
Abstract
Major depressive disorder (MDD) often begins during adolescence, a critical developmental period during which nearly 50% of lifetime cases emerge. Despite its high prevalence and impact, objective diagnostic biomarkers for adolescent MDD remain limited, particularly those related to gut microbiota. Our study examined potential co-diagnostic biomarkers from peripheral blood and fecal samples in adolescents with MDD. We enrolled drug-naïve adolescents with first-episode MDD (n = 46, aged ≤18 years, 71.74% female) and age-/sex-matched healthy controls (HCs, n = 44). The levels of tight junction proteins (Claudin-5, Zonulin, FABP) and inflammatory biomarkers (IL-6, IL-8, TNF-α, and CRP) were markedly elevated in the plasma of adolescents with MDD, indicating gut barrier dysfunction and systemic inflammation. The microbiome in MDD patients exhibited a lower Firmicutes-to-Bacteroidetes ratio. At the genus level, Intestinimonas and Barnesiella were significantly enriched, while Dialister and Collinsella were considerably reduced. Integrating Collinsella abundance with tight junction proteins and inflammatory markers significantly improved diagnostic performance, achieving an area under the curve (AUC) of 0.964. Moreover, Collinsella negatively correlated with sex, Claudin-5, and TNF-α. Claudin-5 was strongly associated with short-chain fatty acids (SCFAs)-related pathways, including alanine, aspartate, glutamate metabolism, D-glutamine and D-glutamate metabolism, and autophagy regulation. Treatment of Caco-2 cells with propionate and butyrate confirmed the regulatory effects of SCFAs on tight junction biomarkers. These findings suggest the interplay between gut dysbiosis, barrier dysfunction, and inflammation in adolescent MDD and support microbiota-host biomarkers as a promising strategy for improving MDD diagnostic precision.
Similar content being viewed by others


Introduction
Depression is a highly prevalent and recurrent disorder that severely affects adolescents, with approximately 50% of cases occurring during this developmental stage [1]. It is recognized as the leading cause of disability and mortality among adolescents, with estimates suggesting that up to 20% of this demographic will experience major depressive disorder (MDD) before reaching adulthood [2]. Furthermore, individuals with a history of adolescent MDD are roughly three times more likely to develop MDD in adulthood [3]. In China, the prevalence of MDD among children and adolescents aged 6–16 years was reported at 2.0% [4], while the lifetime prevalence of MDD in adolescents in the United States rose from 8.1–15.8% between 2009 and 2019 [5]. Despite being one of the most pressing global public health challenges, MDD often remains undiagnosed and inadequately treated [6]. This issue can be primarily attributed to its complex pathogenesis, the variability of individual experiences, and the stigma surrounding mental health [7]. Therefore, identifying objective diagnostic biomarkers and elucidating potential mechanisms underlying early-stage MDD is particularly important in adolescence.
The microbiota-gut-brain (MGB) axis has been proposed to explain the crosstalk between the brain and intestinal flora [8]. Gut microbiota has been linked to several neuropsychiatric disorders via the MGB axis, including Parkinson’s disease [9], autism [10], bipolar disorder [11], and MDD [11]. Fecal transplantation trials have induced depressive-like behaviors in rodents, revealing a causal role for gut microbiota in MDD onset [12]. Dysbiosis may modulate the immune system and trigger cytokine production [13], disrupt the intestinal and blood-brain barrier (BBB) permeability [14], and activate microglia in the central nervous system [15], potentially contributing to clinical depression [16]. Thus, gut microbiota may influence depressive symptoms by modulating both peripheral and central immune systems [17]. Gut microbiome disturbances in adolescents with MDD have previously been explored in a small-scale cohort with 16S rRNA sequencing [18]. However, metagenomics may offer deeper insights into microbial diversity, community structure, and metabolic functional potential. To date, the specific role of gut microbiota in immunoregulation in adolescent MDD remains poorly understood.
Short-chain fatty acids (SCFAs), a class of beneficial microbial metabolites, have been shown to influence peripheral immune function by altering the permeability of the intestine and the BBB [19]. They are essential for maintaining epithelial barrier integrity, modulating monocyte and macrophage antimicrobial and inflammatory activities, and exerting anti-inflammatory and tolerogenic effects on lymphocytes [20]. Particularly, butyrate has been identified to increase the expression of tight junction proteins, such as occludin and Claudin-1 [21]. It also enhances oxygen consumption in intestinal epithelial cells, stabilizes hypoxia-inducible factor (HIF) expression [22], and promotes intestinal barrier repair by inducing the expression of tight junction biomarkers [23]. Additionally, butyrate inhibits the synthesis of pro-inflammatory mediators, such as IL-6, IL-12, and nitric oxide, in response to lipopolysaccharide (LPS), potentially contributing to intestinal immune tolerance [24]. Collectively, these findings support the homeostatic and anti-inflammatory role of SCFAs, particularly butyrate, in intestinal epithelium regulation [20]. However, the strength of the evidence linking SCFAs to alterations in cytokine profiles and gut microbiota composition in the context of adolescent MDD remains unclear.
Considering the critical roles of SCFAs and the fluctuating abundance of specific intestinal microbiota in adolescents with MDD, we hypothesized that particular microbiota compositions may be associated with peripheral inflammation and tight junction biomarkers in adolescent MDD. To test this, we employed metagenomics and 16S rRNA sequencing to identify potential co-diagnostic biomarkers from blood and fecal samples. In addition, we conducted SCFA interventions in Caco-2 cells to assess the regulatory effects of propionate and butyrate on tight junction proteins. The study highlights a previously unrecognized connection between key gut microbes and epithelial barrier biomarkers, offering insights into novel diagnostic approaches for adolescent depression.
Methods
Participants enrollment
A total of 46 drug-naïve patients with first-episode MDD aged ≤18 were recruited from the Department of Psychiatry at the First Affiliated Hospital of Chongqing Medical University. All patients met the Diagnostic and Statistical Manual of Mental Disorders-version 5 (DSM-5) criteria for MDD, as assessed by two experienced psychiatrists. Depression and anxiety severity were evaluated using the 17-item Hamilton Rating Scale for Depression (HAMD-17) and the Hamilton Anxiety Rating Scale (HAMA). The following exclusion criteria were applied to patients with MDD: (1) current or lifetime diagnosis of neurological diseases or other psychiatric disorders such as epilepsy, anxiety disorder, autism, attention-deficit/hyperactivity disorder (ADHD) or obsessive-compulsive disorder (OCD); (2) current presence of acute or chronic physical illnesses; (3) a family history of neurological or psychiatric diseases, such as Parkinson’s disease, schizophrenia, and neurodevelopmental disorders; (4) abnormal results from routine blood examinations; (5) a history of alcohol and/or drug abuse; (6) use of antibiotics, probiotics, prebiotics, or synbiotics for three consecutive days within the past month; (7) girls who are pregnant or nursing; (8) participation in other clinical drug trials within the previous year.
Forty-four age- and sex-matched health controls (HCs) were recruited from the First Affiliated Hospital of Chongqing Medical University’s Medical Center. They were classified as participants according to the following criteria: (1) HAMD-17 and HAMA total scores both <7; (2) no personal history of depression or any other major psychiatric disorder; (3) no prior use of antidepressant medication; (4) no history of self-harm or suicide. The same exclusion criteria applied to the MDD group were also used for the HC group. This study was approved by the Ethics Committee of Chongqing Medical University (approval number: 2020-864).
Sample collection of blood and fecal
For each enrolled participant, 2 mL of fasting venous blood samples were collected between 8:00 a.m. and 12:00 p.m. in ethylene diamine tetraacetic acid (EDTA) tubes. Plasma was separated by centrifugation at 3000 × g at 4 °C for 10 min and stored at −80 °C for subsequent measurement of tight junction and inflammatory biomarkers. Stool samples were collected in sterile centrifuge tubes within 24 h following questionnaire assessments and blood collection, then immediately stored at −80 °C.
Tight junction and inflammatory marker measurements
According to our previous research [25], the plasma levels of tight junction and inflammatory markers, including Zonulin (JL14924), Claudin-5(JL53306), fatty acid-binding protein (FABP, JL20052), LPS (JL12996), IL-6 (JL14113), IL-8 (JL19291), TNF-α (JL10208), and CRP (JL13865), were detected by commercial enzyme-linked immunosorbent assay (ELISA) kits (Shanghai Jianglai Biotechnology, Shanghai, China), following the manufacturer’s instructions. Each sample was measured in triplicate, and the average value was used for analysis. Briefly, 0.1 mL of diluted antibody (50 ng/mL) was added to the reaction hole and incubated overnight at 4 °C. Then, 0.05 mL of diluted sample and freshly diluted enzyme-labeled antibody were added and incubated at 37 °C for 60 min. Next, 0.1 mL of 3,3’,5,5’-tetramethylbenzidine (TMB) substrate solution was added and incubated at 37 °C for 10–30 min. Finally, 0.05 mL of 2 M sulfuric acid was added to stop the reaction, and the optical density (OD) was measured at 450 nm using a microplate reader. The data were analyzed using R (version 4.2.2), and a P-value < 0.05 was considered statistically significant.
Fecal DNA extraction
The microbiota composition of 46 MDD patients and 44 HCs was determined by shotgun metagenomic sequencing and 16S rRNA analysis. Total microbial genomic DNA was extracted using the PF Mag-Bind Stool DNA Kit (Omega Bio-Tek, Georgia, USA) per the manufacturer’s instructions. The concentration and purity of the isolated DNA were determined using a Synergy HTX microplate reader and a NanoDrop 2000, respectively. The integrity of genomic DNA was evaluated by electrophoresis on a 1% agarose gel.
Metagenomic sequencing analysis
Extracted DNA was fragmented to an average size of about 400 bp using a Covaris M220 ultrasonicator (Gene Company Limited, China) for paired-end library preparation. Libraries were constructed using the NEXTFLEX Rapid DNA-Seq Kit (Bioo Scientific, Austin, TX, USA). Paired-end sequencing was performed on an Illumina NovaSeq platform at Majorbio Bio-Pharm Technology (Shanghai, China) using NovaSeq reagent kits. Quality control (QC) of reads was performed using BBDuk (v39.01) for adapter removal (https://sourceforge.net/projects/bbmap/), quality trimming, and filtering. The human reference genome GRCh38.p13, downloaded from the NCBI FTP site, was indexed by BBSplit to remove host-derived reads. Low-complexity sequences (entropy < 0.01) were filtered out.
Following QC, taxonomic profiling was done using MetaPhlAn v4.1.1 with default settings for relative abundance estimation [26]. To improve the resolution of unassigned taxa, profiling was repeated using the unclassified-estimation option. The enriched biological functions were determined using HUMAnN3 v3.9 with default parameters [27], using the ChocoPhlAn and UniRef databases [28], and grouping sequences at 90% identity. HUMAnN gene families (UniRef90) were subsequently collapsed into Kyoto Encyclopedia of Genes and Genomes (KEGG) ortholog (KO) groups for downstream pathway analysis.
16S rRNA sequence analysis
To complement metagenomic profiling, 16S rRNA sequencing was included to identify robust genus-level biomarkers that are resilient to platform-specific biases. The hypervariable V3-V4 region of the bacterial 16S rRNA gene was amplified using primer pairs 338 F (5’-ACTCCTACGGGAGGCAGCAG-3’) and 806 R (5’-GGACTACHVGGGTWTCTAAT-3’) on a T100 Thermal Cycler (BIO-RAD, USA). The PCR reaction mixture, including 4 μL 5 × FastPfu buffer, 2 μL 2.5 mM dNTPs, 0.8 μL each primer (5 μM), 0.4 μL FastPfu polymerase, 0.2 μL BSA, 10 ng of template DNA, and ddH2O to a final volume of 20 µL. PCR amplification cycling conditions were as follows: initial denaturation at 95 °C for 3 min, followed by 25 cycles of denaturing at 95 °C for 30 s, annealing at 55 °C for 30 s, and extension at 72 °Cfor 45 s, and single extension at 72 °C for 10 min, and end at 4 °C. The PCR product was extracted from a 2% agarose gel and purified. Then, it was quantified using a Synergy HTX microplate reader (Biotek, USA). Purified amplicons were pooled in equimolar amounts and then subjected to paired-end sequencing (300 bp) using an Illumina NextSeq 2000 platform (Illumina, San Diego, USA). The sequencing was performed by Majorbio Bio-Pharm Technology Co., Ltd. (Shanghai, China) following their standard protocols.
Quality control was performed using FastQC v0.11.9. The raw sequences were subsequently processed with DADA2 (R dada2 v1.26.0) [29], which included initial quality trimming, error rate estimation, read pair merging, chimeric sequence removal, and construction of the community data matrix. Taxonomy was assigned to amplicon sequence variants (ASVs) using the SILVA database v138.1. Each genus was generated by agglomerating all ASVs assigned to that genus using the tax glom function in phyloseq [30].
Abundance analyses
Abundance analyses were performed using genus- and pathway-level profiles derived from shotgun metagenomic data. The relative proportions of phyla and genera within each sample and across groups were analyzed and visualized using the microbial R package (v0.20.20, https://github.com/guokai8/microbial). This package was also employed to evaluate alpha diversity (Observed, Chao1, Shannon, Simpson, InvSimpson, and Fisher) between the MDD and HC groups. Beta diversity in overall microbial compositions between groups was formally tested using permutational multivariate ANOVA (PERMANOVA) via the adonis2 function in the vegan R package (v2.6-4, https://CRAN.R-project.org/package=vegan).
To identify features significantly associated with MDD status, differential abundance analysis was performed at both the genus and KEGG pathway levels using MaAsLin2 (v1.12.0) [31] and ANCOM-BC (v2.0.2) [32] R packages. Both tools were run with default parameters, except that microbial genera present in fewer than 5% of samples were excluded to ensure a minimum analytic sample size (N = 5, based on 90 samples × 5%). Statistical significance for association with MDD was defined as concordance between ANCOM-BC and MaAsLin2 analysis, with both methods yielding a P-value < 0.05.
Integrated biomarker identification and classification modeling
For metagenomic sequencing data, to identify potential microbial biomarkers at the genus level that distinguish adolescents with MDD from HCs, we employed a random forest classifier (R randomForest v4.7-1.1). The model was trained on genus-level relative abundance profiles, with group label (MDD vs. HC) as the response variable. Feature importance was assessed using the mean decrease in Gini index, which quantifies each genus’s contribution to classification accuracy. Genera were ranked accordingly, and the top genera were selected for downstream analysis.
To identify the most informative clinical biomarkers for integration with microbiome data, least absolute shrinkage and selection operator (LASSO) regression (R glmnet 4.1-7), a regularization method for prediction and feature selection, was applied while minimizing overfitting. The identified biomarkers were then combined with the top microbial genera to construct an integrative prediction model. This combined feature set was used to train a second random forest classifier, and model performance was evaluated using receiver operating characteristic (ROC) and area under the curve (AUC) metrics. Finally, Spearman’s rank correlation coefficient analysis was conducted to examine associations among clinical characteristics, tight junctions and inflammatory biomarkers, and KEGG pathways.
Treatment of Caco2 cells with acetate and propionate
5 × 105 Caco2 cells were seeded into a 12-well plate and incubated for 24 h or 48 h. Cells were then treated with 5 mM, 10 mM, and 20 mM propionate (Sigma, #S5436) and 1 mM, 2 mM, and 5 mM butyrate (Sigma, #S303410), followed by an incubation for an additional 24 or 48 h in a cell culture incubator, respectively. Total RNA was extracted using an RNAsimple Total RNA Isolation Kit (Tiangen, #DP419). For LPS treatment, cells were treated with 20 μg/mL LPS alone or in combination with 20 mM propionate or 5 mM butyrate, and incubated for 24 or 48 h. RNA was again isolated using the same kit. Subsequently, total RNA was reversely transcribed using the HiScript III 1st Strand cDNA Synthesis Kit (Vazyme, #R312-01). Gene expression was quantified by reverse transcription-polymerase chain reaction (RT-PCR) with gene -specific primers of ribosomal protein lateral stalk subunit (RPLP0), claudin-5 (CLDN5), and tight junction protein 1 (TJP1). qPCR primers were as follows: RPLP0-qF GCAGCATCTACAACCATGAA, RPLP0-qR GCAGATGGATCAGCCAAGAA; CLDN5-qF ATGTGGCAGGTGACCGCCTTC, CLDN5-qR CGAGTCGTACACTTTGCACTGC; TJP1-qF CATCTCCAGTCCCTTACCTTTC, TJP1-qR TCTGCTGGCTTGTTTCTCTAC.
Statistical analysis
Demographic characteristics of the participants were carried out using IBM SPSS (version 29.0.1.0, Chicago, IL, USA). The normality of distribution and homogeneity of variance were determined using the Kolmogorov-Smirnov and Levene’s tests, respectively. Continuous variables were presented as the mean and standard deviation and tested using the Mann-Whitney U test. Categorical variables were expressed as frequencies and percentages, and comparisons were made using the Chi-Square Test. Correlations were considered statistically significant at P-value < 0.05. For comparisons involving multiple experimental cell groups, one-way ANOVA followed by Dunnett’s post-hoc test was conducted using GraphPad Prism 10 (San Diego, CA, USA). A P-value ˂ 0.05 was considered statistically significant.
Results
Elevated tight junction and inflammatory biomarkers in adolescent MDD
We enrolled 46 adolescents diagnosed with MDD (33 females and 13 males) and 44 HCs (25 females and 19 males). Participants’ demographics and clinical characteristics are summarized in Table 1, and the study flow diagram is presented in Fig. 1. No statistically significant differences were observed between the MDD and HCs groups in terms of gender, age, or body mass index (BMI). The MDD group scored significantly higher on both HAMD-17 and HAMA than the HC group (P-value < 0.001), as expected by the diagnostic criteria used for group assignment.

1 Participants and sample collection outlined the recruitment of the subjects (healthy controls [HC], n = 44; major depressive disorder [MDD], n = 46), and the categories of biological samples collected; (2) Sample test and data analysis specified the methodologies and biomarkers used for sample analysis; (3) Cell experiment and validation describe the in vitro experiments aimed at validating the regulatory effects of short-chain fatty acids (SCFAs) on tight junction proteins.
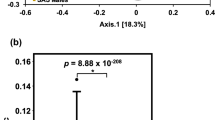
Quantification of tight junction proteins and inflammatory biomarkers revealed substantial elevations in Claudin-5, Zonulin, FABP, and LPS levels in the MMD group relative to controls. This disrupted gut barrier profile was accompanied by significantly elevated levels of pro-inflammatory cytokines IL-6, IL-8, TNF-α, and CRP (Fig. 2, Table 1). Together, these findings strongly indicated the presence of gut barrier disruption and systemic inflammatory responses in adolescent MDD.

Enzyme-linked immunosorbent assay (ELISA) results showing the levels of tight junction proteins (Claudin-5, Zonulin, FABP, and LPS) and inflammatory biomarkers (IL-6, IL-8, TNF-α, and CRP) in peripheral blood plasma samples. All measured biomarkers were significantly increased in the MDD group compared to the healthy controls (HCs). The P-value < 0.05 was considered statistically significant (***, P-value < 0.001).
Dysbiotic gut microbiota profile in adolescent MDD
To examine microbiome alterations associated with adolescent MDD, we analyzed fecal metagenomic sequencing data. Although alpha-diversity metrics, including the Chao index, Fisher index, and the number of detected species, were lower in patients with MDD compared to HCs, these differences did not reach statistical significance (Fig. 3A). In contrast, beta-diversity analysis revealed a substantial distinction in microbial composition between groups, as determined by PERMANOVA with Bray-Curtis dissimilarity (P-value = 0.001, Fig. 3B).

The index of α-diversity A and the PCoA of β-diversity (B) between MDDs and HCs, ns, not significant. C The bacterial taxa at the phylum level with a significant decrease in the F/B ratio, ***, P-value < 0.001. D MaAsLin2 and ANCOM-BC tests were used to explore shared microbiota discrepancies at the genus level, P-value < 0.05. E The bar plot showed the 40 shared differential genus taxa (except 10 genus taxa with only numbers). The colors of the bar represented the MDD (red) and HC (blue) groups. The right dotplot showed the up-regulated (pink) and down-regulated (blue) genera, respectively.
At the phylum level, the MDD group exhibited a marked increase in Bacteroidetes and a notable decrease in Firmicutes, resulting in a significantly lower Firmicutes/Bacteroidetes ratio (P-value < 0.001, Fig. 3C), an imbalance frequently associated with inflammatory states. In total, 40 genera showed differential abundance, including 25 increased and 15 decreased genera (P-value < 0.05, Fig. 3D and Table S1). Specifically, Intestinimonas, Barnesiella, Lawsonibacter, and Parasutterella were significantly increased in the MDD group, while Haemophilus, Dialister, Turicibacter, Agathobaculum, Hungatella, Collinsella, Allisonella, and Solobacterium were substantially reduced (Fig. 3E). These alterations highlighted dysbiotic microbial signature in adolescent MDD.
Diagnostic potential of the combination of target gut microbiota and biomarkers in adolescent MDD
To identify robust microbial biomarkers less susceptible to technical variation, we conducted cross-platform validation of genus-level signatures using 16S rRNA sequencing alongside metagenomic data. Collinsella, Dialister, Phascolarctobacterium and Barnesiella were identified in both datasets (Fig. 4A). Among them, Collinsella and Dialister were consistently decreased in adolescent MDD (Collinsella: 16S rRNA sequencing, P-value < 0.001; metagenomics, P-value < 0.001; Dialister: 16S rRNA sequencing, P-value < 0.001; metagenomics, P-value = 0.020).

A The barplot displayed four shared differential bacteria taxa at the genus level. **, P-value < 0.01; ***, P-value < 0.001; ns, not significant. B Random forest analysis was used to predict specific bacterial genus biomarkers. C The receiver operating characteristic (ROC) curves of single bacteria, biomarkers, and combined models for MDD-HCs.
We next performed random forest classification based on metagenomic profiles to identify bacterial genera with predictive value for adolescent MMD. Collinsella, Candidatus_Saccharibacteria_unclassified, and Parabacteroides emerged as top-ranked candidates and could be promising predictive taxa for adolescent MDD (Fig. 4B). ROC analysis showed AUCs of 0.74, 0.72, and 0.72, respectively (Fig. 4C). In comparison, individual biomarkers, Claudin-5, FABP, CRP, IL-6, IL-8, and TNF-α, yielded AUCs ranging from 0.70–0.81 (Fig. 4C).
Using LASSO regression, we identified six key potential biomarkers (Table S3) and integrated them with the top three genera identified from random forest analysis to construct a composite diagnostic model. Notably, the combined model incorporating Collinsella, Candidatus_Saccaribacteria_unclassified, and Parabacteroides achieved an AUC of 0.96, 0.97, and 0.97, respectively (Fig. 4C), demonstrating a high level of diagnostic accuracy for distinguishing adolescent MDD cases from HCs. These results suggest that integrating host biomarkers with gut microbial signatures may enhance the diagnostic precision for adolescent MDD and support the development of precise and effective microbiota-based diagnostic strategies.
Interconnections between SCFAs-related gut microbiota, tight junction proteins, and pathways
To further investigate the enhanced diagnostic performance provided by the combined model, we conducted a correlation analysis to examine the relationships among clinical features, tight junction proteins, inflammatory biomarkers, and target microbiota. The analysis revealed significant positive correlations among the network of tight junction proteins and inflammatory biomarkers (Fig. 5A). In contrast, Collinsella exhibited notably negative correlations with markers such as Claudin-5 and TNF-α. We also assessed the association between selected pathways and SCFAs through a systematic literature review. For instance, Claudin-5 was negatively associated with downregulated SCFA-related pathways, including alanine, aspartate, and glutamate metabolism [33], D-Glutamine and D-glutamate metabolism [34], and regulation of autophagy [35] (Fig. 5B).

A Spearman correlation among two target bacteria, clinical characteristics, and plasma biomarkers. The colors of the lines represent the negative (blue) and positive (red) correlations, respectively. The line types indicate the significance level of Spearman correlation, with P-value < 0.05 (solid line) and P-value > 0.05 (dashed line). B Spearman correlation between differential KEGG pathways and plasma biomarkers. The color coding of the boxes is used to highlight significant correlations: pink boxes represent KEGG pathways associated with SCFA that significantly correlate with Claudin-5, while blue boxes represent KEGG pathways associated with SCFA that significantly correlate with Zonulin, FABP, and LPS. *, P-value < 0.05; **, P-value < 0.01; ***, P-value < 0.001.
Other tight junction proteins, such as Zonulin, FABP, and LPS, were also negatively correlated with other SCFAs-related pathways, including D-Alanine metabolism [36], glycerolipid metabolism [37], and pyruvate metabolism [38, 39] (Fig. 5B). These findings underscore that elevated levels of tight junction proteins might be linked to SCFA-producing gut microbes and related KEGG pathways in adolescent MDD. Given the decreased abundance of Collinsella, a genus known for its SCFAs production, this indicated a potential reduction in SCFA levels in MDD. Furthermore, the observed negative correlation between Collinsella and Claudin-5 also suggested that SCFAs might modulate the expression of barrier integrity markers.
The regulation of SCFAs on tight junction proteins in Caco-2 Cells
It is well established that patients with depression exhibit a lower abundance of SCFA-producing gut microbes and that the serum levels of SCFAs are reduced in depressed rodents and associated with cytokine expression and neuroinflammation [40, 41]. Collinsella, a relatively novel genus, has been implicated in SCFA biosynthesis [42]. To investigate the regulatory effects of SCFAs on intestinal mucosal tight junction biomarkers, specifically CLDN5 and TJP1, we treated Caco-2 cells with propionate and butyrate. Our results demonstrated that propionate treatment (20 mM) for 24 h significantly increased transcriptional levels of both CLDN5 (P-value < 0.001) and TJP1 (P-value < 0.001), with a consistent dose-dependent trend (Fig. 6A and B). Extending the treatment to 48 h further enhanced the expression of CLDN5 (P-value < 0.001) and TJP1 (P-value < 0.001).

Reverse transcription-polymerase chain reaction (RT-PCR) analysis to investigate the expression of CLDN5 and tight junction protein 1 (TJP1) by propionate treatment (A) and butyrate treatment (B) for 24 h and 48 h. C The transcriptional expression of CLDN5 and TJP1 by lipopolysaccharide (LPS), LPS + P (propionate), and LPS + B (butyrate) treated for 24 h and 48 h. Different colors represent different concentrations of propionate and butyrate treatment. Error bars represent standard deviation (SD) from three independent experiments (n = 6). One-way ANOVA analysis followed by Dunnett’s multiple comparison test, with P-value ˂ 0.05 indicating statistically significant. ns, not significant; *, P-value < 0.05; **, P-value < 0.01; ***, P-value < 0.001.
Similarly, butyrate treatment (5 mM) for 24 h significantly upregulated CLDN5 (P-value < 0.001) and TJP1 (P-value < 0.05), with a similar dose-dependent pattern. Notably, after 48 h of butyrate treatment, CLDN5 and TJP1 levels remained significantly elevated compared to the control group (P-value < 0.001), suggesting that TJP1 responds more slowly or requires higher SCFA concentration than CLDN5. To mimic MDD-associated gut inflammation, we introduced LPS stimulation and combined it with SCFA treatment. Under these conditions, propionate and butyrate continued to significantly elevate transcription levels of CLDN5 and TJP1 (Fig. 6C). These results reinforce the role of SCFAs, produced by key gut microbes like Collinsella, in mediating the intestinal barrier function under inflammatory stress, and align with our broader observations linking SCFA-producing microbiota to gut barrier integrity in adolescent MDD.
Discussion
Our study revealed substantial differences in plasma levels of tight junction and inflammatory biomarkers between adolescents with MDD and HCs. Elevated levels of tight junction biomarkers (Zonulin, Claudin-5, FABP, and LPS) and inflammatory markers (IL-6, IL-8, TNF-α, and CRP) in MDD patients suggest a link between compromised intestinal permeability, systemic inflammation, and the pathogenesis of MDD. The observed microbiome alterations, characterized by a decreased Firmicutes/Bacteroidetes ratio and particular shifts in bacterial genera, highlight the complex role of gut microbiota in psychiatric disorders. The integration of gut microbiota data with tight junction proteins and inflammatory biomarkers resulted in improved predictive capabilities for adolescent MDD. The notable improvement in predictive power underscores the promise of these markers in developing diagnostic tools and personalized treatment strategies. KEGG pathway analysis indicated that increased levels of tight junction biomarkers may be associated with SCFA-producing gut microbes and related pathways in adolescent MDD, offering insights into how gut microbiota alterations might affect metabolic processes relevant to depression. Our cell experiments further validated that SCFAs were sufficient to modulate the transcriptional level of tight junction proteins, including CLDN5 and TPJ1.
The gut microbiota impacts various physiological and pathological aspects via neuronal, hormonal, and immunological pathways in adolescent MDD [11]. Based on previous research [43], we chose MaAsLin2 and ANCOM-BC to compensate for the limitations of relying on a single method. The most consistent inter-tool methods, ANCOM-II and ALDEx2, were based on non-rarefied data, but rare-based MaAsLin2 produced the most consistent results across datasets of the same phenotype [43]. Collinsella, a relatively novel bacterium in the context of depression, has been reported to be decreased in patients with MDD [18, 44]. Acetic acid and butyric acid are the primary fermentation metabolites of the genus Collinsella. The genomic characterization of Collinsella aerofaciens strains isolated from healthy human gut microbiota further revealed their genetic capacity for bile acid metabolism [45], a functional feature that may modulate host immune responses and facilitate gut-brain axis communication [46]. Integrating these findings with our research, we propose that reduced abundance of Collinsella is associated with diminished SCFA production, potentially undermining intestinal barrier integrity [20]. This compromised barrier function may exacerbate the leakage of harmful gut-derived substances [47], thereby aggravating the pathogenesis of depression. The prioritization of Collinsella in our discussion reflects both its significant alteration in this study and established mechanistic links to depression pathogenesis in these studies. Future investigations should expand focus to other SCFA-producing taxa identified, which may help further prospective and future study.
SCFAs are among the most abundant metabolites generated by the gut microbiota during the fermentation of non-digestible dietary fibers [48], with acetic acid, propionic acid, and butyric acid accounting for 95% of total SCFA production [49]. Blautia, Clostridium spp., Bifidobacterium spp., and Bacteroides spp. have been reported to produce acetic acid, while Coprococcus, Bacteroidetes, and Ruminococcus are associated with propionic acid production [38]. The human gut microbiota, including Lactobacillus, Bifidobacterium, Lachnospiraceae, and Ruminococcaceae, produce butyric acid by converting butyryl-CoA into butyrate and acetyl-CoA [37]. It is noteworthy that SCFA producers exhibit metabolic interactions; for example, the acetic acid formed by Bacteroidetes can be further transformed into butyric acid by Firmicutes [38]. In our work, Collinsella, along with tight junction and inflammatory biomarkers, demonstrated predictive capacity for adolescent MDD, underlining its potential in developing diagnostic tools. However, the precise role of the genus Collinsella and its underlying mechanisms in adolescent MDD remain to be elucidated.
SCFAs play a crucial role in regulating mucosal immunity by targeting both immune and non-immune cells, helping maintain epithelial barrier function at mucosal surfaces [20]. Previous clinical research has demonstrated that patients with depression exhibit significantly reduced levels of SCFAs in plasma [17]. Furthermore, an open-label, randomized trial revealed that supplementation with omega-3 polyunsaturated fatty acids indirectly elevated the levels of gut microbial SCFAs by boosting the abundance of SCFA-producing bacteria (e.g., Bifidobacterium, Roseburia, and Lactobacillus) [50]. Additionally, SCFAs protect gut barrier integrity by preventing bacterial and viral invasion [51]. Acetate, a key microbiome-derived SCFA, plays crucial roles in driving microglia maturation and regulating the homeostatic metabolic state [52]. Prebiotics treatment has been shown to reverse depression and anxiety symptoms in high-fat diet mice [53]. This improvement resulted from reduced dysbiosis, increased acetate-producing bacteria, and lower intestinal permeability, decreasing chronic inflammation [53]. Prebiotics also modulated the gut-brain axis, increasing brain acetate and reducing pro-inflammatory cytokines, thus enhancing neuronal proliferation and survival in the hippocampus and prefrontal cortex [53]. Acetic acid can mediate inflammatory responses by recruiting immune cells to protect the gut barrier [54].
In addition, butyric acid directly increases the expression of Claudin-1 and occludin, potentially linked to the activation of AMPK [55]. Valenzano et al. reported that treatment with butyric acid (5 mM) led to a 376% increase in Claudin-7 levels [56]. Moreover, butyric acid further enhances the expression of Claudin-3 and Claudin-4 by stimulating AKT signaling pathways [57]. Consistent with these findings, we further confirmed the regulatory effects of propionic acid and butyric acid on other tight junction proteins (CLDN5 and TJP1). This regulatory effect aligns with our microbiome findings, where reduced SCFA-producing bacteria in adolescents with MDD may contribute to compromised gut barrier function, underscoring a potential microbial pathway influencing MDD pathology through barrier dysfunction. Although SCFAs have exhibited a promising barrier-modulating effect in this study, their efficacy in clinical treatment for adolescent MDD remains to be further explored.
Despite the strengths of this study, there are several limitations to consider. Firstly, the sample size is relatively small to maintain consistency between blood and fecal samples, and fecal analysis for tight junction proteins was not performed. Secondly, this study lacked systemic assessment of common comorbidities(e.g., irritable bowel syndrome), herbal medication, and lifestyle confounding factors such as physical activity, dietary patterns, and sleep quality, all of which may interact with gut microbiota. Thirdly, the causal relationships among tight junction proteins, SCFAs, and the target genus Collinsella require further validation with more robust evidence. Finally, while our findings suggest increased sensitivity of microbiota integrating tight junction and inflammatory biomarkers in adolescent MDD (as indicated by the training and test subsets, Figure S1), the specificity of these predictive models still requires additional evaluation.{Citation} Potential overfitting also needs to be addressed by replicating the study with larger datasets. In summary, future studies should incorporate expanded sample collection and analysis to address these limitations and enhance the robustness of the findings.
In summary, our study observed elevated levels of tight junction proteins and inflammatory biomarkers in adolescents with MDD, indicating compromised gut barrier integrity and increased systemic inflammation. Microbiome analysis revealed significant dysbiosis, particularly a reduction in SCFA-producing bacteria such as Collinsella, potentially contributing to this barrier dysfunction. Integrating microbiota data with these biomarkers markedly improved diagnostic accuracy for MDD, highlighting a promising avenue for more precise and effective diagnostic strategies. Additionally, in vitro experiments demonstrated that SCFAs, including propionic and butyric acids, enhanced tight junction integrity, further supporting the relevance of gut barrier health in adolescent MDD.
Data availability
The metagenome and 16S rRNA data generated in this study are publicly accessible in the NCBI database under accession number PRJNA1186998 and PRJNA1190316. The analysis code has been deposited in the GitHub repository at https://github.com/guokai8/MDD_metagenome.
Code availability
The metagenome and 16S rRNA data generated in this study are publicly accessible in the NCBI database under accession number PRJNA1186998 and PRJNA1190316. The analysis code has been deposited in the GitHub repository at https://github.com/guokai8/MDD_metagenome.
References
Korczak DJ, Westwell-Roper C, Sassi R. Diagnosis and management of depression in adolescents. Cmaj. 2023;195:E739–e746.
Dwyer JB, Stringaris A, Brent DA, Bloch MH. Annual Research Review: Defining and treating pediatric treatment-resistant depression. J Child Psychol Psychiatry. 2020;61:312–32.
Johnson D, Dupuis G, Piche J, Clayborne Z, Colman I. Adult mental health outcomes of adolescent depression: a systematic review. Depress Anxiety. 2018;35:700–16.
Li F, Cui Y, Li Y, Guo L, Ke X, Liu J, et al. Prevalence of mental disorders in school children and adolescents in China: diagnostic data from detailed clinical assessments of 17,524 individuals. J Child Psychol Psychiatry. 2022;63:34–46.
Daly M. Prevalence of depression among adolescents in the U.S. From 2009–2019: analysis of trends by sex, race/ethnicity, and income. J Adolesc Health. 2022;70:496–9.
Miller L, Campo JV. Depression in adolescents. N Engl J Med. 2021;385:445–9.
Cui L, Li S, Wang S, Wu X, Liu Y, Yu W, et al. Major depressive disorder: hypothesis, mechanism, prevention and treatment. Signal Transduct Target Ther. 2024;9:30.
Morais LH, HLt Schreiber, Mazmanian SK. The gut microbiota-brain axis in behaviour and brain disorders. Nat Rev Microbiol. 2021;19:241–55.
Tan AH, Lim SY, Lang AE. The microbiome-gut-brain axis in Parkinson disease - from basic research to the clinic. Nat Rev Neurol. 2022;18:476–95.
Yap CX, Henders AK, Alvares GA, Wood DLA, Krause L, Tyson GW, et al. Autism-related dietary preferences mediate autism-gut microbiome associations. Cell. 2021;184:5916–5931.e5917.
McGuinness AJ, Davis JA, Dawson SL, Loughman A, Collier F, O’Hely M, et al. A systematic review of gut microbiota composition in observational studies of major depressive disorder, bipolar disorder and schizophrenia. Mol Psychiatry. 2022;27:1920–35.
Zheng P, Zeng B, Zhou C, Liu M, Fang Z, Xu X, et al. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry. 2016;21:786–96.
Wastyk HC, Fragiadakis GK, Perelman D, Dahan D, Merrill BD, Yu FB, et al. Gut-microbiota-targeted diets modulate human immune status. Cell. 2021;184:4137–4153.e4114.
Serna-Rodríguez MF, Bernal-Vega S, de la Barquera JAO, Camacho-Morales A, Pérez-Maya AA. The role of damage associated molecular pattern molecules (DAMPs) and permeability of the blood-brain barrier in depression and neuroinflammation. J Neuroimmunol. 2022;371:577951.
Alexandrov PN, Hill JM, Zhao Y, Bond T, Taylor CM, Percy ME, et al. Aluminum-induced generation of lipopolysaccharide (LPS) from the human gastrointestinal (GI)-tract microbiome-resident Bacteroides fragilis. J Inorg Biochem. 2020;203:110886.
Ortega MA, Alvarez-Mon MA, García-Montero C, Fraile-Martinez O, Guijarro LG, Lahera G, et al. Gut microbiota metabolites in major depressive disorder-deep insights into their pathophysiological role and potential translational applications. Metabolites. 2022;12:50.
Liu P, Liu Z, Wang J, Wang J, Gao M, Zhang Y, et al. Immunoregulatory role of the gut microbiota in inflammatory depression. Nat Commun. 2024;15:3003.
Zhou M, Fan Y, Xu L, Yu Z, Wang S, Xu H, et al. Microbiome and tryptophan metabolomics analysis in adolescent depression: roles of the gut microbiota in the regulation of tryptophan-derived neurotransmitters and behaviors in human and mice. Microbiome. 2023;11:145.
Fock E, Parnova R. Mechanisms of blood-brain barrier protection by microbiota-derived short-chain fatty acids. Cells. 2023;12:657.
Mann ER, Lam YK, Uhlig HH. Short-chain fatty acids: linking diet, the microbiome and immunity. Nat Rev Immunol. 2024;24:577–95.
Fachi JL, Felipe JS, Pral LP, da Silva BK, Corrêa RO, de Andrade MCP, et al. Butyrate protects mice from clostridium difficile-induced colitis through an HIF-1-dependent mechanism. Cell Rep. 2019;27:750–761.e757.
Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, et al. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. 2015;17:662–71.
Pral LP, Fachi JL, Corrêa RO, Colonna M, Vinolo MAR. Hypoxia and HIF-1 as key regulators of gut microbiota and host interactions. Trends Immunol. 2021;42:604–21.
Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA. 2014;111:2247–52.
Wu H, Wang J, Teng T, Yin B, He Y, Jiang Y, et al. Biomarkers of intestinal permeability and blood-brain barrier permeability in adolescents with major depressive disorder. J Affect Disord. 2023;323:659–66.
Blanco-Míguez A, Beghini F, Cumbo F, McIver LJ, Thompson KN, Zolfo M, et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat Biotechnol. 2023;41:1633–44.
Beghini F, McIver LJ, Blanco-Míguez A, Dubois L, Asnicar F, Maharjan S, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife. 2021;10:e65088.
Suzek BE, Wang Y, Huang H, McGarvey PB. Wu CH. UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics. 2015;31:926–32.
Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13:581–3.
McMurdie PJ, Holmes S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One. 2013;8:e61217.
Mallick H, Rahnavard A, McIver LJ, Ma S, Zhang Y, Nguyen LH, et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput Biol. 2021;17:e1009442.
Lin H, Peddada SD. Analysis of compositions of microbiomes with bias correction. Nat Commun. 2020;11:3514.
Facchin S, Bertin L, Bonazzi E, Lorenzon G, De Barba C, Barberio B, et al. Short-chain fatty acids and human health: from metabolic pathways to current therapeutic implications. Life. 2024;14:559.
Wang M, Wan J, Rong H, He F, Wang H, Zhou J, et al. Alterations in gut glutamate metabolism associated with changes in gut microbiota composition in children with autism spectrum disorder. mSystems. 2019;4:e00321–18.
Wang G, Qin S, Chen L, Geng H, Zheng Y, Xia C, et al. Butyrate dictates ferroptosis sensitivity through FFAR2-mTOR signaling. Cell Death Dis. 2023;14:292.
Harshaw NS, Meyer MD, Stella NA, Lehner KM, Kowalski RP, Shanks RMQ. The short-chain fatty acid propionic acid activates the rcs stress response system partially through inhibition of d-alanine racemase. mSphere. 2023;8:e0043922.
Wu T, Shen M, Yu Q, Chen Y, Chen X, Yang J, et al. Cyclocarya paliurus polysaccharide improves metabolic function of gut microbiota by regulating short-chain fatty acids and gut microbiota composition. Food Res Int. 2021;141:110119.
Turco L, Opallo N, Buommino E, De Caro C, Pirozzi C, Mattace Raso G, et al. Zooming into gut dysbiosis in parkinson’s disease: new insights from functional mapping. Int J Mol Sci. 2023;24:9777.
Om H, Chand U, Kushawaha PK. Human anaerobic microbiome: a promising and innovative tool in cancer prevention and treatment by targeting pyruvate metabolism. Cancer Immunol Immunother. 2023;72:3919–30.
Cheng J, Hu H, Ju Y, Liu J, Wang M, Liu B, et al. Gut microbiota-derived short-chain fatty acids and depression: deep insight into biological mechanisms and potential applications. Gen Psychiatr. 2024;37:e101374.
Xiao W, Li J, Gao X, Yang H, Su J, Weng R, et al. Involvement of the gut-brain axis in vascular depression via tryptophan metabolism: a benefit of short chain fatty acids. Exp Neurol. 2022;358:114225.
Qin P, Zou Y, Dai Y, Luo G, Zhang X, Xiao L. Characterization a novel butyric acid-producing bacterium collinsella aerofaciens subsp. shenzhenensis subsp. nov. Microorganisms. 2019;7:78.
Nearing JT, Douglas GM, Hayes MG, MacDonald J, Desai DK, Allward N, et al. Microbiome differential abundance methods produce different results across 38 datasets. Nat Commun. 2022;13:342.
Chen HM, Chung YE, Chen HC, Liu YW, Chen IM, Lu ML, et al. Exploration of the relationship between gut microbiota and fecal microRNAs in patients with major depressive disorder. Sci Rep. 2022;12:20977.
Bag S, Ghosh TS, Das B. Complete genome sequence of collinsella aerofaciens isolated from the gut of a healthy Indian subject. Genome Announc. 2017;5:e01361–17.
Jia M, Fan Y, Ma Q, Yang D, Wang Y, He X, et al. Gut microbiota dysbiosis promotes cognitive impairment via bile acid metabolism in major depressive disorder. Transl Psychiatry. 2024;14:503.
Welcome MO. Cellular mechanisms and molecular signaling pathways in stress-induced anxiety, depression, and blood-brain barrier inflammation and leakage. Inflammopharmacology. 2020;28:643–65.
Zhang S, Zhao J, Xie F, He H, Johnston LJ, Dai X, et al. Dietary fiber-derived short-chain fatty acids: A potential therapeutic target to alleviate obesity-related nonalcoholic fatty liver disease. Obes Rev. 2021;22:e13316.
Kaji I, Karaki S, Kuwahara A. Short-chain fatty acid receptor and its contribution to glucagon-like peptide-1 release. Digestion. 2014;89:31–36.
Watson H, Mitra S, Croden FC, Taylor M, Wood HM, Perry SL, et al. A randomised trial of the effect of omega-3 polyunsaturated fatty acid supplements on the human intestinal microbiota. Gut. 2018;67:1974–83.
Guo B, Zhang J, Zhang W, Chen F, Liu B. Gut microbiota-derived short chain fatty acids act as mediators of the gut-brain axis targeting age-related neurodegenerative disorders: a narrative review. Crit Rev Food Sci Nutr. 2025;65:265–86.
Erny D, Dokalis N, Mezö C, Castoldi A, Mossad O, Staszewski O, et al. Microbiota-derived acetate enables the metabolic fitness of the brain innate immune system during health and disease. Cell Metab. 2021;33:2260–2276.e2267.
Paiva IHR, Maciel LM, Silva RSD, Mendonça IP, Souza JRB, Peixoto CA. Prebiotics modulate the microbiota-gut-brain axis and ameliorate anxiety and depression-like behavior in HFD-fed mice. Food Res Int. 2024;182:114153.
van der Hee B, Wells JM. Microbial Regulation of Host Physiology by Short-chain Fatty Acids. Trends Microbiol. 2021;29:700–12.
Miao W, Wu X, Wang K, Wang W, Wang Y, Li Z, et al. Sodium Butyrate Promotes Reassembly of Tight Junctions in Caco-2 Monolayers Involving Inhibition of MLCK/MLC2 Pathway and Phosphorylation of PKCβ2. Int J Mol Sci. 2016;17:1696.
Valenzano MC, DiGuilio K, Mercado J, Teter M, To J, Ferraro B, et al. Remodeling of tight junctions and enhancement of barrier integrity of the CACO-2 intestinal epithelial cell layer by micronutrients. PLoS One. 2015;10:e0133926.
Yan H, Ajuwon KM. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS One. 2017;12:e0179586.
Funding
This work was supported by STI2030-Major Projects (2022ZD0212900), the National Natural Science Foundation of China (82271565), the Natural Science Foundation of Chongqing, China (CSTB2024NSCQ-LZX0060), and Chongqing Medical University Program for Youth Innovation in Future Medicine (W0045) to XZ.
Author information
Authors and Affiliations
Contributions
Concept, design, and supervision of the study: XZ; data acquisition, analysis, interpretation: XL, AG, MX, JL, HW, ZZ, FH, YH, TL, and JH; drafting manuscript: XL and AG; revised the manuscript: JH, KG, and XY; drafting display items: XL and AG. All authors read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
competing interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ethics approval and consent to participant
This study was performed in accordance with the guidelines of Chongqing Medical University and was approved by the University’s Ethics Committee (approval number: 2020-864). Informed consent was obtained from all participants prior to the study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Geng, A., Xia, M. et al. Alterations in short-chain fatty acid-associated gut microbiota and tight junction integrity in adolescent major depressive disorder. Transl Psychiatry 16, 11 (2026). https://doi.org/10.1038/s41398-025-03743-3
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41398-025-03743-3
