
Abstract
Allogeneic stem cell transplantation (SCT) is a curative treatment in myelodysplastic syndromes (MDS). We performed a retrospective single center study of all patients with newly diagnosed higher risk (HR) MDS (IPSS-R > 3.5 points) between 2000 and 2023. We identified 2045 patients with HR-MDS, of which 427 (21%) underwent SCT. The median post-SCT overall survival (OS) was 23.9 months, progression-free survival (PFS) was 14.4 months, 5-year cumulative incidence (CI) of relapse was 37%, and 5-year CI of treatment-related mortality (TRM) was 25%. There were no significant differences in OS, PFS, CI of relapse, or CI of TRM between age categories. Survival improved over the observation period (median 11.8 months in 2000–2010, 28.2 months in 2011–2016, and 40.2 months in 2017–2023; p = 0.01). By multivariate analysis, TP53 status was the most important predictor of post-SCT OS. Patients with TP53-wild type MDS had an OS of 69% at 5 years (HR for death 0.32 with SCT, p < 0.001). Patients with TP53 mutations had poor outcomes, with OS of 9.1 months in monoallelic (HR for death 0.88 with SCT, p = 0.69) and 6.8 months in biallelic (HR for death 0.76 with SCT, p = 0.14). SCT can lead to excellent long-term survival in TP53-wild type HR MDS.
Similar content being viewed by others


Introduction
The myelodysplastic syndromes (MDS) are a group of heterogeneous clonal bone marrow disorders that increase in incidence with older age. MDS is characterized by cytopenias, bone marrow dysplasia, and a variable propensity for transformation to acute myeloid leukemia (AML), with clinical behavior ranging from an indolent disorder to highly aggressive disease [1]. MDS is frequently treated with growth factors, luspatercept, immunomodulators, chemotherapy, and/or hypomethylating agents (HMAs) depending on disease risk [2, 3]. None of these therapies are curative in this patient population.
Allogeneic hematopoietic stem cell transplantation (SCT) is currently the only curative treatment modality in MDS. Given the significant toxicities associated with SCT and advanced median age of patients with MDS, the procedure is generally reserved for younger/fit patients with higher risk MDS [4]. Historically, SCT has been rarely performed in older patients due to the toxicity of myeloablative conditioning (MAC). Advances in conditioning regimens, notably reduced-intensity conditioning (RIC), and improved supportive measures, have expanded the use of SCT to older patients or those with comorbidities. Earlier series of RIC SCT in patients with MDS showed modest results, with 3-year overall survival (OS) in the 20% range [5]. In a landmark retrospective study, the European Group for Blood and Marrow Transplantation (EBMT) evaluated 1333 patients aged 50 years and above who underwent SCT for MDS. SCT was associated with a 4-year overall survival (OS) of 32% with RIC and 30% with MAC. Non-relapse mortality (NRM) and relapse rates at 4 years were 32% and 41% with RIC, and 44% and 33% with MAC, respectively [6]. In a biologic assignment study spanning 2014–2018 comparing RIC SCT to best supportive care or HMA therapy based on donor availability, Nakamura and colleagues described a 3-year OS rate of 48% in the donor arm [7]. Therefore, outcomes in MDS treated with SCT are improving over time.
To better understand the role of SCT at our center, we evaluated outcomes in a large and well characterized cohort of patients with higher-risk MDS (HR-MDS), with specific focus on the clinical and genetic factors influencing SCT-related outcomes.
Subjects and methods
Study design and patients
This was a single-center retrospective study. We identified all patients with newly diagnosed MDS (per the World Health Organization 2016 criteria) [8] and higher risk ( > 3.5 points) by the Revised International Prognostic Scoring System (IPSS-R) [2] presenting to the University of Texas MD Anderson Cancer Center (Houston, TX) between January 2000 and March 2023. Patients with chronic myelomonocytic leukemia (CMML) were excluded. Retrospective chart and database review were used to collect baseline characteristics, treatment history, and outcomes. This study was approved by the Institutional Review Board (IRB) and conducted in accordance to the Declaration of Helsinki.
Baseline cytogenetic and next-generation sequencing (NGS) data were collected when available. Depending on the year of diagnosis, NGS was performed using a panel of either 28, 53, or 81 genes recurrently mutated in myeloid disorders with a sensitivity of 2% in a Clinical Laboratory Improvement Amendments (CLIA) certified laboratory as previously described [9]. Biallelic TP53 mutations were defined as a single mutation with variant allele frequency (VAF) ≥ 50%, 2 mutations, or a single mutation with concomitant 17p deletion. All other TP53 mutations were considered monoallelic. Patients were stratified using the IPSS-R and the Molecular International Prognostic Scoring System (IPSS-M) [2, 3].
Stem cell transplantation and outcome definitions
SCT conditioning regimens with busulfan area under the curve (AUC) ≥ 20,000 μmol/min or ≥16,000 μmol/min with concomitant thiotepa were considered MAC [10, 11]. All other regimens were classified as RIC. The Glucksberg-Seattle criteria and NIH criteria were used to grade acute and chronic graft-versus-host disease (GVHD), respectively [12, 13].
Pre-SCT responses were coded as per the International Working Group (IWG) 2023 definitions [14]. Depending on the analysis, OS was defined as the time from diagnosis or from SCT day 0 to death from any cause. Progression-free survival (PFS) was defined as the time from SCT day 0 to MDS relapse or death from any cause, whichever occurred first. Patients who did not experience an event were censored on the date of last follow-up. Patients with persistent MDS noted on the first post-SCT bone marrow were considered a relapse on day 30 for the purpose of PFS. Treatment-related mortality (TRM) was defined as death from any cause following SCT without prior relapse of MDS.
Statistical analyses
The Fisher’s exact, chi-squared, Wilcoxon–Mann–Whitney, and Kruskal-Wallis tests were used to compare baseline variables. The Kaplan-Meier method was used to estimate OS and PFS. Differences between groups were evaluated using the log-rank test. Cumulative incidence analyses were performed using MDS relapse and TRM as competing risks, with differences between groups evaluated using Gray’s test. To compare OS in patients undergoing SCT versus those who did not, landmark analyses were performed using the median time to SCT as the landmark and excluding patients in the no-SCT group who were older than the oldest transplanted patient. Univariate and multivariate analyses were performed using Cox proportional hazards regression, with SCT as a time-dependent covariate where applicable. Statistical analyses were performed using GraphPad Prism (v. 10.3.1) and R (v. 4.2.2).
Results
Patient and treatment characteristics
Between January 6th 2000 and March 2nd 2023, we identified 2045 consecutive patients presenting to our institution with untreated higher risk MDS. Of these, 427 (21%) underwent SCT during the observation period. The baseline characteristics stratified by receipt of SCT are shown in Table 1. When compared to patients who did not undergo SCT, transplanted patients were significantly younger, had better performance status, had higher baseline hemoglobin and platelet counts, had lower baseline absolute neutrophil counts, and had more MDS-EB2. Complex cytogenetics and TP53 mutations were significantly more common in the patients who did not undergo SCT. We noted a high frequency of TP53 mutations (43%; 424/987) in the overall cohort, likely reflecting a referral bias to a large academic center. Of the TP53-mutated cases, 30% (126/424) were monoallelic and 70% (298/424) were biallelic. SCT was performed progressively more frequently as the observation period advanced (16% SCT rate in 2000–2010, 22% in 2011–2016, and 26% in 2017–2023; Supplementary Fig. 1).
The most common frontline therapies received were HMAs (44%; 898/2045), intensive chemotherapy (8%; 154/2045), venetoclax-based regimens (6%; 113/2045), low-intensity chemotherapy (3%; 71/2045), immunomodulatory drugs (IMiD’s; 1%; 27/2045), and others (5%; 94/2045). The frontline regimen consisted of best supportive care (including growth factors) or was unknown/missing in 34% (688/2045) of patients.
The SCT-related characteristics of the transplanted patients (n = 427) stratified by age at time of SCT are shown in Table 2. The most common source of stem cells was peripheral blood from a matched unrelated donor. RIC was more frequent with increasing age. In terms of GVHD prophylaxis, tacrolimus was used in 97% (415/427) of patients and 42% (179/427) received post-transplant cyclophosphamide. Post-SCT maintenance therapy was administered in 19% (81/427) of patients. Maintenance consisted of an HMA alone in 83% (67/81), HMA plus venetoclax in 16% (13/81), and venetoclax alone in 1% (1/81) of patients.
General outcomes in the full cohort of HR-MDS
With a median follow-up time of 80.6 months, the median OS from diagnosis for the full cohort (irrespective of receipt of SCT) was 15.1 months. OS was 32% at 2 years and 15% at 5 years (Supplementary Fig. 2A). Overall, 25% (516/2045) of patients transformed to acute myeloid leukemia (AML). The cumulative incidence of AML at 2 and 5 years (with death as a competing risk) was 24% and 29%, respectively (Supplementary Fig. 2B).
Outcomes in patients undergoing allogeneic stem cell transplantation
We next examined outcomes in patients who underwent SCT, excluding those who transformed to AML prior to SCT (n = 362 transplanted as MDS). Engraftment was nearly universal (341/347 evaluable, 98%) and occurred at a median of 14 days (range 5–34). Post-SCT, 325/362 (90%) patients attained morphologic remission of MDS, 17/362 (5%) had persistent MDS, 8/362 (2%) had early death, and 12/362 (3%) were non-evaluable. Full donor chimerism was achieved in 291/328 (89%) patients and mixed chimerism in 37/328 (11%) patients. 4-week, 8-week, and 100-day mortality were 2%, 5%, and 12%, respectively. The median number of inpatient days during the first 100 days post-SCT was 24 (range 8–98). Acute GVHD of any grade occurred in 211/346 (61%) patients while grade 3/4 acute GVHD occurred in 45/346 (13%) patients. Chronic GVHD occurred in 93/299 (31%) patients. Of these, chronic GVHD was limited in 14/93 (15%) patients and extensive in 79/93 (85%) patients.
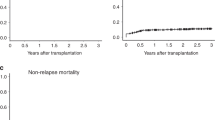
In the patients who underwent SCT (n = 362), the median OS from SCT day 0 was 23.9 months (5-year 40%), the median PFS was 14.4 months (5-year 38%), the cumulative incidence of relapse at 5 years was 37%, and the cumulative incidence of TRM at 5 years was 25%. When stratified by patient age at the time of SCT, post-SCT OS, PFS, cumulative incidence of relapse, and cumulative incidence of TRM were not significantly different between age categories (Fig. 1A–D). There were no significant differences in the cumulative incidences of either relapse or TRM when stratified by conditioning intensity (MAC versus RIC; Fig. 1E, F).

A OS from SCT day 0 stratified by age at time of SCT. B PFS from SCT day 0 stratified by age at time of SCT. C Cumulative incidence of relapse from SCT day 0 stratified by age at time of SCT. D Cumulative incidence of TRM from SCT day 0 stratified by age at time of SCT. E Cumulative incidence of relapse from SCT day 0 stratified by conditioning intensity. F Cumulative incidence of TRM from SCT day 0 stratified by conditioning intensity. CI cumulative incidence, MAC myeloablative conditioning, MDS myelodysplastic syndrome, OS overall survival, PFS progression-free survival, RIC reduced-intensity conditioning, SCT stem cell transplantation, TRM treatment-related mortality.
OS by year of transplantation gradually improved over the observation period (median 11.8 months in 2000–2010, 28.2 months in 2011–2016, 40.2 months in 2017–2023, p = 0.01; Fig. 2A). This was driven by a reduction in both the cumulative incidences of relapse and TRM in the more recent years (5-year relapse 42% in 2000–2010 vs 35% in 2017–2023; 5-year TRM 34% in 2000–2010 vs 24% in 2017–2023; Supplementary Fig. 3A-B).

A OS from SCT day 0 stratified by year of transplant. B OS from SCT day 0 stratified by IPSS-R. C OS from SCT day 0 stratified by TP53 mutation status. D OS from SCT day 0 stratified by immediate pre-SCT bone marrow blast %. E OS from SCT day 0 stratified by pre-SCT cytoreductive therapy. F OS from SCT day 0 stratified by pre-SCT IWG 2023 response. CR complete remission, CRh complete remission with partial hematologic recovery, CRL complete remission with limited count recovery, HI hematologic improvement, HMA hypomethylating agent, IC intensive chemotherapy, IPSS-R revised International Prognostic Scoring System, IWG International Working Group, NR not reached, OS overall survival, PR partial remission, SCT stem cell transplantation, VEN venetoclax.
When stratified by IPSS-R, patients with intermediate and high risk disease had similar OS following SCT (median 140.7 versus 103.1 months, p = 0.60), with 5-year OS in the 59% to 61% range (Fig. 2B). In contrast, patients with very high risk by IPSS-R had significantly shorter OS post SCT when compared to all other risk categories combined (median OS 8.2 vs 123.1 months, p < 0.0001; Fig. 2B). A similar finding was noted when stratifying patients by IPSS-M. IPSS-M very low, low, moderate low, moderate high, and high risk all had similar OS post SCT (median not reached for all, p = 0.54) whereas very high risk had significantly shorter OS compared to the other risk categories combined (median OS 10.5 months vs not reached, p < 0.0001; Supplementary Fig. 4). This appeared to be largely driven by enrichment of TP53 mutations and complex cytogenetics in patients with very high risk by IPSS-R or IPSS-M. Indeed, the incidence of TP53 mutations was 56/91 (62%) in IPSS-R very high risk and 43/70 (61%) in IPSS-M very high risk. The incidence of complex cytogenetics was 119/160 (74%) in IPSS-R very high risk and 42/70 (60%) in IPSS-M very high risk. When stratified by TP53 status, the median post-SCT OS was not reached in TP53-wild type, 9.1 months in monoallelic TP53-mutated, and 6.8 months in biallelic TP53-mutated (p < 0.0001; Fig. 2C). The median VAF of the TP53-mutated cases undergoing SCT was 33% (range 2–93%). When stratified by cytogenetic complexity, the median post-SCT OS was 123.1 months in non-complex and 8.3 months in complex cytogenetics (p < 0.0001; Supplementary Fig. 5A). Patients with complex cytogenetics in the absence of TP53 mutations, although rare (n = 24), had post-SCT OS comparable to patients with neither complex cytogenetics nor TP53 mutations (median OS NR vs NR, 5-year OS 61% vs 70%, p = 0.41; Supplementary Fig. 5B).
When examining post-SCT OS based on the immediate pre-SCT bone marrow blast percentage, patients with blasts of 0–4% and 5–9% had similar outcomes (median OS 27.2 vs 31.8 months, p = 0.58). Patients with 10–19% blasts had significantly worse post-SCT OS compared to patients with 0–9% blasts (median OS 7.1 vs 28.1 months, p = 0.0012; Fig. 2D). Therefore, a 10% pre-SCT blast cutoff separated patients into two significantly different survival categories. To evaluate the effect of pre-SCT cytoreductive therapy, we selected a subset of patients who proceeded to SCT following either a single line of therapy or no therapy (n = 314). Compared to patients treated with HMA, those treated with intensive chemotherapy were younger, had higher bone marrow blast percentages, and had less frequent complex cytogenetics and TP53 mutations (Supplementary Table 1). Post-SCT OS was 22.8 months with no therapy, 15.3 months with HMA, NR with intensive chemotherapy, and 34.8 months with venetoclax-based regimens. There was no significant difference in post-SCT OS between patients treated with HMA (median 15.3 months) and those proceeding to SCT without therapy (median 22.8 months, p = 0.80). Compared to patients treated with HMA, post-SCT OS was significantly longer with intensive chemotherapy (median NR vs 15.3 months, p = 0.019) and numerically longer with venetoclax-based cytoreduction (median 34.8 vs 15.3 months, p = 0.061; Fig. 2E). In response-evaluable patients (n = 205), there was no significant difference in post-SCT OS when stratified by pre-SCT IWG 2023 response (Fig. 2F). Twenty of 40 (50%) non-responders by IWG 2023 had residual blasts in the 5–9% range prior to SCT while 8/40 (20%) had blast clearance below 5% without count recovery (previously marrow CR by IWG 2006).
Univariate and multivariate analyses for post-transplantation overall survival
We next performed an exploratory univariate analysis to examine factors affecting post-SCT OS in patients who underwent SCT in the untransformed MDS state (Supplementary Table 2). Patient-related factors associated with worse OS following SCT included increasing age (HR for death 1.13 per 10 years, p = 0.023) and increasing HCT-CI (HR 1.15 per point increase, p < 0.001) while increasing Karnofsky Performance Status was associated with improved OS (HR 0.74 per 10 point increase, p < 0.001). In terms of disease-related variables, adverse factors were therapy related MDS (HR 1.50, p = 0.005), increasing bone marrow ring sideroblast percentage (HR 1.02 per 1% increase, p < 0.001), IPSS-R very high risk (HR 3.68, p < 0.001), IPSS-M very high risk (HR 3.14, p = 0.001), abnormal 3q (HR 2.32, p = 0.033), chromosome 7 abnormalities (HR 2.21, p = 0.011), complex cytogenetics (HR 5.29 for 3 abnormalities, p < 0.001; HR 6.06 for >3 abnormalities, p < 0.001), TP53 mutations (HR 5.29 for monoallelic, p < 0.001; HR 6.93 for biallelic, p < 0.001), and immediate pre-SCT blasts of 10–19% (HR 2.10, p = 0.001). Conversely, normal diploid cytogenetics (HR 0.30, p < 0.001) and ASXL1 mutations (HR 0.16, p = 0.002) were associated with improved OS. In terms of treatment-related factors, increasing SCT donor age (HR 1.12 per 10 years, p = 0.03) was associated with worse OS while pre-SCT intensive chemotherapy (HR 0.50 compared to HMA, p = 0.020) and post-SCT cyclophosphamide (HR 0.71, p = 0.026) were associated with improved OS. Pre-SCT venetoclax-based therapy was associated with a non-statistically significant improvement in post-SCT OS (HR 0.51 compared to HMA, p = 0.063).
By multivariate analysis considering age, TP53 allelic status, therapy-related MDS, IPSS-R very high risk, HCT-CI, post-SCT cyclophosphamide, year of SCT, and pre-SCT therapy (n = 199), the factors significantly associated with shorter OS were TP53-mutated status (monoallelic HR 3.72, 95% CI 1.77–7.78, p < 0.001; biallelic HR 4.39, 95% CI 2.58–7.45, p < 0.001), IPSS-R very high risk (HR 1.72, 95% CI 1.06–2.79, p = 0.027), and increasing HCT-CI (HR 1.14, 95% CI 1.03–1.27, p = 0.014). The use of venetoclax-based regimens pre-SCT was associated with a hazard ratio for death of 0.58 (95% CI 0.26–1.26) but did not meet statistical significance (p = 0.20; Table 3). When the multivariate analysis was repeated excluding all patients with TP53 mutations (n = 124), increasing HCT-CI was significantly associated with worse post-SCT OS (HR 1.33, 95% CI 1.09–1.62, p = 0.005) while post-SCT cyclophosphamide was associated with improved OS (HR 0.25, 95% CI 0.09–0.70, p = 0.008; Supplementary Table 3).
SCT versus no-SCT analyses
To evaluate the benefit of SCT, we performed a multivariate analysis in the full cohort using a Cox proportional hazards model considering SCT as a time-dependent covariate. In parallel, we performed a landmark analysis to compare OS from diagnosis in patients who did or did not undergo SCT (baseline characteristics for the landmark analysis are shown in Supplementary Table 4). These analyses were stratified by TP53 status. In the patients who underwent SCT, the median time from MDS diagnosis to SCT day 0 was 6.3 months in TP53-wild-type, 5.9 months in monoallelic TP53-mutated, and 6.3 months in biallelic TP53-mutated. SCT was associated with a marked benefit in TP53-wild type patients by both multivariate analysis (HR for death 0.32, 95% CI 0.22–0.48, p < 0.001) and landmark analysis (median OS NR with SCT versus 24.1 months without, 5-year OS 69% versus 13%, p < 0.0001; Fig. 3A). In patients with monoallelic TP53 mutations, there was no significant benefit of SCT on OS by both the multivariate analysis (HR 0.88, 95% CI 0.47–1.65, p = 0.694) and landmark analysis (median OS 17.1 months with SCT versus 15.3 months without, 5-year OS 25% vs 0%, p = 0.17; Fig. 3B). This analysis was limited by small patient numbers. Only 3 patients with monoallelic TP53 mutations had a VAF below 10%, precluding a dedicated analysis in this subset. In patients with biallelic TP53 mutations, no significant benefit of SCT on OS was observed in the multivariate analysis (HR 0.76, 95% CI 0.53–1.09, p = 0.137). However, by landmark analysis, a marginal but statistically significant improvement in OS was noted in the patients with biallelic TP53 mutations who underwent SCT (median OS 14.6 months with SCT versus 12.6 months without, 5-year OS 5% vs 1%, p = 0.038; Fig. 3C).

Multivariate analysis by Cox proportional hazards model with SCT as time-dependent covariate (left) and landmark analysis (right) for overall survival in patients who did or did not receive SCT in A TP53 wild-type patients, B TP53 monoallelic mutated patients, and C TP53 biallelic mutated patients. Overall survival is calculated from the date of MDS diagnosis. BM bone marrow, CG cytogenetics, HR hazard ratio, IPSS-R revised International Prognostic Scoring System, NR not reached, SCT stem cell transplantation, t-MDS therapy-related MDS.
Outcomes in patients undergoing allogeneic stem cell transplantation following transformation to secondary acute myeloid leukemia
Sixty-five of 427 (15%) transplanted patients in this cohort proceeded to SCT following transformation of MDS to secondary AML (sAML). The median OS from SCT day 0 in these patients was 15.3 months (2-year OS 45%, 5-year 40%; Supplementary Fig. 6A). The median PFS from SCT day 0 was 9.9 months (2-year PFS 41%, 5-year 40%; Supplementary Fig. 6B). The cumulative incidence of relapse was 34% at 2 years and 36% at 5 years (Supplementary Fig. 6C). The cumulative incidence of TRM was 25% at 2 years and 25% at 5 years (Supplementary Fig. 6D). Of note, 21/22 (95%) patients with available testing in the transplanted sAML group were TP53 wild-type, indicating SCT was almost never performed in TP53-mutated MDS following transformation to sAML.
Discussion
SCT is the only curative treatment modality in MDS but its application is challenging due to patient age and comorbidity burden. The development of RIC regimens has expanded the use of SCT to older patient populations and outcomes have improved in recent years. However, the optimal selection of patients who stand to benefit the most from SCT, as well as the impact of pre-SCT therapy and response, are incompletely understood. Here, we examined a large contemporary cohort of patients with HR-MDS undergoing SCT to identify the most important clinical and genetic factors associated with post-SCT outcomes.
In this study, patients with MDS proceeding to SCT had a median OS from SCT of 23.9 months, with 40% of patients being alive at 5 years. TP53 mutational status was the most important predictor of outcome. SCT was curative in the majority of TP53 wild type patients (median OS NR, 5-year OS 69%), and SCT vs no SCT analyses revealed a significant and marked benefit with SCT in this population. Conversely, patients with TP53 mutations derived a much smaller benefit from SCT. In TP53 monoallelic mutated cases, SCT was associated with a median post-SCT OS of only 9.1 months but with a plateau of around 20% survival at 5 years, indicating a subset of these patients experience long term disease control following SCT. Baranwal et al. recently identified patients with TP53 VAF’s below 10% as having particularly favorable outcomes compared to other TP53-mutated cases [15]. Our cohort included too few patients with monoallelic VAF’s below 10% to allow for a dedicated analysis. Patients with biallelic TP53 alterations had the worst outcomes, with median post-SCT OS of only 6.8 months and only 5% survival at 5 years. In these patients, SCT was associated with a statistically significant improvement in OS over no SCT (median 14.6 vs 12.6 months) by landmark analysis. However, the clinical relevance of such a modest improvement is unclear. This fundamental role of TP53 mutations in driving outcomes is consistent with observations by others [16, 17]. Our results are overall consistent with those of Versluis et al. who noted a modest but statistically significant benefit with SCT in TP53-mutated MDS [18]. A thorough discussion of the risks versus benefits of SCT is warranted in patients with TP53-mutated MDS given the limited impact on survival and low chance of cure combined with the potential toxicity of SCT and immediate effect on quality of life in older adults [19]. Emerging evidence suggests the bone marrow in TP53-mutated myeloid neoplasms displays immune checkpoint dysregulation, lower numbers of cytotoxic and helper T cells, and increased immunosuppressive regulatory T cells, which may explain the poor response to an immune-based therapy such as SCT [20]. Novel therapies, ideally targeting these immune defects, should be explored in TP53-mutated disease.
We noted a progressive improvement in post-SCT OS over the observation period (2000–2023), reflecting advances in patient and donor selection, GVHD prophylaxis (post-SCT cyclophosphamide), general supportive care, and conditioning regimens such as pharmacokinetic (PK)-guided fractionated busulfan which allows delivery of myeloablative conditioning with reduced toxicity [10]. We did not observe any significant differences in post-SCT OS, PFS, cumulative incidence of relapse, or cumulative incidence of TRM across the different age categories of patients. This suggests that patients should not be denied SCT based on age alone, although we acknowledge that patients aged 70 years and above who underwent SCT represented a highly selected subset. “Physiological” age, evaluated based on the assessment of comorbidities, functional status, and frailty, as opposed to “biological” (chronological) age may allow for improved selection of older candidates for SCT [21, 22]. Furthermore, “prehabilitation”, consisting of a resistance exercise training program in anticipation of SCT, has been shown to be feasible and may allow for improvement in functional status prior to SCT and enhanced post-SCT recovery and outcomes [23].
The optimal pre-SCT therapy remains an important unanswered question in the field. Intensive chemotherapy, although associated with the best outcomes in our study, is not feasible in most patients with MDS given age and comorbidities. The intensive chemotherapy-treated population was also younger and likely enriched for more favorable disease biology. HMAs are the most widely used therapy prior to SCT. Although these agents induce clinical response and prolong survival in higher risk MDS, they are unable to eliminate the MDS hematopoietic stem and progenitor cell (HSPC) clone [24, 25]. The BCL-2 inhibitor venetoclax has been shown to induce apoptosis in the HSPC compartment of higher risk MDS [25, 26]. Therefore, venetoclax-based combinations, typically with an HMA, may result in deeper responses prior to SCT and improved OS following SCT. In our study, the receipt of venetoclax prior to SCT was associated with trends toward improved post-SCT OS. This finding should be considered preliminary and hypothesis-generating as it was observed in a small number of patients and was not statistically significant by multivariate analysis (HR for death 0.58, p = 0.2). The phase 3 VERONA study evaluating the addition of venetoclax to azacitidine in HR-MDS was recently reported to have failed to meet its primary endpoint of overall survival, calling into question the utility of venetoclax in HR-MDS [27]. However, the use of venetoclax specifically as a pre-SCT cytoreductive therapy represents a different context that warrants evaluation in larger patient cohorts. In terms of responses, formal IWG 2023 responses pre-SCT were not significantly associated with post SCT OS. This was likely related to a large proportion (50%) of non-responders who had residual blasts in the 5–9% range, a group we showed had similar outcomes to patients with 0–4% blasts pre-SCT.
Our study has several important limitations. First, our cohort spans over two decades, during which MDS therapy and SCT conditioning have evolved, in addition to general advances in supportive care measures. For these reasons, our cohort is heterogeneous across time and treatment approaches. The patients included in this study were treated in a single large academic center with a high volume of SCT procedures. Our results may not be applicable to lower volume centers. Our patient population included a high proportion of cases with therapy-related MDS, likely reflecting referral bias to a tertiary cancer center, and may not be fully representative of a broader HR-MDS population. We explored the impact of multiple variables on outcomes, including small subsets, increasing the chance for type I error. Therefore, associations identified in a limited number of patients should be considered preliminary and confirmed in other cohorts. In addition, transplant versus no-transplant analyses have selection bias favoring the inclusion of fitter and/or better responding patients within the transplant arms. Therefore, the results of our landmark and time-dependent multivariate analyses must be interpreted with caution. Finally, it remains that many patients with MDS are not candidates for SCT either due to extremes of age, comorbidities, or lack of financial/social resources. Alternative and novel approaches to therapy beyond SCT are needed in this population.
In conclusion, allogeneic SCT represents an effective, curative approach in select patients with MDS without TP53 mutations, yielding 5-year OS rates of 69%. Exploring ways to improve the accessibility and safety of SCT in this population as well as identifying the optimal pre-SCT therapy represent important avenues for further research. Alternative approaches should be investigated in patients with TP53 mutations.
Data availability
The data are not publicly available in order to protect patient confidentiality. Reasonable requests for de-identified data should be directed to the corresponding author.
References
Ogawa S. Genetics of MDS. Blood. 2019;133:1049–59.
Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Sole F, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454–65.
Bernard E, Tuechler H, Greenberg PL, Hasserjian RP, Arango Ossa JE, Nannya Y, et al. Molecular international prognostic scoring system for myelodysplastic syndromes. NEJM Evid. 2022;1:EVIDoa2200008.
Cutler CS, Lee SJ, Greenberg P, Deeg HJ, Perez WS, Anasetti C, et al. A decision analysis of allogeneic bone marrow transplantation for the myelodysplastic syndromes: delayed transplantation for low-risk myelodysplasia is associated with improved outcome. Blood. 2004;104:579–85.
Laport GG, Sandmaier BM, Storer BE, Scott BL, Stuart MJ, Lange T, et al. Reduced-intensity conditioning followed by allogeneic hematopoietic cell transplantation for adult patients with myelodysplastic syndrome and myeloproliferative disorders. Biol Blood Marrow Transpl. 2008;14:246–55.
Lim Z, Brand R, Martino R, van Biezen A, Finke J, Bacigalupo A, et al. Allogeneic hematopoietic stem-cell transplantation for patients 50 years or older with myelodysplastic syndromes or secondary acute myeloid leukemia. J Clin Oncol. 2010;28:405–11.
Nakamura R, Saber W, Martens MJ, Ramirez A, Scott B, Oran B, et al. Biologic Assignment Trial of Reduced-Intensity Hematopoietic Cell Transplantation Based on Donor Availability in Patients 50-75 Years of Age With Advanced Myelodysplastic Syndrome. J Clin Oncol. 2021;39:3328–39.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391–405.
Ok CY, Singh R, Luthra R, Hatfield D, Floyd K, Loghavi S, et al. Endleukemia Assay v1: Enabling NGS-Based Comprehensive Routine Molecular Profiling of Leukemias in Routine Clinical Care. Blood. 2017;130:2679.
Popat UR, Pasvolsky O, Bassett R Jr, Mehta RS, Olson A, Chen J, et al. Myeloablative fractionated busulfan for allogeneic stem cell transplant in older patients or patients with comorbidities. Blood Adv. 2023;7:6196–205.
Popat UR, Mehta RS, Bassett R, Alousi AM, Alatrash G, Bashir Q, et al. Optimizing myeloablative fractionated busulfan, fludarabine and thiotepa regimen: results of two parallel cohorts in a phase 2 prospective clinical trial. Blood. 2021;138:1802.
Thomas ED, Storb R, Clift RA, Fefer A, Johnson L, Neiman PE, et al. Bone-marrow transplantation (second of two parts). N Engl J Med. 1975;292:895–902.
Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National institutes of health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: i. the 2014 diagnosis and staging working group report. Biol Blood Marrow Transpl. 2015;21:389–401.e1.
Zeidan AM, Platzbecker U, Bewersdorf JP, Stahl M, Ades L, Borate U, et al. Consensus proposal for revised International Working Group 2023 response criteria for higher-risk myelodysplastic syndromes. Blood. 2023;141:2047–61.
Baranwal A, Langer KJ, Gannamani V, Rud D, Cibich A, Saygin C, et al. Factors associated with survival after allogeneic transplantation for myeloid neoplasms harboring TP53 mutations. Blood Adv. 2025;9:3395–407.
Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson MD, et al. Prognostic Mutations in Myelodysplastic Syndrome after Stem-Cell Transplantation. N Engl J Med. 2017;376:536–47.
Bejar R, Stevenson KE, Caughey B, Lindsley RC, Mar BG, Stojanov P, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem-cell transplantation. J Clin Oncol. 2014;32:2691–8.
Versluis J, Saber W, Tsai HK, Gibson CJ, Dillon LW, Mishra A, et al. Allogeneic hematopoietic cell transplantation improves outcome in myelodysplastic syndrome across high-risk genetic subgroups: genetic analysis of the blood and marrow transplant clinical trials network 1102 study. J Clin Oncol. 2023;41:4497–510.
Newcomb R, Johnson PC, Cronin K, Choe JJ, Holmbeck K, Nabily A, et al. Quality of life, physical functioning, and psychological distress of older adults undergoing hematopoietic stem cell transplantation. Transpl Cell Ther. 2023;29:387.e1–e7.
Sallman, McLemore DA, Aldrich AL AF, Komrokji RS, McGraw KL, Dhawan A, et al. TP53 mutations in myelodysplastic syndromes and secondary AML confer an immunosuppressive phenotype. Blood. 2020;136:2812–23.
Holmes HM, Des Bordes JK, Kebriaei P, Yennu S, Champlin RE, Giralt S, et al. Optimal screening for geriatric assessment in older allogeneic hematopoietic cell transplantation candidates. J Geriatr Oncol. 2014;5:422–30.
Artz AS. Biologic vs physiologic age in the transplant candidate. Hematol Am Soc Hematol Educ Program. 2016;2016:99–105.
Potiaumpai M, Caru M, Mineishi S, Naik S, Zemel BS, Schmitz KH. IMPROVE-BMT: a pilot randomized controlled trial of prehabilitation exercise for adult hematopoietic stem cell transplant recipients. J Clin Med. 2024;13:2052.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10:223–32.
Ganan-Gomez I, Yang H, Ma F, Montalban-Bravo G, Thongon N, Marchica V, et al. Stem cell architecture drives myelodysplastic syndrome progression and predicts response to venetoclax-based therapy. Nat Med. 2022;28:557–67.
Jilg S, Reidel V, Muller-Thomas C, Konig J, Schauwecker J, Hockendorf U, et al. Blockade of BCL-2 proteins efficiently induces apoptosis in progenitor cells of high-risk myelodysplastic syndromes patients. Leukemia. 2016;30:112–23.
Abbvie. AbbVie Provides Update on VERONA Trial for Newly Diagnosed Higher-Risk Myelodysplastic Syndromes [updated June 16, 2025. Available from: https://news.abbvie.com/2025-06-16-AbbVie-Provides-Update-on-VERONA-Trial-for-Newly-Diagnosed-Higher-Risk-Myelodysplastic-Syndromes.
Acknowledgements
This work was supported in part by the University of Texas MD Anderson Cancer Center Support Grant CA016672 and the University of Texas MD Anderson Cancer Center MDS/AML Moon Shot.
Author information
Authors and Affiliations
Contributions
ABazinet, GG-M, and UP designed the study. ABazinet, ABataller, KC, KJ, IB, MS, WYJ, EK, SU, GGR, JB, RB, JJRS, SP, JC, and UP gathered/provided the data. GG-M, HK, and UP provided ressources. RKS provided correlative molecular data. ABazinet and ABataller analysed the data and performed the statistical analysis. ABazinet and ABataller generated the figures. ABazinet wrote the manuscript. All authors critically reviewed the analysis and manuscript.
Corresponding author
Ethics declarations
Competing interests
KS: Honoraria: Otsuka, Amgen, Chugai Pharma. Consulting or Advisory Role: Novartis, Pfizer, Takeda. Research Funding: Novartis, Enliven Therapeutics. Travel, Accommodations, Expenses: Otsuka, Amgen. GMB: Research Funding: IFM Therapeutics, Takeda. TK: Honoraria: CURE. Consulting or Advisory Role: Novartis, Jazz Pharmaceuticals, Pfizer, AbbVie/Genentech, Agios, Daiichi Sankyo/UCB Japan, Liberum, Sanofi, Servier, Pinot Bio, Sellas Life Sciences, Bristol Myers Squibb/Celgene. Research Funding: Bristol Myers Squibb, Celgene, Amgen, BiolineRx, Incyte, Genentech/AbbVie, Pfizer, Jazz Pharmaceuticals, AstraZeneca, Astellas Pharma, Ascentage Pharma, Genfleet Therapeutics, Cyclacel, Pulmotech, cellenkos, Glycomimetics, Astex Pharmaceuticals, Iterion Therapeutics, Delta-Fly Pharma, Regeneron, Sellas Life Sciences, Abcuro, DAAN Biotherapeutics. GB: Consulting or Advisory Role: Argenx, PTC Therapeutics, BiolineRx, BioTheryX, Nkarta, Treadwell Therapeutics, Novartis, Catamaran Bio, Takeda, AbbVie. Research Funding: Incyte, GlaxoSmithKline, Cyclacel, BiolineRx, MedImmune, Lilly, Oncoceutics, Ryvu Therapeutics, Janssen Scientific Affairs, Bristol Myers Squibb, AbbVie, Novartis, AstraZeneca, Mundipharma Research, PTC Therapeutics, BioTheryX, XBiotech, Arvinas, Astex Pharmaceuticals, TCR2 Therapeutics, Nkarta, Nkarta, Treadwell Therapeutics, Cellestia Biotech. CD: Honoraria: AbbVie, GlaxoSmithKline, Servier, Genentech/AbbVie, Astellas Pharma, Bristol Myers Squibb Foundation, Schrodinger, Daiichi Sankyo/Astra Zeneca, Ellipses Pharma. Consulting or Advisory Role: AbbVie, AstraZeneca, Genmab, Molecular Partners, Servier, Schrodinger, Rigel, Ryvu Therapeutics. Research Funding: AbbVie, Cleave Therapeutics, Foghorn Therapeutics, Immune-Onc Therapeutics, Jazz Pharmaceuticals, Servier, Loxo/Lilly, Astex Pharmaceuticals, Bristol Myers Squibb/Celgene, BeiGene, Remix Therapeutics. NS: Honoraria: Amgen. Consulting or Advisory Role: AstraZeneca, NGM Biopharmaceuticals, Pfizer, Takeda, Novartis, Amgen. Research Funding: Takeda, Astellas Pharma, Stemline Therapeutics. NP: Consulting or Advisory Role: Blueprint Medicines, Pacylex, Immunogen, Bristol Myers Squibb, Clearview Healthcare Partners, Astellas Pharma, CTI BioPharma Corp, AbbVie, Aptitude Health, Aplastic Anemia and MDS International Foundation, CancerNet, CareDx, Celgene, Cimeio Therapeutics, Curio Science, Dava Oncology, EUSA Pharma, Harborside Press, Imedex, Intellisphere, Intellisphere, Magdalen Medical Publishing, Medscape, Menarini Group, Neopharm, Novartis, OncLive, Patient Power, Peerview, Pharmaessentia, Physicans’ Education Resource. Research Funding: US DOD. EJ: Consulting or Advisory Role: Pfizer, Takeda, Amgen, AbbVie, Bristol Myers Squibb, Adaptive Biotechnologies, Genentech, Autolus, Ascentage Pharma, Taiho Pharmaceutical, Novartis, TargetRX, Terns Pharmaceuticals. Research Funding: Pfizer, Adaptive Biotechnologies, Ascentage Pharma Group, AbbVie, Takeda, Johnson & Johnson/Janssen, Jazz Pharmaceuticals, Terns Pharmaceuticals, Taiho Pharmaceutical, Novartis, Amgen, TGRX. Travel, Accommodations, Expenses: Pfizer, Aptitude Health, Autolus, Nucleus Global, ONVIV, MD Education, HMP Education, Daiichi Sankyo Nordics. ND: Consulting or Advisory Role: Celgene, Agios, Jazz Pharmaceuticals, Pfizer, AbbVie, Astellas Pharma, Daiichi Sankyo, Novartis, Bristol Myers Squibb, Amgen, Immunogen, Genentech, Servier, Syndax, Trillium Therapeutics, Gilead Sciences, Arog, Shattuck Labs, Kite, a Gilead company, Stemline/Menarini. Research Funding: Bristol Myers Squibb, Pfizer, Immunogen, Genentech, AbbVie, Astellas Pharma, Servier, Daiichi Sankyo, Gilead Sciences, Amgen, Trillium Therapeutics, Hanmi, Trovagene, FATE Therapeutics, Novimmune, Glycomimetics, Kite, a Gilead company. FR: Honoraria: Amgen, Pfizer, Astellas Pharma, Celgene, Agios, AbbVie/Genentech, AstraZeneca, Bristol Myers Squibb, Takeda, Jazz Pharmaceuticals, Novartis, Syros Pharmaceuticals, Taiho Pharmaceutical, Syndax. Consulting or Advisory Role: Amgen, Astellas Pharma, Celgene, Jazz Pharmaceuticals, Agios, AbbVie/Genentech, Bristol Myers Squibb, AstraZeneca, Taiho Oncology, Syros Pharmaceuticals, Certara Inc. Research Funding: Bristol Myers Squibb, Amgen, Macrogenics, Xencor, Selvita, Cellerant, Astex Pharmaceuticals, Taiho Oncology, Syros Pharmaceuticals, Prelude Therapeutics, Biomea Fusion, Astellas Pharma. HK: Honoraria: AbbVie, Amgen, Pfizer, Ascentage Pharma Group, Ipsen, KAHR Medical, Novartis, Shenzhen Target Rx, Daiichih-Sankyo, Immunogen. Consulting or Advisory Role: AbbVie. Research Funding: Amgen, Bristol Myers Squibb, Novartis, AbbVie, Immunogen, Jazz Pharmaceuticals, Ascentage Pharma, Daiichi Sankyo/Lilly. GGM: Honoraria: Curis, Bristol Myers Squibb/Celgene, Taiho Oncology, Ascentage Pharma, Servier, Keros Therapeutics. Consulting or Advisory Role: Bristol Myers Squibb, Taiho Oncology, Curis, Ascentage Pharma, Servier, Keros Therapeutics. Research Funding: Novartis, AbbVie, Bristol Myers Squibb, Genentech, Curis, Rigel, Taiho Oncology, Stemline Therapeutics, Zentalis, Chordia Therapeutics, Jazz Pharmaceuticals. The remaining authors declare no competing interests.
Ethics approval and consent to participate
All methods were performed in accordance with the relevant guidelines and regulations. This study was approved by the MD Anderson Institutional Review Board (protocol # 2024-0341). The need for informed consent was waived due to the retrospective, non-interventional nature of this study.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bazinet, A., Bataller, A., Chien, K. et al. Impact of allogeneic stem cell transplantation in patients with higher risk myelodysplastic syndromes. Blood Cancer J. 16, 48 (2026). https://doi.org/10.1038/s41408-026-01479-x
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41408-026-01479-x
