
Abstract
The human brain, a pinnacle of biological complexity, comprises a diverse array of cell types that regulate cognition and maintain neural homeostasis. Advances in single-cell transcriptomics have revolutionized neuroscience by enabling high-resolution molecular profiling, revealing unprecedented insights into cellular heterogeneity, lineage dynamics, and disease-associated states. Large-scale brain-mapping initiatives have identified numerous novel cell types, yet their functional roles in health and disease remain poorly understood. This review synthesizes current knowledge of brain cell diversity, from neurogenesis to pathological states, and highlights key gene markers that define cellular identity and function. By integrating insights from single-cell transcriptomics, we explore how cellular diversity shapes brain function and contributes to disease mechanisms, providing a foundation for future research and translational applications.
Similar content being viewed by others


Introduction
The human brain, representing the epitome of biological complexity, comprises a diverse array of cell types that orchestrate cognition, behavior, and neural homeostasis. Over the past two million years, the brain has undergone rapid evolutionary changes, marked by significant expansion and specialization, particularly in the prefrontal cortex (PFC). These adaptations underpin advanced cognitive abilities such as language, decision-making, and memory. Despite these advancements, the cellular and molecular mechanisms driving brain function and disease remain incompletely understood.
A key distinction in human brain evolution lies not merely in size but in its neuronal organization and connectivity. Although Neanderthals possessed larger cranial capacities, Homo sapiens exhibit a significantly higher neuronal count and more intricate cortical architecture, highlighting the importance of coordinated brain organization in cognitive evolution. The human cerebral cortex, for instance, contains approximately 16.3 billion neurons [1], far surpassing those of chimpanzees (7.4 billion) [2] and mice (13.7 million) [3]. Comparative studies reveal both conserved and species-specific neuronal features, particularly in circuits governing complex behaviors such as language and higher-order cognition [4, 5]. Understanding this cellular diversity—spanning molecular identity, connectivity, and plasticity—is crucial for unravelling brain function in normal and disease states.
Recent breakthroughs in single-cell transcriptomics have revolutionized neuroscience by enabling high-resolution molecular profiling of individual cells [6]. This technology has unveiled unprecedented insights into cellular heterogeneity, lineage dynamics, and disease-associated states. Large-scale brain-mapping initiatives like the NIH’s BRAIN Initiative have identified hundreds of novel cell types [7], yet their functional roles in health and disease remain poorly characterized. For instance, the discovery of human-specific basal radial glia (bRG) subtypes has reshaped our understanding of cortical expansion, yet their role in neurodevelopmental disorders like autism spectrum disorder (ASD) remains underexplored.
In this review, we synthesize current knowledge of brain cell diversity, from neurogenesis to pathological states, and highlight key gene markers that define cellular identity and function. By integrating insights from single-cell transcriptomics, we explore how cellular diversity shapes brain function and contributes to disease mechanisms. This work provides a framework for future research and translational advancements, offering new avenues to address the complexities of the human brain in health and disease.
Cellular diversity in neurodevelopment and disease
Neurogenesis and brain cell plasticity
The human brain’s complexity arises from its diverse array of neurons and glial cells, which orchestrate complex behaviors and cognitive functions (Box 1). Neurogenesis, the process of generating new neurons from neural progenitors [8], is crucial for constructing functional neural circuits (Fig. 1). While embryonic and postnatal neurogenesis establish foundational neural networks critical for complex behaviors, adult neurogenesis persists in specialized niches like the hippocampal dentate gyrus and olfactory bulbs, integrating new neurons into existing circuits to enhance memory and cognitive plasticity [9]. Recent studies have shown that adult hippocampal neurogenesis is modulated by environmental enrichment and physical exercise, with deficits linked to depression and age-related cognitive decline [10, 11]. This process underscores the brain’s remarkable plasticity, enabling adaptation to environmental stimuli and experiences. However, disruptions in neurogenesis have been linked to neurological and psychiatric disorders, such as Alzheimer’s disease and depression, highlighting its dual role in health and pathology.

The left panel shows fetal neurogenesis in the developing neocortex, highlighting progenitor zones (ventricular, subventricular, intermediate) and migrating neurons. The middle panel depicts mature cortical layers with excitatory and inhibitory neurons. The right panel presents neurodevelopmental and neurodegenerative disorders linked to neuronal imbalances. Figure created using BioRender.com.

Neocortical evolution
The evolution of the human neocortex, responsible for higher cognitive functions, has seen significant changes in neural stem and progenitor cell (NSPC) populations. Comparative studies across mammals reveal a notable increase in basal progenitors and basal radial glia (bRG) in humans compared to species like mice, which lack these cell types [12,13,14,15]. These bRG subtypes, characterized by bifurcated basal processes, are absent in non-human primates (NHPs) such as macaques, suggesting evolutionary adaptations that drive cortical complexity. For example, Florio et al. [16] showed that human bRG cells exhibit prolonged proliferative capacity compared to mouse counterparts, enabling the generation of additional cortical layers [16]. Beyond progenitors, human-specific microglia in the dorsolateral prefrontal cortex (dlPFC) specialize in synaptic pruning and maintenance, diverging from the immune-focused roles seen in NHPs [17]. Additionally, astrocytes and oligodendrocyte progenitor cells show pronounced human-specific gene expression, enabling enhanced intercellular communication critical for higher cognition. Human astrocytes express distinct calcium signaling pathways that enhance their ability to modulate neuronal activity, a feature absent in chimpanzees [18].
These evolutionary innovations not only enhance cognitive abilities but also reveal vulnerabilities. Changes in human-specific neural stem cell populations, such as basal radial glia (bRG) and intermediate progenitors, have contributed to the complexity of the human cortex. However, disruptions in these cell types are associated with neurological disorders, including epilepsy, intellectual disability, and cortical malformations, highlighting the delicate balance between evolutionary advantages and developmental risks [19]. Additionally, enhanced astrocyte-microglia interactions, while crucial for brain function, may exacerbate neuroinflammatory responses in aging and Alzheimer’s disease [20, 21]. Importantly, emerging evidence indicates that human astrocytes and microglia display distinct molecular, morphological, and functional features compared to other species, suggesting that these interactions may have unique characteristics in the human brain that influence disease susceptibility [22, 23]. The interplay between evolutionary adaptations and disease susceptibility underscores the importance of studying human-specific cell types in both health and pathology. These examples illustrate how evolutionary trade-offs predispose the human brain to neurodevelopmental and neurodegenerative disorders. To understand the mechanisms underlying these pathologies, researchers are increasingly leveraging cutting-edge technologies, such as single-cell transcriptomics and multi-omics integration, to resolve cellular heterogeneity with unprecedented precision. In the following section, we explore how these breakthroughs are decoding the molecular architecture of brain cells and bridging evolutionary insights with clinical translation.
Cellular dynamics using single-cell technologies
Single-cell RNA sequencing and spatial transcriptomics
Brain transcriptome atlases, such as the Allen Human Brain Atlas, BrainSpan Atlas, and datasets from PsychENCODE have become invaluable tools for deciphering gene expression patterns across different brain regions and developmental stages in mammals [24,25,26]. Early studies revealed variations in messenger ribonucleic acid (mRNA) transcript abundance between human and NHP brains, providing crucial insights into the evolution of the primate brain transcriptome [27,28,29]. Furthermore, alternative splicing (AS) enhances transcriptome complexity, with the adult human brain exhibiting a higher AS prevalence, contributing significantly to evolutionary diversity [30, 31]. While traditional bulk RNA sequencing (RNA-seq) has identified differentially expressed genes (DEGs) in various diseases, its limitation lies in capturing cellular nuances and potential false positives due to sample variability.
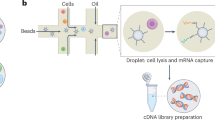
Recent breakthroughs in next-generation sequencing (NGS), single-cell isolation techniques, and molecular barcoding have revolutionized the generation of cDNA libraries from individual cells. The initial single-cell transcriptome analysis using NGS focused on early developmental stages, with subsequent innovations such as droplet-based microfluidic methods—including Drop-seq and inDrop—greatly enhancing the scalability and efficiency of single-cell RNA sequencing (scRNA-seq) [32]. Figure 2a outlines a representative scRNA-seq workflow, from sample preparation and RNA capture to primary, secondary, and tertiary computational analyses. While this workflow is commonly illustrated, we include it here to contextualize the analytical layers discussed later in this review. These high-throughput techniques enable the simultaneous capture and barcoding of thousands of single cells, significantly improving resolution and throughput. Extensive reviews have detailed the experimental workflow (Fig. 2a) of these technologies [33]. Moreover, the integration of single-cell technologies with rapid advancements in computational tools has ushered in a transformative era in this field. Supplementary Table 1 provides a compilation of various scRNA-seq techniques and commonly used bioinformatics tools for analysis.

a Workflow of scRNA-seq experiment. Schematic representation outlining the sequential steps involved in scRNA-seq, including single-cell isolation, cDNA synthesis, library preparation, sequencing, and subsequent data analysis. b Applications of scRNA-seq. Illustration highlighting diverse applications of scRNA-seq, ranging from cell type identification to dissecting cellular heterogeneity in complex biological systems. Figure created with BioRender.com.
Obtaining fresh human brain tissue for single-cell gene expression studies poses significant challenges. Single-nucleus RNA sequencing (snRNA-seq) has emerged as a viable solution, analyzing frozen post-mortem samples to characterize cellular diversity and isolate nuclei, particularly from complex cell types such as neurons [34]. Widely adopted in brain research, snRNA-seq enables the analysis of archived clinical materials in brain banks. While nuclei datasets may lack certain transcripts, snRNA-seq consistently replicates single-cell studies. Despite challenges, rapid advancements in scRNA-seq protocols and bioinformatics tools are expanding capabilities, enabling researchers to address questions beyond the scope of bulk transcriptomics.
Recent advancements in scRNA-seq have profoundly enhanced our understanding of the human brain and beyond. This technique has identified novel cell types and subtypes [35], characterized rare cell populations [36], and provided insights into evolutionary dynamics [37]. It has influenced established differentiation hierarchies, revealing insights into random allelic gene expression, developmental changes, and responses to stimuli [38]. Moreover, scRNA-seq distinguishes between normal and abnormal cells and identifies cell types linked to various pathological conditions [39]. Figure 2b illustrates its wide-ranging applications, including its role in uncovering disease-specific molecular mechanisms, potentially leading to new drug targets and biomarkers. Despite its maturity, the transformative impact of scRNA-seq lies in its ability to manage vast datasets, positioning it as a cornerstone of single-cell resolution technology in biomedical research and a potent source of untapped knowledge.
The influx of scRNA-seq data has led to a paradigm shift in classifying brain cell types. Initiatives like the Human Cell Atlas have realized the goal of comprehensively characterizing every human cell. Although, not yet widely applied in the studies summarized in this review, emerging integrative methods, such as combining biocytin staining (for neuronal morphology) [40], patch-seq (linking transcriptomics with electrophysiology) [41], and multiplexed error-robust fluorescence in situ hybridization MERFISH (spatial transcriptomics) [42], are enhancing the interpretability of single-cell data by connecting molecular signatures with cellular function and spatial context. These methods represent future directions that may further refine brain cell type classification and disease understanding. Furthermore, integrating single-projection neuron morphology with transcriptomic profiles confirms that molecular signatures align with major neuronal projection types, refining our understanding of brain circuits and functional organization.
Multi-omics integration
Building on these advances, cross-modal investigations are essential for achieving comprehensive single-cell anatomical mapping and refining cell type classifications in both healthy and pathological brain tissues. The potential of scRNA-seq extends beyond classification to investigating cellular responses to genetic perturbations through techniques like perturb-seq [43], which systematically introduces genetic modifications and captures their transcriptomic consequences at single-cell resolution. To gain deeper insights into cellular states, researchers are increasingly adopting single-cell multi-omics, which integrates transcriptomic data with proteomic, metabolomic, or chromatin accessibility information. For instance, cellular indexing of transcriptomes and epitopes by sequencing (CITE-seq) combines scRNA-seq with protein-marker detection using oligonucleotide-conjugated antibodies, allowing simultaneous analysis of gene expression and surface protein levels [44]. These multi-omics approaches hold immense potential for unraveling the molecular networks governing individual brain cells, enabling deeper investigations into neurodevelopment, pathology, and evolution.
While these insights have significantly advanced our understanding of human cellular dynamics, a fundamental question arises: how do these processes compare across species? To address this, recent studies have leveraged single-cell technologies for systematic cross-species comparisons, revealing both conserved and human-specific features of brain organization.
Novel cell types revealed by single-cell transcriptomics
In addition to the well-characterized bRG, recent scRNA-seq and snRNA-seq studies have identified previously unrecognized or novel brain cell populations. Among these, rosehip neurons represent a distinct subtype of layer 1 GABAergic interneurons, characterized by dense axonal arborization and restricted expression of canonical interneuron markers [45]. These cells have been identified exclusively in the human cortex and are hypothesized to modulate local microcircuit activity through targeted inhibitory signaling. Human-specific astrocyte subtypes have also been defined, marked by elevated expression of calcium signaling genes and transcriptional programs implicated in synaptic support and modulation, suggesting expanded functional roles relative to rodent astrocytes [27]. In neurodegenerative conditions, reactive hybrid glial populations—co-expressing astrocytic and oligodendrocyte precursor cell (OPC) markers—have been detected in both epilepsy and Alzheimer’s disease, implicating them in neuroinflammatory processes and gliosis [46]. Furthermore, scRNA-seq analyses have delineated distinct microglial subpopulations with developmentally restricted neuroprotective or pro-inflammatory transcriptional signatures, highlighting their dynamic and context-dependent roles in immune surveillance and neural remodeling [47]. During mid-gestational cortical development, transitional excitatory neuronal populations with unique and time-specific gene expression profiles have also been identified, underscoring critical windows of circuit formation [48]. Collectively, these findings exemplify the utility of single-cell transcriptomic technologies in refining brain cell taxonomies and uncovering novel cellular phenotypes and states relevant to both normal brain function and pathology.
Cross-species atlases
Recent advancements in single-cell technologies have revolutionized our ability to assess gene expression in thousands of individual cells simultaneously, facilitating detailed cross-species comparisons. This has enabled researchers to discern how differences in the diversity and abundance of cell types, rather than merely transcriptional variations, might underlie species-specific brain functions. Integrated transcriptomic and epigenomic analyses within the primary motor cortex (M1) of humans, NHPs, and mice have exemplified this progress, leading to the creation of comprehensive reference datasets like the Azimuth reference dataset [49,50,51]. This dataset consolidates maps from various single-cell references, including those relevant to brain data. Figure 3 represents the taxonomy of different cells derived from the primary motor cortex (M1) of human, marmoset, and mice. This comprehensive analysis, involving over 450,000 nuclei, has unveiled a multimodal, hierarchical classification of approximately 100 unique cell types in each species (Supplementary Table 2). These cell types exhibit distinct marker gene expression patterns and specific sites of accessible chromatin.

Circular dendrogram depicting primary motor cortex cell type classification [50]. Cell-type relationships were modelled using single-cell transcriptomic and epigenomic data from the BRAIN Initiative Cell Census Network (BICCN) study (PMID: 34616075), which profiled the primary motor cortex in humans, marmosets, and mice. Hierarchical structure was derived from curated parent–child relationships and node metadata (e.g., species, cell type annotations, proportions) provided in Supplementary Table 2. The directed graph was constructed using the igraph R package and visualized as a circular dendrogram using ggraph, employing a dendrogram layout optimized for tree structures. Node labels were radially aligned, and diagonal curved edges illustrate branching hierarchy. Species-specific cell types are distinguished by color (Human, Marmoset, Mouse), and additional attributes are visualized using a Viridis colormap. Cluster proportions represent the relative abundance of each neuronal or non-neuronal population. This figure highlights the cross-species conservation and diversity of cortical cell types in the primary motor cortex.
To delve deeper into human brain cellular composition and complexity, the National Institutes of Health’s (NIH) Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Initiative—Cell Census Network (BICCN)—has united diverse laboratories from multiple disciplines. Their collective mission is to systematically identify, characterize, and map every cell type within the brains of humans, NHPs, and rodents, encompassing molecular, electrophysiological, and morphological attributes. Siletti initiated the foundational work for the atlas by meticulously sequencing the RNA of over 3 million individual cells drawn from 106 distinct regions spanning the entirety of the human brain [52]. This analysis characterized 461 overarching classifications of brain cell types (Supplementary Table 3), encompassing an impressive array of over 3000 distinct subtypes.
Human-specific regulation
Despite significant efforts in characterizing brain cell types, we have yet to determine the role of human-specific genes and the specific brain cells they regulate. A comparative analysis of humans and our closest evolutionary relatives, chimpanzees and gorillas, reveals shared brain cell types with notable differences in the genes that regulate them [53]. These variations include genes associated with neuronal connections and neural circuit formation, suggesting that the enhancement of cognitive abilities during evolution may result from the adaptation of similar cell types in brain circuitry or the fine-tuning of their functions. Building upon the BICCN atlas introduced earlier, recent studies using single-cell approaches have explored regional and cell-type–specific expression patterns in greater detail. For example, using data from 75 adult donors, one study examined the middle temporal gyrus and reported inter-individual variability in the abundance and gene expression profiles, particularly in deep-layer glutamatergic neurons and microglia [54]. Discrepancies in cell type annotations across the studies shown in Fig. 4 arise from methodological variability in sc-RNA and sn-RNA sequencing workflows. For instance, studies on the human cortex report varying neuronal and glial subtypes due to differences in tissue source (e.g., frozen post-mortem samples in Siletti et al. vs. fresh fetal cortex in Zhu et al.) [52, 55], sequencing platform (e.g., SMART-seq, 10x Chromium, Drop-seq), and developmental stage (adult vs. prenatal). Analytical approaches also differ, including clustering algorithms (e.g., Louvain vs. Leiden), dimensionality reduction techniques, marker gene prioritization, and the integration of spatial transcriptomics. For example, Siletti et al. used whole-brain snRNA-seq to classify broad neuronal groups such as deep-layer corticothalamic neurons; Zhu et al. profiled fetal cortex to define early progenitors; Jorstad et al. applied MERFISH to resolve laminar excitatory and inhibitory neurons; and Johansen et al. used Leiden clustering to identify fine-grained cortical interneuron subtypes [52, 54,55,56]. These methodological and biological variations underscore the importance of interpreting reported taxonomies within their experimental context and highlight the ongoing need for standardized pipelines in cross-study comparisons.

Comprehensive representation of 11 studies identifying diverse brain cell types within the human brain. A collection of papers published in Science, Science Advances, and Science Translational Medicine by the NIH’s BRAIN Initiative – Cell Census Network (BICCN) presents a large-scale, multi-omics analysis of human brain cell types. Figure created using BioRender.com.
A comprehensive collection of twenty-one studies has compiled extensive single-cell datasets from both developing and adult brains in humans, NHPs, and mice [52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72]. The list of all brain cell types in NHPs and mice are presented in Supplementary Tables 4, 5, and 6. Figure 4 summarizes a synopsis of ten studies that have identified diverse brain cell types within the human brain. These studies have culminated in the creation of a human brain atlas, comprising over 3000 cell types, and offering insights into their locations and functions at different developmental stages. The proposed taxonomies and representative specimens for mammalian brain cell types will serve as a fundamental reference framework for comparative research across different species. The proposed taxonomies and representative specimens, many of which have emerged from BICCN and the broader BICAN ecosystem, now serve as a fundamental reference framework for comparative research across species.To further contextualize the diversity illustrated in Fig. 4, we provide an overview of key features from each contributing study. Siletti et al. [52] conducted large-scale snRNA-seq across 106 post-mortem adult human brain regions, defining over 3000 transcriptomically distinct cell types, including deep-layer excitatory neurons and specialized glial populations [52]. Zhu et al. [55] employed droplet-based scRNA-seq on fresh fetal cortex samples, identifying early progenitor populations such as excitatory neuron fetal-early and intermediate progenitor cells. Jorstad et al. (2023) combined high-depth scRNA-seq with MERFISH spatial transcriptomics to delineate laminar neuron subtypes—including L2/3 intratelencephalic neurons, chandelier cells, and somatostatin-expressing interneurons—in the middle temporal gyrus.
Johansen et al. [54] applied snRNA-seq coupled with Leiden clustering to classify fine-grained excitatory and inhibitory neuron subtypes in the adult cortex, such as L6 intratelencephalic Car3 neurons and SST-Chodl interneurons. Velmeshev et al. [71] profiled prenatal human cortex samples to identify early neurodevelopmental populations, including dorsal and ventral radial glia and excitatory neurons [71]. Ament et al. [73] focused on cerebellar cell diversity, highlighting granule neurons, Bergmann glia, and immune populations such as microglia and T cells. Kim et al. [66] profiled the developing thalamus, tracing progenitor cell trajectories and identifying temporally restricted neural precursors [73]. Tian et al. [69] created a cross-regional, multimodal brain atlas integrating snRNA-seq and epigenomic data, revealing distinct cortical, hippocampal, and cerebellar populations [69]. Finally, Li et al. [60] combined chromatin accessibility (ATAC-seq) and transcriptomic data to define conserved and divergent cell types across human, macaque, and mouse brains, identifying key neuronal markers including FOXP2, PVALB, and SST [60]. Together, these studies illustrate how technical, biological, and computational variables contribute to differences in reported brain cell taxonomies.
Moreover, comparative studies using scRNA-seq have illuminated neurogenic trajectories across species, offering insights into the regional and temporal differences in brain circuit formation and maturation. For instance, comparative analyses in NHPs and humans have highlighted conserved aspects of neuronal development alongside species-specific differences in oligodendrocyte maturation [74]. These findings emphasize the importance of cross-species comparisons in understanding neurodevelopmental processes. A notable example of human-specific genetic regulation involves ARHGAP11B, a gene associated with cortical expansion in humans. Dysregulation of ARHGAP11B has been linked to autism-related macrocephaly, highlighting its role in neurodevelopment [75]. Additionally, in neurodegenerative conditions like Alzheimer’s disease, hyperreactive microglia exacerbate neurotoxic inflammation [20], contributing to disease progression. These instances illustrate how evolutionary adaptations in brain cell types and circuits may confer cognitive advantages but also predispose the human brain to specific neurodevelopmental and neurodegenerative disorders. In the next section, we will delve into how disruptions in these specialized cell types and circuits further drive these pathologies.
Neurodevelopmental and neurodegenerative disorders
Brain connectivity is shaped by both genetic and environmental factors, which begins during embryonic development and continues through young adulthood. Disruptions in these processes contribute to neurodevelopmental disorders (NDDs), while neurodegenerative diseases (NDs), involve progressive neuronal loss and cognitive decline. Recent research has increasingly focused on identifying the specific brain cell types involved in these conditions, with particular attention to the unique molecular and cellular features of the human brain that may predispose individuals to these disorders.
Neurodevelopmental disorders
Neurodevelopmental disorders (NDDs) encompass a diverse range of conditions, including ASD, epileptic encephalopathy, and intellectual disabilities (IDs). These disorders are closely linked to disruptions in early neurodevelopmental processes, with genetic factors playing a central role in their pathogenesis [76,77,78,79].
ASD is characterized by deficits in social interaction and communication. scRNA-seq studies have shown that ASD-associated genes are enriched in specific cell types during development, particularly in mid-fetal projection neurons of the PFC and motor cortex [80]. A key pathological feature of ASD is an imbalance in excitatory/inhibitory (E/I) circuits, with downregulated genes in excitatory neurons and upregulated genes in glial cells [81]. Further integrative analyses of ASD-related variants using single-cell transcriptomes have revealed distinct expression patterns in both neuronal and non-neuronal subtypes, emphasizing the spatiotemporal specificity of gene regulation during development [82].
Epileptic encephalopathy, characterized by recurrent seizures and cognitive impairment, has also been studied using single-cell transcriptomics. Changes in gene expression across different neuronal subtypes, particularly in interneurons such as basket and chandelier cells, have been identified, implicating them in seizure initiation and propagation [83]. Additional investigations in epilepsy patients have uncovered transcriptomic disparities in various neuronal subtypes [84]. In temporal lobe epilepsy (TLE), snRNA-seq has identified a hybrid glial population with features of both reactive astrocytes and oligodendrocyte precursor cells, contributing to the pathological process [85].
Intellectual Disability (ID) is marked deficits in cognitive and adaptive behaviors, was traditionally studied through neuronal morphological changes. However, recent findings highlight the regulatory role of astrocytes in neural development [86]. Gene sequencing in ID patients reveals that many ID-related genes are predominantly expressed in astrocytes, rather than neurons [87, 88]. Fragile X Syndrome (FXS), a common inherited form of ID and ASD, has been investigated using scRNA-seq, revealing cell type-specific transcriptomic alterations that disrupt core functional processes, particularly in neurons [89].
Neurodegenerative diseases
Neurodegenerative diseases (NDs) are progressive conditions affecting the CNS, characterized by the gradual loss of neurons. Single-cell transcriptomic approaches have been instrumental in unraveling the cellular and molecular mechanisms underlying these diseases, shedding light on how subtle gene expression changes contribute to disease progression.
Parkinson’s Disease (PD) is primarily characterized by the loss of dopaminergic neurons and α-synuclein aggregation. Recent studies combining scRNA-seq with immunofluorescence analysis of the nigrostriatal pathway have identified microglial abnormalities in the midbrain, suggesting a broader cellular involvement in PD beyond dopaminergic neuron loss [90]. Post-mortem transcriptomic analyses have further linked PD risk variants to neuron- and microglia-specific genes, highlighting the role of neuroinflammation in disease progression [91].
Alzheimer’s Disease (AD), the most common neurodegenerative disorder, characterized by progressive cognitive decline, affects a wide range of cell types. scRNA-seq studies in AD patients have revealed widespread transcriptional changes, with downregulated genes in excitatory and inhibitory neurons and upregulation in astrocytes, microglia, and oligodendrocytes [92]. These findings have been consistent across studies investigating the prefrontal and entorhinal cortices, highlighting the critical role of glial cells in disease progression [93].
Huntington’s Disease (HD) primarily affects projection neurons in the striatum. Transcriptomic analyses of post-mortem HD brains have identified high expression of metallothionein (MT) genes in astrocytes, suggesting a neuroprotective response during disease progression [94]. Amyotrophic Lateral Sclerosis (ALS) presents with heterogeneity in both clinical manifestations and genetic underpinnings. scRNA-seq studies of ALS mouse brainstem samples have shown significant transcriptomic changes in inflammation, stress response, neurogenesis, and synaptic organization pathways [95]. Further studies on degenerating motor neurons from ALS patients have identified disrupted transcriptional networks, offering insights into ALS-specific disease mechanisms [96].
A compiled list of cell types associated with various disorders, based on analyses of published studies, is presented in Table 1. Collectively, these findings highlight the significance of integrating mechanistic insights into clinical frameworks. Longitudinal studies tracking aging and disease trajectories offer a promising approach to bridging the gap between molecular discoveries and therapeutic advancements.
Concluding remarks and future perspectives
The advent of single-cell transcriptomics has profoundly transformed our understanding of brain cell diversity, providing unprecedented resolution into the molecular and cellular mechanisms underlying neural development, function, and disease. By resolving the molecular profiles of individual cells, this technology has revealed the critical links between genetic regulation, cellular behavior, and neurological disorders. These insights are not only refining our classification of brain cell types but are also unveiling previously unrecognized cellular vulnerabilities that may serve as therapeutic targets.
Looking forward, the integration of multi-omics approaches—transcriptomics, epigenomics, proteomics, and metabolomics—will be pivotal to unravel the complex regulatory networks governing brain cell function and disease. Spatial transcriptomics will further refine our ability to map neural circuits at subcellular resolution, bridging the gap between molecular identity and anatomical connectivity. Concurrently, advances in computational modeling and artificial intelligence (AI) will enhance the integration of vast datasets, enabling precise predictions of cellular behaviors and disease trajectories.
Comparative cross-species analyses will remain instrumental in distinguishing evolutionarily conserved molecular programs from human-specific adaptations, shedding light on the genetic and cellular innovations that underlie higher cognitive functions. Future research will likely elucidate how species-specific gene regulation influences neuronal connectivity, synaptic plasticity, and cognition. Furthermore, single-cell and single-nucleus transcriptomics will facilitate longitudinal studies, enabling dynamic tracking of cellular changes across developmental stages, aging, and disease progression.
To bridge the gap between molecular discovery and clinical application, experimental validation using high-throughput CRISPR-based functional screens, brain organoids, and advanced in vivo models will be crucial. These approaches will enable precise dissection of disease-associated pathways at the level of specific cell types, paving the way for targeted interventions. By leveraging these technological advancements, neuroscience is poised to uncover fundamental principles of brain organization and function. This progress not only promises to deepen our understanding of human brain complexity but also holds immense potential for developing transformative therapies for neurological and psychiatric disorders, ultimately improving patient outcomes.
References
Silbereis J, Pochareddy S, Zhu Y, Li M, Neuron NS-, 2016 undefined. The cellular and molecular landscapes of the developing human central nervous system. CellComJC Silbereis, S Pochareddy, Y Zhu, M Li, N SestanNeuron, 2016•cellCom 2016. https://doi.org/10.1016/j.neuron.2015.12.008.
Collins CE, Turner EC, Sawyer EK, Reed JL, Young NA, Flaherty DK, et al. Cortical cell and neuron density estimates in one chimpanzee hemisphere. Proc Natl Acad Sci USA. 2016;113:740–5. https://doi.org/10.1073/PNAS.1524208113
Herculano-Houzel S, Mota B, Lent R. Cellular scaling rules for rodent brains. Proc Natl Acad Sci USA. 2006;103:12138–43. https://doi.org/10.1073/PNAS.0604911103
Khanna AR, Muñoz W, Kim YJ, Kfir Y, Paulk AC, Jamali M, et al. Single-neuronal elements of speech production in humans. Nature. 2024;626:603–10. https://doi.org/10.1038/s41586-023-06982-w
Leonard MK, Gwilliams L, Sellers KK, Chung JE, Xu D, Mischler G, et al. Large-scale single-neuron speech sound encoding across the depth of human cortex. Nature. 2023;626:593–602. https://doi.org/10.1038/s41586-023-06839-2
Aldridge S, Teichmann SA. Single cell transcriptomics comes of age. Nat Commun. 2020;11:1–4. https://doi.org/10.1038/s41467-020-18158-5
Penisson M, Ladewig J, Belvindrah R, Francis F. Genes and mechanisms involved in the generation and amplification of basal radial glial cells. Front Cell Neurosci. 2019;13:381 https://doi.org/10.3389/FNCEL.2019.00381
Ming GL, Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687 https://doi.org/10.1016/J.NEURON.2011.05.001
Boldrini M, Fulmore CA, Tartt AN, Simeon LR, Pavlova I, Poposka V, et al. Human hippocampal neurogenesis persists throughout aging. Cell Stem Cell. 2018;22:589–99.e5. https://doi.org/10.1016/j.stem.2018.03.015
Eisch AJ, Petrik D. Depression and hippocampal neurogenesis: a road to remission? Science. 2012;338:72 https://doi.org/10.1126/SCIENCE.1222941
Khalil MH. The BDNF-Interactive model for sustainable hippocampal neurogenesis in humans: synergistic effects of environmentally-mediated physical activity, cognitive stimulation, and mindfulness. Int J Mol Sci. 2024;25:12924 https://doi.org/10.3390/IJMS252312924
Hansen DV, Lui JH, Parker PRL, Kriegstein AR. Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature. 2010;464:554–61. https://doi.org/10.1038/NATURE08845
Betizeau M, Cortay V, Patti D, Pfister S, Gautier E, Bellemin-Ménard A, et al. Precursor diversity and complexity of lineage relationships in the outer subventricular zone of the primate. Neuron. 2013;80:442–57. https://doi.org/10.1016/j.neuron.2013.09.032
Fietz SA, Kelava I, Vogt J, Wilsch-Bräuninger M, Stenzel D, Fish JL, et al. OSVZ progenitors of human and ferret neocortex are epithelial-like and expand by integrin signaling. Nat Neurosci. 2010;13:690–9. https://doi.org/10.1038/NN.2553
Pollen AA, Nowakowski TJ, Chen J, Retallack H, Sandoval-Espinosa C, Nicholas CR, et al. Molecular identity of human outer radial glia during cortical development. Cell. 2015;163:55 https://doi.org/10.1016/J.CELL.2015.09.004
Florio M, Albert M, Taverna E, Namba T, Brandl H, Lewitus E, et al. Human-specific gene ARHGAP11B promotes basal progenitor amplification and neocortex expansion. Science. 2015;347:1465–70. https://doi.org/10.1126/SCIENCE.AAA1975
Mallya AP, Wang HD, Lee HNR, Deutch AY. Microglial pruning of synapses in the prefrontal cortex during adolescence. Cereb Cortex. 2019;29:1634–43. https://doi.org/10.1093/CERCOR/BHY061
Wang F, Smith NA, Xu Q, Fujita T, Baba A, Matsuda T, et al. Astrocytes modulate neural network activity by Ca 2+-dependent uptake of extracellular K +. Sci Signal 2012;5. https://doi.org/10.1126/SCISIGNAL.2002334/SUPPL_FILE/5_RA26_SM.PDF.
Klingler E, Francis F, Jabaudon D, Cappello S. Mapping the molecular and cellular complexity of cortical malformations. Science. 2021;371:eaba4517 https://doi.org/10.1126/SCIENCE.ABA4517/ASSET/30CE620B-4299-4DF8-9AF3-C7A0EDEF631E/ASSETS/GRAPHIC/371_ABA4517_F5.JPEG.
Singh D. Astrocytic and microglial cells as the modulators of neuroinflammation in Alzheimer’s disease. J Neuroinflammation. 2022;19:1–15. https://doi.org/10.1186/S12974-022-02565-0
Gudkov SV, Burmistrov DE, Kondakova EV, Sarimov RM, Yarkov RS, Franceschi C, et al. An emerging role of astrocytes in aging/neuroinflammation and gut-brain axis with consequences on sleep and sleep disorders. Ageing Res Rev. 2023;83:101775 https://doi.org/10.1016/J.ARR.2022.101775
Oberheim NA, Takano T, Han X, He W, Lin JHC, Wang F, et al. Uniquely hominid features of adult human astrocytes. J Neurosci. 2009;29:3276–87. https://doi.org/10.1523/JNEUROSCI.4707-08.2009
Geirsdottir L, David E, Keren-Shaul H, Weiner A, Bohlen SC, Neuber J, et al. Cross-Species single-cell analysis reveals divergence of the primate microglia program. Cell. 2019;179:1609–22.e16. https://doi.org/10.1016/j.cell.2019.11.010
Li M, Santpere G, Kawasawa YI, Evgrafov OV, Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science. 2018;362:eaat7615 https://doi.org/10.1126/SCIENCE.AAT7615/SUPPL_FILE/AAT7615_LI_TABLES_S1_TO_S16.XLSX.
Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, Szafer A, et al. Transcriptional landscape of the prenatal human brain. Nature. 2014;508:199–206. https://doi.org/10.1038/nature13185
Hawrylycz MJ, Lein ES, Guillozet-Bongaarts AL, Shen EH, Ng L, Miller JA, et al. An anatomically comprehensive atlas of the adult human brain transcriptome. Nature. 2012;489:391–9. https://doi.org/10.1038/nature11405.
Sousa AMM, Zhu Y, Raghanti MA, Kitchen RR, Onorati M, Tebbenkamp ATN, et al. Molecular and cellular reorganization of neural circuits in the human lineage. Science. 2017;358:1027–32. https://doi.org/10.1126/SCIENCE.AAN3456/SUPPL_FILE/AAN3456_SOUSA_SM.PDF.
Bakken TE, Miller JA, Ding SL, Sunkin SM, Smith KA, Ng L, et al. Comprehensive transcriptional map of primate brain development. Nature. 2016;535:367. https://doi.org/10.1038/NATURE18637
Pollen AA, Bhaduri A, Andrews MG, Nowakowski TJ, Meyerson OS, Mostajo-Radji MA, et al. Establishing cerebral organoids as models of human-specific brain evolution. Cell. 2019;176:743. https://doi.org/10.1016/J.CELL.2019.01.017
Gueroussov S, Gonatopoulos-Pournatzis T, Irimia M, Raj B, Lin ZY, Gingras AC, et al. An alternative splicing event amplifies evolutionary differences between vertebrates. Science. 2015;349:868–73. https://doi.org/10.1126/SCIENCE.AAA8381.
Recinos Y, Bao S, Wang X, Phillips BL, Yeh YT, Weyn-Vanhentenryck SM, et al. Lineage-specific splicing regulation of MAPT gene in the primate brain. Cell Genomics. 2024;4:100563. https://doi.org/10.1016/J.XGEN.2024.100563
Klein AM, Macosko E. InDrops and Drop-seq technologies for single-cell sequencing. Lab Chip. 2017;17:2540–1. https://doi.org/10.1039/C7LC90070H
Haque A, Engel J, Teichmann SA, Lönnberg T. A practical guide to single-cell RNA-sequencing for biomedical research and clinical applications. Genome Med. 2017;9:1–12. https://doi.org/10.1186/S13073-017-0467-4/TABLES/1
Slyper M, Porter CBM, Ashenberg O, Waldman J, Drokhlyansky E, Wakiro I, et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat Med. 2020;26:792–802. https://doi.org/10.1038/s41591-020-0844-1
Eze UC, Bhaduri A, Haeussler M, Nowakowski TJ, Kriegstein AR. Single-cell atlas of early human brain development highlights heterogeneity of human neuroepithelial cells and early radial glia. Nat Neurosci. 2021;24:584–94. https://doi.org/10.1038/s41593-020-00794-1
Grün D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–5. https://doi.org/10.1038/nature14966
Krienen FM, Goldman M, Zhang Q CH, del Rosario R, Florio M, Machold R, et al. Innovations present in the primate interneuron repertoire. Nature. 2020;586:262–9. https://doi.org/10.1038/s41586-020-2781-z
Marques S, van Bruggen D, Vanichkina DP, Floriddia EM, Munguba H, Väremo L, et al. Transcriptional convergence of oligodendrocyte lineage progenitors during development. Dev Cell. 2018;46:504–17.e7. https://doi.org/10.1016/J.DEVCEL.2018.07.005
Nagy C, Maitra M, Tanti A, Suderman M, Théroux JF, Davoli MA, et al. Single-nucleus transcriptomics of the prefrontal cortex in major depressive disorder implicates oligodendrocyte precursor cells and excitatory neurons. Nat Neurosci. 2020;23:771–81. https://doi.org/10.1038/s41593-020-0621-y
Tan S, Mo X, Qin H, Dong B, Zhou J, Long C, et al. Biocytin-Labeling in whole-cell recording: electrophysiological and morphological properties of pyramidal neurons in CYLD-Deficient mice. Molecules. 2023;28:4092. https://doi.org/10.3390/MOLECULES28104092/S1
van den Hurk M, Erwin JA, Yeo GW, Gage FH, Bardy C. Patch-Seq protocol to analyze the electrophysiology, morphology and transcriptome of whole single neurons derived from human pluripotent stem cells. Front Mol Neurosci. 2018;11:261. https://doi.org/10.3389/FNMOL.2018.00261
Xia C, Fan J, Emanuel G, Hao J, Zhuang X. Spatial transcriptome profiling by MERFISH reveals subcellular RNA compartmentalization and cell cycle-dependent gene expression. Proc Natl Acad Sci USA. 2019;116:19490–9. https://doi.org/10.1073/PNAS.1912459116/SUPPL_FILE/PNAS.1912459116.SD15.XLSX
Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853–66.e17. https://doi.org/10.1016/J.CELL.2016.11.038
Stoeckius M, Hafemeister C, Stephenson W, Houck-Loomis B, Chattopadhyay PK, Swerdlow H, et al. Large-scale simultaneous measurement of epitopes and transcriptomes in single cells. Nat Methods. 2017;14:865. https://doi.org/10.1038/NMETH.4380
Boldog E, Bakken TE, Hodge RD, Novotny M, Aevermann BD, Baka J, et al. Transcriptomic and morphophysiological evidence for a specialized human cortical GABAergic cell type. Nat Neurosci. 2018;21:1185. https://doi.org/10.1038/S41593-018-0205-2
Habib N, McCabe C, Medina S, Varshavsky M, Kitsberg D, Dvir-Szternfeld R, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701–6. https://doi.org/10.1038/s41593-020-0624-8
Masuda T, Sankowski R, Staszewski O, Böttcher C, Amann L, Sagar, et al. Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 2019;566:388–92. https://doi.org/10.1038/s41586-019-0924-x
Polioudakis D, de la Torre-Ubieta L, Langerman J, Elkins AG, Shi X, Stein JL, et al. A single-cell transcriptomic atlas of human neocortical development during mid-gestation. Neuron. 2019;103:785–801.e8. https://doi.org/10.1016/j.neuron.2019.06.011
Callaway EM, Dong HW, Ecker JR, Hawrylycz MJ, Huang ZJ, Lein ES, et al. A multimodal cell census and atlas of the mammalian primary motor cortex. Nature. 2021;598:86–102. https://doi.org/10.1038/s41586-021-03950-0
Bakken TE, Jorstad NL, Hu Q, Lake BB, Tian W, Kalmbach BE, et al. Comparative cellular analysis of motor cortex in human, marmoset and mouse. Nature. 2021;598:111–9. https://doi.org/10.1038/s41586-021-03465-8
Hao Y, Hao S, Andersen-Nissen E, Mauck WM, Zheng S, Butler A, et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–87.e29. https://doi.org/10.1016/J.CELL.2021.04.048
Siletti K, Hodge R, Mossi Albiach A, Lee KW, Ding SL, Hu L, et al. Transcriptomic diversity of cell types across the adult human brain. Science. 2023;382:eadd7046. https://doi.org/10.1126/SCIENCE.ADD7046/SUPPL_FILE/SCIENCE.ADD7046_MDAR_REPRODUCIBILITY_CHECKLIST.PDF.
Jorstad NL, Song JHT, Exposito-Alonso D, Suresh H, Castro-Pacheco N, Krienen FM, et al. Comparative transcriptomics reveals human-specific cortical features. Science. 2023;382:eade9516. https://doi.org/10.1126/SCIENCE.ADE9516/SUPPL_FILE/SCIENCE.ADE9516_DATA_S1.ZIP.
Johansen N, Somasundaram S, Travaglini KJ, Yanny AM, Shumyatcher M, Casper T, et al. Interindividual variation in human cortical cell type abundance and expression. Science. 2023;382:eadf2359. https://doi.org/10.1126/SCIENCE.ADF2359/SUPPL_FILE/SCIENCE.ADF2359_TABLES_S1_TO_S7.ZIP.
Zhu K, Bendl J, Rahman S, Vicari JM, Coleman C, Clarence T, et al. Multi-omic profiling of the developing human cerebral cortex at the single-cell level. Sci Adv. 2023;9:eadg3754. https://doi.org/10.1126/SCIADV.ADG3754/SUPPL_FILE/SCIADV.ADG3754_SM.V1.PDF
Jorstad NL, Close J, Johansen N, Yanny AM, Barkan ER, Travaglini KJ, et al. Transcriptomic cytoarchitecture reveals principles of human neocortex organization. Science. 2023;382:eadf6812. https://doi.org/10.1126/SCIENCE.ADF6812/SUPPL_FILE/SCIENCE.ADF6812_TABLES_S1_TO_S13.ZIP.
Costantini I, Morgan L, Yang J, Balbastre Y, Varadarajan D, Pesce L, et al. A cellular resolution atlas of Broca’s area. Sci Adv. 2023;9:eadg3844. https://doi.org/10.1126/SCIADV.ADG3844
Krienen FM, Levandowski KM, Zaniewski H, del Rosario RCH, Schroeder ME, Goldman M, et al. A marmoset brain cell census reveals regional specialization of cellular identities. Sci Adv. 2023;9:eadk3986. https://doi.org/10.1126/SCIADV.ADK3986
Han X, Guo S, Ji N, Li T, Liu J, Ye X, et al. Whole human-brain mapping of single cortical neurons for profiling morphological diversity and stereotypy. Sci Adv. 2023;9:eadf3771. https://doi.org/10.1126/SCIADV.ADF3771
Li YE, Preissl S, Miller M, Johnson ND, Wang Z, Jiao H, et al. A comparative atlas of single-cell chromatin accessibility in the human brain. Science. 2023;382:eadf7044. https://doi.org/10.1126/SCIENCE.ADF7044.
Chiou KL, Huang X, Bohlen MO, Tremblay S, DeCasien AR, O’Day DR, et al. A single-cell multi-omic atlas spanning the adult rhesus macaque brain. Sci Adv. 2023;9:eadh1914. https://doi.org/10.1126/SCIADV.ADH1914
Wilbers R, Metodieva VD, Duverdin S, Heyer DB, Galakhova AA, Mertens EJ, et al. Human voltage-gated Na + and K + channel properties underlie sustained fast AP signaling. Sci Adv. 2023;9:eade3300. https://doi.org/10.1126/SCIADV.ADE3300
Komiyama T. Diversity of primate brain cells unraveled. Sci Adv. 2023;9:eadl0650. https://doi.org/10.1126/SCIADV.ADL0650
Wilbers R, Galakhova AA, Driessens SLW, Heistek TS, Metodieva VD, Hagemann J, et al. Structural and functional specializations of human fast-spiking neurons support fast cortical signaling. Sci Adv. 2023;9:eadf0708. https://doi.org/10.1126/SCIADV.ADF0708
Rózsa M, Tóth M, Oláh G, Baka J, Lákovics R, Barzó P, et al. Temporal disparity of action potentials triggered in axon initial segments and distal axons in the neocortex. Sci Adv. 2023;9:eade4511 https://doi.org/10.1126/SCIADV.ADE4511
Kim CN, Shin D, Wang A, Nowakowski TJ. Spatiotemporal molecular dynamics of the developing human thalamus. Science. 2023;382:eadf9941. https://doi.org/10.1126/SCIENCE.ADF9941.
Chartrand T, Dalley R, Close J, Goriounova NA, Lee BR, Mann R, et al. Morphoelectric and transcriptomic divergence of the layer 1 interneuron repertoire in human versus mouse neocortex. Science. 2023;382:eadf0805. https://doi.org/10.1126/SCIENCE.ADF0805.
Lee BR, Dalley R, Miller JA, Chartrand T, Close J, Mann R, et al. Signature morphoelectric properties of diverse GABAergic interneurons in the human neocortex. Science. 2023;382:eadf6484. https://doi.org/10.1126/SCIENCE.ADF6484.
Tian W, Zhou J, Bartlett A, Zeng Q, Liu H, Castanon RG, et al. Single-cell DNA methylation and 3D genome architecture in the human brain. Science. 2023;382:eadf5357. https://doi.org/10.1126/SCIENCE.ADF5357.
Micali N, Ma S, Li M, Kim S-K, Mato-Blanco X, Sindhu SK, et al. Molecular programs of regional specification and neural stem cell fate progression in macaque telencephalon. Science. 2023;382:eadf3786. https://doi.org/10.1126/SCIENCE.ADF3786.
Velmeshev D, Perez Y, Yan Z, Valencia JE, Castaneda-Castellanos DR, Wang L, et al. Single-cell analysis of prenatal and postnatal human cortical development. Science. 2023;382:eadf0834. https://doi.org/10.1126/SCIENCE.ADF0834.
Braun E, Danan-Gotthold M, Borm LE, Lee KW, Vinsland E, Lönnerberg P, et al. Comprehensive cell atlas of the first-trimester developing human brain. Science. 2023;382:eadf1226. https://doi.org/10.1126/SCIENCE.ADF1226.
Ament SA, Cortes-Gutierrez M, Herb BR, Mocci E, Colantuoni C, McCarthy MM. A single-cell genomic atlas for maturation of the human cerebellum during early childhood. Sci Transl Med. 2023;15:eade1283. https://doi.org/10.1126/SCITRANSLMED.ADE1283/SUPPL_FILE/SCITRANSLMED.ADE1283_MDAR_REPRODUCIBILITY_CHECKLIST.PDF
Zhu Y, Sousa AMM, Gao T, Skarica M, Li M, Santpere G, et al. Spatiotemporal transcriptomic divergence across human and macaque brain development. Science. 2018;362:eaat8077. https://doi.org/10.1126/SCIENCE.AAT8077
Heide M, Huttner WB, El Ghouzzi V, Planck M. Human-Specific genes, cortical progenitor cells, and microcephaly. Cells. 2021;10:1209. https://doi.org/10.3390/CELLS10051209
Doi M, Usui N, Shimada S. Prenatal environment and neurodevelopmental disorders. Front Endocrinol. 2022;13:860110. https://doi.org/10.3389/FENDO.2022.860110/BIBTEX
Uddin M, Tammimies K, Pellecchia G, Alipanahi B, Hu P, Wang Z, et al. Brain-expressed exons under purifying selection are enriched for de novo mutations in autism spectrum disorder. Nat Genet. 2014;46:742–7. https://doi.org/10.1038/NG.2980
Akter H, Rahman MM, Sarker S, Basiruzzaman M, Islam MM, Rahaman MA, et al. Construction of copy number variation landscape and characterization of associated genes in a Bangladeshi cohort of neurodevelopmental disorders. Front Genet. 2023;14:955631. https://doi.org/10.3389/FGENE.2023.955631
Safizadeh Shabestari SA, Nassir N, Sopariwala S, Karimov I, Tambi R, Zehra B, et al. Overlapping pathogenic de novo CNVs in neurodevelopmental disorders and congenital anomalies impacting constraint genes regulating early development. Hum Genet. 2023;142:1201–13. https://doi.org/10.1007/S00439-022-02482-5
Wang P, Zhao D, Lachman HM, Zheng D. Enriched expression of genes associated with autism spectrum disorders in human inhibitory neurons. Transl Psychiatry. 2018;8:13. https://doi.org/10.1038/S41398-017-0058-6
Velmeshev D, Schirmer L, Jung D, Haeussler M, Perez Y, Mayer S, et al. Single-cell genomics identifies cell type–specific molecular changes in autism. Science. 2019;364:685–9. https://doi.org/10.1126/SCIENCE.AAV8130/SUPPL_FILE/AAV8130_VELMESHEV_SM.PDF.
Nassir N, Bankapur A, Samara B, Ali A, Ahmed A, Inuwa IM, et al. Single-cell transcriptome identifies molecular subtype of autism spectrum disorder impacted by de novo loss-of-function variants regulating glial cells. Hum Genomics. 2021;15:1–16. https://doi.org/10.1186/S40246-021-00368-7/FIGURES/6
Magloire V, Mercier MS, Kullmann DM, Pavlov I. GABAergic interneurons in seizures: investigating causality with optogenetics. Neuroscientist. 2018;25:344 https://doi.org/10.1177/1073858418805002
Pfisterer U, Petukhov V, Demharter S, Meichsner J, Thompson JJ, Batiuk MY, et al. Identification of epilepsy-associated neuronal subtypes and gene expression underlying epileptogenesis. Nat Commun. 2020;11:5038. https://doi.org/10.1038/S41467-020-18752-7
Pai B, Tome-Garcia J, Cheng WS, Nudelman G, Beaumont KG, Ghatan S, et al. High-resolution transcriptomics informs glial pathology in human temporal lobe epilepsy. Acta Neuropathol Commun. 2022;10:1–19. https://doi.org/10.1186/S40478-022-01453-1/FIGURES/6
Cresto N, Pillet LE, Billuart P, Rouach N. Do astrocytes play a role in intellectual disabilities? Trends Neurosci. 2019;42:518–27. https://doi.org/10.1016/J.TINS.2019.05.011
Nassir N, Sati I, Al Shaibani S, Ahmed A, Almidani O, Akter H, et al. Detection of copy number variants and genes by chromosomal microarray in an Emirati neurodevelopmental disorders cohort. Neurogenetics. 2022;23:137–49. https://doi.org/10.1007/S10048-022-00689-2
Lelieveld SH, Reijnders MRF, Pfundt R, Yntema HG, Kamsteeg EJ, De Vries P, et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat Neurosci. 2016;19:1194–6. https://doi.org/10.1038/NN.4352
Donnard E, Shu H, Garber M. Single cell transcriptomics reveals dysregulated cellular and molecular networks in a fragile X syndrome model. PLoS Genet. 2022;18:e1010221. https://doi.org/10.1371/JOURNAL.PGEN.1010221
Uriarte Huarte O, Kyriakis D, Heurtaux T, Pires-Afonso Y, Grzyb K, Halder R, et al. Single-Cell transcriptomics and in situ morphological analyses reveal microglia heterogeneity across the nigrostriatal pathway. Front Immunol. 2021;12:639613. https://doi.org/10.3389/FIMMU.2021.639613/FULL
Smajić S, Prada-Medina CA, Landoulsi Z, Ghelfi J, Delcambre S, Dietrich C, et al. Single-cell sequencing of human midbrain reveals glial activation and a Parkinson-specific neuronal state. Brain. 2022;145:964–78. https://doi.org/10.1093/brain/awab446
Mathys H, Davila-Velderrain J, Peng Z, Nature FG-, 2019 undefined. Single-cell transcriptomic analysis of Alzheimer’s disease. NatureComH Mathys, J Davila-Velderrain, Z Peng, F Gao, S Mohammadi, JZ Young, M Menon, L HeNature, 2019•natureCom n.d.
Grubman A, Chew G, Ouyang J, Sun G, … XC-N, 2019 undefined. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. NatureComA Grubman, G Chew, JF Ouyang, G Sun, XY Choo, C McLean, RK Simmons, S BuckberryNature Neuroscience, 2019•natureCom n.d.
Al-Dalahmah O, Sosunov AA, Shaik A, Ofori K, Liu Y, Vonsattel JP, et al. Single-nucleus RNA-seq identifies Huntington disease astrocyte states n.d. https://doi.org/10.1186/s40478-020-0880-6.
Liu W, Venugopal S, Majid S, Ahn I, … GD-N of, 2020 undefined.. Single-cell RNA-seq analysis of the brainstem of mutant SOD1 mice reveals perturbed cell types and pathways of amyotrophic lateral sclerosis. Elsevier n.d.
Namboori SC, Thomas P, Ames R, Hawkins S, Garrett LO, Willis CRG, et al. Single-cell transcriptomics identifies master regulators of neurodegeneration in SOD1 ALS iPSC-derived motor neurons. Stem Cell Rep. 2021;16:3020–35. https://doi.org/10.1016/J.STEMCR.2021.10.010
Jäkel S, Dimou L. Glial cells and their function in the adult brain: a journey through the history of their ablation. Front Cell Neurosci. 2017;11:235525. https://doi.org/10.3389/FNCEL.2017.00024/BIBTEX
Swanson OK, Maffei A. From hiring to firing: activation of inhibitory neurons and their recruitment in behavior. Front Mol Neurosci. 2019;12:168. https://doi.org/10.3389/FNMOL.2019.00168
Ophir O, Shefi O, Lindenbaum O. Classifying neuronal cell types based on shared electrophysiological information from humans and mice. Neuroinformatics. 2024;22:473. https://doi.org/10.1007/S12021-024-09675-5
Acknowledgements
This work was supported, in whole or in part, by the Al Jalila Foundation, internal grant award from Mohammed Bin Rashid University of Medicine and Health Sciences (MBRU) – College of Medicine (MBRU-CM-RG2024-15); Al Jalila Foundation Research Grants (AJF2023-103; AJF2023-207).
Author information
Authors and Affiliations
Contributions
AS and MU conceived the study. AS, BB, NN, HA, ZS, and MU performed data curation from literature and various databases. AS, BB, BKB, MWS, RK, BC, HTB, and MU contributed to writing the manuscript. AS, BB, and MU worked on the figures and tables.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Soorajkumar, A., Balan, B., Nassir, N. et al. Mapping human brain cell type origin and diseases through single-cell transcriptomics. Transl Psychiatry 15, 349 (2025). https://doi.org/10.1038/s41398-025-03562-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03562-6

{kind=link}
{kind=link}
{kind=link}