and chromosome 8p23.1 (hg19 location chr8: 7242715–12483982) genomic regions from the FDR fitting procedures because complex correlations in regions with intricate LD can bias FDR estimation.
Lead SNPs definition
Functional mapping and annotation (FUMA) [39] (http://fuma.ctglab.nl/) was used to define lead SNPs with an EUR-LD reference panel of the 1000 Genomes Project [40]. For each conditional (FDR < 0.01) or conjunctional (FDR < 0.05) analysis, we defined all significant SNPs with LD r2 < 0.6 as independent significant SNPs, from which we selected SNPs with LD r2 < 0.1 as lead SNPs. To define distinct genomic loci, we merged loci within 250 kb into one locus and selected the most significant SNP as the lead SNP of the merged locus. Of all SNPs identified by the conjunctional analysis (conjFDR < 0.05), we defined those with an LD r2 < 0.1 as lead SNPs and named them as SNPs shared by SCZ and AD. Similarly, we defined lead SNPs identified by SCZ conditioned on AD (condFDR <0.01) as SCZ-related SNPs and those identified by AD conditioned on SCZ (condFDR <0.01) as AD-related SNPs. Of the SCZ-related SNPs, those showing an LD r2 < 0.1 with AD-related SNPs and shared SNPs were defined as SCZ-only SNPs. Of the AD-related SNPs, those showing an LD r2 < 0.1 with SCZ-related SNPs and shared SNPs were defined as AD-only SNPs. Lastly, the genetic signatures shared by SCZ and AD were defined as conjFDR < 0.05 and exhibiting the same effect direction between patients with SCZ and AD.
Functional annotation
The three categories of lead SNPs (SCZ-only SNPs, AD-only SNPs, shared SNPs with the same effect direction) were annotated using FUMA v1.4.1 (https://fuma.ctglab.nl), an online platform for functional annotations of genetic variants [39]. With FUMA, we used annotate variation (ANNOVAR) to determine the functional consequences of the identified SNPs by mapping them to genes and regulatory elements [41]. We calculated combined annotation dependent depletion (CADD) scores to assess deleteriousness and pathogenicity of each SNP [42]. We assessed the regulatory potential of the identified SNPs using RegulomeDB, which integrates the available data to prioritize SNPs that may involve in gene regulation by affecting regulatory elements [43]. We also assessed the minimum chromatin state to predict the consequence of each SNP [44].
All the candidate SNPs in the genomic loci having an LD r2 ≥ 0.6 with one of the shared lead SNPs were aligned to genes by three gene-mapping methods. The first method involved positional gene mapping, where SNPs were linked to genes according to the physical proximity around a 10-kb window in the human reference assembly (GRCh37/hg19). The second method was the expression quantitative trait locus (eQTL) mapping, where SNPs were mapped to genes based on the expression level of a gene is affected by one SNP across 54 human tissue types, utilizing data from the Genotype-Tissue Expression (GTEx) dataset [45]. Significant eQTLs were determined at FDR P < 0.05. The third strategy was chromatin interaction mapping, establishing connections between SNPs and genes through three-dimensional DNA-DNA interactions (FDR P < 1 × 10−6) [39]. Based on the genes identified by either of three strategies, we conducted gene enrichment analyses using ToppGene (https://toppgene.cchmc.org/), which is a reliable tool to perform comprehensive biological pathway analyses [46]. In this analysis, all genes encoding protein were considered as the background genes, and the final results were adjusted for multiple testing (FDR P < 0.05).
Colocalization with gene expression in brain tissue
The coloc package in R [47] (https://chr1swallace.github.io/coloc) was used to identify the Bayesian colocalizations of the lead SNPs shared by SCZ and AD with eQTLs of six brain tissues (cortex, anterior cingulate cortex, frontal cortex, hippocampus, caudate, and putamen) from GTEx [45]. For each identified lead SNP shared by SCZ and AD, we selected SNPs within 250 kb centered on the lead SNP from the original GWASs for SCZ or AD. For each pair of disorders and brain tissues, we calculated the posterior probability (PP) for a SNP being associated with one or both traits by evaluating five possible hypotheses: H0, no association with either trait; H1 or H2, association with only one trait; H3, association with both traits driven by distinct causal variants; and H4, association with both traits driven by a shared causal variant. A higher PP for H4 (PP.H4) means a higher probability of a shared causal variant between traits. Here, we considered evidence for colocalization significant when PP.H4 > 0.80. Shared colocalization should meet this criterion in both SCZ-GWAS and AD-GWAS.
Results
Genetic overlap and correlation between SCZ and AD
Based on the GWAS summary statistics for SCZ and AD, we conducted LDSC analysis and found a modest but statistically significant genetic correlation between SCZ and AD (rg = 0.097; P = 0.026). The conditional Q-Q plots showed pleiotropic enrichment between SCZ and AD. The conditional Q-Q figures of SCZ showed consistent leftward deflection for subsets of variants with higher significance in AD when SCZ was conditioned on AD and vice versa (Fig. 1), indicating substantial polygenic overlap between SCZ and AD.

Conditional Q-Q figures of nominal versus empirical -log10 P values in AD below the conventional GWAS significant threshold of P < 5 × 10−8 with SCZ A and vice versa B, at different levels of significance P ≤ 1, P ≤ 0.1, P ≤ 0.01, P ≤ 0.001, P ≤ 0.0001, separately, are shown. The purple lines indicate all SNPs. The gray lines indicate the null hypothesis. Increasing leftward deflection from the gray line for SNPs with higher significance in the conditional phenotype indicates polygenic overlap. AD Alzheimer’s disease, SCZ schizophrenia.
The shared and distinct genetic signatures between SCZ and AD
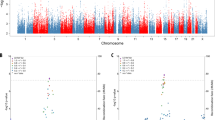
At conjFDR < 0.05, we identified 16 lead SNPs associated with both SCZ and AD (Table 1), and the Manhattan plots of conjunctional analyses of SCZ and AD are shown in Fig. 2A. Considering the direction of effect (Beta value), 8 of 16 lead SNPs (50.00%) with concordant association directions in both patients with SCZ and AD were defined as the genetic signatures shared by SCZ and AD, and their genomic distributions are shown in Fig. 3A. All the 8 shared genetic signatures by SCZ and AD were not reached the genome-wide significance (P < 5 × 10−8) in the previous GWASs for both disorders [15, 17]. For example, rs12979278 (chr19) in the MAMSTER gene was identified as a SNP shared by SCZ and AD, but it cannot survive at the genome-wide significance threshold (P < 5 × 10−8) in both SCZ-GWAS (P = 1.07 × 10−5) and AD-GWAS (P = 5.75 × 10−5) (Fig. 3D).

A Manhattan plot shows the -log10 transformed conjFDR values for each SNP on the y axis and chromosomal positions along the x axis. The lilac horizontal line represents the threshold for significant shared SNPs (conjFDR < 0.05) between SCZ and AD. B, C Manhattan plots shows the -log10 transformed condFDR values for SCZ conditioned on AD (B), and vice versa (C). The y-axis shows the -log10 transformed condFDR values for each SNP, while the x-axis represents the position of chromosomes. The lilac horizontal lines represent the threshold for conditionally significant SNPs (conjFDR < 0.05). Abbreviations: AD Alzheimer’s disease, FDR false discovery rate, SCZ schizophrenia.

A The ideogram shows the chromosomal distribution of 268 SCZ-only, 125 AD-only, and 8 shared genetic signatures, visualizing genomic regions of overlap or divergence. B-D. B. The regional plots show an example of the SCZ-only SNPs. The rs2867655 is significantly associated with SCZ conditioned on AD (condFDR = 0.008), but it is not significant (P = 1.12 × 10−7) in SCZ-GWAS. C The regional plots show an example of the AD-only SNPs. The rs17783630 is significantly associated with AD conditioned on SCZ (condFDR = 0.004), but it is not significant (P = 6.38 × 10−7) in AD-GWAS. D The regional plots show an example (rs12979278, conjFDR = 0.024) of SNPs shared by SCZ and AD in the conjFDR analysis, but it is significant in neither SCZ-GWAS (P = 1.07 × 10−5) nor AD-GWAS (P = 5.75 × 10−5). AD Alzheimer’s disease, condFDR conditional false discovery rate, conjFDR conjunctional false discovery rate, GWAS genome-wide association study, SCZ schizophrenia, SNP single nucleotide polymorphism.
We performed condFDR analyses to improve the discovery of SNPs associated with SCZ or AD. At condFDR <0.01, we found 279 lead SNPs associated with SCZ conditioned on AD (Supplementary Table S2), and 133 lead SNPs associated with AD conditioned on SCZ (Supplementary Table S3). The results of the conditional analyses for SCZ and AD were shown using Manhattan plots Fig. 2B, C. Of the 279 condFDR-identified SNPs for SCZ, 268 SNPs were defined as SCZ-only genetic signatures after excluding SNPs showing LD (r2 ≥ 0.1) with any of the 133 condFDR-identified SNPs for AD and 16 conjFDR-identified shared SNPs. Of these 268 SCZ-only lead SNPs, 78 were considered as novel because they could not survive at P < 5 × 10−8 in the original GWAS for SCZ and did not show LD (r2 ≥ 0.6) with any SNPs in the reported 287 distinct loci of SCZ [17]. For example, rs2867655 (chr20) was identified as a SCZ-only SNP that could not survive at the genome-wide significance threshold (P < 5 × 10−8) in SCZ-GWAS (Fig. 3B).With the same strategy we also defined 125 SNPs as AD-only genetic signatures and 55 novel AD-only SNPs [15]. For example, rs17783630 (chr14) was identified as AD-only SNP (Fig. 3C). The distinct genetic signatures for SCZ and AD are provided in Supplementary Tables S2–3 and their genomic distributions are shown in Fig. 3A.
Functional annotations
Functional annotations were conducted for 268 SCZ-only, 125 AD-only, and 8 shared genetic signatures using FUMA. Most SNPs were located in the intron (49% in SCZ-only, 49% in AD-only, and 25% in shared groups) and intergenic (28% in SCZ-only, 22% in AD-only, and 25% in shared groups) regions of protein-coding genes (Fig. 4A). Employing a cutoff of a minimum chromatin state < 8, we identified that 90% of SCZ-only SNPs, 97% of AD-only SNPs, and 100% of shared SNPs were situated within open chromatin regions (Fig. 4B). Among these SNPs, 6 SCZ-only SNPs (rs3098974, rs12492391, rs12671676, rs9668929, rs6499508, rs133367) exhibited RegulomeDB scores of 1f, while 4 AD-only SNPs displayed RegulomeDB scores of 1b (rs13230744), 1d (rs17783630, rs2455069), and 1f (rs6557994) (Fig. 4C), suggesting that they may exert regulatory control over gene expression by affecting transcription factor binding [43]. Furthermore, CADD scores above 12.37 were observed for 21 SCZ-only SNPs and 2 AD-only SNPs (Fig. 4D), indicating their potential deleterious effects [42]. For example, rs2867655 (chr20; CADD = 22.1; Fig. 3B) was one of the most deleterious SCZ-only SNPs. This SNP acts as an eQTL for PTPRT, playing an essential role in neuronal development and the regulation of synaptic formation [48]. The novel SNP rs17783630 (chr14; minimum chromatin state = 2; RegulomeDB = 1d; Fig. 3C) was a regulatory AD-only SNP, which is an eQTL and mQTL of SLC24A4, playing a crucial role in neuronal development and being associated with pathological diagnosis of AD [49]. Additionally, the shared novel SNP rs12979278 (chr19; minimum chromatin state = 1; RegulomeDB = 4; Fig. 3D) is another regulatory SNP, an eQTL and mQTL of MAMSTR in prefrontal cortex. Dysfunction of MAMSTR may lead to increased microglia sensitivity to immune stimulation, a common feature in the pathological AD and SCZ [50, 51].

A The functional consequences of the 268 SCZ-only (green), 125 AD-only (blue), and 8 shared (red) SNPs. B The minimum chromatin state of the three groups of SNPs. The scores of 1–7 suggest open chromatin states. C The RegulomeDB scores of the three groups of SNPs. The lower scores mean higher likelihood of having regulatory functions. D The distribution of CADD scores of the three groups of SNPs. The scores above 12.37 indicate deleterious. AD Alzheimer’s disease, CADD combined annotation dependent depletion, SCZ schizophrenia, SNP single nucleotide polymorphism.
We used three different gene-mapping strategies to map all the candidate SNPs in the genomic loci having an LD r2 ≥ 0.6 with the 8 genetic signatures shared by SCZ and AD, and obtained 124 unique genes (Supplementary Table S4). And we found there were mainly enriched for the biological process of cell-cell adhesion and molecular function of calcium ion binding (Supplementary Table S5).
Shared colocalizations with eQTLs of brain tissues
Coloc was utilized to detect the shared genetic colocalizations between SCZ and AD with eQTLs from 6 brain tissues at the shared SNPs. The shared colocalizations with brain eQTLs were defined as PP.H4 > 0.8 in both SCZ and AD. We found that SCZ and AD had a shared colocalization (SCZ: PP.H4 = 0.900; AD: PP.H4 = 0.973) at rs6494036 (chr15) with the SAFB like transcription modulator (SLTM) expression in putamen (Supplementary Table S6).
Discussion
In this study, we conducted the cond/conjFDR analysis to identify SNPs associated with SCZ or AD and to discover SNPs shared by both disorders. We found 268 SCZ-only SNPs, 125 AD-only SNPs, and 8 SNPs shared by SCZ and AD. The shared genetic signatures between SCZ and AD may explain for the clinical symptoms shared by both disorders and hold the potential to generate novel hypotheses regarding the relationship between these two disorders and may offer valuable insights for clinical practices.
Although GWASs have identified many common genetic variants associated with SCZ or AD [14,15,16,17, 52], only 1 genome-wide significant SNP (rs6504163) was shared by both disorders, which may be biased by the fact that the sample sizes of current SCZ-GWASs and AD-GWASs are far from enough to discover all SNPs associated with each disorder. Consistent with the marginally significant positive genetic correlation (rg = 0.08, P = 0.044) between SCZ and AD [27], we also found a modest genetic correlation (rg = 0.097; P = 0.026) between these two disorders. However, the LDSC-based genetic correlation analysis may underestimate the genetic sharing between two disorders when some shared SNPs have opposite directions of effects. These indicate that more suitable approaches may discover more SNPs shared by SCZ and AD.
By conducting the condFDR analysis to enhance the ability of genetic discovery, one contribution of this study is the identification of 78 novel loci for SCZ and 55 novel loci for AD compared to their original GWASs [15, 17], improving our understanding of the genetic architectures of these two disorders. For example, rs2867655 (chr20) was identified as a new risk SNP for SCZ, a nonsynonymous exonic variant in PTPRT. This gene encodes a member of the type IIB receptor-type protein tyrosine phosphatase family, which is exclusively expressed in the central nervous system [53, 54]. PTPRT plays a crucial role in the regulation of synaptic formation and neuronal development [48], providing additional evidence for the involvement of the two biological processes in schizophrenia [55]. In line with the reported risk SNPs in MS4A6E and CD33 in previous AD-GWASs [14, 56, 57], we found two novel exonic nonsynonymous variants for AD: rs12798157 (chr11) in MS4A6E and rs2455069 (chr19) in CD33. CD33 encodes a sialic-acid-binding immunoglobulin-like lectin family, which is implicated in cell adhesion, endocytosis, and immune regulation [58]. MS4A6E encodes a member of the membrane-spanning 4A gene family, which also affects the immune system [59]. These findings provide further evidence for the role of immune dysfunction in AD.
The most important contribution of this study is the discovery of eight lead SNPs shared by SCZ and AD, which may explain for the psychotic symptoms and cognitive decline shared by both disorders and inform therapeutic strategies that are potentially effective for both disorders. For instance, rs6504163 in ACE has been identified as the only shared SNP between GWASs for SCZ and AD [15, 17]. We confirmed the shared association of ACE with these two disorders by identifying a new shared SNP (rs4308) in this gene. The angiotensin-converting enzyme (ACE), a critical component of renin–angiotensin system (RAS), has been associated with both SCZ and AD [60,61,62,63,64,65,66]. In the postmortem parietal cortex samples, the levels of ACE2 protein and mRNA were higher in patients with a neuropathological diagnosis of AD compared to healthy controls, and brain soluble ACE2 levels were associated with cognitive decline and the accumulation of soluble amyloid-β peptides (Aβ) and insoluble phospho-tau [66]. Similarly, patients with SCZ also showed increased ACE levels in cerebrospinal fluid [63] and serum [64] as well as increased plasmatic ACE activity [65]. The increased plasmatic ACE activity was also correlated with cognitive decline in SCZ patients [65]. These findings indicate that the increased ACE levels may account for the shared cognitive decline between SCZ and AD, and that drugs such as ACE inhibitors or angiotensin receptor antagonists (ARAs) may benefit for cognitive improvement in both disorders [62]. The rs12979278 in MAMSTR was another lead SNP shared by SCZ and AD, and MAMSTR stimulates the expression of Myocyte Enhancer Factor-2C (MEF2C). MEF2C plays a crucial role in neurogenesis and synapse development [67] and has been associated with cognitive decline and risk for neuropsychiatric disorders including SCZ and AD [68,69,70]. These findings indicate that the MEF2C deficiency may also account for the shared cognitive decline between SCZ and AD, informing the therapeutic potential of targeting MEF2C to improve cognitive function in both disorders. However, the mechanisms underlying the associations between SNPs and psychotic symptoms shared by SCZ and AD remain largely unknown, deserving further investigation.
We also found that SCZ and AD showed a shared colocalization with expression of SAFB-like transcription modulator (SLTM) in the putamen at the shared locus with rs6494036 (chr15) as the lead SNP. SLTM induces apoptosis [71] and regulates GLI factor binding to chromatin and contributes to the transcriptional outcomes of Sonic hedgehog (SHH) signaling [71]. The SHH pathway affects neurogenesis and neural patterning during the development of the central nervous system, and the dysregulation of SHH signaling in the brain contributes to neuropsychiatric disorders including SCZ and AD [72,73,74]. These findings indicate that the SLTM expression in the putamen may account for the shared symptoms between SCZ and AD, and targeting SLTM expression and its downstream SHH pathway are potentially effective for both disorders.
Several limitations should be considered when interpreting our findings. First, the potential confounders related to population stratification may influence our results. Second, the use of summary-level data rather than individual-level data may reduce the precise of our analyses. Third, the sample sizes of SCZ-GWAS and AD-GWAS are far from enough to discover all SNPs associated with each disorder, which may limit the ability to identify SNPs shared by SCZ and AD. Lastly, the SCZ-GWAS and AD-GWAS summary statistics were derived from meta-analysis of many original studies, and thus the inconsistency in diagnostic criteria for disorders across studies may influence our findings.
In sum, our study provides novel insight into the pleiotropic enrichment between SCZ and AD, discovering eight shared genetic architectures with a concordant allelic effect direction. Future studies should elucidate whether and how the shared genetic variants causally lead to the comorbidity and shared symptoms between SCZ and AD. Through targeting the shared pathophysiological pathways, new therapeutic strategies should be developed to improve clinical outcomes of both disorders, especially for AD patients with psychotic symptoms.
Data availability
GWAS summary statistics were publicly available. All analyses in this study were performed using publicly available code (https://github.com/alexploner/cfdr.pleio). No custom code was developed or used in this study.
Code availability
GWAS summary statistics were publicly available. All analyses in this study were performed using publicly available code (https://github.com/alexploner/cfdr.pleio). No custom code was developed or used in this study.
References
Patterson C. World Alzheimer Report 2018. The state of the art of dementia research: new frontiers. 2018. Alzheimer's Disease International.
Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE, et al. Alzheimer’s disease. Lancet. 2021;397:1577–90. https://doi.org/10.1016/S0140-6736(20)32205-4
Sweet RA, Nimgaonkar VL, Devlin B, Jeste DV. Psychotic symptoms in Alzheimer disease: evidence for a distinct phenotype. Mol Psychiatry. 2003;8:383–92. https://doi.org/10.1038/sj.mp.4001262
Murray PS, Kumar S, Demichele-Sweet MA, Sweet RA. Psychosis in Alzheimer’s disease. Biol Psychiatry. 2014;75:542–52. https://doi.org/10.1016/j.biopsych.2013.08.020
Ropacki SA, Jeste DV. Epidemiology of and risk factors for psychosis of Alzheimer’s disease: a review of 55 studies published from 1990 to 2003. Am J Psychiatry. 2005;162:2022–30. https://doi.org/10.1176/appi.ajp.162.11.2022
Sweet RA, Bennett DA, Graff-Radford NR, Mayeux R, National Institute on Aging Late-Onset Alzheimer’s Disease Family Study G. Assessment and familial aggregation of psychosis in Alzheimer’s disease from the National Institute on Aging Late Onset Alzheimer’s Disease Family Study. Brain. 2010;133:1155–62. https://doi.org/10.1093/brain/awq001
Seltman HJ, Mitchell S, Sweet RA. A bayesian model of psychosis symptom trajectory in Alzheimer’s disease. Int J Geriatr Psychiatry. 2016;31:204–10. https://doi.org/10.1002/gps.4326
McCutcheon RA, Marques TR, Howes OD. Schizophrenia-an overview. JAMA Psychiatry. 2020;77:201–10. https://doi.org/10.1001/jamapsychiatry.2019.3360
Saha S, Chant D, Welham J, McGrath J. A systematic review of the prevalence of schizophrenia. PLoS Med. 2005;2:e141 https://doi.org/10.1371/journal.pmed.0020141
Jeste DV, Finkel SI. Psychosis of Alzheimer’s disease and related dementias. Diagnostic criteria for a distinct syndrome. Am J Geriatr Psychiatry. 2000;8:29–34. https://doi.org/10.1097/00019442-200002000-00004
Schutte MJL, Linszen MMJ, Marschall TM, Ffytche DH, Koops S, van Dellen E, et al. Hallucinations and other psychotic experiences across diagnoses: a comparison of phenomenological features. Psychiatry Res. 2020;292:113314 https://doi.org/10.1016/j.psychres.2020.113314
Ohi K, Fujikane D, Shioiri T. Genetic overlap between schizophrenia spectrum disorders and Alzheimer’s disease: current evidence and future directions - an integrative review. Neurosci Biobehav Rev. 2024;167:105900 https://doi.org/10.1016/j.neubiorev.2024.105900
Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003;60:1187–92. https://doi.org/10.1001/archpsyc.60.12.1187
Wightman DP, Jansen IE, Savage JE, Shadrin AA, Bahrami S, Holland D, et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat Genet. 2021;53:1276–82. https://doi.org/10.1038/s41588-021-00921-z
Jansen IE, Savage JE, Watanabe K, Bryois J, Williams DM, Steinberg S, et al. Genome-wide meta-analysis identifies new loci and functional pathways influencing Alzheimer’s disease risk. Nat Genet. 2019;51:404–13. https://doi.org/10.1038/s41588-018-0311-9
Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. https://doi.org/10.1038/nature13595
Trubetskoy V, Pardinas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–8. https://doi.org/10.1038/s41586-022-04434-5
Li Z, Chen J, Yu H, He L, Xu Y, Zhang D, et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat Genet. 2017;49:1576–83. https://doi.org/10.1038/ng.3973
Smeland OB, Frei O, Shadrin A, O’Connell K, Fan CC, Bahrami S, et al. Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet. 2020;139:85–94. https://doi.org/10.1007/s00439-019-02060-2
Smeland OB, Shadrin A, Bahrami S, Broce I, Tesli M, Frei O, et al. Genome-wide association analysis of Parkinson’s disease and schizophrenia reveals shared genetic architecture and identifies novel risk loci. Biol Psychiatry. 2021;89:227–35. https://doi.org/10.1016/j.biopsych.2020.01.026
Bahrami S, Shadrin A, Frei O, O’Connell KS, Bettella F, Krull F, et al. Genetic loci shared between major depression and intelligence with mixed directions of effect. Nat Hum Behav. 2021;5:795–801. https://doi.org/10.1038/s41562-020-01031-2
Fominykh V, Shadrin AA, Jaholkowski PP, Bahrami S, Athanasiu L, Wightman DP, et al. Shared genetic loci between Alzheimer’s disease and multiple sclerosis: crossroads between neurodegeneration and immune system. Neurobiol Dis. 2023;183:106174 https://doi.org/10.1016/j.nbd.2023.106174
Wang H, Zhao Q, Zhang Y, Ma J, Lei M, Zhang Z, et al. Shared genetic architecture of cortical thickness alterations in major depressive disorder and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2024;135:111121 https://doi.org/10.1016/j.pnpbp.2024.111121
Liu M, Wang L, Zhang Y, Dong H, Wang C, Chen Y, et al. Investigating the shared genetic architecture between depression and subcortical volumes. Nat Commun. 2024;15:7647 https://doi.org/10.1038/s41467-024-52121-y
Madhusoodanan S, Shah P. Management of psychosis in patients with Alzheimer’s disease: focus on aripiprazole. Clin Interv Aging. 2008;3:491–501. https://doi.org/10.2147/cia.s3351
Cummings JL, Devanand DP, Stahl SM. Dementia-related psychosis and the potential role for pimavanserin. CNS Spectr. 2022;27:7–15. https://doi.org/10.1017/S1092852920001765
Wingo TS, Liu Y, Gerasimov ES, Vattathil SM, Wynne ME, Liu J, et al. Shared mechanisms across the major psychiatric and neurodegenerative diseases. Nat Commun. 2022;13:4314 https://doi.org/10.1038/s41467-022-31873-5
Brainstorm C, Anttila V, Bulik-Sullivan B, Finucane HK, Walters RK, Bras J, et al. Analysis of shared heritability in common disorders of the brain. Science. 2018;360:eaap8757 https://doi.org/10.1126/science.aap8757
Price AL, Weale ME, Patterson N, Myers SR, Need AC, Shianna KV, et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet. 2008;83:132–5. https://doi.org/10.1016/j.ajhg.2008.06.005
Kulski JK, Shiina T, Dijkstra JM. Genomic diversity of the major histocompatibility complex in health and disease. Cells. 2019;8:1270 https://doi.org/10.3390/cells8101270
Trowsdale J, Knight JC. Major histocompatibility complex genomics and human disease. Annu Rev Genomics Hum Genet. 2013;14:301–23. https://doi.org/10.1146/annurev-genom-091212-153455
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41. https://doi.org/10.1038/ng.3406
Andreassen OA, Thompson WK, Dale AM. Boosting the power of schizophrenia genetics by leveraging new statistical tools. Schizophr Bull. 2014;40:13–7. https://doi.org/10.1093/schbul/sbt168
Schork AJ, Wang Y, Thompson WK, Dale AM, Andreassen OA. New statistical approaches exploit the polygenic architecture of schizophrenia-implications for the underlying neurobiology. Curr Opin Neurobiol. 2016;36:89–98. https://doi.org/10.1016/j.conb.2015.10.008
Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209. https://doi.org/10.1016/j.ajhg.2013.01.001
Nichols T, Brett M, Andersson J, Wager T, Poline JB. Valid conjunction inference with the minimum statistic. Neuroimage. 2005;25:653–60. https://doi.org/10.1016/j.neuroimage.2004.12.005
Smeland OB, Bahrami S, Frei O, Shadrin A, O’Connell K, Savage J, et al. Genome-wide analysis reveals extensive genetic overlap between schizophrenia, bipolar disorder, and intelligence. Mol Psychiatry. 2020;25:844–53. https://doi.org/10.1038/s41380-018-0332-x
Bahrami S, Steen NE, Shadrin A, O’Connell K, Frei O, Bettella F, et al. Shared genetic loci between body mass index and major psychiatric disorders: a Genome-Wide Association study. JAMA Psychiatry. 2020;77:503–12. https://doi.org/10.1001/jamapsychiatry.2019.4188
Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun. 2017;8:1826 https://doi.org/10.1038/s41467-017-01261-5
Genomes Project C, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526:68–74. https://doi.org/10.1038/nature15393
Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164 https://doi.org/10.1093/nar/gkq603
Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310–5. https://doi.org/10.1038/ng.2892
Boyle AP, Hong EL, Hariharan M, Cheng Y, Schaub MA, Kasowski M, et al. Annotation of functional variation in personal genomes using RegulomeDB. Genome Res. 2012;22:1790–7. https://doi.org/10.1101/gr.137323.112
Roadmap Epigenomics C, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–30. https://doi.org/10.1038/nature14248
Consortium G T, Laboratory D A, Coordinating Center -Analysis Working G, Statistical Methods groups-Analysis Working G, Enhancing G g, Fund N I H C. et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–13. https://doi.org/10.1038/nature24277
Chen J, Bardes EE, Aronow BJ, Jegga AG. ToppGene Suite for gene list enrichment analysis and candidate gene prioritization. Nucleic Acids Res. 2009;37:W305–311. https://doi.org/10.1093/nar/gkp427
Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 2014;10:e1004383 https://doi.org/10.1371/journal.pgen.1004383
Lee JR. Protein tyrosine phosphatase PTPRT as a regulator of synaptic formation and neuronal development. BMB Rep. 2015;48:249–55. https://doi.org/10.5483/bmbrep.2015.48.5.037
Yu L, Chibnik LB, Srivastava GP, Pochet N, Yang J, Xu J, et al. Association of brain DNA methylation in SORL1, ABCA7, HLA-DRB5, SLC24A4, and BIN1 with pathological diagnosis of Alzheimer disease. JAMA Neurol. 2015;72:15–24. https://doi.org/10.1001/jamaneurol.2014.3049
Zhang L, Silva TC, Young JI, Gomez L, Schmidt MA, Hamilton-Nelson KL, et al. Epigenome-wide meta-analysis of DNA methylation differences in prefrontal cortex implicates the immune processes in Alzheimer’s disease. Nat Commun. 2020;11:6114 https://doi.org/10.1038/s41467-020-19791-w
Dietz AG, Goldman SA, Nedergaard M. Glial cells in schizophrenia: a unified hypothesis. Lancet Psychiatry. 2020;7:272–81. https://doi.org/10.1016/S2215-0366(19)30302-5
Lam M, Chen CY, Li Z, Martin AR, Bryois J, Ma X, et al. Comparative genetic architectures of schizophrenia in East Asian and European populations. Nat Genet. 2019;51:1670–8. https://doi.org/10.1038/s41588-019-0512-x
Besco J, Popesco MC, Davuluri RV, Frostholm A, Rotter A. Genomic structure and alternative splicing of murine R2B receptor protein tyrosine phosphatases (PTPkappa, mu, rho and PCP-2). BMC Genomics. 2004;5:14 https://doi.org/10.1186/1471-2164-5-14
Lim SH, Kwon SK, Lee MK, Moon J, Jeong DG, Park E, et al. Synapse formation regulated by protein tyrosine phosphatase receptor T through interaction with cell adhesion molecules and Fyn. EMBO J. 2009;28:3564–78. https://doi.org/10.1038/emboj.2009.289
Jauhar S, Johnstone M, McKenna PJ. Schizophrenia. Lancet. 2022;399:473–86. https://doi.org/10.1016/S0140-6736(21)01730-X
Hollingworth P, Harold D, Sims R, Gerrish A, Lambert JC, Carrasquillo MM, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat Genet. 2011;43:429–35. https://doi.org/10.1038/ng.803
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43:436–41. https://doi.org/10.1038/ng.801
Crocker PR, Paulson JC, Varki A. Siglecs and their roles in the immune system. Nat Rev Immunol. 2007;7:255–66. https://doi.org/10.1038/nri2056
Zuccolo J, Bau J, Childs SJ, Goss GG, Sensen CW, Deans JP. Phylogenetic analysis of the MS4A and TMEM176 gene families. PLoS ONE. 2010;5:e9369 https://doi.org/10.1371/journal.pone.0009369
Hu J, Igarashi A, Kamata M, Nakagawa H. Angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide (A beta); retards A beta aggregation, deposition, fibril formation; and inhibits cytotoxicity. J Biol Chem. 2001;276:47863–8. https://doi.org/10.1074/jbc.M104068200
Kehoe PG, Miners S, Love S. Angiotensins in Alzheimer’s disease - friend or foe? Trends Neurosci. 2009;32:619–28. https://doi.org/10.1016/j.tins.2009.07.006
Kehoe PG, Passmore PA. The renin-angiotensin system and antihypertensive drugs in Alzheimer’s disease: current standing of the angiotensin hypothesis? J Alzheimers Dis. 2012;30(Suppl 2):S251–268. https://doi.org/10.3233/JAD-2012-111376
Wahlbeck K, Ahokas A, Miettinen K, Nikkila H, Rimon R. Higher cerebrospinal fluid angiotensin-converting enzyme levels in neuroleptic-treated than in drug-free patients with schizophrenia. Schizophr Bull. 1998;24:391–7. https://doi.org/10.1093/oxfordjournals.schbul.a033334
Baskan NM, Basaran A, Yenilmez C, Kurt H, Ozdemir F, Gunes HV, et al. Investigation of association between angiotensin-converting enzyme gene insertion/deletion polymorphism frequency in Turkish patients with schizophrenia. Genet Test Mol Biomarkers. 2010;14:753–7. https://doi.org/10.1089/gtmb.2010.0064
Gadelha A, Vendramini AM, Yonamine CM, Nering M, Berberian A, Suiama MA, et al. Convergent evidences from human and animal studies implicate angiotensin I-converting enzyme activity in cognitive performance in schizophrenia. Transl Psychiatry. 2015;5:e691 https://doi.org/10.1038/tp.2015.181
Reveret L, Leclerc M, Emond V, Tremblay C, Loiselle A, Bourassa P, et al. Higher angiotensin-converting enzyme 2 (ACE2) levels in the brain of individuals with Alzheimer’s disease. Acta Neuropathol Commun. 2023;11:159 https://doi.org/10.1186/s40478-023-01647-1
Deczkowska A, Matcovitch-Natan O, Tsitsou-Kampeli A, Ben-Hamo S, Dvir-Szternfeld R, Spinrad A, et al. Mef2C restrains microglial inflammatory response and is lost in brain ageing in an IFN-I-dependent manner. Nat Commun. 2017;8:717 https://doi.org/10.1038/s41467-017-00769-0
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45:1452–8. https://doi.org/10.1038/ng.2802
Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74:691–705. https://doi.org/10.1016/j.neuron.2012.03.026
Mitchell AC, Javidfar B, Pothula V, Ibi D, Shen EY, Peter CJ, et al. MEF2C transcription factor is associated with the genetic and epigenetic risk architecture of schizophrenia and improves cognition in mice. Mol Psychiatry. 2018;23:123–32. https://doi.org/10.1038/mp.2016.254
Zhang Z, Zhan X, Kim B, Wu J. A proteomic approach identifies SAFB-like transcription modulator (SLTM) as a bidirectional regulator of GLI family zinc finger transcription factors. J Biol Chem. 2019;294:5549–61. https://doi.org/10.1074/jbc.RA118.007018
Boyd PJ, Cunliffe VT, Roy S, Wood JD. Sonic hedgehog functions upstream of disrupted-in-schizophrenia 1 (disc1): implications for mental illness. Biol Open. 2015;4:1336–43. https://doi.org/10.1242/bio.012005
Parashar A, Jha D, Mehta V, Chauhan B, Ghosh P, Deb PK, et al. Sonic hedgehog signalling pathway contributes in age-related disorders and Alzheimer’s disease. Ageing Res Rev. 2024;96:102271 https://doi.org/10.1016/j.arr.2024.102271
Yang C, Qi Y, Sun Z. The role of sonic hedgehog pathway in the development of the central nervous system and aging-related neurodegenerative diseases. Front Mol Biosci. 2021;8:711710 https://doi.org/10.3389/fmolb.2021.711710
Acknowledgements
The authors would like to thank all the investigators and participants for sharing the related GWAS summary statistics data.
Funding
This work was supported by the Natural Science Foundation of China (81971599, 81771818, 82030053, 82001797, 82072001), Tianjin Natural Science Foundation (24ZXGQSY00040, 19JCYBJC25100), and Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-001A).
Author information
Authors and Affiliations
Contributions
Chunshui Yu and Huaigui Liu designed the study. Huaigui Liu, Yingying Xie, and Yuan Ji analyzed the data. Huaigui Liu, Yingying Xie, Yuan Ji, Wen Qin, Feng Liu and Chunshui Yu wrote the manuscript. Yujing Zhou, Jiayuan Xu, Jie Tang, Nana Liu, Hao Ding contributed to the analysis. Chunshui Yu, Feng Liu and Wen Qin co-supervised this work. All authors critically reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
All the datasets in our study are publicly available GWAS summary statistics, and hence no additional ethical approval and participants’ consent was required. Detailed ethical approval and participants’ consent can be found in the original GWAS publications.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, H., Xie, Y., Ji, Y. et al. Identification of genetic architecture shared between schizophrenia and Alzheimer’s disease. Transl Psychiatry 15, 150 (2025). https://doi.org/10.1038/s41398-025-03348-w
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03348-w
