
Abstract
Non-viral engineering can ease CAR-T cell production and reduce regulatory and cost requirements. We utilized Sleeping Beauty transposon to engineer donor-derived anti-CD19.CD28.OX40.CD3zeta T cells differentiated in cytokine-induced killer (CARCIK-CD19) for B-cell precursor acute lymphoblastic leukemia (BCP-ALL) patients relapsed after allogeneic hematopoietic stem cell transplantation (alloHSCT). We report the results of CARCIK-CD19 observed in 36 patients (4 children and 32 adults) treated according to the final recommended dose. Cytokine release syndrome of grade 2 or lower occurred in 15 patients, ICANS grade 2 in 1 patient, and late-onset peripheral neurotoxicity of grade 3 in 2 patients. GVHD never occurred after treatment with allogeneic CARCIK-CD19. Complete remission was achieved by 30 out of 36 patients (83.3%), with MRD negativity in 89% of responders. With a median follow-up of 2.2 years, the 1-year overall survival was 57.0%, and event-free survival was 32.0%. The median duration of response at 1 year was 38.6%. CAR-T cells expanded rapidly after infusion and remained detectable for over 2 years. Integration site analysis after infusion showed a high clonal diversity. These data demonstrated that SB-engineered CAR-T cells are safe and induce durable remission in heavily pretreated patients with BCP-ALL relapsed after alloHSCT. Trial registration: The phase 1/2 and phase II trials are registered at www.clinicaltrials.gov as NCT#03389035 and NCT#05252403.
Similar content being viewed by others


Introduction
Chimeric antigen receptor (CAR) T cells targeting the B-cell specific CD19 antigen are licensed for the treatment of relapsed/refractory (r/r) B-cell precursor acute lymphoblastic leukemia (BCP-ALL) in children and young adults, after the achievement of remarkable clinical response characterized by a remission rate of 70–90% and overall survival (OS) of 60–75% at one year [1]. In the adult population, substantial toxicities have hindered the application of CD19 CAR-T cell therapy [2], which has only recently been approved [3, 4]. To date, FDA/EMA-approved therapies are produced in centralized manufacturing facilities from patient-derived apheresis material through genetic engineering with viral vectors [5] with standardized procedures in compliance with good manufacturing practices (GMP) [6]. Despite viral vectors have demonstrated a decades-long safety and efficacy, regulatory requirements and time-consuming processes necessitating producer cell lines limit their production to specialized biosafety level (BSL) 2 manufacturing facilities, impacting their availability and the final cost of the cell product. Furthermore, patient-derived cell products may experience difficulties in achieving the target cell dose, particularly in late-stage patients who are often lymphodepleted and typically have circulating blasts in their blood [7].
We recently reported an approach to engineered human T cells without the need for viral vectors [8, 9] by using the recombinant DNA-based Sleeping Beauty (SB) transposon [10]. The SB vector allows for stable expression of the transgene by exploiting transposition from a donor DNA to an acceptor site within the genome [11]. This avoids the need for producer cell lines and instead exploits mRNA and DNA-based vectors that can be produced in large quantities for multiple patients, easing the process of obtaining clinical-grade CAR-T cells [12]. We therefore evaluated non-viral CD19 CAR-T cells in BCP-ALL patients who relapsed after alloHSCT. CAR-T cells were produced in-house from alloHSCT donor cells to avoid using heavily-pretreated patient’s lymphocytes. To decrease the likelihood of inducing graft-versus-host disease (GVHD), CAR-T cells were differentiated into effector CD3+ cells according to the cytokine-induced killer cell (CIK) protocol [13]. Preliminary results during the dose-escalation part of a phase I/II trial demonstrated a remarkable safety profile associated with anti-leukemic activity using high doses of non-viral allogeneic cells [14].
Here we report the results of the 36 adult and pediatric patients with BCP-ALL who received the final recommended dose of SB-engineered CD19 CAR-T cells, also referred to as CARCIK-CD19, within a phase 1/2 (FT01CARCIK), compassionate use (FT02CARCIK) and phase 2 (FT03CARCIK) studies with a median follow-up of 2.2 years.
Subjects and methods
Study design and patients
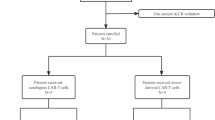
Thirty-six consecutive pediatric and adult patients with BCP-ALL relapsed after alloHSCT, treated with the final recommended dose of CARCIK-CD19 cells (>7.5 × 106 transduced CARCIK-CD19 cells/kg), were analysed. Fifteen patients were treated within the FT01CARCIK study, 6 within a compassionate base after approval by the Agenzia Italiana del Farmaco (AIFA) (FT02CARCIK) and 15 within the phase 2 FT03CARCIK study. The patients were treated in 2 sites in Italy, the Pediatric Clinic of the University of Milano-Bicocca/Fondazione IRCCS San Gerardo dei Tintori for pediatric patients and at the Papa Giovanni XXIII Hospital Bergamo for adult patients. Detailed methodology for the single-arm, multicenter, FT01CARCIK study (NCT#03389035) was previously reported [14] and it is superimposable to that of the study FT02CARCIK and FT03CARCIK (NCT#05252403) (Supplementary Methods). The inclusion criteria were modified after FT01CARCIK where patients were eligible with a morphological disease in the bone marrow (BM, >5% CD19 positive blasts) at study entry and without active graft-versus-host disease (GVHD) and adequate organ function (see details in Appendix) to enroll also patients presenting with at least 1% molecular disease at polymerase chain reaction (PCR) or flow cytometry. After lymphodepletion with Fludarabine (30 mg/m2/day × 4 days) and Cyclophosphamide (500 mg/m2/day × 2 days), patients received a single CARCIK-CD19 infusion. A second infusion was permitted when persistence of residual disease after one month from the first infusion in FT03CARCIK. Bridging therapy was allowed before CAR-T cell infusion.
Objectives and endpoints
The objectives were to define the safety and tolerability of CARCIK-CD19 in pediatric and adult patients with BCP-ALL relapsed after alloHSCT, as well as the rate of overall remission (ORR), duration of response (DOR), event-free survival (EFS) and OS. ORR includes complete remission (CR) and CR with incomplete blood count recovery (CRi). DOR was defined as the time from CR to relapse, or death due to any cause, whichever occurred first. EFS was from the date of the first CARCIK-CD19 infusion to the earliest of the following events: treatment failure (i.e., no CR), relapse, or death from any cause. OS was the time from the date of the first CARCIK-CD19 infusion to death due to any reason. Patients were censored at the last follow-up in case no events occurred and to new anticancer therapies (including alloHSCT) in DOR and EFS. Relapse was defined by the recurrence of more than 5% of lymphoblasts in the peripheral blood or in the BM and/or by the presence of extramedullary disease. Measurable residual disease (MRD) was assessed by validated multiparametric flow cytometry and PCR for leukemia-specific immunoglobulin (IG)/T cell receptor (TR) gene rearrangements.
Integration site retrieval and sequencing
A comprehensive integration site (IS) analysis for biosafety evaluation was performed on pre-infusion products, blood and bone marrow samples of the 15 patients enrolled in the FT01 clinical trial and treated with the final recommended dose. The retrieval of Sleeping Beauty IS was performed by a sonication-based linker mediated (SLiM)-PCR method, as previously reported [14]. The fragmented DNA was split in 3 technical replicates and subjected to end repair and 3’ adenylation using the NEBNext® Ultra™ DNA Library Prep Kit for Illumina® (New England Biolabs, Ipswich, MA.), and then ligated (DNA Technologies ligation kit, Skokie, IL) to linker cassettes containing an 8 nucleotide sequence barcode used for sample identification. All PCR primers were synthetized at Integrated DNA Technologies (Coralville, Iowa). Barcoded Linker cassettes were generated as previously reported [14]. Primers are reported in Supplementary Table 5. Each library was sequenced on the Illumina NovaSeq sequencer (illumine, San Diego, CA.) using SP500 flow cells with the 2 × 250 paired end mode. All sequence data obtained in the IS analysis are available at the NCBI’s SRA database (PRJNA1229450): https://www.ncbi.nlm.nih.gov/sra/PRJNA1229450.
Statistics
Categorical variables were summarized by counts and percentages, while continuous ones were described with measures of location (i.e., arithmetic or geometric mean, median) and variability (i.e., range, SD, coefficient of variation). The remission rate estimates were reported with 95% exact Clopper-Pearson Confidence Intervals (CI), while the time to event endpoints were described at specific time-points using the Kaplan–Meier estimator with the Greenwood standard error (SE) and the corresponding 95%CI. Between groups comparisons on time to event endpoints were performed by means of the log-rank-test, while for continuous variables non parametric tests were performed (i.e., Mann-Whitney test for 2 groups and Kruskal-Wallis test for more than 2 groups). The tests were two-sided at a significant level of 5%. Statistical analyses were performed with SAS 9.4 and R 4.3.1, except those on GMP manufacture and CAR-T persistence, which were performed with GraphPad Prism 9.0.
Ethics approval and consent to participate
The phase I/II study (FT01CARCIK), the compassionate base (FT02CARCIK) and the phase 2 (FT03CARCIK) were performed in accordance with the protocols of the EMA, approved by the Italian Regulatory Central Authorities (AIFA) and by the local ethics committees (COMITATO ETICO BRIANZA CE150179 and COMITATO ETICO BERGAMO CE150180), and registered (EudraCT 2017-00900-38 and ClinicalTrials.gov NCT03389035 for FT01CARCIK, EudraCT 2020-005025-85 and ClinicalTrials.gov NCT#05252403 for FT03CARCIK). The studies were approved at each study site by the local Institutional Review Board, and conducted in accordance with the Declaration of Helsinki. All patients or their guardians provided written informed consent.
Results
Characteristics of product and patients
From February 2019 to December 2023, 36 BCP-ALL patients refractory or relapsed after alloHSCT pooled from the 3 studies (15 Phase 1/2, 6 compassionate, 15 Phase 2 patients) were treated with the final recommended dose of CARCIK-CD19 cells. CAR-T cells were manufactured in-house by electroporation with SB plasmids expressing a CD19 third-generation CAR incorporating the CD28 and OX40 costimulatory domains. The cellular source consisted of 50 ml of peripheral blood from the previous transplant donor, collected after the screening of patients. Successful production of the cell product was achieved for all patients enrolled. The final cell product consisted of CD3+ cells (mean 98.3%, range 82.9–99.8%). The mean transduction efficiency (percent of CD3+ cells expressing the CD19 CAR) was 33.5% (range, 5.0–62.4%, Supplementary Table 1). Patients were required to be no longer under immunosuppression and to have no evidence of GVHD and central nervous system (CNS) involvement at screening. The median age was 39 years (range, 1–67 years, Table 1). Eleven patients had previously received blinatumomab (31%), 2 patients Inotuzumab (6%) and 5 patients received both (14%). The median number of prior lines of therapies was 3 (range, 1–8) with a median time interval from prior alloHSCT to relapse of 7 months (range, 1–168). Nine patients (25%) were enrolled into this study after failing a second alloHSCT. The median BM blast count at enrollment was 24% (range, 4–100%) and 0% (range, 0–80%) after lymphodepletion. Seven patients (19%) presented extramedullary diseases, 3 without BM involvement (all enrolled into the compassionate protocol) and 4 with a concomitant BM involvement. Before the CARCIK-CD19 cell infusion, 5 patients (14%) had previously experienced acute GVHD, 1 patient chronic GVHD (3%) and 1 patient both (3%). Eleven patients (31%) were BCR-ABL positive (Ph+ ALL) and failed at least two TK inhibitors. Thirty-five patients received bridging therapy before lymphodepleting chemotherapy to control disease progression. Among them, 12 patients (33%) received Inotuzumab, 21 (62%) received low-dose chemotherapy with or without steroids or radiotherapy, and 2 patients ponatinib (3%). The median time from enrollment to the infusion of CARCIK-CD19 cells was 76 days (range 46–288). This duration included the time required to obtain the cellular source from the donor, approximately three weeks for cell product manufacturing, followed by an additional 7 to 10 days for the release after production. In some cases, this timeline could be extended due to the need for patients to recover from complications related to their disease, such as infections, or the completion of intrathecal therapy to address central nervous system involvement before the infusion.
Safety of treatment
Cytokine release syndrome (CRS) occurred in 15 patients (42%), with grade 1 in 8 patients and grade 2 in the remaining 7 patients (19%) (Table 2). No grade 3 or 4 CRS occurred. As defined by Lee et al. [15] grade 1 CRS was treated with antipyretic and/or i.v. fluids, while patients with grade 1 CRS persisting more than 48 hours and those with grade 2 CRS, also received tocilizumab (n = 9). None of the patients who experienced CRS needed admission to the intensive care unit. The median first onset of CRS symptoms after CARCIK-CD19 infusion occurred on day 2 (range 1–11 days). Only one patient (3%) experienced immune effector cell-associated neurotoxicity syndrome (ICANS) grade 2 recovered with the administration of dexamethasone. Two patients (6%) experienced a late onset peripheral neuropathy of the lower limbs (at day +44 and +61, respectively), characterized by walking difficulties of grade 3 according to CTCAE. The diagnostic work-up, including MRI, lumbar puncture, and the search of onconeural antigens did not reveal a clear causative event. These patients received steroids with only a minimal benefit, then started monthly cycles of high-dose immunoglobulins and physiotherapy which led to a slow improvement of neurological symptoms, but not a complete recovery after 6 months. GVHD did not occur after treatment with donor-derived CARCIK-CD19. Ten patients had infectious complications, with 5 pneumonia (2 fungal pneumonia), 1 upper respiratory infection, 1 sepsis due to Gram-negative bacteria, 1 enterocolitis, and 2 neutropenic fever. Fourteen out of 30 patients (47%) in remission at day 28 showed neutropenia of grade 3 or 4. Long-lasting severe neutropenia (at day 90) was documented in 7 out of 21 (33%) evaluable patients still in remission (only 1 with grade 4). Thrombocytopenia of grade ≥3 was reported in 15 out of 30 (50%) patients in CR at day 28 with persistence of severe thrombocytopenia at day 90 in 8 out of 21 evaluable patients (38%) still in remission.
Efficacy of treatment
The primary endpoint of the overall response rate as defined by the Phase II part of the study (ORR) was achieved by 30 out of the 36 pooled patients from the 3 studies (83%, 95%CI = 67–94%). Twenty-four out of 28 evaluable patients (86%) achieved MRD-negative or positive-not-quantifiable (PNQ) CR. Patients with ≥5% BM blasts (median 25%, range 14–80%) post lymphodepletion showed an inferior CR as compared to patients with <%5 (62% vs 100%, p = 0.0046). We observed higher response rates (100%) in 12 patients receiving anti-CD22 therapy as bridging therapy before the cellular infusion. There were not significant differences in CR rate between Ph positive and negative patients (Table 3).
Of 30 patients achieving CR, 18 (60%) relapsed, including 16 with a CD19+ disease (89%), and 2 with CD19-negative/dim BCP-ALL [11]% (Table 4). Among the 12 patients who did not experience a relapse after CARCIK-CD19 infusion, 3 patients (25%) underwent consolidation with a second alloHSCT within seven months after infusion and remain alive and disease-free ( +3.9 and +4.0 years, and +2 months). Additionally, 6 patients (50%) are still alive and disease-free without additional new therapies (1 with CAR-T cells circulating after 40 months), while 3 (25%) died in CR (1 due to sepsis and 1 due to hyporexia and ascites and 1 due to epilepsy with lumbar puncture negative for disease). Among the relapsed patients, 3 (17%) are alive in CR after salvage therapies for relapse, 2 (11%) died in a new CR after salvage treatment, and 1 (6%) patient is alive with active disease. The remaining 12 (67%) patients died due to disease progression, as did 5 out of 6 non-responders. One non-responder patient died in CR after additional therapies due to sepsis. Overall, as of the data cutoff, 13 of the 36 (36%) patients were alive, and 12 (33%) in ongoing CR with a median follow-up of 1.8 years (range 2.8 months-4.1years) (Table 4). Of interest, all 7 patients with involvement of extramedullary sites at the enrollment achieved CR. One patient remained in CR after 1.7 years, while the remaining 6 patients relapsed. CAR-T cells were detected in the pleural effusion of one patient with pleural localization and in the cerebrospinal fluid of a patient who had a history of CNS infiltration.
After a median follow-up of 2.2 years, the 1-year OS was 57.0% (95%CI 42.6–76.2%), with a median duration of 12.3 months (95%CI 6.3–23.3 months). EFS at 1 year was 32.0% (95%CI 18.9–54.4%), with a median duration of 4.9 months (95%CI 3.2–11.6 months) (Fig. 1A). The median DOR of the 30 patients who achieved CR was 8.1 months (95%CI 3.1–16.2), with a 1-year DOR of 38.6% (95%CI 23.3–64.1%) (Fig. 1B). Unlike previous clinical trials of CD19 CAR in BCP-ALL, our cohort included 25% of patients who relapsed after a second alloHSCT. Interestingly, in a subanalysis of 34 out of 36 patients with available blast level after lymphodepletion, patients with prior one alloHSCT and a lower blast level post-lymphodepletion (<5%) had a better EFS and OS at 1 year (47.5% (95%CI 26.3–86% and 80.5% (95% CI 73.4-100), respectively) than patients with >1 prior alloHSCT or a higher burden of disease (20.6% (95% CI 7.7–54.9% and 35.3% (95% CI 18.5–67.2), respectively p = 0.0373 and p = 0.0055) (Fig. 1C). It is worth noting that the median OS for patients with blast <5% after lymphodepletion was about 24 months and this result was independent of the bridging therapy used. The difference in prognosis was mainly driven by blast after lymphodepletion, which resulted the only factor associated with ORR, EFS and OS (p = 0.0046, p = 0.0352 and p = 0.0396, respectively). A tendency of better outcomes was observed among Ph+ patients, probably as the consequence of a pre-emptive treatment with TKIs in case of molecular relapse (Table 3).

A OS, and EFS, in the 36 patients who received CARCIK-CD19 at the recommended dose. Kaplan–Meier estimates of OS and EFS. EFS was estimated censoring at treatment shift and all estimates are reported with the corresponding SE. B DOR in the 30 patients who received CARCIK-CD19 and achieved CR. Kaplan–Meier estimates of DOR. DOR was estimated censoring at treatment shift and all estimates are reported with the corresponding SE. C OS among 34 patients who received CARCIK-CD19 according to number of prior alloHSCT and bone marrow burden post-lymphodepletion.
CAR-T cell expansion, persistence and B-cell aplasia
CAR-T cell expansion was observed in all patients (Fig. 2A). The median peak expansion was 38.1 cells (range, 0.3-2283.2) per μl (Supplementary Table 2). The median time to maximal expansion in blood was 10 days (range, 7–28). CAR-T cell expansion was associated with CRS occurrence in terms of area under the curve (AUC) and peak of expansion (p = 0.0015 and 0.0004, respectively, Fig. 2B, C). No other meaningful associations were observed, including the achievement of response or tumor burden (Fig. 2D, E and Supplementary Fig. 1A–C). The median AUC from day 0 to day 28 was 280 cells (range, 3–24,572) per μl (Supplementary Table 2). Interestingly, the type of donor used to manufacture cells did not influence CAR-T cell expansion (Fig. 2F). CAR-T cells showed durable persistence, being detectable by flow cytometry for more than 2 years with a median of 165 days (range, 15–660) post-infusion and quantifiable in 23 out of 36 evaluated patients at the last follow-up (Fig. 2A). Engrafted CAR-T cells were also found to control the emergence of healthy B-cell progenitors and to maintain B-cell aplasia in one patient where we observed peaks of expansions at late follow-ups (150 and 450 days) after infusion (Fig. 2G). Loss of B-cell aplasia was observed in 14 of 30 (47%) responder patients at 3 months and in 18 (61%) at 6 months (Fig. 2H).

A CAR-T cells in peripheral blood by flow cytometry. B–C AUC-d28 (B) and Cmax-d28 (C) according to the severity of CRS. D AUC-d28 in patients who experienced CR or NR after CAR-T cell treatment. E AUC-d28 in patients with BM blasts post lymphodepletion less than or higher than 5%. F AUC-d28 in patients treated with CAR-T cells manufactured from identical sibling donors (ISD), haploidentical donors (Haplo), and matched unrelated donors (MUD). G Normal B-cell and CAR-T cell engraftment in the peripheral lood of patient 5 at different time points by flow cytometry. H B-cell recovery in CR and NR patients.
Elevation of cytokines in the serum of treated patients was observed, with a peak between day 7 and day 10 post-infusion (Supplementary Fig. 2A). Peak values were generally low in most patients (Supplementary Table 3) and levels above 1000 pg/ml were rarely detected. High peaks of IFN-γ and IL-6 were associated with grade 2 CRS (Supplementary Fig. 2B). We found no correlation between cytokine elevation and increased tumor burden at enrollment or after lymphodepletion.
Integration Site analysis of SB modified CARCIK-CD19 and peripheral blood of treated patients
To evaluate the biosafety of SB engineering, as first-in-human use of non-viral technology, we performed a comprehensive IS analysis of the genomic DNA extracted from the medicinal product and from the PB and BM of the 15 patients enrolled in the FT01 trial, harvested at different time points after infusion. A total of 42,508,099 raw reads passed our quality test and were associated to the barcodes used to tag the Sonication Linker Mediated (SLiM)-PCR performed on 45 samples. A total of 16,852,624 IS reads were identified and led to an overall number of 95,718 unique ISs (Supplementary Table 4). The genome wide distribution profile of SB-engineered T cells showed no preference for gene dense regions and transcriptional start site, and no differences between IS retrieved in the batch and in circulating CAR-T cells (Supplementary Fig. 3A). The distribution of IS showed no preference for promoter regions (Supplementary Fig. 3B). We observed enriched gene ontology terms in the medicinal product, mostly related to T-cell biology, due to SB preference for accessible chromatin regions (Supplementary Fig. 4). We identified genes targeted at a frequency significantly higher than expected, also named sporadic common IS, in single patients and targeting nearby genes by a relatively small number of IS (3 to 5). None of them was classified as a cancer related gene (Supplementary Figs. 5, 6). To observe potential clonal expansions caused by insertional mutagenesis, we computed accurately the clonal abundance. Based on this analysis, transduced CARCIK-CD19 cells in vitro showed a highly polyclonal repertoire. After infusion, a sizeable amount of IS present in the cell product persisted overtime (Fig. 3A and Supplementary Fig. 7). No signs of expansion have been observed in any patient. Moreover, we analyzed the clonal population diversity by the Shannon (H) index [16] (Fig. 3B), which was very high in the cellular product and early after infusion, but considerably reduced at later time points in association with CAR-T cell contraction. Finally, we determined IS frequency into introns, exons or intergenic regions in the cell products and post-infusion (Fig. 3C). SB insertion maintained the characteristic low bias towards genomic regions over time [17, 18]. We can conclude that SB transposon has no bias for regulatory and exonic regions and the close-to-random insertion profile is maintained post-infusion with absence of clonal dominance.

A Clonal abundance as percentage of genomes with a specific IS over the total genomes represented over time in the cell product (time 0) and in the peripheral blood post infusion of patients 9 and 13; ribbons connect tracked clones between two consecutive time points. Below each plot, the ten most abundant clones annotated with the closest gene are reported. B Diversity index (Shannon entropy) computed for each time points in the peripheral blood. Each line represents a different patient. C Percentage of IS located in exonic, intronic, and intergenic genomic regions overtime in the cell product, in the BM, and in the peripheral blood post infusion.
Discussion
High costs and regulatory requirements associated with the use of viral vectors limit accessibility to CAR-T cell therapy and impact healthcare spending [19]. In addition, the production of autologous CAR-T cells for patients who are heavily pretreated or have rapidly progressive disease may be a challenge. CARCIK-CD19 is an anti-CD19 CAR-T cell product manufactured using the non-viral SB transposon vector from the previous transplant donor, the first to demonstrate achievement of remission in clinical trials using SB technology [14]. In this extended analysis including 36 patients, we confirmed that allogeneic CARCIK-CD19 is extremely safe and able to induce long-term remission in a group of heavily pre-treated patients all relapsed after one or even two alloHSCT. These findings support the value of using donor-derived allogeneic T cells and a non-viral technologies for T-cell-engineered immunotherapies. CARCIK-CD19 was successfully manufactured for all the patients we enrolled into this study. We reported manageable toxicity with a low incidence of CRS even in patients with high disease burden, which is associated to an increased incidence of adverse events [20]. This is particularly relevant for the adult population, where some studies reported revised adverse event management strategy [43], and a fractionated dose scheduling [2, 21, 22] to mitigate the toxicity. In this study, CRS was only of grade 1 or 2 and reversible in all cases and neurotoxicity was rare, presenting as a late-onset peripheral neuropathy of grade 3 without a clear causative event in 2 patients. Late-onset peripheral neuropathies are a rare side effect previously reported in a few patients treated with anti-BCMA CAR-T cells, but not with anti-CD19 CAR-T cells [23]. We did not find any evidence supporting a direct role of CARCIK-CD19 in mediating this late-occurring peripheral neuropathy. This side effect may not be related to the cell therapy program, but rather to the previous pharmacological treatments or to the leukemia itself, as both patients subsequently relapsed. Importantly, favorable safety data were associated with low cytokine elevation compared to other CAR-T cell products but similar expansion and persistence. The low toxicity and need for higher doses may be related to the prevalence of CD8+ CD56+ effector memory cells which is in keeping with the data obtained using cord blood-derived allogeneic NK cells [13]. This is also in line with previous studies, demonstrating that ex vivo expanded T cells [24] and combined CD28-costimulated CAR and allogeneic TCR activation [25] can be associated with a manageable risk of GVHD. Indeed, GVHD was not observed after CARCIK-CD19 infusion, despite some patients experiencing GVHD after the previous transplant from the same donor. Similar to our strategy but using conventional viral engineering, donor-derived CD19 CAR-T cells have been explored in pediatric [26] and adult [27] BCP-ALL patients relapsing post-alloHSCT, reporting mild GVHD episodes. In addition, CD19 CAR-T cells from universal allogeneic donors, obtained by genome editing of the TCRα constant chain (TRAC) [28], showed a promising safety profile in adult [29] and pediatric [30] BCP-ALL patients with a low risk of inducing GVHD. As frequently reported for patients treated with CAR-T [31], we observed an incidence of prolonged cytopenia of grade >3 after 90 days from infusion in a third of cases. This might be due to the severity of the disease, the multiple previous lines of treatments, and poor hematopoietic reconstitution after alloHSCT and lymphodepletion. Therefore, our data demonstrated a remarkable safety profile of donor-derived CARCIK-CD19 in adult and pediatric patients with BCP-ALL relapsing after alloHSCT.
Although the study included patients with highly refractory diseases, CARCIK-CD19 showed a relevant therapeutic potential with an overall response in 83% of patients, with an MRD negativity in 86% of responders. Currently, two anti-CD19 CAR-T cell products are available for r/r BCP-ALL, tisagenlecleucel (tisa-cel) for pediatric and young adult patients [1] and brexucabtagene autoleucel (brexu-cel) for patients aged 18 and older [3]. Tisa-cel in the Eliana study was reported to have 81% of overall response rate [1]. Long-term follow-up of the ELIANA study showed 5-year EFS and OS rates of 42% and 55%, respectively [32]. It is worth noting that this study included mostly pediatric patients (83%), that is known to have superior EFS compared with the adult population [33, 34]. The real-world analysis confirmed similar results [35]. The single-arm ZUMA-3 trial evaluated KTE-X19 in the largest population of adults with r/r BCP-ALL. The overall CR rate was 71% with RFS of 58% at 6 months and OS of 71% at 12 months [3]. In a recent analysis of 78 patients who received brexu-cel in ZUMA-3 (23 phase 1 patients and 55 phase 2 patients), the treatment benefit was higher in patients in earlier lines of therapy and without prior exposure to alloHSCT [36]. Although patients enrolled in our studies showed more unfavorable characteristics compared to previous studies, such as an increased number of prior lines of therapy, extramedullary involvements, and all were enrolled as post-alloHSCT relapses, our results appeared largely comparable, showing excellent CR rates with the majority of patients being molecular remissions. We observed slightly lower EFS (44% at 6 months) and OS (57% at 12 months) compared to other studies [2,3,4, 20, 36, 37] but this might be due to the inclusion of patients with more unfavorable characteristics, as also demonstrated by our subanalysis showing an OS of 85% at 12 months in patients who had previously undergone a single alloHSCT and with low (<5%) BM burden post-lymphodepletion. Since the most important predicting factor for response was < vs > 5% blasts after lymphodepletion, it would have been important to collect more detailed information about the percentage of blasts pre-bridging, post-bridging, and then pre-and post-lymphodepletion. However, per study protocol, the total amount of bone marrow evaluation was limited for ethical reasons similar to other studies including pediatric patient. Patients who had experienced a relapse after alloHSCT have been recently reported to have a poorer prognosis in a sub-analysis of the Zuma-3 study [36]. A third autologous CAR-T product, obecabtagene autoleucel (obe-cel), was recently approved by FDA, and, in the coming months, will be available using a novel anti-CD19 scFV conferring a fast target binding off-rate. Data from the 153 patients enrolled in Phase 1b/ 2 trial demonstrated a CR/CRi rate of 77%, a favorable safety profile, high persistence, and durable responses in r/r adult ALL patients with EFS of 65% at 6 months and OS of 61% at 12 months [4].
The presence of extramedullary BCP-ALL is predictive of inferior outcomes after CAR-T cell therapy [20, 38]. In our study, we observed responses in all patients with extramedullary diseases, with one patient achieving a long-term remission. Experimental data with CIK cells support the notion that these cells might have a peculiar capability to infiltrate large tumor masses and extramedullary disease [24, 39]. We observed the highest response rates (100%) in the patients receiving anti-CD22 therapy as bridging therapy before the cellular infusion, suggesting that a sequential administration of dual antigen targeting might increase the overall response rate. This observation is in keeping with a similar positive preliminary experience in patients receiving co- or sequential administration of CAR-T targeting CD19 and CD22 [21, 40,41,42]. In this context, we have identified BAFF-R as a target for relapsing BCP-ALL which now we are combining with anti-CD22 CAR as a multi-targeting approach [43]. In this study using the CD28 and OX40 costimulatory domains, CD19-negative relapses were observed but with less frequency compared to CD19+ relapses, similar to data reported with adult patients treated with the CD28-costimulated KTE-X19 [3].
Virus-free technologies are rapidly evolving to reduce high costs and increase accessibility of gene therapy manufactured with viral vectors [11]. DNA-transposons require only nucleic acid components as starting materials, providing greater biosafety, sustainability and regulatory benefits. Compared to CRISPR/Cas9 editing, they can mobilize large portions of DNA, which allows multiplexed engineering within a single vector [44]. Moreover, transposases of the Tc1/mariner superfamily do not generate open double-strand breaks because they act through DNA hydrolysis and transesterification [45]. However, their use in clinical trials is at early stages and limited information are available on their efficacy and safety. In the present study, SB-engineered CAR-T cell expansion showed pharmacokinetic properties comparable to those reported for adult patients treated with commercial products [46]. The expansion was associated with CRS incidence, in agreement with previous observation [4]. Notably, the long-term persistence of functional CAR-T cells was evidenced by the maintenance of B-cell aplasia associated with detecting CAR-T cells in peripheral blood by flow cytometry. This data compares favorably to the CD28-containing CAR construct [47]. Incorporation of the OX40 costimulatory domain, which provides signals favouring late proliferation, in a CD28-based CAR architecture may have helped sustain the response [48], although it could predispose to excessive stimulation [49]. Importantly, we carefully balanced the activity of the transposase to achieve a integrated vector copy number per cell lower than five, and no adverse events associated with SB-engineering were observed. Conversely, malignant transformation has been recently reported in patients treated with CAR-T cells generated with PiggyBac transposon [50]. We found that SB transposon’s absence of bias towards transcriptional start sites and genomic regions are maintained in circulating CAR-T cells even long-term after infusion. Insertion within oncogenes and tumor suppressor genes, as CBL and TET2, may promote clonal expansion [51], as reported in retroviral and lentiviral vector-based anti-CD22 [52], and anti-CD19 CAR-T cells [53]. Despite previous reports have suggested that the effects of insertional mutagenesis in T cells are pervasive, leading to expansion of clones with integration into genes involved in cell-signaling and chromatin modification [54], we did not observe any signs of clonal dominance. This is very relevant after the recent warning on the risk of secondary and primary malignancies following viral CAR-T cell therapies [55, 56]. After infusion, circulating CAR-T cells showed a highly clonal repertoire and diversity, and a stable clonal composition at early time points in most of the treated patients. A repertoire associated with a small number of clones at late time points was observed, as previously reported in decade-long persistent CAR-T cells [57].
Finally, the safety and the activity of this allogeneic CAR-T cell product and the reduced cost of its production pave the way for the potential use of this cell therapy in a preemptive setting for patients after alloHSCT with evidence of measurable residual disease at the time of conditioning regimen. In fact, many clinical trials have shown that the indication for allogeneic transplant in first complete remission can now be restricted to patients with very high-risk features, such as those who fail to achieve a molecular remission [58,59,60]. The ability to increase the proportion of patients who achieve a robust molecular remission will further reduce in the near future the use of allogeneic transplantation in first remission [61]. Nonetheless, allogeneic transplants will remain a salvage and potentially curative option for very high-risk patients in first or subsequent remission. For this reason, the preemptive use of donor-derived allogeneic CARCIK-CD19 cells may represent a safe and effective option to increase the activity of the allogeneic transplant procedure. The use of this approach is warranted in our future research plan.
In conclusion, our analysis demonstrated sustained disease eradication in a proportion of patients with relapsed or refractory BCP-ALL in the absence of severe toxicities, and provides support for the clinical benefit of using CARCIK-CD19. Based on these results, CARCIK-CD19 is currently being evaluated in patients with relapsed/refractory B-cell NHL or CLL in a phase 1/2 trial (NCT05869279).
Data availability
Data generated or analyzed during this study are included in the article and Supplementary Material. The sequencing data generated in this study and code used for analysis are available at the NCBI’s SRA database (PRJNA1229450): https://www.ncbi.nlm.nih.gov/sra/PRJNA1229450.
References
Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N. Engl J Med. 2018;378:439–48.
Frey NV, Shaw PA, Hexner EO, Pequignot E, Gill S, Luger SM, et al. Optimizing chimeric antigen receptor t-cell therapy for adults with acute lymphoblastic leukemia. J Clin Oncol. 2020;38:415–22.
Shah BD, Ghobadi A, Oluwole OO, Logan AC, Boissel N, Cassaday RD, et al. KTE-X19 for relapsed or refractory adult B-cell acute lymphoblastic leukaemia: phase 2 results of the single-arm, open-label, multicentre ZUMA-3 study. Lancet. 2021;398:491–502.
Roddie C, Sandhu KS, Tholouli E, Logan AC, Shaughnessy P, Barba P, et al. Obecabtagene autoleucel in adults with B-Cell acute lymphoblastic leukemia. N Engl J Med. 2024;391:2219–30.
Levine BL, Miskin J, Wonnacott K, Keir C. Global manufacturing of CAR T cell therapy. Mol Ther Methods Clin Dev. 2017;4:92–101.
Wang X, Riviere I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol Ther Oncolytics. 2016;3:16015.
Magnani CF, Biondi A, Biagi E. Donor-derived CD19-targeted T cells in allogeneic transplants. Curr Opin Hematol. 2015;22:497–502.
Magnani CF, Mezzanotte C, Cappuzzello C, Bardini M, Tettamanti S, Fazio G, et al. Preclinical efficacy and safety of CD19CAR cytokine-induced killer cells transfected with sleeping beauty transposon for the treatment of acute lymphoblastic leukemia. Hum Gene Ther. 2018;29:602–13.
Biondi A, Magnani CF, Tettamanti S, Gaipa G, Biagi E. Redirecting T cells with Chimeric Antigen Receptor (CAR) for the treatment of childhood acute lymphoblastic leukemia. J Autoimmun. 2017;85:141–52.
Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of sleeping beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–10.
Moretti A, Ponzo M, Nicolette CA, Tcherepanova IY, Biondi A, Magnani CF. The past, present, and future of non-viral CAR T Cells. Front Immunol. 2022;13:867013.
Magnani CF, Tettamanti S, Alberti G, Pisani I, Biondi A, Serafini M, et al. Transposon-Based CAR T cells in acute leukemias: where are we going? Cells;9. 2020.
Introna M, Lussana F, Algarotti A, Gotti E, Valgarsdottir R, Mico C, et al. Phase II Study Of Sequential Infusion of DLI and cytokine induced killer cells for patients relapsed after alloHSCT. Biol Blood Marrow Transplant. 2017;23:2070–8.
Magnani CF, Gaipa G, Lussana F, Belotti D, Gritti G, Napolitano S, et al. Sleeping beauty-engineered CAR T cells achieve antileukemic activity without severe toxicities. J Clin Investig. 2020;130:6021–33.
Lee DW, Gardner R, Porter DL, Louis CU, Ahmed N, Jensen M, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95.
Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151.
Vigdal TJ, Kaufman CD, Izsvak Z, Voytas DF, Ivics Z. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J Mol Biol. 2002;323:441–52.
Gogol-Döring A, Ammar I, Gupta S, Bunse M, Miskey C, Chen W, et al. Genome-wide profiling reveals remarkable parallels between insertion site selection properties of the MLV retrovirus and the piggyBac transposon in primary human CD4(+) T cells. Mol Ther. 2016;24:592–606.
Vokinger KN, Avorn J, Kesselheim AS. Sources of innovation in gene therapies - approaches to achieving affordable prices. N. Engl J Med. 2023;388:292–5.
Park JH, Riviere I, Gonen M, Wang X, Senechal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N. Engl J Med. 2018;378:449–59.
Frey NV, Gill S, Hwang W-T, Luger SM, Martin ME, McCurdy SR, et al. CART22-65s co-administered with huCART19 in adult patients with relapsed or refractory ALL. Blood. 2021;138:469.
Roddie C, Dias J, O’Reilly MA, Abbasian M, Cadinanos-Garai A, Vispute K, et al. Durable responses and low toxicity after fast off-rate CD19 chimeric antigen receptor-T therapy in adults with relapsed or refractory B-Cell acute lymphoblastic leukemia. J Clin Oncol. 2021;39:3352–63.
Bishop MR. Late complications and long-term care of adult CAR T-cell patients. Hematology. 2024;2024:109–15.
Nishimura R, Baker J, Beilhack A, Zeiser R, Olson JA, Sega EI, et al. In vivo trafficking and survival of cytokine-induced killer cells resulting in minimal GVHD with retention of antitumor activity. Blood. 2008;112:2563–74.
Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017;23:242–9.
Del Bufalo F, De Angelis B, Caruana I, Del Baldo G, De Ioris MA, Serra A, et al. GD2-CART01 for relapsed or refractory high-risk neuroblastoma. N. Engl J Med. 2023;388:1284–95.
Aldoss I, Khaled SK, Wang Y, Wang X, Palmer J, Clark MC, et al. Donor-derived CD19-targeted chimeric antigen receptor T cells in adult transplant recipients with relapsed/refractory acute lymphoblastic leukemia. Blood Cancer J. 2023;13:107.
Bonini C, Chapuis AG, Hudecek M, Guedan S, Magnani C, Qasim W. Genome editing in engineered T cells for cancer immunotherapy. Hum Gene Ther. 2023;34:853–69.
Benjamin R, Jain N, Maus MV, Boissel N, Graham C, Jozwik A, et al. UCART19, a first-in-class allogeneic anti-CD19 chimeric antigen receptor T-cell therapy for adults with relapsed or refractory B-cell acute lymphoblastic leukaemia (CALM): a phase 1, dose-escalation trial. Lancet Haematol. 2022;9:e833–e43.
Ottaviano G, Georgiadis C, Gkazi SA, Syed F, Zhan H, Etuk A, et al. Phase 1 clinical trial of CRISPR-engineered CAR19 universal T cells for treatment of children with refractory B cell leukemia. Sci Transl Med. 2022;14:eabq3010.
Fried S, Avigdor A, Bielorai B, Meir A, Besser MJ, Schachter J, et al. Early and late hematologic toxicity following CD19 CAR-T cells. Bone Marrow Transpl. 2019;54:1643–50.
Laetsch TW, Maude SL, Rives S, Hiramatsu H, Bittencourt H, Bader P, et al. Three-year update of tisagenlecleucel in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia in the ELIANA Trial. J Clin Oncol. 2023;41:1664–9.
Myers RM, Taraseviciute A, Steinberg SM, Lamble AJ, Sheppard J, Yates B, et al. Blinatumomab nonresponse and high-disease burden are associated with inferior outcomes after CD19-CAR for B-ALL. J Clin Oncol. 2022;40:932–44.
Dourthe ME, Baruchel A. CAR T-cells in acute lymphoblastic leukemia: current results. Bull Cancer. 2021;108:S40–S54.
Pasquini MC, Hu ZH, Curran K, Laetsch T, Locke F, Rouce R, et al. Real-world evidence of tisagenlecleucel for pediatric acute lymphoblastic leukemia and non-Hodgkin lymphoma. Blood Adv. 2020;4:5414–24.
Shah BD, Cassaday RD, Park JH, Houot R, Oluwole OO, Logan AC, et al. Impact of prior therapies and subsequent transplantation on outcomes in adult patients with relapsed or refractory B-cell acute lymphoblastic leukemia treated with brexucabtagene autoleucel in ZUMA-3. J Immunother cancer. 2023;11:e007118.
Grover P, Veilleux O, Tian L, Sun R, Previtera M, Curran E, et al. Chimeric antigen receptor T-cell therapy in adults with B-cell acute lymphoblastic leukemia. Blood Adv. 2022;6:1608–18.
Hay KA, Gauthier J, Hirayama AV, Voutsinas JM, Wu Q, Li D, et al. Factors associated with durable EFS in adult B-cell ALL patients achieving MRD-negative CR after CD19 CAR T-cell therapy. Blood. 2019;133:1652–63.
Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311:1780–4.
Wang T, Tang Y, Cai J, Wan X, Hu S, Lu X, et al. Coadministration of CD19- and CD22-Directed Chimeric Antigen receptor T-Cell therapy in childhood B-Cell acute lymphoblastic leukemia: a single-arm, multicenter, phase II Trial. J Clin Oncol. 2022;41:1670–83.
Pan J, Zuo S, Deng B, Xu X, Li C, Zheng Q, et al. Sequential CD19-22 CAR T therapy induces sustained remission in children with r/r B-ALL. Blood. 2020;135:387–91.
Ceolin V, Brivio E, van Tinteren H, Rheingold SR, Leahy A, Vormoor B, et al. Outcome of chimeric antigen receptor T-cell therapy following treatment with inotuzumab ozogamicin in children with relapsed or refractory acute lymphoblastic leukemia. Leukemia. 2023;37:53–60.
Turazzi N, Fazio G, Rossi V, Rolink A, Cazzaniga G, Biondi A, et al. Engineered T cells towards TNFRSF13C (BAFFR): a novel strategy to efficiently target B-cell acute lymphoblastic leukaemia. Br J Haematol. 2018;182:939–43.
Magnani CF, Myburgh R, Brunn S, Chambovey M, Ponzo M, Volta L, et al. Anti-CD117 CAR T cells incorporating a safety switch eradicate human acute myeloid leukemia and hematopoietic stem cells. Mol Ther Oncolytics. 2023;30:56–71.
Izsvak Z, Ivics Z. Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther. 2004;9:147–56.
Mueller KT, Maude SL, Porter DL, Frey N, Wood P, Han X, et al. Cellular kinetics of CTL019 in relapsed/refractory B-cell acute lymphoblastic leukemia and chronic lymphocytic leukemia. Blood. 2017;130:2317–25.
Lee DW, Kochenderfer JN, Stetler-Stevenson M, Cui YK, Delbrook C, Feldman SA, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28.
Pule MA, Straathof KC, Dotti G, Heslop HE, Rooney CM, Brenner MK. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–41.
Hombach AA, Rappl G, Abken H. Arming cytokine-induced killer cells with chimeric antigen receptors: CD28 outperforms combined CD28-OX40 “super-stimulation. Mol Ther. 2013;21:2268–77.
Micklethwaite KP, Gowrishankar K, Gloss BS, Li Z, Street JA, Moezzi L, et al. Investigation of product-derived lymphoma following infusion of piggyBac-modified CD19 chimeric antigen receptor T cells. Blood. 2021;138:1391–405.
Cesana D, Santoni de Sio FR, Rudilosso L, Gallina P, Calabria A, Beretta S, et al. HIV-1-mediated insertional activation of STAT5B and BACH2 trigger viral reservoir in T regulatory cells. Nat Commun. 2017;8:498.
Shah NN, Zhu F, Schneider D, Taylor C, Krueger W, Worden A, et al. Results of a phase I study of bispecific anti-CD19, anti-CD20 chimeric antigen receptor (CAR) modified T cells for relapsed, refractory, non-Hodgkin lymphoma. JCO. 2019;37:2510.
Fraietta JA, Nobles CL, Sammons MA, Lundh S, Carty SA, Reich TJ, et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature. 2018;558:307–12.
Nobles CL, Sherrill-Mix S, Everett JK, Reddy S, Fraietta JA, Porter DL, et al. CD19-targeting CAR T cell immunotherapy outcomes correlate with genomic modification by vector integration. J Clin Investig. 2020;130:673–85.
Levine BL, Pasquini MC, Connolly JE, Porter DL, Gustafson MP, Boelens JJ, et al. Unanswered questions following reports of secondary malignancies after CAR-T cell therapy. Nat Med. 2024;30:338–41.
Elsallab, Ellithi M, Lunning MA M, D’Angelo, Ma C, Perales MA J, et al. Second primary malignancies after commercial CAR T-cell therapy: analysis of the FDA adverse events reporting system. Blood. 2024;143:2099–105.
Melenhorst JJ, Chen GM, Wang M, Porter DL, Chen C, Collins MA, et al. Decade-long leukaemia remissions with persistence of CD4(+) CAR T cells. Nature. 2022;602:503–9.
Bassan R, Chiaretti S, Della Starza I, Spinelli O, Santoro A, Paoloni F, et al. Pegaspargase-modified risk-oriented program for adult acute lymphoblastic leukemia: results of the GIMEMA LAL1913 trial. Blood Adv. 2023;7:4448–61.
Ribera JM, Morgades M, Ciudad J, Montesinos P, Esteve J, Genescà E, et al. Chemotherapy or allogeneic transplantation in high-risk Philadelphia chromosome-negative adult lymphoblastic leukemia. Blood. 2021;137:1879–94.
Gökbuget N, Boissel N, Chiaretti S, Dombret H, Doubek M, Fielding A, et al. Diagnosis, prognostic factors, and assessment of ALL in adults: 2024 ELN recommendations from a European expert panel. Blood. 2024;143:1891–902.
Litzow MR, Sun Z, Mattison RJ, Paietta EM, Roberts KG, Zhang Y, et al. Blinatumomab for MRD-negative acute lymphoblastic leukemia in adults. N. Engl J Med. 2024;391:320–33.
Acknowledgements
This work was supported by grants from Associazione Italiana Ricerca contro il Cancro (AIRC) IG 2017:20564, AIRC/Cancer Research UK (CRUK)/Fundación Científica de la Asociación Española Contra el Cáncer (FC AECC) 22791, AIRC 5 × 1000 Immunity in Cancer Spreading and Metastasis (grant 21147) to AB and AR, Bando AIFA (TRS-2019-00002022), the Ministero della Salute Research project on CAR-T cells for hematological malignancies and solid tumors conducted under the aegis of Alliance Against Cancer network to AB. Italian PNRR CN3 “National Center for Gene Therapy and Drugs based on RNA Technology” and LSH-TA Ecosistema innovativo della Salute to AB; AIRC IG grant (2023 29175) to GC, and Fondazione Regionale per la Ricerca Biomedica (FRRB, Regione Lombardia), Project N°CP2_10/2018 “Plagencell”. The authors would like to thank Riccardo and Donatella Ruschi and the “Amici di Duccio” association, the “Quelli che con Luca” Association, “Comitato Maria Letizia Verga,” the Fondazione Benedetta è la vita ONLUS, Associazione Italiana Lotta alla Leucemia, Linfoma e Mieloma (AIL) sezione Paolo Belli Bergamo, and “Comitato Stefano Verri” for support.
Funding
Associazione Italiana Ricerca contro il Cancro (AIRC); Cancer Research UK (CRUK), Fundación Científica de la Asociación Española Contra el Cáncer (FC AECC); Ministero della salute; Ministero dell’Università e della Ricerca; Regione Lombardia.
Author information
Authors and Affiliations
Contributions
FL and CFM contributed equally to this work. FL acted as coinvestigator, main clinical referent for the patients treated in the clinical trials, and wrote the paper. CFM acted as the study scientific leader who developed the cellular product, collected and analyzed the data, and wrote the paper. SG, G Risca, and MGV designed the clinical trial and performed the statistical analyses. FL, CFM, G Gaipa, A Balduzzi, GD, A Rambaldi, and A Biondi conceptualized and designed the trial. A Rambaldi and A Biondi were the principal investigators of the study and revised the paper. A Balduzzi and FL were the coinvestigators and revised the paper. GD was the clinical study manager and MP was the data manager. DB, BC, CB, GMB, BR, CM, ST, CC and EG performed experiments. GP, GS, and FB performed IS analysis and analyzed data. GC, EM, JG, and MI designed experiments. FL, G Gritti, SN, AM, SF, AG, G Rizzuto are part of the medical team who has followed patients, collected clinical data, revised the manuscript and gave the final approval before manuscript submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lussana, F., Magnani, C.F., Galimberti, S. et al. Donor-derived CARCIK-CD19 cells engineered with Sleeping Beauty transposon in acute lymphoblastic leukemia relapsed after allogeneic transplantation. Blood Cancer J. 15, 54 (2025). https://doi.org/10.1038/s41408-025-01260-6
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-025-01260-6
This article is cited by
-
Emerging CAR immunotherapies: broadening therapeutic horizons beyond cancer
Clinical and Experimental Medicine (2025)
-
Dual targeting of solid tumors using cytokine-induced killer cells modified with a CAR anti-tenascin C and a secretable EGFRxCD3 bispecific antibody
Cancer Immunology, Immunotherapy (2025)
