
Abstract
Cytogenetic abnormalities and recurrent gene mutations are central to the biology and prognosis of myelodysplastic syndromes/neoplasms (MDS); however, the significance of patients without detectable genomic lesions remains unclear. In this retrospective analysis of 758 patients with primary MDS, the participants were stratified according to the presence of gene mutations and/or cytogenetic abnormalities. Approximately twenty percent lacked both abnormalities and were characterized by younger age, female predominance, lower IPSS-R/IPSS-M risk, and the most favorable survival outcomes. Bulk RNA sequencing further revealed that mutation-negative/normal-karyotype MDS was characterized by immune-metabolic transcriptional programs, whereas genomically altered disease exhibited inflammatory, proliferative, and stress-response signatures, indicating a biologically distinct low-risk state. Therefore, patients without genomic abnormalities appear to constitute a biologically distinct subgroup of MDS. To refine risk assessment beyond IPSS-M systems, a multivariable Cox-based nomogram was developed incorporating age, ferritin level >800 ng/mL, lactate dehydrogenase level >200 U/L, bone marrow fibrosis, and IPSS-M. The proposed model outperformed IPSS-M alone and remained robust following bootstrap validation, allowing individualized 12- and 36-month survival estimation. These findings underscore the limitations of relying solely on molecular assays for risk stratification and support the integration of clinical, genomic, and model-based predictors to improve classification and guide individualized MDS management.
Similar content being viewed by others


Introduction
Myelodysplastic syndromes/neoplasms (MDS) are a heterogeneous group of clonal myeloid disorders characterized by peripheral cytopenias—such as anemia, neutropenia, or thrombocytopenia—arising from dysplastic hematopoietic cells [1, 2]. The disease exhibits substantial clinical and genetic heterogeneity, resulting in variable prognoses and therapeutic challenges. Cytogenetic abnormalities and recurrent somatic mutations play central roles in MDS pathogenesis and are integral to the diagnostic classification and prognostic assessment [3]. In 2022, two updated classification systems were introduced: the International Consensus Classification (ICC) [4] and the 5th edition of the World Health Organization classification (WHO-2022) [5]. Although these contemporary frameworks differ considerably in terminology and entity definitions, both place strong emphasis on incorporating cytogenetic abnormalities and gene mutations into diagnostic criteria.
Cytogenetic aberrations are identified in ~40–50% of patients with primary MDS, whereas myeloid-related gene mutations detectable by next-generation sequencing (NGS) are observed in more than 70% of patients [6,7,8,9]. Consequently, only 10–30% of patients lack identifiable cytogenetic or molecular abnormalities. In such cases, distinguishing MDS from idiopathic cytopenia of undetermined significance remains challenging and frequently depends on meticulous morphological evaluation of bone marrow (BM) hematopoietic cells. Despite the established importance of genomic and cytogenetic features, the clinical characteristics and prognostic outcomes of patients harboring cytogenetic abnormalities or at least one somatic mutation—compared with those of individuals without detectable molecular lesions—remain underexplored. Therefore, the present study aimed to investigate the clinical and transcriptomic profiles of patients with MDS with or without molecular abnormalities. The prognostic implications of integrating cytogenetic and mutational status in patients with MDS were also evaluated.
Materials and methods
Patient cohort
Data from 758 consecutive patients with primary MDS who were diagnosed and treated at the National Taiwan University Hospital (NTUH) were retrospectively analyzed. Patients with a history of chemotherapy, radiotherapy, or prior hematologic malignancies were excluded to avoid the distinct mutational landscapes and clinical outcomes associated with secondary MDS [10, 11]. In patients without detectable mutations, cytogenetic abnormalities, or excess of blasts, an MDS diagnosis was established based on evidence of persistent or progressive cytopenia(s) in conjunction with significant BM dysplasia. The study was approved by the NTUH Research Ethics Committee (approval numbers: 201709072RINC, 202109078RINB, and 20220705RINB). Written informed consent was obtained from all participants in accordance with the Declaration of Helsinki.
BM assessment and cytogenetic analysis
BM cellularity and fibrosis grade were assessed using reticulin staining and independently confirmed by board-certified hemato-pathologists [12]. Cytogenetic studies were performed on BM aspirates and interpreted according to the International System for Human Cytogenetic Nomenclature [13, 14].
Targeted NGS
NGS was conducted using the TruSight Myeloid Sequencing Panel (Illumina, San Diego, CA, USA) on a HiSeq platform (Illumina), covering 54 myeloid neoplasm–associated genes [7, 14,15,16] (Supplementary Table 1). Five genes (ETNK1, GNB1, NF1, PPM1D, and PRPF8) included in the molecular International Prognosis Scoring System (IPSS-M) model were not analyzed, and TP53 copy-neutral loss of heterozygosity was not assessed. Library preparation and sequencing were conducted according to the manufacturer’s protocols, yielding a median read depth of 10,550×. Somatic variants were annotated using COSMIC v86, dbSNP v151, ClinVar, PolyPhen-2, and SIFT, with pathogenicity interpreted as described in previous studies [16, 17]. Owing to the limitations of NGS, FLT3 internal tandem duplications were detected by polymerase chain reaction (PCR) followed by fluorescence capillary electrophoresis, whereas KMT2A partial tandem duplications were confirmed using PCR and Sanger sequencing [18].
Bulk RNA sequencing of BM cells
Bulk RNA sequencing was performed on BM mononuclear cells obtained from 129 patients with MDS at diagnosis. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Hilden, Germany) in accordance with the manufacturer’s protocol. RNA quality and integrity were assessed with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA), and only samples with RNA integrity number (RIN) of ≥7.0 were selected for library preparation. Libraries were constructed using the TruSeq Stranded mRNA Library Prep Kit (Illumina) and sequenced on the Illumina NovaSeq 6000 platform to generate 150-bp paired-end reads, achieving an average depth of 40 million reads per sample. Raw sequencing reads were subjected to quality control using FastQC, trimmed using Trimmomatic, and aligned to the GRCh38 human reference genome using the STAR aligner. Gene-level counts were quantified with featureCounts, and transcript abundances were normalized to transcripts per million for subsequent downstream differential expression and pathway enrichment analyses [19].
Statistical analysis
Overall survival (OS) was defined as the interval from diagnosis to death from any cause, whereas leukemia-free survival (LFS) was defined as the interval from diagnosis to progression to acute myeloid leukemia (AML) or death from any cause. Time to disease progression was defined as the interval from initial diagnosis to the first documented event of disease advancement, including transition from low-blast MDS to MDS with excess blasts (EB), progression from MDS-EB1 to MDS-EB2, or progression from any MDS subtype to AML. Categorical variables were compared using the χ² or Fisher’s exact test, whereas continuous variables were compared using the Mann–Whitney U test or Kruskal–Wallis test, as appropriate.
Maximally selected rank statistics were applied to determine the optimal cutoff points for continuous variables. This method systematically evaluates all potential thresholds and selects the value that maximizes the standardized two-sample linear rank statistic, thereby identifying the cutoff that yields the greatest separation between outcome groups [20, 21]. For both univariable and multivariable analyses, Cox proportional hazards models were applied. Variables with clinical relevance or statistical significance in the univariable analysis were included in the multivariable model. A prognostic nomogram was subsequently developed based on the final multivariable Cox proportional hazards model. The regression coefficients derived from the multivariable Cox model were converted into a point-based scoring system to generate individualized risk scores. For each patient, the total score corresponded to the predicted OS probability at predefined time points, as mapped through the baseline survival function. Model calibration was assessed by comparing predicted versus observed survival using bootstrapped calibration curves, and discriminative performance was quantified by Harrell’s C-index with internal validation via bootstrapping [22]. The final nomogram was graphically rendered for clinical application.
Survival curves were estimated by the Kaplan–Meier method and compared using the log-rank test. All p-values were two-sided, and a p-value of <0.05 was considered significant. All analyses were performed using IBM SPSS Statistics v23 and R statistical language v4.5.1 for Windows.
Results
Demographic features and genetic profiles
Patient characteristics are summarized in Table 1. The median age at diagnosis was 67.4 years, with a male predominance (63.6%). Based on mutational and cytogenetic profiles, patients were categorized into four groups: normal karyotype without gene mutations (n = 155, 20.4%), normal karyotype with gene mutations only (n = 299, 39.4%), cytogenetic abnormalities without gene mutations (n = 62, 8.2%), and concomitant cytogenetic abnormalities with gene mutations (n = 242, 31.9%).
Compared with all other groups harboring either cytogenetic abnormalities or gene mutations, patients with normal karyotype and no detectable gene mutations were more frequently female, younger, had a lower BM blast percentage, and were more frequently classified as very low or low risk according to the revised International Prognosis Scoring System (IPSS-R) or IPSS-M (all p < 0.05). Conversely, patients with concomitant cytogenetic abnormalities and gene mutations exhibited more severe neutropenia than those without cytogenetic abnormalities; moreover, they had higher BM blast percentages compared with the other three groups.
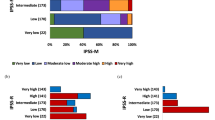
Although gene mutations are not included in the IPSS-R scoring, patients with a normal karyotype and gene mutations had significantly higher proportions in the high/very high-risk categories of both IPSS-R and IPSS-M compared with those with a normal karyotype but no detectable gene mutations (IPSS-R: 34.4% vs. 6.5%; IPSS-M: 49.6% vs. 6.2%; both p < 0.001). Most patients with gene mutations only were classified as low/intermediate risk by IPSS-R (62.2%) but as high or very high risk by IPSS-M (49.6%). By contrast, patients with cytogenetic abnormalities only demonstrated a more heterogeneous distribution across IPSS-R and IPSS-M risk categories (Supplementary Fig. 1).
According to the WHO-2016 classification, most patients with a normal karyotype and no gene mutations were classified as having MDS with single-lineage dysplasia (SLD) or multilineage dysplasia (MLD) (n = 125, 80.6%). Moreover, significantly fewer patients in this group exhibited excess blasts compared with the other three groups (all pairwise p < 0.05). Under the WHO-2022 classification, 52.3% of patients with a normal karyotype and no gene mutations were classified as having hypoplastic MDS, followed by MDS with low blasts (27.9%). In the ICC classification, most patients without any molecular lesions were categorized as having MDS, not otherwise specified (NOS), with SLD (42.6%) or MLD (43.2%). Among the mutated TP53 subtypes in both WHO-2022 and ICC, over 90% of patients harbored concurrent gene mutations and cytogenetic abnormalities (Table 2 and Supplementary Fig. 2).
The karyotypic and mutational landscapes were further examined in relation to the presence or absence of concomitant gene mutations and cytogenetic abnormalities. Karyotypic analysis demonstrated that patients harboring concomitant gene mutations demonstrated a markedly higher frequency of deletion 7q or monosomy 7 and were overrepresented in the very poor cytogenetic risk category as defined by the IPSS-R criteria (Supplementary Table 2). Mutational profiling revealed distinct patterns between the groups. Patients with gene mutations only were significantly enriched for DNMT3A, TET2, ASXL1, BCOR, BCORL1, SRSF2, SF3B1, and NPM1 mutations (all p < 0.05), whereas those with both gene mutations and cytogenetic abnormalities more frequently carried JAK2 and TP53 mutations (Supplementary Table 3 and Supplementary Fig. 3). These findings highlight a functional divergence in mutational landscapes, suggesting that patients without cytogenetic abnormalities predominantly acquire mutations in epigenetic regulators and splicing genes, whereas those with concomitant abnormalities more often harbor alterations in tumor suppressors and signaling pathway drivers (Supplementary Fig. 4).
With regard to treatment, patients with a normal karyotype and no detectable gene mutations were less frequently administered hypomethylating agents (7.2%). The majority received supportive care alone (53.6%) or alternative therapies (33.3%), including anti-thymocyte globulin, cyclosporine, danazol, or erythropoiesis-stimulating agents (Supplementary Fig. 5).
Survival analysis
Survival analysis across these four groups demonstrated that patients without any molecular lesions had the most favorable outcomes (LFS: 218.6 months and OS: 250.7 months). By contrast, patients with concomitant gene mutations and cytogenetic abnormalities exhibited the poorest prognosis (LFS: 10.4 months and OS: 16.0 months, Fig. 1). Patients with only gene mutations or only cytogenetic abnormalities exhibited comparable outcomes (LFS: 28.7 vs. 48.1 months, p = 0.197; OS: 37.1 vs. 48.2 months, p = 0.395). To investigate the prognostic impact of the interplay between genetic and cytogenetic alterations, outcomes were analyzed across subgroups stratified by mutational and cytogenetic status. Patients with good or intermediate karyotypes without gene mutations had significantly better survival compared with those harboring poor karyotypes (median LFS: 85.3 and 70.3 vs. 19.1 months and OS: 85.3 and 77.3 vs. 26.7 months, p = 0.008 and 0.002, respectively). Among patients with gene mutations, outcomes in the good and intermediate karyotype groups were comparable; however, both groups exhibited markedly better survival compared with those with poor karyotypes (median LFS: 21.2 and 16.8 vs. 6.1 months and OS: 27.9 and 33.0 vs. 8.6 months, all p < 0.001). Notably, in the presence of gene mutations, a favorable karyotype failed to mitigate the adverse prognostic impact associated with genetic lesions (Fig. 2).

Kaplan–Meier analysis of a leukemia-free survival (LFS) and b overall survival (OS) in patients categorized into four groups: normal karyotype (NK) without gene mutations (n = 155), gene mutations only (n = 299), cytogenetic abnormalities only (n = 62), and concomitant gene mutations and cytogenetic abnormalities (n = 242).

Survival outcomes for a LFS and b OS across subgroups stratified by mutational status and IPSS-R cytogenetic risk categories. Subgroups include: GK (very good/good), IK (intermediate), and PK (poor/very poor) karyotypes, with or without concomitant gene mutations.
To evaluate the prognostic impact of mutational burden, patients were stratified according to the number of mutated genes. Kaplan–Meier analyses demonstrated a pronounced stepwise association between increasing mutation number and adverse outcomes (both p < 0.001). For LFS, patients without mutations (n = 217) had a median LFS of 185.5 months, which was significantly longer than that of patients harboring one mutation (32.2 months), two to three mutations (16.6 months), four to five mutations (12.4 months), or more than five mutations (4.5 months) (Fig. 3). A similar stepwise decline was observed for OS. Patients without mutations had a median OS of 218.6 months, whereas those with one, two to three, four to five, and more than five mutations had median OS durations of 42.6, 27.6, 22.0, and 13.4 months, respectively (Fig. 3). These findings underscored that the cumulative number of gene mutations represented a strong adverse prognostic factor, with both LFS and OS progressively declining as mutational burden increased.

Kaplan–Meier curves for a LFS and b OS stratified by the cumulative number of gene mutations (0, 1, 2–3, 4–5, and 5).
Next, survival outcomes were subsequently evaluated across IPSS-R and IPSS-M categories according to the presence of gene mutations and/or cytogenetic abnormalities. When stratified by cytogenetic and molecular status, patients with MDS who had a normal karyotype and no detectable gene mutations consistently exhibited the most favorable clinical outcomes. As shown in Supplementary Fig. 6, these double-negative patients demonstrated a markedly longer time to disease progression and superior OS compared with all other subgroups carrying gene mutations and/or cytogenetic abnormalities, regardless of stratification by the IPSS-R or IPSS-M. By contrast, patients with higher risk scores experienced a stepwise decline in survival (both p < 0.001). Further stratification revealed that this favorable outcome persisted across all IPSS-R or IPSS-M risk categories. Even among patients classified as high or very high risk, the double-negative subset exhibited a median time to progression that was not reached and prolonged OS (Supplementary Figs. 7 and 8). Despite the incorporation of molecular risk features, the IPSS-M model did not attenuate the survival advantage observed in patients with normal karyotypes and no mutations. To evaluate whether existing prognostic models could further discriminate outcomes within this indolent subgroup, the analyses were restricted to patients with MDS who had a normal karyotype and no detectable mutations. Neither IPSS-R nor IPSS-M successfully differentiated outcomes, as LFS and OS remained uniformly prolonged across all strata, with no significant differences (IPSS-R: p = 0.861 and 0.963; IPSS-M: p = 0.481 and 0.646, respectively) (Supplementary Fig. 9).
Analysis based on the ICC further corroborated these findings at the diagnostic-entity level. Patients with double-negative MDS exhibited the longest survival and the slowest disease progression, outperforming all ICC-defined categories, including MDS with mutated SF3B1, MDS, NOS with SLD/MLD, and MDS with EB (Supplementary Fig. 10).
Transcriptional differences analysis by bulk RNA sequencing
Patients from the three groups—those harboring cytogenetic abnormalities only, gene mutations only, or both gene mutations and cytogenetic abnormalities—exhibited comparable transcriptomic profiles (data not shown). Consequently, these groups were combined into a single cohort for comparison with patients lacking both gene mutations and cytogenetic abnormalities. Comparative transcriptomic profiling revealed distinct gene expression patterns in patients without gene mutations or cytogenetic abnormalities. Volcano plot analysis demonstrated that this mutation/karyotype-negative subgroup was characterized by relative upregulation of CD177, CTSE, LCN2, CAMP, and OR6N1, genes associated with neutrophil function, antigen presentation, and innate immune defense (Fig. 4a). Conversely, transcripts such as ZNF662, OBSL1, CPXM1, ZNF219, and KRT8 were downregulated in this subgroup, highlighting transcriptional programs that distinguished patients lacking molecular or cytogenetic abnormalities from those harboring genomic alterations (Fig. 4a).

a Volcano plot depicting differentially expressed genes between patients lacking molecular/cytogenetic abnormalities and those with genomic alterations. b Gene set enrichment analysis showing pathways enriched in genomically altered disease versus mutation-negative disease.
Gene set enrichment analysis revealed pronounced transcriptional differences between patients harboring gene mutations or cytogenetic abnormalities and those without detectable abnormalities. Pathways associated with immune activation and inflammation, including tumor necrosis factor alpha (TNFα) signaling via NF-κB, inflammatory response, interferon-γ response, interferon-α response, complement activation, interleukin (IL)2–STAT5 signaling, and IL6–JAK–STAT3 signaling, were markedly enriched in patients harboring genetic or cytogenetic abnormalities (all nominal p < 0.05, Fig. 4b). Moreover, KRAS signaling, hypoxia, and mitotic spindle pathways were upregulated in this group, reflecting enhanced proliferative and stress-response programs. Conversely, patients without detectable abnormalities demonstrated relative enrichment of oxidative phosphorylation, apical junction, apical surface, and early estrogen response pathways, consistent with preserved metabolic and structural epithelial programs (Fig. 4b). Subgroup comparative analysis showed that, compared with patients lacking both gene mutations and cytogenetic abnormalities, those with gene mutations only displayed significant upregulation of the heme metabolism pathway (Supplementary Fig. 11a). Patients harboring intermediate- or poor-risk cytogenetic abnormalities demonstrated marked activation of the differentiating T lymphocyte (Supplementary Fig. 11b) and TNF-α- NF-κB signaling (Supplementary Fig. 11c) pathways, accompanied by downregulation of the reactome C4 and C2 activators pathway (Supplementary Fig. 11d). By contrast, patients with concurrent gene mutations and cytogenetic abnormalities demonstrated significant upregulation of the AML cluster signature (Supplementary Fig. 11e) and suppression of the DNA repair pathway (Supplementary Fig. 11f).
Discriminative performance of the nomogram versus IPSS-M
Patients without detectable gene mutations or cytogenetic abnormalities exhibited survival outcomes comparable with those categorized as very low or low risk by IPSS-M. Accordingly, these double-negative patients were consolidated with the very low- and low-risk IPSS-M groups. On univariable analysis, age, male sex, ferritin level >800 ng/mL, lactate dehydrogenase (LDH) level >200 U/L, BM fibrosis, and higher IPSS-M risk strata were all significantly associated with inferior LFS and OS, whereas higher absolute lymphocyte count (ALC) and absolute monocyte count (AMC) were identified as protective factors (Supplementary Table 4). Variables demonstrating statistical relevance were subsequently included in the multivariable model. In the adjusted analysis, age (per year increase), ferritin level >800 ng/mL, LDH level >200 U/L, BM fibrosis, and the refined IPSS-M category remained independently associated with OS. Notably, patients in the highest IPSS-M tier exhibited more than a ninefold increased risk of mortality compared with those in the very low/low-risk/double-negative group (hazard ratio: 9.984; 95% confidence interval (CI): 7.112–14.015, p < 0.001). By contrast, male sex, ALC, AMC, and transfusion dependency did not retain significance after statistical adjustment (Table 3).
Using the five independent predictors identified in the multivariable Cox model, a point-based nomogram was constructed (Fig. 5), achieving substantially improved prognostic discrimination compared with IPSS-M alone. The nomogram yielded a Harrell’s C-index of 0.773, exceeding the performance of IPSS-M alone (C=0.738). Bootstrap internal validation demonstrated a bias-corrected 95% CI of 0.740–0.808, supporting the robustness and reproducibility of the model. The point-based scale further enabled individualized estimation of 12- and 36-month survival, facilitating bedside interpretability and providing potential decision support for risk-aligned treatment planning.

A clinical-genomic prognostic nomogram developed using a multivariable Cox-based model incorporating age, ferritin level (800 ng/mL), LDH level (200 U/L), bone marrow fibrosis, and refined IPSS-M categories. The model allows for the individualized estimation of 12- and 36-month survival probabilities.
Discussion
In the present study, the clinical characteristics, WHO-2016, WHO-2022, and ICC classifications, risk stratification by IPSS-R and IPSS-M, and clinical outcomes were systematically compared between patients with primary MDS lacking cytogenetic abnormalities or gene mutations and those harboring molecular lesions (either mutations or karyotypic abnormalities).
In this large cohort, 20.4% of patients with primary MDS lacking cytogenetic abnormalities or gene mutations were diagnosed through meticulous BM morphological assessment performed by experienced hematologists and hematopathologists at a tertiary medical center. Patients initially diagnosed with MDS but subsequently later determined to have secondary cytopenias from other causes or those in whom cytopenias resolved spontaneously were excluded. Patients with mutation- and karyotype-negative MDS exhibited distinct clinical features, tended to be younger, were predominantly women, and were more commonly classified into lower-risk IPSS-R or IPSS-M groups.
Furthermore, the prognostic implications of gene mutations and cytogenetic abnormalities were evaluated. Although both exerted significant effects on survival, cytogenetic abnormalities demonstrated a more heterogeneous influence. Previous studies have firmly established the prognostic relevance of specific chromosomal abnormalities and recurrent gene mutations in MDS [7, 16, 23,24,25,26,27]. Nevertheless, the interplay between cytogenetic lesions and gene mutations remains incompletely defined. The prognostic impact of certain gene mutations appears to depend on the underlying karyotype. For example, TP53 alterations are associated with dismal outcomes across multiple malignancies [28,29,30,31,32]. Their prognostic consequences in myeloid neoplasms are influenced by allelic status and cytogenetic context [33, 34]. Similarly, SF3B1 mutations can be further stratified into molecular subgroups (SF3B15q, SF3B1α, and SF3B1β) based on co-mutational patterns and the presence of del(5q) [7]. Additionally, Ravindran et al. demonstrated that the prognosis of patients with isolated del(20q) varied according to accompanying gene mutations [35].
Previously, Wei Wang et al. specifically examined patients with primary MDS carrying a normal karyotype. In their analysis, FLT3 and CEBPA mutations were identified as independent prognostic factors for OS in both the training set and the International Working Group for the Prognosis of MDS validation cohort, whereas TP53 mutations were not significantly associated with outcomes. These findings underscored the importance of integrating karyotypic context when assessing the prognostic significance of individual gene mutations in MDS [36]. In another study, Wang et al. evaluated 317 patients with MDS, among whom 35 had no detectable somatic mutations and exhibited either a normal karyotype or an isolated Y. Within this subset, no significant differences in OS or AML-free survival were observed between patients with and without gene mutations, suggesting that, in the context of normal or –Y karyotypes, the prognostic impact of gene mutations may be attenuated. This observation raises the possibility that alternative mechanisms, such as epigenetic dysregulation or transcriptional reprogramming, may play a more central role in disease biology and clinical outcomes in this subgroup [37].
The findings of this study demonstrated that patients lacking any molecular lesions exhibited the most favorable outcomes. Notably, the presence of a favorable karyotype [38] did not mitigate the adverse prognostic effect of pathogenic mutations, underscoring the dominant role of molecular alterations in determining disease outcomes. Additionally, even in the absence of mutations, poor or very poor karyotypes were sufficient to predict poor outcomes, underscoring the synergistic adverse effect of cytogenetic and genetic abnormalities on disease progression. Survival analyses performed using the IPSS-R, IPSS-M, and ICC frameworks underscored the remarkable prognostic stability of MDS patients with normal karyotypes and no detectable gene mutations, a group that consistently demonstrated excellent outcomes regardless of the risk stratification system applied. Although IPSS-M effectively captures prognostic heterogeneity, the presence of gene mutations and/or cytogenetic abnormalities within risk subgroups provides additional refinement of outcome prediction. The inability of both IPSS-R and IPSS-M to further discriminate outcomes within the double-negative population underscored the potential need for refined models to delineate risk within this exceptionally low-risk population. These patients likely represent a distinct subset of early-stage, indolent myeloid disorders characterized by minimal genomic instability and low leukemic transformation potential.
Our transcriptomic analysis provides evidence for the biological heterogeneity underlying MDS, specifically delineating a spectrum of disease states linked to cytogenetic and molecular features. Notably, the subgroup of patients lacking both genetic and cytogenetic abnormalities exhibited a unique transcriptional signature characterized by the upregulation of immune and host-defense genes. This suggests a preserved immune-surveillance capacity and metabolic integrity, reflected by the enrichment of oxidative phosphorylation and apical junction pathways, potentially contributing to their more favorable clinical course. Conversely, MDS patients harboring genomic lesions displayed transcriptional reprogramming indicative of disease-driving stress and clonal evolution. The enrichment of inflammatory and proliferative programs, coupled with specific pathway activations, underscores the molecular basis of aggressive disease biology. Specifically, the presence of combined gene mutations and cytogenetic abnormalities correlated with the suppression of DNA repair pathways and the strong enrichment of the AML cluster program, highlighting a state of heightened genomic instability and a significant propensity for leukemic transformation. This molecular continuum, ranging from preserved equilibrium in double-negative MDS to progressive inflammatory activation and repair failure in genetically complex disease, provides a crucial framework for understanding the mechanisms driving adverse clinical outcomes and underscores the need for molecular-based risk stratification in MDS management.
Finally, a new risk prediction model was developed. Although 26.9% of patients with double-negative MDS were initially classified as having moderate low risk by IPSS-M, survival analysis demonstrated outcomes comparable to the very low- and low-risk groups, leading to their downstaging. Several routinely measured variables were associated with outcome at the univariable level; after adjustment, age, ferritin>800 ng/mL, LDH > 200 U/L, BM fibrosis, and refined IPSS-M remained independently prognostic. Elevated ferritin levels reflect chronic transfusion burden and iron-driven oxidative or inflammatory injury, which has been repeatedly associated with inferior survival and leukemic progression [39, 40]. Likewise, high LDH levels may indicate proliferative or cytolytic tumor kinetics. BM fibrosis has also emerged as a mechanistically distinct adverse feature linked to inferior survival and increased transfusion requirements [41,42,43]. On this basis, a parsimonious nomogram was constructed, demonstrating superior discriminatory capacity compared with IPSS-M alone and stability on bootstrap validation, enabling individualized survival probability estimation rather than reliance on categorical risk grouping.
This study is limited by its retrospective design, treatment heterogeneity, and the relatively small number of patients with available transcriptomic data. As RNA sequencing was performed in only a subset of the cohort, the statistical power of subgroup analyses was limited, potentially underrepresenting the full spectrum of transcriptomic diversity among distinct molecular and cytogenetic subgroups. Consequently, although the observed pathway differences provide valuable biological insights, these findings should be interpreted with caution and validated in larger, independent cohorts with comprehensive multi-omic profiling. Moreover, TP53 copy-neutral loss of heterozygosity was not assessed, and rare genomic lesions outside the targeted panel may have been missed. The risk prediction model also required further external validation. Although the nomogram improves prognostication, its impact on clinical decision-making was not formally evaluated. Future studies with independent cohorts and decision-analytic evaluation will be needed to determine whether this model provides reclassification gains that are clinically actionable. Despite these limitations, this study is one of the largest cohorts systematically comparing clinical characteristics, diagnostic classification according to the WHO-2022 and ICC, risk stratification, and outcomes between patients with and without genomic alterations. To our knowledge, it is also the first to profile differentially expressed genes at the transcriptomic level across these subgroups, providing novel insight that alternative mechanisms may drive transcriptional reprogramming in mutation- or karyotype-negative MDS, warranting further investigation and validation in independent populations.
In conclusion, this study underscores the biological and clinical distinctiveness of mutation-negative, cytogenetically normal MDS, highlighting the limitations of relying solely on genetic and cytogenetic testing for diagnosis and risk stratification. Importantly, morphologic dysplasia and persistent cytopenia remain sufficient to establish an MDS diagnosis in the absence of molecular lesions. Future studies incorporating genomic and transcriptomic profiling, validated in independent cohorts, are warranted to refine classification systems and enhance personalized management of MDS.
Data availability
The datasets generated during and/or analyzed during this study are available from the corresponding author upon reasonable request.
References
Karel D, Valburg C, Woddor N, Nava VE, Aggarwal A. Myelodysplastic neoplasms (MDS): the current and future treatment landscape. Curr Oncol. 2024;31:1971–93.
Tefferi A, Vardiman JW. Myelodysplastic syndromes. N Engl J Med. 2009;361:1872–85.
Sekeres MA, Taylor J. Diagnosis and treatment of myelodysplastic syndromes: a review. JAMA. 2022;328:872–80.
Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International consensus classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022;140:1200–28.
Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022;36:1703–19.
Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, García-Manero G, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496–506.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. 2022;1:EVIDoa2200008.
Lee WH, Lin CC, Tsai XC, Tien FM, Lo MY, Kuo YY, et al. Epidemiology, treatment outcomes, and prognosis of myelodysplastic syndromes/neoplasms in Taiwan: real-world insights and trends. Clin Lymphoma Myeloma Leuk. 2025;25:e336–e47.e14.
Gill H, Yim R, Lee P, Tsai XC, Li VWK, Leung GMK, et al. A clinico-genomic prognostic model for primary myelodysplastic neoplasm in Asia. Blood Cancer J. 2025;15:128.
Singhal D, Wee LYA, Kutyna MM, Chhetri R, Geoghegan J, Schreiber AW, et al. The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia. 2019;33:2842–53.
Kuzmanovic T, Patel BJ, Sanikommu SR, Nagata Y, Awada H, Kerr CM, et al. Genomics of therapy-related myeloid neoplasms. Haematologica. 2020;105:e98–e101.
Lee WH, Lin CC, Tsai CH, Tien FM, Lo MY, Tseng MH, et al. Comparison of the 2022 World Health Organization classification and international consensus classification in myelodysplastic syndromes/neoplasms. Blood Cancer J. 2024;14:57.
International Standing Committee on Human Cytogenomic Nomenclature M-JJHRJMSSKG. ISCN 2020, an International System for Human Cytogenomic Nomenclature 2020: recommendations of the International Standing Committee on Human Cytogenomic Nomenclature, including revised sequence-based cytogenomic nomenclature developed in collaboration with the Human Genome Variation Society (HGVS) Sequence Variant Description Working Group, 2020.
Lee WH, Tsai MT, Tsai CH, Tien FM, Lo MY, Tseng MH, et al. Validation of the molecular international prognostic scoring system in patients with myelodysplastic syndromes defined by international consensus classification. Blood Cancer J. 2023;13:120.
Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127:29–41.
Lee WH, Lin CC, Tsai CH, Tseng MH, Kuo YY, Liu MC, et al. Effect of mutation allele frequency on the risk stratification of myelodysplastic syndrome patients. Am J Hematol. 2022;97:1589–98.
Tsai CH, Tang JL, Tien FM, Kuo YY, Wu DC, Lin CC, et al. Clinical implications of sequential MRD monitoring by NGS at 2 time points after chemotherapy in patients with AML. Blood Adv. 2021;5:2456–66.
Lee WH, Lin CC, Tsai CH, Tien FM, Lo MY, Ni SC, et al. Clinico-genetic and prognostic analyses of 716 patients with primary myelodysplastic syndrome and myelodysplastic syndrome/acute myeloid leukemia based on the 2022 International Consensus Classification. Am J Hematol. 2023;98:398–407.
Yao CY, Lin CC, Wang YH, Kao CJ, Tsai CH, Hou HA, et al. Kinome expression profiling improves risk stratification and therapeutic targeting in myelodysplastic syndromes. Blood Adv. 2024;8:2442–54.
Zheng BW, Huang W, Liu FS, Zhang TL, Wang XB, Li J, et al. Clinicopathological and prognostic characteristics in spinal chondroblastomas: a pooled analysis of individual patient data from a single institute and 27 studies. Global Spine J. 2023;13:713–23.
Laska E, Meisner M, Wanderling J. A maximally selected test of symmetry about zero. Stat Med. 2012;31:3178–91.
Steyerberg EW, Harrell FE Jr, Borsboom GJ, Eijkemans MJ, Vergouwe Y, Habbema JD. Internal validation of predictive models: efficiency of some procedures for logistic regression analysis. J Clin Epidemiol. 2001;54:774–81.
Haase D, Germing U, Schanz J, Pfeilstöcker M, Nösslinger T, Hildebrandt B, et al. New insights into the prognostic impact of the karyotype in MDS and correlation with subtypes: evidence from a core dataset of 2124 patients. Blood. 2007;110:4385–95.
Schanz J, Cevik N, Fonatsch C, Braulke F, Shirneshan K, Bacher U, et al. Detailed analysis of clonal evolution and cytogenetic evolution patterns in patients with myelodysplastic syndromes (MDS) and related myeloid disorders. Blood Cancer J. 2018;8:28.
Solé F, Luño E, Sanzo C, Espinet B, Sanz GF, Cervera J, et al. Identification of novel cytogenetic markers with prognostic significance in a series of 968 patients with primary myelodysplastic syndromes. Haematologica. 2005;90:1168–78.
Maurya N, Mohanty P, Dhangar S, Panchal P, Jijina F, Mathan SLP, et al. Comprehensive analysis of genetic factors predicting overall survival in myelodysplastic syndromes. Sci Rep. 2022;12:5925.
Lee WH, Lin CC, Wang YH, Yao CY, Kuo YY, Tseng MH, et al. Distinct genetic landscapes and their clinical implications in younger and older patients with myelodysplastic syndromes. Hematol Oncol. 2023;41:463–73.
Schaafsma E, Takacs EM, Kaur S, Cheng C, Kurokawa M. Predicting clinical outcomes of cancer patients with a p53 deficiency gene signature. Sci Rep. 2022;12:1317.
Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, et al. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007;357:2552–61.
Abel HJ, Oetjen KA, Miller CA, Ramakrishnan SM, Day RB, Helton NM, et al. Genomic landscape of TP53-mutated myeloid malignancies. Blood Adv. 2023;7:4586–98.
Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7.
Voskarides K, Giannopoulou N. The role of TP53 in adaptation and evolution. Cells. 2023;12:512.
Badar T, Nanaa A, Atallah E, Shallis RM, Craver EC, Li Z, et al. Prognostic impact of “multi-hit” versus “single-hit” TP53 alteration in patients with acute myeloid leukemia: results from the Consortium on Myeloid Malignancies and Neoplastic Diseases. Haematologica. 2024;109:3533–42.
Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes. Nat Med. 2020;26:1549–56.
Ravindran A, He R, Ketterling RP, Jawad MD, Chen D, Oliveira JL, et al. The significance of genetic mutations and their prognostic impact on patients with an incidental finding of isolated del(20q) in bone marrow without morphologic evidence of a myeloid neoplasm. Blood Cancer J. 2020;10:7.
Wang W, Zhang Y, Yang W, Han Y, Jiang L, Liu X, et al. Mutation landscape of normal karyotype myelodysplastic syndromes and their prognostic impact. Am J Hematol. 2024;99:E51–e4.
Wang SA, Ok CY, Kim AS, Lucas F, Morgan EA, Thakral B, et al. Myelodysplastic syndromes with no somatic mutations detected by next-generation sequencing display similar features to myelodysplastic syndromes with detectable mutations. Am J Hematol. 2021;96:E420–e3.
Schanz J, Tüchler H, Solé F, Mallo M, Luño E, Cervera J, et al. New comprehensive cytogenetic scoring system for primary myelodysplastic syndromes (MDS) and oligoblastic acute myeloid leukemia after MDS derived from an international database merge. J Clin Oncol. 2012;30:820–9.
Malcovati L, Porta MG, Pascutto C, Invernizzi R, Boni M, Travaglino E, et al. Prognostic factors and life expectancy in myelodysplastic syndromes classified according to WHO criteria: a basis for clinical decision making. J Clin Oncol. 2005;23:7594–603.
Armand P, Kim HT, Cutler CS, Ho VT, Koreth J, Alyea EP, et al. Prognostic impact of elevated pretransplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood. 2007;109:4586–8.
Della Porta MG, Malcovati L, Boveri E, Travaglino E, Pietra D, Pascutto C, et al. Clinical relevance of bone marrow fibrosis and CD34-positive cell clusters in primary myelodysplastic syndromes. J Clin Oncol. 2009;27:754–62.
Melody M, Al Ali N, Zhang L, Ramadan H, Padron E, Sallman D, et al. Decoding bone marrow fibrosis in myelodysplastic syndromes. Clin Lymphoma Myeloma Leuk. 2020;20:324–8.
Jain AG, Zhang L, Bennett JM, Komrokji R. Myelodysplastic syndromes with bone marrow fibrosis: an update. Ann Lab Med. 2022;42:299–305.
Acknowledgements
We acknowledge the services provided by the Department of Laboratory Medicine and Medical Research, National Taiwan University Hospital, and the Tai-Chen Cell Therapy Center. We also extend our appreciation to the DNA Sequencing Core of the First Core Laboratory, National Taiwan University College of Medicine, Taiwan, for their invaluable assistance.
Funding
This work was partially supported by grants from the Hematology Society of Taiwan; the Ministry of Science and Technology, Taiwan (MOST 109-2314-B-002-213); the National Science and Technology Council, Taiwan (NSTC 114-2314-B-002 -286 -MY2, 111-2314-B-002-122-MY3, 114-2314-B-002-256-MY3 and 114-2314-B-002-256-MY3); the Ministry of Health and Welfare, Taiwan (MOHW 107-TDU-B-211-114009 and 111-TDU-B-221-114001); and the National Key Area International Cooperation Alliance through the University Academic Alliance in Taiwan (UAAT)–Kyushu-Okinawa Open University (KOOU) Medicine and Life Sciences Integrative Program. The funding agencies had no role in the study design, data acquisition, data analysis, manuscript preparation, or the decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
WHL was responsible for data collection and management, statistical analysis and interpretation, literature review, and manuscript drafting. XCHT, YYK, and MHT conducted next-generation sequencing data collection and interpretation. MCL performed cytogenetic analyses. HCL, YSC and CYY carried out transcriptomic analyses. CCL, FMT, MYL, YLP, WCC, MY, and HFT contributed to data collection and management. SCY and CTY performed the grading of bone marrow fibrosis. BSK coordinated the execution of the study, analyzed and interpreted the data, and contributed to manuscript writing and revision. HAH conceived and designed the study, coordinated its execution, analyzed and interpreted the data, and contributed to manuscript writing and revision. All the authors have reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Lee, WH., Hou, HA., Lin, CC. et al. Prognostic implications of genetic and transcriptomic abnormalities in MDS according to IPSS-R, IPSS-M, and the International Consensus Classification. Blood Cancer J. 16, 34 (2026). https://doi.org/10.1038/s41408-026-01456-4
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-026-01456-4
