
Abstract
Schizophrenia (SCZ), bipolar disorder (BD), and major depressive disorder (MDD) share common genetic and environmental risk factors, with immune-inflammatory dysregulation playing a crucial role in their pathophysiology. However, further research is needed to clarify causal relationships. We conducted a Mendelian randomization (MR) study using summary statistics from large-scale genome-wide association studies (GWAS) of European ancestry as instrumental variables to assess the association between 95 circulating inflammatory proteins and the risk of SCZ, BD, and MDD. Initially, a bidirectional two-sample MR analysis was performed to clarify the direction of causality. Subsequently, multivariable MR analysis was employed to investigate whether the effects of different circulating proteins on psychiatric disorders exhibit complex overlap or interrelated effects. Finally, colocalization analysis was applied to determine whether circulating proteins share causal genetic variants with psychiatric disorders. To enhance the robustness of our findings, a series of sensitivity analyses were conducted, including tests for horizontal pleiotropy, heterogeneity, and rigorous quality control procedures. Discovery analyses were based on data from the Psychiatric Genomics Consortium (PGC), while validation was performed using data from the FinnGen biobank. Meta-analysis was used to integrate results from multiple data sources. Evidence strength was evaluated based on consistency across MR, replication, and colocalization. Forward univariable MR and meta-analysis revealed that elevated circulating C-reative protein (CRP) (OR = 0.93, Pmeta = 0.013) and fractalkine(CX3CL1) (OR = 0.92, PIVW = 0.040) significantly reduced the risk of SCZ, whereas Delta and Notch-like epidermal growth factor-related receptor (DNER) (OR = 1.07, Pmeta = 0.007) and eukaryotic translation initiation factor 4E-binding protein 1 (EIF4EBP1)(OR = 1.09, PIVW = 0.015) was associated with an increased risk of SCZ. For MDD, fibroblast growth factor 23 (FGF23) (OR = 0.97, Pmeta = 0.015) and interleukin-20 (IL20) (OR = 0.96, PIVW = 0.028) significantly reduced the risk, whereas tumor necrosis factor ligand superfamily member 12 (TNFSF12) was associated with an increased risk of MDD (OR = 1.02, Pmeta = 0.039). In BD, elevated circulating levels of C-X-C motif chemokine 5 (CXCL5) (OR = 0.95, PIVW = 0.024), TRANCE (OR = 0.95, Pmeta = 0.016), and CC motif chemokine ligand 7 (CCL7) (OR = 0.94, Pmeta = 0.023) were significantly associated with reduced risk, suggesting potential protective effects. In multivariable MR analysis, after adjusting for other inflammatory protein levels, CRP showed a significant protective effect on SCZ (OR = 0.91, PIVW = 0.008), FGF23 demonstrated a significant protective effect on MDD (OR = 0.96, PIVW = 0.038), and TNFSF12 significantly increased the risk of MDD (OR = 1.03, PIVW = 0.032). Colocalization analysis provided strong evidence for shared genetic variants between DNER and SCZ (PH4 = 0.89), with the rs35975053 locus being the most significant. This MR study suggests potential causal links between inflammatory proteins and psychiatric disorders. CRP and CX3CL1 were protective for SCZ, while DNER and EIF4EBP1 increased risk. FGF23 and IL20 were protective for MDD, whereas TNFSF12 increased risk. CXCL5, TRANCE, and CCL7 were suggestively protective for BD. The shared causal genetic variants between DNER and SCZ suggest a mechanism underlying neuropsychiatric pathogenesis. Further experimental studies are warranted to elucidate the underlying biological mechanisms and identify novel diagnostic biomarkers and therapeutic targets.
Similar content being viewed by others
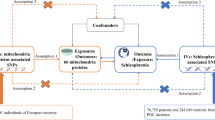

Introduction
Schizophrenia (SCZ), bipolar disorder (BD), and major depressive disorder (MDD) are major contributors to the global burden of disease—MDD affects over 3400 individuals per 100,000, BD impacts nearly 40 million people worldwide, and SCZ leads to considerable disability due to its early onset and chronic course [1]. These disorders share overlapping genetic and environmental mechanisms. For instance, the liability to BD due to common genetic variants has a genetic correlation (rg) of approximately 0.7 with SCZ and around 0.45 with MDD [2]. Liability to SCZ and MDD also overlap, with rg of approximately 0.4 [3]. Furthermore, adverse environmental factors, including infections and psychological stress, especially in early life, may lead to dysregulated immune-inflammatory responses, which are recognized as significant contributors to the development of these disorders [4].
The complex interaction between genetic predispositions and environmental factors can exacerbate immune disturbances, characterized by disruption in the homeostatic balance between pro- and anti-inflammatory cytokines, further influencing neurodevelopment and increasing the risk of SCZ, BD and MDD [5]. Meta-analysis also indicated that there were similarities in the pattern of blood cytokine alterations in these major psychiatric disorders during acute and chronic phases of illness, raising the possibility of common underlying pathways for immune dysfunction [6]. Although some prospective longitudinal studies have found elevated inflammatory markers are associated with subsequent risk of psychotic disorders [7,8,9,10,11], suggesting inflammation may be a causative factor of illness rather than a consequence, residual confounding could still be an issue. For example, mental disorders are often comorbid with chronic physical conditions, such as hypertension, cardiovascular disease, metabolic disease, as well as asthma, whereas they are all associated with chronic inflammation [12, 13].
Mendelian randomization (MR) is an important genetic epidemiological tool employed for causal inference. This method fundamentally relies on the utilization of genetic variants as instrumental variables, which are inherited by offspring at the moment of fertilization. This inheritance process mitigates potential biases inherent in traditional observational studies [14]. Consequently, researchers can more accurately evaluate the causal relationships between modifiable exposure factors, such as lifestyle or biomarkers, and health outcomes. In recent years, some studies have utilized MR to explore the association between inflammatory biomarkers and psychiatric disorders. For instance, genetically-predicted soluble interleukin-6 receptor (sIL-6R) [15, 16] and IL-6 [17] were reported to be risk factors for both SCZ and MDD. Previous observational studies have shown that the levels of tumor necrosis factor (TNF) increased in patients with SCZ [6, 18, 19], MR analysis also suggested a causal link of TNF with SCZ [20]. However, there were some inconsistent results, such as C reactive protein (CRP). Meta-analytic observational evidence showed that patients with SCZ and MDD had higher CRP levels than controls [21, 22], which seems to have achieved a consensus, while MR studies did not find evidence for a reciprocal relationship between CRP levels and these two disorders [17, 23], suggesting further research is required to enable a more definitive conclusion. For BD, recent MR studies have suggested potential causal links between circulating inflammatory proteins (e.g., ISG15, MLN, and CD40) and reduced disease risk, supporting the involvement of immune-related pathways in its pathogenesis [24, 25]. However, these findings remain inconsistent, and further research is needed to clarify robust inflammatory biomarkers and their clinical implications in BD [26].
Additionally, recent genome-wide association study (GWAS) meta-analyses have been performed to identify the genetic basis underlying a broader range of circulating cytokines [27, 28], beyond a specific set of cytokines, providing an opportunity to gain a more thorough comprehension of the potential associations between inflammation-related proteins and mental disorders. So far, there have been a few univariable MR (UVMR) studies investigating the causal relationships of 41/91 cytokines and chemokines detected in blood samples with SCZ, BD or MDD [29,30,31]. Nevertheless, inflammatory proteins exhibit a highly intricate and interconnected network of actions with multiple overlapping functions, necessitating the use of multivariable MR (MVMR) analysis.
We hypothesize that circulating inflammatory proteins have a direct effect on the risk of psychiatric disorders. In this study, we employed two-sample MVMR and colocalization analyses to comprehensively investigate the associations between genetic predispositions to 95 circulating inflammation-related biomarkers and SCZ, BD, as well as MDD. To further enhance the robustness of our findings, we integrated bidirectional MR estimates from different data sources of psychiatric disorders through meta-analyses.
Materials and methods
Study design
As shown in Fig. 1, MR analysis is based on three fundamental assumptions to ensure the reliability of its findings: (1) a strong association between genetic variants and the exposure (relevance assumption); (2) genetic variants are independent of potential confounders that could influence the relationship between the exposure and the outcome (independence assumption); and (3) genetic variants affect the outcome solely through their influence on the exposure (exclusion restriction assumption).

Colored results represent key findings.
Following the Strengthening the Reporting of Observational Studies in Epidemiology Using MR (STROBE-MR) guidelines, we used summary-level data from large-scale GWAS of 95 circulating inflammatory proteins and three major psychiatric disorders. SNPs were selected as instrumental variables based on prior evidence linking these proteins to neuroinflammation, immune–brain communication, and neurodevelopment. Discovery analyses were conducted using GWAS data from the Psychiatric Genomics Consortium (PGC), with validation in the FinnGen biobank (https://r11.finngen.fi/) and meta-analysis integrating both datasets. Bayesian colocalization (COLOC) analyses were further applied to assess whether shared genetic variants influence both protein levels and psychiatric disorder risk. All participants in the original GWAS provided informed consent, and all data were obtained from published studies and public resources, requiring no additional ethical approval.
GWASs of circulating inflammatory proteins
The dataset of 95 circulating inflammatory protein levels used in this study was obtained from three studies (Table 1). A genome-wide protein quantitative trait loci (pQTL) study utilized the Olink Target platform to measure 91 plasma proteins in 14,824 participants, identifying their genetic determinants [28]. CRP data were derived from the GWAS by Ligthart et al. [32], which analyzed data from over 200,000 participants across 88 studies. Additionally, data for IL-1 beta, IL6RA, and IL6RB were sourced from a GWAS by Sun et al. [33], which examined genome-wide associations for 2994 plasma proteins in 3301 individuals. Details on the abbreviations, full names, and sample sizes of the 95 circulating inflammatory proteins were provided in Table S1.
GWASs of psychiatric disorders
This study focused on the causal relationships between circulating inflammatory protein levels and three psychiatric disorders. To ensure robust MR analysis, we utilized large-scale publicly available GWAS datasets while minimizing potential biases from sample overlap. To achieve this, we included only GWAS datasets derived from European ancestry cohorts of distinct study populations. These datasets included summary statistics for SCZ [34], BD [2], and MDD [35] from the PGC as well as corresponding data from the FinnGen biobank (Table 1). All GWAS summary statistics were obtained from publicly available sources.
Instruments selection
To satisfy the three core assumptions of MR analysis, we implemented a rigorous instrumental variable (IV) selection process. First, we identified single nucleotide polymorphisms (SNPs) strongly associated with the exposure (P < 1 × 10⁻⁵). Second, to account for linkage disequilibrium (LD), we used PLINK software (https://www.cog-genomics.org/plink/2.0/) to perform clumping, retaining only independent SNPs within a 1 Mb window (r² threshold = 0.001), using the 1000 Genomes European sample data as the reference panel. Finally, SNPs significantly associated with the outcome (P < 5 × 10⁻⁵) were excluded to minimize potential confounding effects.
Data harmonization and quality control
Before conducting MR analysis, exposure and outcome data were harmonized by removing palindromic SNPs (e.g., A/T or G/C alleles) to ensure that IVs were aligned to the same DNA strand. Additionally, unmatched variants between exposure and outcome datasets were excluded. A core principle of MR analysis is the use of genetic variants as instrumental variables to infer causal relationships between exposure and outcome. To ensure the reliability and robustness of the genetic instruments, several quality control measures were implemented. First, Cochran’s Q test was performed in the inverse-variance weighted (IVW) model to assess heterogeneity, with outliers (P < 0.05) removed and an I² value threshold of ≤25% set to control for heterogeneity. The MR-Egger model was further assessed using Rucker’s Q′ test to detect horizontal pleiotropy, where P > 0.05 indicated no significant pleiotropic effects. Second, F-statistics were calculated to assess the strength of IVs, with an F-statistic >10 suggesting a low risk of weak instrument bias in MR analyses [36]. Finally, SNPs were filtered using the Steiger test to minimize reverse causality and reinforce the exclusion restriction assumption [37].
Bidirectional two-sample MR analyses
To investigate the causal relationship between circulating inflammatory proteins and psychiatric disorders, we conducted bidirectional two-sample MR analyses. In the forward MR analysis, 95 circulating inflammatory protein levels were used as exposures, with three psychiatric disorders as outcomes. Given that psychiatric disorders may also influence circulating inflammatory protein levels, reverse MR analysis was performed using psychiatric disorders as exposures and inflammatory proteins as outcomes. As the IVW method provides precise causal estimates when all IVs are valid, we employed the random-effects IVW approach as the primary analysis in UVMR and the multivariable IVW (MV-IVW) method in MVMR. However, IVW estimates may be biased in the presence of horizontal pleiotropy [38]. To assess the robustness of our findings under different assumptions, we additionally applied complementary MR methods: the weighted median, which yields consistent estimates when up to 50% of IVs are invalid; the weighted mode, which provides unbiased estimates when the largest cluster of IVs is valid; the simple mode, which is robust when the most frequent causal effect estimate among IVs represents the true effect; and MR-Egger, which can detect and correct for directional pleiotropy even if all IVs are affected. The causal effect was quantified using odds ratios (OR) and 95% confidence intervals (CI), representing the change in psychiatric disorder risk associated with a one-unit increase or decrease in circulating inflammatory protein levels.
Sensitivity analyses
We conducted a series of sensitivity analyses to validate the MR results. First, the MR-PRESSO global test was applied to assess horizontal pleiotropy (P < 0.05 indicates the presence of pleiotropy) [39]. Second, MR-Egger regression was used to detect potential bias from directional pleiotropy, with the intercept value representing the average pleiotropic effect of all genetic variants; a non-zero intercept (P < 0.05) indicates the presence of directional pleiotropy [40]. Additionally, a leave-one-out analysis was performed, where one IV was removed at a time, and the causal relationship was re-estimated to determine if any single SNP was driving the causal inference [38].
Validation analysis
Additionally, we performed bidirectional two-sample MR analyses using data from the FinnGen biobank, with three psychiatric disorders as outcomes, to further validate the relationship between circulating inflammatory proteins and psychiatric disorders. The results from both data sources, analyzed using the IVW method, were then combined in a meta-analysis (fixed-effects model applied when I² ≤ 50%). The combined P-values were subsequently adjusted for false discovery rate (FDR) to account for multiple comparisons.
Multivariate MR
We conducted multivariable MR analysis to examine whether the significant effects of circulating inflammatory proteins on psychiatric disorders are confounded or mediated by other inflammatory proteins. In this analysis, we adjusted for the genetic associations of circulating inflammatory proteins that showed significant results (PIVW < 0.05) in the univariable MR analysis based on the PGC data, by including them as covariates.
Colocalization analysis
Colocalization analysis was used to examine the shared local genetic architecture between specific circulating inflammatory proteins and psychiatric disorders, assessing whether they share the same causal variants [41]. The analysis provides posterior probabilities for each hypothesis (H₀, H₁, H₂, H₃, and H₄). We set the prior probabilities for SNPs associated with circulating inflammatory proteins (P₁) and psychiatric disorders (P₂) to 1 × 10⁻⁵, with a gene window of 50k. Strong evidence of colocalization was considered if PH₄ ≥ 0.8, medium colocalization was defined as 0.5 < PH₄ < 0.8, and lower evidence was supported for the remaining cases [42]. All data preprocessing and statistical analyses were conducted using R version 4.4.1.
Results
Forward MR of circulating inflammatory proteins on psychiatric disorders
As shown in Table 2 and Table S2, in the forward MR analysis based on PGC data, we identified seven circulating inflammatory proteins that may have causal relationships with SCZ. Among them, CX3CL1 (PIVW = 0.040, OR = 0.92, 95% CI: 0.85, 1.00), TNFB (PIVW = 0.037, OR = 0.96, 95% CI: 0.93, 1.00), TNFSF14 (PIVW = 0.036, OR = 0.96, 95% CI: 0.92, 1.00), and CRP (PIVW = 0.006, OR = 0.92, 95% CI: 0.86, 0.97) showed protective effects. EIF4EBP1 (PIVW = 0.015, OR = 1.09, 95% CI: 1.02, 1.17), DNER (PIVW = 0.02, OR = 1.07, 95% CI: 1.01, 1.13), and IL6RA (PIVW = 0.025, OR = 1.03, 95% CI: 1.00, 1.06) were associated with increased disease risk. CXCL (PIVW = 0.024, OR = 0.95, 95% CI: 0.90, 0.99) may have a protective causal relationship with MDD. FGF23 (PIVW = 0.009, OR = 0.96, 95% CI: 0.93, 0.99) and IL20 (PIVW = 0.030, OR = 0.96, 95% CI: 0.92, 1.00) may also have protective causal relationships with MDD. ARTN (PIVW = 0.026, OR = 1.03, 95% CI: 1.00, 1.06) and TNFSF12 (PIVW = 0.049, OR = 1.03, 95% CI: 1.00, 1.05) significantly increased MDD risk. CXCL5 (PIVW = 0.024, OR = 0.95, 95% CI: 0.90, 0.99) was found to potentially have a protective causal relationship with BD.
Reverse MR of psychiatric disorders on circulating inflammatory proteins
As shown in Table 3 and Table S3, the reverse MR analysis based on PGC data identified potential causal relationships between two psychiatric disorders and five circulating inflammatory proteins. SCZ was associated with increased levels of NTF3 (PIVW = 0.035, OR = 1.03, 95% CI: 1.00, 1.05) and decreased levels of IL20RA (PIVW = 0.021, OR = 0.97, 95% CI: 0.94, 1.00). MDD was associated with increased levels of CCL19 (PIVW = 0.008, OR = 1.11, 95% CI: 1.03, 1.20) and SCF (PIVW = 0.033, OR = 1.09, 95% CI: 1.01, 1.17), as well as decreased levels of FGF19 (PIVW = 0.043, OR = 0.93, 95% CI: 0.86, 1.00).
Validation analysis
We incorporated GWAS data from the FinnGen biobank for both forward and reverse analyses, and conducted a meta-analysis of the results using the IVW method from both data sources. As showed in Fig.2 and Fig.3, the results showing potential causal relationships (P value < 0.05), with detailed findings provided in Tables S2 and S3. We identified that CX3CL1 (Pmeta = 0.012, OR = 0.91, 95% CI: 0.84, 0.98), IL1A (Pmeta = 0.043, OR = 0.93, 95% CI: 0.84, 0.98), and CRP (Pmeta = 0.013, OR = 0.93, 95% CI: 0.84, 0.98) significantly reduced the risk of SCZ. We also found that DNER (Pmeta = 0.007, OR = 1.07, 95% CI: 1.02, 1.13), EIF4EBP1 (Pmeta = 0.019, OR = 1.08, 95% CI: 1.01, 1.15), CCL23 (Pmeta = 0.030, OR = 1.04, 95% CI: 1.00, 1.09), TNFRSF11B (Pmeta = 0.032, OR = 1.07, 95% CI: 1.01, 1.15), and GDNF (Pmeta = 0.036, OR = 1.06, 95% CI: 1.00, 1.12) significantly increased the risk of SCZ. We also observed that TRANCE (Pmeta = 0.016, OR = 0.95, 95% CI: 0.91, 0.99) and CCL7 (Pmeta = 0.023, OR = 0.94, 95% CI: 0.89, 0.99) significantly reduced the risk of BD. We also found that FGF23 (Pmeta = 0.015, OR = 0.97, 95% CI: 0.94, 0.99), CD40 (Pmeta = 0.018, OR = 0.97, 95% CI: 0.95, 1.00), IL20 (Pmeta = 0.028, OR = 0.97, 95% CI: 0.95, 1.00), ADA (Pmeta = 0.034, OR = 0.99, 95% CI: 0.97, 1.00), and SULT1A1 (Pmeta = 0.043, OR = 0.98, 95% CI: 0.96, 1.00) were significantly reduced the risk of MDD. VEGFA (Pmeta = 0.009, OR = 1.03, 95% CI: 1.01, 1.05) and TNFSF12 (Pmeta = 0.039, OR = 1.02, 95% CI: 0.97, 1.00) were significantly increased the risk of MDD. In the reverse MR analysis, only CRP (Pmeta = 0.015, OR = 1.04, 95% CI: 1.01, 1.07) was significantly associated with an increased risk of MDD. The meta-analysis results of bidirectional MR were not significant after FDR correction.

Triangles or squares represent odds ratios (ORs); lines indicate 95% confidence intervals (CIs).

Triangles or squares represent odds ratios (ORs); lines indicate 95% confidence intervals (CIs).
Sensitivity analyses
We performed a series of sensitivity analyses to assess the robustness of the causal relationships identified through bidirectional MR. First, the causal directions inferred from the bidirectional MR results were consistent. Second, heterogeneity testing based on the IVW model revealed substantial heterogeneity in most of the causal relationships between psychiatric disorders and their respective outcomes (Table S4). Third, MR-Egger intercept analysis indicated horizontal pleiotropy in the prediction of SCZ risk by CCL23, DNER, FGF5, IL6RA, and TNFB, and in the prediction of MDD risk by ARTN and IL17C, based on the PGC data. Similarly, horizontal pleiotropy was observed for SCZ risk genetically predicted by TNFB and MDD risk genetically predicted by CD40 in the FinnGen dataset. MR-PRESSO analysis identified several outliers, and after excluding these outliers, the results remained stable (Table S5). In the reverse MR analysis, MR-Egger intercept analysis did not detect horizontal pleiotropy. However, MR-PRESSO analysis identified horizontal pleiotropy for the relationship between CRP and MDD, but after removing the outlier, the results remained stable (Table S6). Finally, leave-one-out analysis was conducted to further validate the significant MR results. The analysis indicated that no single SNP drove the causal estimates (Fig. S1 and Fig. S2). Overall, the sensitivity analyses confirmed the robustness and reliability of the causal relationships identified in bidirectional MR analysis.
Multivariate MR
Based on the results from the univariable MR analysis using data from PGC, we conducted a MVMR analysis, using the MV-IVW method as the primary approach, and included circulating inflammatory proteins with P IVW < 0.05 as covariates (Fig. 4). For SCZ, after adjusting for EIF4EBP1, CX3CL1, DNER, TNFB, TNFSF14, and IL6RA, CRP demonstrated a significant protective effect (P MV-IVW = 0.008, OR = 0.91, 95% CI: 0.85, 0.98). For MDD, after adjusting for ARTN and IL20, FGF23 showed a significant protective effect (P MV-IVW = 0.038, OR = 0.96, 95% CI: 0.92, 1.00), while TNFSF12 significantly increased the risk of MDD (P MV-IVW = 0.032, OR = 1.03, 95% CI: 1.00, 1.06).

MVMR adjusting for significant UVMR proteins as covariates; green indicates significant associations in MVMR.
Colocalization analysis
We conducted a colocalization analysis to further assess the likelihood of shared causal genetic variation between psychiatric disorders and inflammatory proteins. We used the exposure and outcome variables from the forward and reverse MR analyses based on the dataset from PGC, with significant results (PIVW < 0.05) treated as specific traits. The results provided strong evidence for shared variation between DNER and SCZ (PH4 = 0.89), with the rs35975053 locus being the most significant (Table S4 and Fig. 5).

Manhattan plots for associations of genetically predicted DNER levels with schizophrenia (SCZ).
Discussion
We performed MR analyses to investigate potential causal relationships between a large array of circulating immunological proteins and three psychiatric disorders, in combination with meta-analyses and colocalization methods to further validate the results. Firstly, both UVMR analyses utilizing the PGC database and meta-analyses integrating PGC with FinnGen biobank suggested that CRP and CX3CL1 might serve as protective factors against SCZ, while enhanced levels of DNER and EIF4EBP1 were correlated with a higher risk of SCZ. In addition, a possible causal inverse relationship was identified between the genetically proxied levels of FGF23, IL20 and the risk of MDD, however, TNFSF12 may increase the risk of developing MDD. Secondly, among the inflammatory proteins mentioned above, MVMR analyses based on the PGC cohort also showed causal effects of CRP for SCZ risk, as well as FGF23 and TNFSF12 for MDD risk, respectively. Furthermore, the strong colocalization support observed for DNER highlighted the probability of shared genetic variants directly impacting both DNER levels and SCZ susceptibility. Notably, sensitivity analyses provided strong support for the robustness and reliability of these results. Additionally, associations such as the protective effect of TNFSF14 and the increased risk linked to IL6RA for SCZ, identified in UVMR, were not replicated in MVMR or colocalization analyses and thus remain suggestive evidence requiring further validation.
CRP, known as an acute-phase protein, has been widely studied as a systemic inflammation biomarker in SCZ. Our findings from UVMR, MVMR and meta-analyses, primarily estimated using the IVW method, are consistent with previous MR studies suggesting a potential protective effect of genetic predisposition to elevated CRP levels on the risk of SCZ [15, 43,44,45,46]. Nevertheless, these genetic findings are inconsistent with previous observations that higher CRP levels were associated with increased SCZ risk, as reported in cross-sectional and population-based longitudinal studies [21, 47,48,49]. These discrepancies are likely attributable to different methodological approaches. It is important to note that MR is designed to estimate the lifetime effects of exposures [50], and genetic variations only partially influence the circulating CRP levels. However, the interpretation of observational data, particularly from studies with single or sparse time-point measurements, is frequently challenged by residual confounders, such as lifestyle heterogeneity, latent infections and comorbid metabolic disturbances, which may obscure the associations between CRP and SCZ. On the other hand, the positive correlation between CRP and SCZ observed in the cross-sectional studies was unlikely due to genetic reverse causality, since no causal effects of liability to SCZ on CRP levels were found by reverse MR analyses in our and other MR studies [17, 44,45,46].
Similarly, Yang et al. identified a significant negative genetic correlation at the whole genome level and discovered 83 loci shared between CRP levels and SCZ [51]. The genetic variants from these shared loci exhibited a pattern of mixed effect directions concerning CRP levels and SCZ risk [51]. Based on these effect directions, the shared SNPs were categorized into “concordant effect” SNPs (ceSNPs) and “discordant effect” SNPs (deSNPs). Their study indicated that genes mapped to deSNPs demonstrated higher expression levels in the human brain and greater enrichment in brain cell types compared to genes mapped to ceSNPs, hence, it is plausible that genes mapped to deSNPs may predominantly affect the relationship between CRP levels and SCZ risk [51]. Furthermore, genetic profiles indicative of enhanced CRP functionality in pathogen defense may reduce the odds of childhood infections. Additionally, CRP-interferon pathways might promote neuroprotection by facilitating glutamate clearance [43, 52, 53], thereby safeguarding neurons from oxidative stress related to excess glutamate [54, 55]. These factors may collectively contribute to the protective role of CRP in mitigating the risk of SCZ. Given the complexity of the relationship between CRP and SCZ, further study is warranted to explore neurobiological consequences of CRP signaling in pathophysiological mechanisms of SCZ from the perspective of dynamic processes.
Although MVMR analyses, which adjusted for several inflammatory proteins, fully attenuated the observed positive association of genetically predicted DNER levels with SCZ risk identified in UVMR estimations, the robust colocalization supported the likelihood of shared genetic variants causally influencing both DNER levels and SCZ risk, especially rs35975053 locus. The recent MR study by Cao et al. also suggested that DNER may increase the risk of developing SCZ, regrettably, they did not perform MVMR analyses [56], while our results indicated that the relationship between them may be affected by other inflammatory proteins in the blood rather than the independent effect of peripheral DNER levels. Meanwhile, despite sensitivity analyses (including MR-PRESSO) being aligned with the main results, horizontal pleiotropy cannot be entirely ruled out in the present study. In addition, the strong colocalization signal further implied that DNER-associated genetic variants may influence SCZ risk through central mechanisms, particularly expression of DNER in brain is significantly higher compared to whole blood DNER [57], specifically expressed in dendrites and cell bodies of postmitotic neurons, acts as a ligand in the Notch signaling pathway and regulates neuron-glia interaction during astrocytogenesis [58, 59], which may be implicated in the pathogenesis of SCZ.
It is worth noting that SNP rs35975053 has been determined to be a chromatin accessibility quantitative trait loci (caQTL) in neurons and glia, and since chromatin status is directly related to the transcriptional activity regulation, the colocalized caQTL signal within a given locus could uncover functional components linking genetic variants to disease mechanisms [60]. More research is needed to get further insights into the specific mechanisms by which DNER affects SCZ.
Regarding MDD, in the forward directions of both meta and MVMR analyses, we found the independent causal effects of genetically determined FGF23 and TNFSF12 concentrations on the illness risk, to our knowledge, which is a previously unidentified finding. FGF Signaling could contribute to axonal branching in serotonin neurons, potentially reducing the risk of depressive and anxiety-related behaviors in animal models [61]. Dysregulation of FGF system transcripts has been observed in the frontal cortical regions of individuals with MDD [62]. FGF23, an endocrine hormone secreted by osteocytes, is essential for maintaining phosphate and vitamin D homeostasis and plays an important role in chronic kidney disease [63]. Knockout of the FGF23 gene in mice has been shown to induce aging-like phenotypes, including reduced lifespan, cognitive impairment, hypogonadism, and impaired growth [63]. Research exploring the relationship between FGF23 and MDD remains limited. One study indicated that lithium augmentation in the treatment of acute depressive patients significantly increased serum FGF23 concentrations following effective treatment, suggesting a potential role for FGF23 in the antidepressant effects of lithium [64]. The other study reported that peripheral FGF23 levels were significantly elevated in women experiencing postpartum depressive symptoms compared to those without such symptoms [65], however, postpartum physiological changes would have made the relationship more complex. FGF23 signaling in hippocampal cells critically modulates neuronal morphology and synaptic density [66], and FGF23-deficient mice develop dose-dependent cognitive impairment [67], while overexpressed FGF23 in mice lead to deficits in spatial memory and learning [68]. Together with our MR results, it is plausible that FGF23 may exsert protective effects against MDD, but only within a certain range of concentrations.
TNFSF12, more commonly known as TWEAK, is a TNF family ligand that mediates central effects through its receptor Fn14 expressed on neurons, astrocytes, microglia, and perivascular endothelial cells [69]. Animal experiments demonstrated that upregulation of TWEAK/Fn14 signaling or intracerebroventricular injection of Fc-TWEAK may lead to the accumulation of inflammatory cells in the choroid plexus, disruption of blood-brain barrier integrity, and increased neuronal damage, concomitantly with obvious depression-like behavior and cognitive impairment [70,71,72]. Conversely, these effects could be significantly ameliorated in Fn14 knockout mice [70, 73]. These studies may help to explain the risk effect of genetically proxied TNFSF12 levels on MDD revealed by our MR analyses. However, observational clinical studies indicated that serum levels of TWEAK were either lower in depressed patients or showed no statistical difference when compared to healthy controls [74, 75], whereas these results may be subject to small sample sizes and confounding factors. Therefore, further exploration of TWEAK/Fn14 pathway implicated in the pathogenesis of MDD is still be required.
Despite lack of replication, clinical findings suggest immune involvement may underpin these associations. In relation to BD, the UVMR estimations utilizing BD GWAS data from the PGC identified genetically determined CXCL5 as a protective factor. Previous reports have documented differences in the genetic architecture of BD according to patient source and clinical subtype (type I or type II). If GWAS analyses are not strictly stratified by subtype, mixed genetic signals may attenuate the observed associations with cytokines [76]. However, this finding was not corroborated by subsequent meta-analyses that incorporated FinnGen data. Furthermore, reverse MR analyses based on the PGC database indicated that increased genetic risk of mental diseases were potentially associated with the levels of five inflammatory regulators, including NTF3 and IL20RA by SCZ, as well as CCL19, SCF, and FGF19 by MDD. These associations were not validated by meta-analyses either, suggesting that these findings should be interpreted with caution. Notably, clinical studies have shown that patients with SCZ and MDD exhibit elevated levels of inflammatory markers, including IL-18, IL-1β, IL-6, TNF-α, and inflammasome-related genes such as NLRP3 and NLRC4, highlighting the potential involvement of inflammation in these disorders [77, 78].
Our study has several advantages. First, we conducted comprehensive MR analyses of the causal associations between 95 circulating inflammatory proteins and SCZ, BD and MDD, reducing the influences of confounding factors prevalent in observational studies and providing a thorough understanding of their relationships. Second, we employed the latest large-scale GWAS of circulating inflammation-related proteins and three psychiatric disorders, and further performed combined meta-analyses of GWAS datasets obtained from PGC and FinnGen to validate the results, yet avoid sample overlap. Third, we also conducted a series of sensitivity analyses to ensure the robustness of the results. Finally, the identification of proteins with independent causal effects offers promising candidates for translation into clinically relevant biomarkers for early risk stratification and as potential targets for inflammation- or neurodevelopment-focused interventions.
However, there are also some limitations in this study. First of all, our analyses were conducted in European-ancestry populations, and thus the applicability of these findings to other racial groups requires validation in multi-ancestry cohorts. Secondly, We employed a lenient significance threshold of P < 1 × 10−5 for the selection of IVs, which may have included false-positive variants, potentially introducing bias. Nevertheless, the F-statistics for all IVs were greater than 10, indicating a reduced possibility of weak instrument variables. Lowering the P-value threshold should have enhanced the overall statistical power for MR analyses and facilitated the detection of subtle associations. Thirdly, we did not correct for multiple testing in our exploratory MR analyses, and none survived FDR correction in the meta-analyses, therefore, these findings can only be regarded as suggestive indications of potential causality. Fourthly, our study is limited by the cross-sectional nature of GAWS data for circulating inflammatory proteins measured in adults, which precludes assessment of critical developmental windows (e.g., childhood or adolescent periods) where immune dysregulation may disproportionately increase psychiatric risk. Future GWAS study with age-stratified cohorts could delineate whether specific vulnerability periods exist, informing targeted preventive strategies. Finally, this study employed peripheral inflammatory proteins as exposures, which may not accurately reflect the neuroinflammatory mechanisms implicated in the pathophysiology of psychiatric disorders. Nonetheless, growing evidence suggests that peripheral inflammation can influence CNS immunity through mechanisms such as blood-brain barrier disruption and microglial activation. For instance, elevated CRP levels have been associated with reduced brain permeability to TSPO radioligands in depression [79], and peripheral immune activation may initiate microglial responses that amplify neuroinflammation [80]. Given the distinct molecular profiles between peripheral and central immune systems, caution is warranted when extrapolating peripheral findings to CNS pathology. Future studies incorporating cerebrospinal fluid or brain-derived inflammatory markers could offer more direct mechanistic insights [34]. Moreover, as with all MR analyses, these results provide genetically proxied evidence that is consistent with a potential causal relationship, and future experimental or longitudinal clinical studies are warranted to further substantiate the observed links.
In conclusion, our study identified potential causal associations between genetically predicted circulating levels of CRP, CX3CL1, DNER, and EIF4EBP1 and an altered risk of SCZ, as well as between FGF23, IL20, and TNFSF12 and the risk of developing MDD. These associations were corroborated by subsequent meta-analyses. Specifically, CRP, FGF23, and TNFSF12 demonstrated independent causal effects. Furthermore, colocalization analyses provided robust evidence of shared causal genetic variants between DNER and SCZ. Further experimental investigations are necessary to validate these findings, elucidate the underlying biological mechanisms, and evaluate their potential as diagnostic markers and therapeutic targets.
Data availability
The datasets analyzed during the current study are available from the corresponding author on reasonable request due to ethical limitations.
References
Ferrari AJ, Santomauro DF, Aali A, Abate YH, Abbafati C, Abbastabar H, et al. Global incidence, prevalence, years lived with disability (ylds), disability-adjusted life-years (dalys), and healthy life expectancy (hale) for 371 diseases and injuries in 204 countries and territories and 811 subnational locations, 1990–2021: a systematic analysis for the global burden of disease study 2021. Lancet. 2024;403:2133–61.
Mullins N, Forstner AJ, O’Connell KS, Coombes B, Coleman JRI, Qiao Z, et al. Genome-wide association study of more than 40,000 bipolar disorder cases provides new insights into the underlying biology. Nat Genet. 2021;53:817–29.
Cross-Disorder Group of the Psychiatric Genomics Consortium, Lee SH, Ripke S, Neale BM, Faraone SV, Purcell SM, et al. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. Nat Genet. 2013;45:984–94.
Lumertz FS, Kestering-Ferreira E, Orso R, Creutzberg KC, Tractenberg SG, Stocchero BA, et al. Effects of early life stress on brain cytokines: a systematic review and meta-analysis of rodent studies. Neurosci Biobehav Rev. 2022;139:104746.
Borsini A, Zunszain PA, Thuret S, Pariante CM. The role of inflammatory cytokines as key modulators of neurogenesis. Trends Neurosci. 2015;38:145–57.
Goldsmith DR, Rapaport MH, Miller BJ. A meta-analysis of blood cytokine network alterations in psychiatric patients: comparisons between schizophrenia, bipolar disorder and depression. Mol Psychiatry. 2016;21:1696–709.
Zeng Y, Chourpiliadis C, Hammar N, Seitz C, Valdimarsdottir UA, Fang F, et al. Inflammatory biomarkers and risk of psychiatric disorders. JAMA Psychiatry. 2024;81:1118–29.
Kappelmann N, Khandaker GM, Dal H, Stochl J, Kosidou K, Jones PB, et al. Systemic inflammation and intelligence in early adulthood and subsequent risk of schizophrenia and other non-affective psychoses: a longitudinal cohort and co-relative study. Psychol Med. 2019;49:295–302.
Allswede DM, Yolken RH, Buka SL, Cannon TD. Cytokine concentrations throughout pregnancy and risk for psychosis in adult offspring: a longitudinal case-control study. Lancet Psychiatry. 2020;7:254–61.
Lamers F, Milaneschi Y, Smit JH, Schoevers RA, Wittenberg G, Penninx B. Longitudinal association between depression and inflammatory markers: results from the Netherlands study of depression and anxiety. Biol Psychiatry. 2019;85:829–37.
Hayes JF, Khandaker GM, Anderson J, Mackay D, Zammit S, Lewis G, et al. Childhood interleukin-6, C-reactive protein and atopic disorders as risk factors for hypomanic symptoms in young adulthood: a longitudinal birth cohort study. Psychol Med. 2017;47:23–33.
Dragioti E, Radua J, Solmi M, Gosling CJ, Oliver D, Lascialfari F, et al. Impact of mental disorders on clinical outcomes of physical diseases: an umbrella review assessing population attributable fraction and generalized impact fraction. World psychiatry. 2023;22:86–104.
Marrie RA, Bernstein CN. Psychiatric comorbidity in immune-mediated inflammatory diseases. World psychiatry. 2021;20:298–9.
Lovegrove CE, Howles SA, Furniss D, Holmes MV. Causal inference in health and disease: a review of the principles and applications of mendelian randomization. J Bone Min Res. 2024;39:1539–52.
Hartwig FP, Borges MC, Horta BL, Bowden J, Davey Smith G. Inflammatory biomarkers and risk of schizophrenia: a 2-sample mendelian randomization study. JAMA Psychiatry. 2017;74:1226–33.
Kelly KM, Smith JA, Mezuk B. Depression and interleukin-6 signaling: a mendelian randomization study. Brain Behav Immun. 2021;95:106–14.
Perry BI, Upthegrove R, Kappelmann N, Jones PB, Burgess S, Khandaker GM. Associations of immunological proteins/traits with schizophrenia, major depression and bipolar disorder: a bi-directional two-sample mendelian randomization study. Brain Behav Immun. 2021;97:176–85.
Zhu S, Zhao L, Fan Y, Lv Q, Wu K, Lang X, et al. Interaction between TNF-alpha and oxidative stress status in first-episode drug-naive schizophrenia. Psychoneuroendocrinology. 2020;114:104595.
Luo Y, He H, Zhang J, Ou Y, Fan N. Changes in serum TNF-alpha, IL-18, and IL-6 concentrations in patients with chronic schizophrenia at admission and at discharge. Compr Psychiatry. 2019;90:82–7.
Ma N, Wang R. Mendelian randomization study on the effect of tumor necrosis factor on schizophrenia. Psychiatr Genet. 2022;32:238–45.
Lestra V, Romeo B, Martelli C, Benyamina A, Hamdani N. Could CRP be a differential biomarker of illness stages in schizophrenia? a systematic review and meta-analysis. Schizophr Res. 2022;246:175–86.
Cakici N, Sutterland AL, Penninx B, Dalm VA, de Haan L, van Beveren NJM. Altered peripheral blood compounds in drug-naive first-episode patients with either schizophrenia or major depressive disorder: a meta-analysis. Brain Behav Immun. 2020;88:547–58.
Galan D, Perry BI, Warrier V, Davidson CC, Stupart O, Easton D, et al. Applying mendelian randomization to appraise causality in relationships between smoking, depression and inflammation. Sci Rep. 2022;12:15041.
Zhao L, Tan L, Liu W, Zhang S, Liao A, Yuan L, et al. The causal relationships between inflammatory proteins, brain structure, and psychiatric disorders: a two-step mendelian randomization analysis. Schizophrenia bulletin. 2024;51:1390–1401.
Hu JJ, Zhang YB, Zheng SF, Chen GR, Lin YX, Kang DZ, et al. The causal relationship between circulating biomarkersand the risk of bipolar disorder: a two-sample mendelian randomization study. J Psychiatr Res. 2023;164:66–71.
Lu T, Forgetta V, Greenwood CMT, Zhou S, Richards JB. Circulating proteins influencing psychiatric disease: a mendelian randomization study. Biol psychiatry. 2023;93:82–91.
Ahola-Olli AV, Wurtz P, Havulinna AS, Aalto K, Pitkanen N, Lehtimaki T, et al. Genome-wide association study identifies 27 loci influencing concentrations of circulating cytokines and growth factors. Am J Hum Genet. 2017;100:40–50.
Zhao JH, Stacey D, Eriksson N, Macdonald-Dunlop E, Hedman AK, Kalnapenkis A, et al. Genetics of circulating inflammatory proteins identifies drivers of immune-mediated disease risk and therapeutic targets. Nat Immunol. 2023;24:1540–51.
Chen X, Yao T, Cai J, Fu X, Li H, Wu J. Systemic inflammatory regulators and 7 major psychiatric disorders: a two-sample mendelian randomization study. Prog Neuropsychopharmacol Biol Psychiatry. 2022;116:110534.
Wang M, Jin G, Cheng Y, Guan SY, Zheng J, Zhang SX. Genetically predicted circulating levels of cytokines and the risk of depression: a bidirectional Mendelian-randomization study. Front Genet. 2023;14:1242614.
Li YT, Zeng X. Circulating inflammatory cytokines influencing schizophrenia: a mendelian randomization study. Front Psychiatry. 2024;15:1417213.
Ligthart S, Vaez A, Võsa U, Stathopoulou MG, de Vries PS, Prins BP, et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet. 2018;103:691–706.
Sun BB, Maranville JC, Peters JE, Stacey D, Staley JR, Blackshaw J, et al. Genomic atlas of the human plasma proteome. Nature. 2018;558:73–9.
Trubetskoy V, Pardiñas AF, Qi T, Panagiotaropoulou G, Awasthi S, Bigdeli TB, et al. Mapping genomic loci implicates genes and synaptic biology in schizophrenia. Nature. 2022;604:502–8.
Howard DM, Adams MJ, Clarke TK, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci. 2019;22:343–52.
Burgess S, Thompson SG. Avoiding bias from weak instruments in mendelian randomization studies. Int J Epidemiol. 2011;40:755–64.
Hemani G, Tilling K, Davey Smith G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 2017;13:e1007081.
Burgess S, Davey Smith G, Davies NM, Dudbridge F, Gill D, Glymour MM, et al. Guidelines for performing mendelian randomization investigations: update for summer 2023. Wellcome open Res. 2019;4:186.
Verbanck M, Chen CY, Neale B, Do R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat Genet. 2018;50:693–8.
Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol. 2015;44:512–25.
Liu B, Gloudemans MJ, Rao AS, Ingelsson E, Montgomery SB. Abundant associations with gene expression complicate GWAS follow-up. Nat Genet. 2019;51:768–9.
Chen J, Xu F, Ruan X, Sun J, Zhang Y, Zhang H, et al. Therapeutic targets for inflammatory bowel disease: proteome-wide mendelian randomization and colocalization analyses. EBioMedicine. 2023;89:104494.
Prins BP, Abbasi A, Wong A, Vaez A, Nolte I, Franceschini N, et al. Investigating the causal relationship of c-reactive protein with 32 complex somatic and psychiatric outcomes: a large-scale cross-consortium mendelian randomization study. PLoS Med. 2016;13:e1001976.
Lin BD, Alkema A, Peters T, Zinkstok J, Libuda L, Hebebrand J, et al. Assessing causal links between metabolic traits, inflammation and schizophrenia: a univariable and multivariable, bidirectional Mendelian-randomization study. Int J Epidemiol. 2019;48:1505–14.
Said S, Pazoki R, Karhunen V, Vosa U, Ligthart S, Bodinier B, et al. Genetic analysis of over half a million people characterises C-reactive protein loci. Nat Commun. 2022;13:2198.
Reay WR, Kiltschewskij DJ, Geaghan MP, Atkins JR, Carr VJ, Green MJ, et al. Genetic estimates of correlation and causality between blood-based biomarkers and psychiatric disorders. Sci Adv. 2022;8:eabj8969.
Fernandes BS, Steiner J, Bernstein HG, Dodd S, Pasco JA, Dean OM, et al. C-reactive protein is increased in schizophrenia but is not altered by antipsychotics: meta-analysis and implications. Mol Psychiatry. 2016;21:554–64.
Metcalf SA, Jones PB, Nordstrom T, Timonen M, Maki P, Miettunen J, et al. Serum C-reactive protein in adolescence and risk of schizophrenia in adulthood: a prospective birth cohort study. Brain Behav Immun. 2017;59:253–9.
Osimo EF, Baxter L, Stochl J, Perry BI, Metcalf SA, Kunutsor SK, et al. Longitudinal association between CRP levels and risk of psychosis: a meta-analysis of population-based cohort studies. NPJ Schizophr. 2021;7:31.
Labrecque JA, Swanson SA. Interpretation and potential biases of mendelian randomization estimates with time-varying exposures. Am J Epidemiol. 2019;188:231–8.
Yang Z, Li D, He Y, Chen X, Li Z. Unrevealing the shared genetic mechanisms underlying C-reactive protein and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2023;126:110785.
Shaked I, Tchoresh D, Gersner R, Meiri G, Mordechai S, Xiao X, et al. Protective autoimmunity: interferon-gamma enables microglia to remove glutamate without evoking inflammatory mediators. J Neurochem. 2005;92:997–1009.
Garg SK, Banerjee R, Kipnis J. Neuroprotective immunity: T cell-derived glutamate endows astrocytes with a neuroprotective phenotype. J Immunol. 2008;180:3866–73.
Javitt DC. Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci. 2010;47:4–16.
Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Bull. 2013;39:120–9.
Cao H, Fu L, Liu D, Baranova A, Zhang F. Mendelian randomization analysis of causal and druggable circulating inflammatory proteins in schizophrenia. Front Psychiatry. 2024;15:1465291.
Pratt HE, Andrews G, Shedd N, Phalke N, Li T, Pampari A, et al. Using a comprehensive atlas and predictive models to reveal the complexity and evolution of brain-active regulatory elements. Sci Adv. 2024;10:eadj4452.
Eiraku M, Hirata Y, Takeshima H, Hirano T, Kengaku M. Delta/notch-like epidermal growth factor (EGF)-related receptor, a novel EGF-like repeat-containing protein targeted to dendrites of developing and adult central nervous system neurons. J Biol Chem. 2002;277:25400–7.
Eiraku M, Tohgo A, Ono K, Kaneko M, Fujishima K, Hirano T, et al. DNER acts as a neuron-specific notch ligand during bergmann glial development. Nat Neurosci. 2005;8:873–80.
Zeng B, Bendl J, Deng C, Lee D, Misir R, Reach SM, et al. Genetic regulation of cell type-specific chromatin accessibility shapes brain disease etiology. Science. 2024;384:eadh4265.
Shimada T, Kohyama K, Yoshida T, Yamagata K. Neuritin controls axonal branching in serotonin neurons: a possible mediator involved in the regulation of depressive and anxiety behaviors via fgf signaling. J Neurosci. 2024;44:e0129232024.
Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, et al. Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci USA. 2004;101:15506–11.
Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. 2013;75:503–33.
Fakhri H, Ricken R, Adli M, Fajol A, Walter M, Foller M, et al. Impact of lithium treatment on FGF-23 serum concentrations in depressive patients. J Clin Psychopharmacol. 2014;34:745–7.
Brann E, Fransson E, White RA, Papadopoulos FC, Edvinsson A, Kamali-Moghaddam M, et al. Inflammatory markers in women with postpartum depressive symptoms. J Neurosci Res. 2020;98:1309–21.
Hensel N, Schon A, Konen T, Lubben V, Forthmann B, Baron O, et al. Fibroblast growth factor 23 signaling in hippocampal cells: impact on neuronal morphology and synaptic density. J Neurochem. 2016;137:756–69.
Laszczyk AM, Nettles D, Pollock TA, Fox S, Garcia ML, Wang J, et al. FGF-23 deficiency impairs hippocampal-dependent cognitive function. eNeuro. 2019;6:0469–18.
Liu P, Chen L, Bai X, Karaplis A, Miao D, Gu N. Impairment of spatial learning and memory in transgenic mice overexpressing human fibroblast growth factor-23. Brain Res. 2011;1412:9–17.
Wiley SR, Cassiano L, Lofton T, Davis-Smith T, Winkles JA, Lindner V, et al. A novel TNF receptor family member binds TWEAK and is implicated in angiogenesis. Immunity. 2001;15:837–46.
Wen J, Doerner J, Weidenheim K, Xia Y, Stock A, Michaelson JS, et al. TNF-like weak inducer of apoptosis promotes blood brain barrier disruption and increases neuronal cell death in MRL/lpr mice. J Autoimmun. 2015;60:40–50.
Stock AD, Wen J, Putterman C. Neuropsychiatric lupus, the blood brain barrier, and the TWEAK/Fn14 pathway. Front Immunol. 2013;4:484.
Wen J, Chen CH, Stock A, Doerner J, Gulinello M, Putterman C. Intracerebroventricular administration of TNF-like weak inducer of apoptosis induces depression-like behavior and cognitive dysfunction in non-autoimmune mice. Brain Behav Immun. 2016;54:27–37.
Wen J, Xia Y, Stock A, Michaelson JS, Burkly LC, Gulinello M, et al. Neuropsychiatric disease in murine lupus is dependent on the TWEAK/Fn14 pathway. J Autoimmun. 2013;43:44–54.
Schmidt FM, Koch J, Nowak C, Holdt LM, Teupser D, Hegerl U, et al. Ligands and receptors of the TNF superfamily are decreased in major depression and during early antidepressant therapy. J Psychiatr Res. 2019;119:116–21.
Karadag H, Saygili G, Yuksel R, Usta MB, Topcuoglu C, Erzin G. Serum tnf- related weak inducer of apoptosis (tweak), tnf- related apoptosis-inducing ligand (trail) levels in patients with bipolar depression, major depression and a healthy control group. Psychiatr Danub. 2021;33:314–9.
O’Connell KS, Koromina M, van der Veen T, Boltz T, David FS, Yang JMK, et al. Genomics yields biological and phenotypic insights into bipolar disorder. Nature. 2025;639:968–75.
Szabo A, O’Connell KS, Ueland T, Sheikh MA, Agartz I, Andreou D, et al. Increased circulating IL-18 levels in severe mental disorders indicate systemic inflammasome activation. Brain, Behav, Immun. 2022;99:299–306.
Dahl J, Ormstad H, Aass HC, Malt UF, Bendz LT, Sandvik L, et al. The plasma levels of various cytokines are increased during ongoing depression and are reduced to normal levels after recovery. Psychoneuroendocrinology. 2014;45:77–86.
Turkheimer FE, Althubaity N, Schubert J, Nettis MA, Cousins O, Dima D, et al. Increased serum peripheral C-reactive protein is associated with reduced brain barriers permeability of TSPO radioligands in healthy volunteers and depressed patients: implications for inflammation and depression. Brain, Behav, Immun. 2021;91:487–97.
Xie X, Luo X, Liu N, Li X, Lou F, Zheng Y, et al. Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J neurochemistry. 2017;141:222–35.
Acknowledgements
We would like to express our sincere gratitude to the Psychiatric Genomics Consortium, the FinnGen Biobank, and the research teams of Jing Hua Zhao, Symen Ligthart, Benjamin B. Sun, and others for providing the publicly available data. We also extend our heartfelt thanks to all the participants involved in the aforementioned studies. Additionally, we acknowledge the support provided by the Beijing Research Ward Demonstration Project.
Funding
Supports were received from the Scientific foundation of Being Huilongguan Hospital (No.LY202501), National Natural Science Foundation of China (82171507), Capital’s Funds for Health Improvement and Research (2022-1-2131), High-level public health technical talent construction project of Beijing Municipal Health Commission (Leading Talent-03-04), and the National Natural Science Foundation of China (No.82001415).
Author information
Authors and Affiliations
Contributions
ZD, SC and YLT designed the study. RML, XHW, BB, YRZ, XW and HBL contributed to the data collection. ZD, SC, and YLT conducted data analysis and interpretation. ZD and SC drafted the manuscript. BB, HBL, BPT, and YLT contributed administrative, technical, or material support. YLT, ZD, GPW, ZZS, ESP, CHH and SC supervised the draft and revision of the manuscript. The corresponding author attests that all the listed authors meet authorship criteria and that no others meeting the criteria have been omitted. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dong, Z., Bi, B., Li, R. et al. Circulating inflammatory proteins associated with risks of schizophrenia, bipolar disorder, and major depressive disorder: a mendelian randomization study. Transl Psychiatry 16, 3 (2026). https://doi.org/10.1038/s41398-025-03738-0
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03738-0
