
Abstract
Osteoarthritis (OA) poses a significant challenge in orthopedics. Inflammatory pathways are regarded as central mechanisms in the onset and progression of OA. Growing evidence suggests that senescence acts as a mediator in inflammation-induced OA. Given the lack of effective treatments for OA, there is an urgent need for a clearer understanding of its pathogenesis. In this review, we systematically summarize the cross-talk between cellular senescence and inflammation in OA. We begin by focusing on the mechanisms and hallmarks of cellular senescence, summarizing evidence that supports the relationship between cellular senescence and inflammation. We then discuss the mechanisms of interaction between cellular senescence and inflammation, including senescence-associated secretory phenotypes (SASP) and the effects of pro- and anti-inflammatory interventions on cellular senescence. Additionally, we focus on various types of cellular senescence in OA, including senescence in cartilage, subchondral bone, synovium, infrapatellar fat pad, stem cells, and immune cells, elucidating their mechanisms and impacts on OA. Finally, we highlight the potential of therapies targeting senescent cells in OA as a strategy for promoting cartilage regeneration.
Similar content being viewed by others

Introduction
The aging population presents a significant and imminent challenge to global society. By 2050, individuals over 60 years old are projected to make up approximately 22% of the global population.1 The aging body becomes increasingly susceptible to age-related chronic diseases, including chronic heart failure, atherosclerosis, chronic obstructive pulmonary disease, diabetes, and osteoarthritis (OA).2,3,4 Due to the gradual loss of internal homeostasis in aging organisms, effective defenses fail to activate in response to internal and external stressors. Senescence, an intricate and interconnected process, manifests universally in biological organisms, regardless of a clear correlation between biological and chronological aging.5
Senescence is marked by a progressive loss of function at the tissue and cellular levels. Aging is driven by the accumulation of senescent cells at the cellular level. First introduced by Leonard Hayflick and Paul Moorhead in 1961, the concept of “senescence” suggests a correlation between cellular senescence and biological aging.6 Notably, senescence can be classified into three types in vivo: acute, embryonic, and chronic senescence.7,8 Unlike the former two, cells entering senescence due to chronic stimuli exert harmful effects on the organism. This review focuses on this form of senescence. Inflammation, the most common source of chronic stimulation, follows an age-related pattern, with longitudinal studies revealing a tendency for chronic low-grade inflammation with age.9 Comparing inflammation-related markers in the lungs of mice at different ages shows a pro-inflammatory shift in the lungs of aged mice.10 A unique inflammatory cell profile in adipose tissue has been identified in aged mice.11 These findings indirectly suggest an link between inflammation and senescent cells, with studies attempting to uncover their causal relationship.12,13 Consequently, the term “inflammsenescence” was coined to describe the heightened pro-inflammatory state during senescence.14
Osteoarthritis (OA), a joint disorder characterized by chronic pain and cartilage deterioration, manifests as a persistent low-grade inflammatory condition.15,16 Current clinical treatments for OA include oral nonsteroidal anti-inflammatory drugs, joint injections of hyaluronic acid, and surgical interventions. However, none can halt the pathological process.17 With an aging population, early personalized prevention is emerging as a key intervention, focusing on avoiding risk factors such as obesity, joint damage, and impaired muscle function.18 However, senescence, an additional risk factor for OA, is difficult to avoid and plays a significant role in the disease process.19 In this review, we summarize the cross-talk between cellular senescence and inflammation in OA (Fig. 1). First, we focus on the mechanisms and hallmarks of cellular senescence, summarizing evidence supporting the correlation between cellular senescence and inflammation. We then discuss the mechanisms underlying the interaction between cellular senescence and inflammation. Additionally, we examine various types of cellular senescence in OA, including senescence in cartilage, subchondral bone, synovium, infrapatellar fat pads, and stem cells. Furthermore, we summarize the senescence of stem and immune cells, elucidating their mechanisms and impacts within OA. Finally, we highlight the potential of targeting senescent cells as a therapeutic approach for OA, offering new strategies to promote cartilage regeneration.

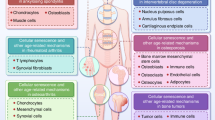
Schematic diagram of the cross-talk between cellular senescence and OA. Characteristic manifestations of joint degeneration are as follows: senescent subchondral bone with decreased bone density, reduced bone mass and thinning, senescent infrapatellar fat pad with inflammatory changes, senescent chondrocytes with decreased function accompanied by increased matrix degradation, senescent synoviocytes with diminished function and chronic synovitis, and senescent stem cells with diminished function and genetic stability. Alterations in senescent cells include telomere attrition, DNA instability, vacuolization of the cytoplasm, endoplasmic reticulum stress, mitochondrial dysfunction and alterations in the cell membrane. The characteristic pathology of OA includes reconstruction of the subchondral bone, synovial hyperplasia, cartilage damage, and inflammatory changes in the infrapatellar fat pad. The inflammatory mediators form a bridge of communication between cellular senescence and OA, and senescent cell-associated secretory phenotypes (SASP) play a critical role in this process
Aging and cellular senescence
Organisms experience an irreversible aging process marked by a gradual loss of physiological integrity, reducing their ability to withstand internal and external stressors and increasing their susceptibility to injury and disease.20 Senescence reflects the culmination of changes over an organism’s lifetime, evidenced by the accumulation of senescent cells in aging tissues.21,22 Therefore, elucidating cellular senescence is crucial for understanding the processes associated with aging and its pathology.
Cellular senescence was first observed around 60 years ago when Leonard Hayflick and Paul Moorhead discovered that human fibroblasts degenerate after approximately 50 passages and a year in culture, a phenomenon termed the “Hayflick limit,” which represents replicative senescence.6 Premature cellular senescence, which occurs before telomere shortening, has also gained attention and includes stress-induced senescence, oncogene-induced senescence in vitro, and tumor suppressor deficiency-induced senescence.23 Regardless of the trigger for cellular senescence, the ultimate outcome is cell cycle arrest.
Hallmarks of aging
Numerous studies have extensively documented the hallmarks of aging, despite some variations. These hallmarks share common characteristics, including time dependence, aging-promoting effects, and potential therapeutic applications.24,25 López-Otín et al. systematically summarized nine hallmarks of aging as early as 2013: DNA instability, telomere attrition, epigenetic alterations, loss of proteostasis, dysregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication.20 Subsequently, two additional hallmarks, chronic inflammation (inflammaging) and ecological dysregulation, were added to highlight the critical role of inflammation in aging, which is the primary focus of this review.26
Characterization of senescent cells
Senescent cells are characterized by altered cell morphology, telomere shortening, and a range of distinctive senescence markers, as shown in Table 1. Another key feature is the shift in secretory phenotype observed in senescent cells, first identified by Krtolica et al.27 through the detection of soluble and insoluble factors secreted by senescent human fibroblasts. This phenomenon, known as the senescence-associated secretory phenotype (SASP), is marked by increased pro-inflammatory mediators (IL-1, IL-6, IL-7, IL-8, IL-18, TNF-α) and matrix metalloproteinases (MMP1, MMP10), significantly influencing the microenvironment (Fig. 2).28,29,30,31 Notably, although SASP maintains senescent cell cycle arrest, its regulation does not appear to be associated with cell cycle arrest.32,33 Unexpectedly, SASP plays a positive role in linking with the immune system by activating immune cells via paracrine effects to clear senescent cells (Fig. 2).34,35 In conclusion, senescent cells secrete a large number of bioactive molecules via SASP, which significantly impact their microenvironment and play key roles in various pathological processes, including tissue aging, chronic diseases, and cancer.

Schematic overview of the role of SASP in senescent cells. Senescent cells secrete interleukins and chemokines, which act on neighboring cells in a paracrine manner. SASP factors act on neighboring cells to induce a late senescent state among young and early senescent cells promoting a senescent microenvironment. In contrast, by activating immune cells to eliminate senescent cells, SASP establishes a connection with the immune system to participate in tissue repair and remodeling
Mechanism of SASP
The SASP in senescent cells is generated and maintained by complex regulatory mechanisms. Key signaling pathways, such as NF-κB, JAK2/3, and p38 MAPK, play pivotal roles in initiating SASP.7,36 Sustained DNA damage, along with GATA4 suppression, has been identified as an inducer of SASP.9,37 Cytoplasmic chromatin fragments in senescent cells trigger SASP by activating the cytoplasmic DNA-sensing cGMP-AMP synthase-STING (cGAS-STING) pathway of innate immunity. The presence of topoisomerase 1-DNA covalent cleavage complexes in cytoplasmic chromatin is essential for this process, suggesting the role of conserved features of innate immunity in senescence.38,39,40,41 Victorelli et al. revealed a mechanism by which mitochondria regulate SASP: in senescent cells, a small fraction of mitochondrial outer membranes is permeabilized, requiring BAX and BAK macropores to release mitochondrial DNA into the cytoplasm.42 Cytoplasmic mitochondrial DNA (mtDNA) then activates the cGAS-STING pathway. Yasuda et al. found that pro-inflammatory cytokine-driven downregulation of EZH2 maintains SASP via demethylation of the H3K27me3 marker in cancer-associated fibroblasts.43 Non-classical monocytes accumulate in aged individuals with elevated plasma TNF-α and IL-8 levels. The highly pro-inflammatory nature of non-classical monocytes may be a manifestation of SASP, induced by elevated levels of phosphorylated NF-κB (p65).44 Senescent cells maintain and regulate SASP through these mechanisms, which play a key role in shaping the tissue microenvironment.
Cross-talk between inflammation and cellular senescence
The impact of cellular senescence on inflammation has been previously discussed. Here, we focus on the reciprocal effects of inflammation on cellular senescence. Inflammation precedes senescence and is a better predictor of senescence onset than telomere length.45 Senescent cells secrete pro-inflammatory mediators, which in turn influence cellular senescence. This phenomenon, occurring when inflammation is excessively regulated, is known as inflammsenescence.13,46,47 Senescent chondrocytes are consistently observed in OA cartilage.29 Although low-grade inflammation may exist in normal joints, the presence of senescent chondrocytes in OA cartilage strongly suggests that inflammation plays a pivotal role in driving cellular senescence. Similarly, anti-inflammatory phenotypes have been found in centenarian cells, where longevity-associated activation of transcription factor 7 is upregulated, indicating that inflammation plays an important role in cellular senescence.48,49 These findings suggest a correlation between inflammatory mediator levels and the expression of senescence markers.50 Accordingly, we discuss the effects of inflammation on cellular senescence, focusing on the opposing aspects of inflammation and anti-inflammation.
Inflammation promotes cellular senescence
Inflammation promotes senescence through pathways such as immune system overstimulation, leading to immunosenescence, tissue degradation, and disruption of stem cell function.51 However, the molecular mechanisms underlying this process remain unclear (Fig. 3). Ribeiro et al. found that, even without a pro-inflammatory lipopolysaccharide attack, indoxyl sulfate alone induced low-grade inflammation via macrophages, while promoting senescence in renal tubular epithelial cells during injury.52 Overexpression of eotaxin-1/CCL11 increases senescence markers such as CDKN2A (p16INK4a) and SERPINE1 in airway epithelial pneumocytes via pro-oxidative and pro-inflammatory pathways.53 Strong evidence clarifying the molecular mechanisms of how inflammation promotes cellular senescence is lacking, and most studies focus on elucidating this mechanism based on inflammatory factor levels. Impaired endothelium produces IL-1β, driving inflammation in the stromal niche and leading to hematopoietic senescence characterized by skewed stem cell differentiation, which can be ameliorated by blocking IL-1β.54 Exposure to pro-inflammatory cytokines IL-6 and IL-8 induces a self-perpetuating senescent microenvironment, increasing breast cancer cell invasiveness.55 An investigation demonstrated that prolonged exposure of MCF cells to IL-6 or IL-8 induced senescence, a process that could be reversed using a neutralizing antibody.56 However, higher concentrations of IL-6 and IL-8 failed to induce cellular senescence; only when cells were moderately damaged or in a near-senescent state could inflammatory factors promote senescence.57 These observations highlight the complexity of the relationship between inflammatory factors and the induction of cellular senescence, demonstrating the multifaceted and tightly regulated nature of this interaction.

Schematic illustration of the mechanisms by which inflammation regulates cellular senescence. DNA damage and telomere attrition are associated with cellular senescence, accompanied by elevated senescence markers. Prolonged exposure of cells to a microenvironment comprising inflammatory factors such as IL 6 or IL 8 transduces signals into the cell interior via receptors on the cell membrane. Increased ROS activate the Nlrp3 inflammatory vesicle, regulate the activation of caspase-1 molecules upstream of inflammatory mediators, and increase the synthesis of SASP factors in senescent cells. Inducible nitric oxide synthase (iNOS) produces large amounts of nitric oxide (NO), increases prostaglandin E2 (PGE2) and cyclooxygenase (COX2) synthesis, further increases matrix metalloproteinase synthesis, and stabilizes DNA damage
Pro-inflammatory factors (e.g., IL-1β and TNF-α) do not act alone in promoting senescence; they stimulate the accumulation of reactive oxygen species (ROS), which synergistically accelerates the deterioration process.43 Notably, Yagi et al. found that ROS play a crucial role in inflammation-induced cellular senescence.58 Cells with telomeric wear in the plasma of young leukemia patients exhibit senescent biology, associated with elevated inflammatory cytokines and ROS-induced telomeric DNA damage.5,59 Knockout of the nfkb1 subunit induces chronic, low-level inflammation, leading to premature aging in mice. The underlying mechanism involves DNA damage, stabilized by increased NF-κB, COX-2, and ROS.60 Although consensus on the precise mechanisms of how inflammation promotes cellular senescence remains elusive, it is widely acknowledged that ROS-mediated DNA damage plays a role.
As a major source of inflammatory mediators in vivo, lipids accelerate inflammation, thereby promoting senescence.61,62,63 Chronic auto-inflammation triggered by adipocytes in RECC1-deficient mice plays a key role in adipose tissue degeneration, leading to premature senescence.64 A negative correlation between plasma lipid levels and telomere length was found in patients with Cushing’s syndrome, with a more pronounced effect in obese patients.65 The strong correlation between lipids and immune senescence is attributed to adipose tissue producing a subpopulation of pro-inflammatory B-cells, which induces the expansion of pro-inflammatory T-cells, accelerating immune senescence.66,67 The inflammatory phenotype of adipose tissue is linked to macrophages. Therefore, limiting macrophage numbers and their antigen presentation function can reduce adipose tissue inflammation to some degree.68,69,70
Anti-inflammatory strategies inhibit cellular senescence
Medication is one of the most common strategies for inhibiting cellular senescence. Cellular senescence can be suppressed by anti-inflammatory drugs. Nonsteroidal anti-inflammatory drugs (NSAIDs) have been shown to rescue telomere dysfunction in mice with premature senescence induced by knockout of the nfkb1 subunit.60 Similarly, the lifespan of genetically heterogeneous wild-type mice was extended by long-term aspirin use.71 Although 17α-estradiol has minimal effects on senescent cells, it significantly extends lifespan in mice by reducing adipose tissue inflammation.72 Schroer et al. recently found that platelet factor 4 (PF4) levels in mouse and human plasma negatively correlate with age.73 They also showed that treating aged male mice with plasma from young mice significantly reduced hippocampal neuroinflammation, ultimately rescuing cognition. Increased hyaluronan levels were observed in several tissues of transgenic mice overexpressing the naked mole rat hyaluronan synthase 2 gene, along with a significant reduction in tissue inflammation. This led to prolonged lifespan and improved health, attributed to the anti-inflammatory properties of macromolecular hyaluronan.74 These conclusions suggest that inflammation levels can be regulated by anti-inflammatory drugs, thereby inhibiting cellular aging. In the later sections of this review, we will summarize the use of medications, including anti-inflammatory drugs.
Exercise or diet can also inhibit cellular senescence by reducing inflammation. Exercise training significantly suppressed inflammatory signaling in the hippocampus and increased Fas- and mitochondria-dependent apoptosis.75 Interestingly, older rats showed elevated levels of inflammatory proteins after swimming exercise alone, which the authors attributed to the intensity of the exercise. A 5-year follow-up showed that anti-inflammatory diets reduced mortality from aging-associated chronic diseases, a mechanism linked to the maintenance of telomere length.76 Furthermore, a large population-based cohort study found an association between pro-inflammatory diets and low-grade inflammation, increasing the risk of chronic diseases.77 These findings suggest that anti-inflammatory approaches may delay or even reverse cellular senescence, offering new avenues for future research aimed at mitigating cellular senescence.
Inflammatory respones in tissue healing and repair
The body’s defense mechanisms initiate inflammation as an adaptive response to harmful stimuli, such as infections and tissue damage.78 The inflammatory response is a tightly regulated and precise process. Upon exposure to a harmful stimulus, the first step is detecting the stimulus via cell surface pattern receptors. These receptors include pathogen-associated molecular patterns (PAMPs) that activate germline-encoded pattern-recognition receptors (PRRs) in immune and nonimmune cells. Danger-associated molecular patterns (DAMPs) are also recognized by PRRs in response to signals released during tissue or cellular injury.79 Several intracellular signaling pathways, including nuclear factor kappa-B (NF-κB) and mitogen-activated protein kinase (MAPK), are activated upon receptor activation.80,81 Activation of inflammatory cells, like macrophages and adipocytes, triggers the release of inflammatory markers, including cytokines (e.g., interleukins, colony-stimulating factors, IFNs, TNFs, TGFs, and chemokines). Additionally, the coordinated network of multiple cell types recruits activated macrophages, monocytes, and other cells to the site of tissue injury or infection.82
Tissue damage from traumatic injury often leads to cell death. Unlike apoptosis, necrosis is more likely to cause cell membrane disruption.83 Inflammatory stimuli include various molecules released from necrotic cells, such as DNA, RNA, histones, and heat shock proteins, collectively known as DAMPs. Additionally, damaged cells release cytokines like interleukin 1a (IL-1a) and interleukin-33 (IL-33), known as alarmins. Alarmins induce immune cell migration, while DAMPs induce immune cell activation. Precise coordination between inflammatory and tissue-specific cells is crucial for restoring injured tissue and maintaining homeostasis in vivo. While the regulatory mechanisms behind this process remain unclear, a well-regulated inflammatory response is essential for tissue repair. The type 2 immune response plays a key role in limiting the reparative component of acute tissue injury. Additionally, a regulated inflammatory response prevents fibrosis. However, if the inflammatory response triggered by tissue injury is uncontrolled, it can lead to fibrosis and impaired function, especially in chronic inflammation.
The molecular and cellular mechanisms of inflammation in OA
Inflammatory responses in OA
Although OA is classified as an aseptic “non-inflammatory” arthropathy, its inflammatory response is complex, extending beyond the cartilage to the subchondral bone, synovial membrane, and infrapatellar fat pads.84,85 In addition to activated macrophages and neutrophils, chondrocytes and fibroblast-like synoviocytes play important roles in the process.86,87 Joint-resident cells, along with immune cells stimulated by DAMP, co-regulate the inflammatory network.88
Cytokines and chemokines, including pro-inflammatory cytokines IL-6, IL-8, IL-15, and IL-33, are secreted by the above cells. The secretion of these pro-inflammatory cytokines increases with DAMP expression. Inflammation-triggering mediators IL-1β and TNF-α are secreted in the early stages of OA.89 TNF-α stimulates TNF receptor 1 (TNFR1) and TNF receptor 2 (TNFR2), activating downstream signaling pathways. Notably, both receptors are expressed in synovial membranes, with TNFR1 strongly inducing proinflammation and TNFR2 capable of eliciting both proinflammatory and anti-inflammatory effects depending on the pathology.90,91,92 These pro-inflammatory factors stimulate the production of large amounts of nitric oxide (NO) by inducible nitric oxide synthase (iNOS), which in turn increases prostaglandin E2 (PGE2) and cyclooxygenase (COX2) synthesis. Meanwhile, PGE2 increases MMP13 production, leading to collagen degradation (Fig. 3).93,94,95 Pro-inflammatory cytokines activate the inflammatory response in surrounding cells, further sustaining SASP. The prolonged presence of SASP, in turn, exacerbates chronic inflammatory responses, creating a feedback loop.
Anti-inflammatory cytokines, including IL-4, IL-10, and IL-37, act as negative regulators, with IL-37 inhibiting M1 polarization and IL-33 promoting it. Rai et al. in their analysis of knee and hip cartilage from OA patients, found that increased IL-37 expression inhibited macrophage conversion to the M1 phenotype, while IL-33 had the opposite effect.88 These findings suggest that interactions between pro-inflammatory and anti-inflammatory cytokines, along with macrophages, play a key role in inflammation-mediated cartilage damage in conditions like OA. Anti-inflammatory factors can inhibit or attenuate inflammatory responses, thereby reducing the secretion of SASP and helping to alleviate chronic inflammation caused by senescent cells. Additionally, anti-inflammatory factors can regulate the survival and function of senescent cells, minimizing their harmful effects on surrounding tissues.
Inflammatory signaling pathways play a crucial role in mediating the inflammatory process in OA. Classical signaling pathways like MAPK, NF-κB, and ERK1/2 are involved, making them targets for drugs aimed at treating inflammation in OA.96,97,98,99,100,101,102,103,104,105 Catabolic factor stimulation activates these pathways, upregulating the expression of inflammatory genes like MMP and ADAMTS.106 The Wnt signaling pathway is also involved in inflammatory processes.107 As upstream regulators, MAPK and NF-κB pathways influence autophagy-mediated cartilage homeostasis. Therefore, regulating the autophagic process may delay OA progression.100 Additionally, a correlation between copper apoptosis-related genes and immune infiltration in OA patients was found through combinatorial analysis of OA transcriptome data.108 Given the interaction between inflammation and cellular senescence, tissues involved in the inflammatory response in OA may undergo cellular senescence.
Cellular senescence in OA
The concept of SASP and the relationship with disease
Age-related SASP contributes to the onset and progression of many senescence-related diseases.109 SASP in senescent cells affects the microenvironment via inflammatory mediators.110 Evidence has shown that numerous inflammatory factors in SASP may induce low-level chronic inflammation in aging tissues and accelerate organ degeneration.
Some scholars have reported that transplanting senescent cells into mice leads to age-related pathological changes and persistent physiological impairment, particularly in secretory function.111 Zeng et al. reported that aging-related kidney injury and inflammation regulate the RIG-I/NF-κB signaling pathway by promoting Klotho downregulation, accelerating aging in mice.112 Bailey-Downs et al. revealed a novel paracrine pathway leading to vascular redox imbalance, suggesting that senescence exacerbates oxidative stress and secondary low-level chronic inflammation in vivo.113 Additionally, several studies have found that clearing senescent cells in vivo provides varying benefits across different disease models.114
Numerous molecular mechanisms regulate SASP. NF-κB enhancer and C/EBP-β transcription factors play pivotal roles in regulating SASP at the mRNA level.115 The transcription factor GATA4 regulates SASP, and its activation depends on DNA damage regulators ATM and ATR, which activate NF-κB to promote SASP and aging. IL-1α has been shown to promote NF-κB signaling and upregulate many SASP genes.115 Many ROS-related factors, such as the ROS protein kinase CD1 axis, are crucial for the induction of IL-8 and IL-6, and thus the regulation of SASP.116 Additionally, MTOR regulates MAP kinase-activated protein kinase 2 (MAPK/APK2) and IL-1α, making it an important regulator of SASP. MAPK/APK2 can be phosphorylated by p38 to inactivate ZFP36L1, contributing to the degradation of pro-inflammatory SASP factors.117
Increasing evidence supports that low-grade systemic and local inflammation play a key role in the pathogenesis of OA.118 Numerous studies have shown that senescence is related to the etiopathogenesis of many age-related diseases, including OA.119 Senescent chondrocytes have been identified in the cartilage of replacement joints. Notably, senescent cells are not restricted to chondrocytes. They are also found in other joint components, including subchondral bone, synovium, stem cells, and the infrapatellar fat pad. Researchers have developed a method to alleviate OA by preventing the aging of chondrocytes and other joint cells.120 This section explores the age-related phenotype of resident joint cells and examines its relationship with OA pathogenesis (Fig. 4).

Inflammation leads to OA through SASP. Inflammation of internal joint tissues induce the hallmarks of senescence in resident cells, which further facilitates SASP (secretion of multiple bioactive factors such as chemokines, and cytokines and growth factors) and the secondary pathophysiological changes (recruitment of inflammatory cells such as macrophages, ROS&RNS, ECM degradation, and subchondral osteosclerosis) to lead to pathologies commonly found in OA (synovitis or hyperplasia, cartilage destruction, and subchondral bone sclerosis). Senescence-mediated SASP triggers or accelerates the process of inflammation-induced arthritis, exerting a cascading amplification effect
Chondrocyte senescence
Chondrocytes are the key resident cells in articular cartilage, crucial for maintaining cartilage function and contributing to OA development. Although chondrocytes have poor self-renewal ability, they retain some proliferative potential during the early stages of tissue repair.121 In vitro, chondrocytes promote cell division and exhibit the ability to form cell clusters.122 While chondrocyte senescence is correlated with OA, the mechanism linking aging and OA remains unclear.123
Several studies suggest that chondrocyte senescence can be triggered by factors such as nutritional deficiency, hypoxia, ROS, DNA damage, protein aggregation, damaged organelles, or intracellular pathogens. Furthermore, chondrocyte senescence and SASP trigger pathological biochemical changes in joint cells, ultimately leading to the onset and progression of OA.124 However, the molecular mechanisms driving chondrocyte senescence and renewal remain unclear. Evidence suggests that chondrocyte senescence can be caused by several factors. Coryell et al. reported that articular chondrocyte senescence is primarily driven by telomere erosion, cyclin-dependent kinase (CDK), and increased senescence-associated heterochromatin.120 Martin et al. studied the link between telomere erosion and senescence in chondrocytes, demonstrating their causal relationship.125 Based on the literature, we conclude that chondrocyte senescence is primarily caused by the following factors: (1) Telomere erosion. Senescent joint cells commonly exhibit telomere erosion, which strongly correlates with articular cartilage degradation. This process is driven by replication-related aging, reduced mitotic activity, and shorter telomere length.126 (2) Decline in mitotic activity. Senescent chondrocytes increase ROS levels via mitochondrial dysfunction and elevated senescence-related heterochromatin, promoting oxidative stress. (3) H-thymidine incorporation assay is a primary method for measuring mitotic activity.126 (4) Cell cycle arrest. Cellular senescence is characterized by a hyporeplicative state termed cell cycle arrest, primarily mediated by the upregulation of p53/p21/p16 pathways. Childs et al. demonstrated that replication arrest is typically triggered by DNA damage or stress signals and executed by activation of the p16 or p53 pathway.7 Loeser et al. reported that aging chondrocytes in OA exhibit intrinsic replicative senescence, known as cell-cycle arrest, primarily dependent on increased expression of p53, p16, p21, and other effectors.126 As a key participant in the DNA damage response (DDR) pathway, the p53 tumor suppressor protein is a crucial regulator of the cell cycle. The accumulation of phosphorylated p53 promotes cyclin-dependent kinase inhibitor (CDKI) activation, eventually leading to cell cycle arrest.127 p21, a recognized marker of senescence, is the downstream CDKI of phosphorylated p53. When p21 binds to CDK2, it inhibits the cell cycle by blocking the transition from G1 to S phase.128,129 Notably, p16 is highly expressed in aged chondrocytes. It induces senescence by binding CDK4 and CDK6, blocking the retinoblastoma protein (Rb), a cell cycle repressor. It mediates responses to cellular stress, such as DNA damage from radiation, telomere shortening, ROS, or oncogenic stress.130 Collectively, these cell-cycle inhibitors trigger apoptosis and senescence, leading to damage and loss of articular cartilage, contributing to OA development.
Numerous studies explore the mechanisms underlying chondrocyte senescence. In addition to the molecules mentioned above, including p53, p16, and p21, which are involved in cell cycle arrest, other key factors contribute to chondrocyte senescence and OA progression. Li et al. found that Sirt6 reduces chondrocyte aging and OA progression by interacting with STAT5, inactivating the IL-15/JAK3/STAT5 pathway.131 Varela-Eirín et al. demonstrated that Cx43 promotes chondrocyte-mesenchymal transition and reduces cellular senescence by increasing Twist-1 nuclear translocation during OA progression.132 Horváth et al. reported that chondrocyte senescence in OA is induced by Sox-9, aggrecan, and Col2a1 suppression, while increased expression of HIF-2α, RunX2, and MMP-13 inhibits the transition to senescence.133 However, the role of senescence in compromising joint stability and function in OA remains unclear, and further investigation is urgently needed.
The extracellular matrix (ECM) provides structural support, creates a favorable environment for various cell types, and influences key cellular processes.134 Changes in senescence-related ECM proteins profoundly affect homeostasis and physiology. Studies show that metabolic disorders and increased ECM catabolism in articular cartilage are key factors in OA development.135 Guo et al. studied the relationship between mitochondrial DNA dysfunction and OA, finding that STING activates the NF-κB signaling cascade to promote senescence, inducing secondary ECM degradation in OA.136 Lu et al. demonstrated that fibroblast growth factor 21 (FGF21) alleviates chondrocyte senescence and ECM impairment in OA via the SIRT1-mTOR signaling pathway.137 FGF21 administration has been shown to alleviate both chondrocyte senescence and ECM catabolism. While the molecular mechanisms driving ECM degradation and OA remain unclear, increased expression of inflammatory mediators from cartilage aging and SASP may be key factors in OA development. Notably, a balance exists between chondrocyte senescence and the metabolic regulation of inflammation, as demonstrated by recent findings. In a seminal study, Arra et al. found that chondrocytes undergo metabolic shifts in inflammatory states involving NF-κB activation, which reprograms cellular glycolysis and lactate dehydrogenase A.97 Lactate dehydrogenase A promotes ROS-induced catabolism. Inflammation and senescence converge on IκB-ζ, a key mediator downstream of NF-κB, regulating RANKL, inflammation, catabolism, and SASP gene expression to program chondrocytes into an “inflammatory phenotype”.123 This indicates a close link between metabolic regulation of cellular senescence and inflammation, suggesting that cellular senescence can be alleviated by correcting metabolic imbalances.138,139
Notably, SASP occurs not only in chondrocytes but also in bone and synovial cells, possibly induced by chondrocyte-centered intercellular communication.140,141 Cellular aging is characterized by DNA damage, mitochondrial dysfunction, and permanent cell cycle arrest, ultimately leading to SASP. SASP leads to the release of pro-inflammatory molecules into neighboring tissues and cells. Studies suggest that chondrocytes stimulate osteocytes and synovial fibroblasts, affecting their limited regenerative potential.20 Thus, we believe that senescent chondrocytes promote OA through intercellular communication, including chronic low-grade inflammation known as “inflammsenescence”. Some studies have explored the mechanisms by which chondrocytes alter their surrounding environment and cells. These findings suggest that the spread of senescence relies heavily on SASP development. Several studies have shown that senescent chondrocytes spread senescence to surrounding tissue through SASP, involving the secretion of cytokines, growth factors, chemokines, and other bioactive factors to influence signaling in an autocrine or paracrine manner. Jeon et al. documented that elevated activation of the IL-6–STAT3 signaling pathway in the synovium of OA patients induces fibroblast aging, indicating bystander effects that lead to secondary aging and SASP in neighboring cells.142 Coppé et al. suggested that SASP manipulates the surrounding microenvironment through paracrine signaling pathways.110 Zhu et al. reported that senescent chondrocytes produce and secrete bioactive molecules, including chemokines, cytokines, matrix-degrading enzymes (MMPs), and growth factors, facilitating cell-cell contact through gap junctions and further inducing neighboring cell aging.143 Collectively, these findings suggest that senescent chondrocytes activate SASP to secrete bioactive factors via paracrine pathways, transforming neighboring microenvironments and exerting systemic effects on the entire joint.
Additionally, evidence suggests that chondrocytes promote intercellular communication by releasing extracellular vesicles (EVs) into the synovial microenvironment, triggering senescence in bystander cells.142,144 Jeon et al. investigated the differences between EVs and SASP.142 They evaluated EVs production from senescent chondrocytes in arthritic cartilage and found a positive correlation between EVs production and the number of senescent cells. EVs are crucial cellular messengers that transfer senescence signals from senescent cells, playing a key role in senescence propagation and age-related OA. Mechanistically, EVs transfer senescence markers to bystander cells and inhibit cartilage regeneration by altering the expression of miR-34a, −92a, −24, −186, and −150. In summary, EVs produced or secreted from synovial fluid and senescent chondrocytes may serve as key mediators of senescence progression and OA pathology.
Senescence propagation occurs in other tissues. Liu et al. showed that bone-marrow adipocytes (BMAds) spread senescence to surrounding bone and bone marrow tissue through SASP, increasing oxylipin synthesis and expression of key senescence genes.145 They also demonstrated that oxylipin and its downstream effector PPARγ induce the expression of senescence-related genes, which in turn promote oxylipin synthesis in BMAds, forming a positive feedback loop. Additionally, Nelson et al. reported that senescent MRC5 fibroblasts induced secondary senescence in bystander fibroblasts through the production and secretion of bioactive factors, including cytokines, MMPs, growth factors, and ROS, via gap junction-mediated cell-cell contact.146 They further demonstrated that continuous exposure to senescent MRC5 fibroblasts induced senescence in neighboring fibroblasts, and that senescent hepatocytes aggregate in vivo. Waters et al. reported that senescent lung fibroblasts (LFs) induced a senescent-like phenotype in non-senescent LFs when exposed to alveolar epithelial cells (AECs) in vitro.147 This study offers a possible explanation for the abnormal abundance of senescent cells in the lungs of patients with idiopathic pulmonary fibrosis. We propose that senescent cells, including chondrocytes and other bone-resident cells, may stimulate secondary senescence and damage the local environment through SASP, senescence-associated EVs, and gap junction-mediated cell-cell contact. These hypotheses require further experimental validation and theoretical support.
Stem cell senescence
In recent years, mesenchymal stem cell (MSC)-based therapy has emerged as a complementary approach to treat OA. MSCs offer advantages such as easy accessibility, simple isolation, favorable proliferation, and multilineage differentiation potential, making them an excellent resource for OA treatment. Rizzo et al. demonstrated that MSCs or MSC-derived EVs combined with senolytic agents regulate intercellular communication, providing targeted therapeutic effects against senescent cells and SASP in OA.148 Based on tissue specificity, MSCs in the joint include synovial MSCs, adipose-derived MSCs, and BMSCs. Viable MSC-based therapies are in preclinical models and clinical treatments for OA, including local injection of MSCs, MSC-derived EVs, and MSC-loaded scaffold implants.
Understanding the mechanisms underlying MSC application in OA treatment is crucial. The immunomodulatory effect of MSCs is key in treating OA. Inflammatory factors released by senescent cells can activate MSCs, which then secrete PGE2, IDO, and NO to inhibit inflammatory cells and alleviate OA.149,150 MSC-derived cytokines regulate the synthesis and breakdown of metabolic factors, inducing anti-inflammatory factor expression in the synovium.151 Additionally, MSC-derived cytokines promote chondrocyte proliferation and ECM synthesis, repairing damaged bone and cartilage.152
MSC senescence also influences the development and progression of OA.153,154,155 Ye et al. suggested that MSC senescence is closely related to organic aging and the occurrence of aging-associated diseases, including OA.156 Čamernik et al.157 demonstrated that MSC depletion and functional decline in subchondral bone may contribute to OA development. Cao et al.158 showed that aging chondrocytes reduce MSCs’ natural potential to differentiate and proliferate, driving apoptosis of senescent chondrocytes and promoting OA. MSC senescence leads to significant changes in cell phenotype, including telomere shortening, altered cell surface markers, epigenetic changes, flattened or enlarged cell morphology, impaired differentiation potential, and decreased proliferation capacity.159 Nevertheless, deeper understanding of stem cell senescence mechanisms in OA is needed, and exploration of anti-aging agents to treat OA is essential.
Synovium senescence
Pathological changes in synovial of OA, such as proliferative and fibrous synovitis, are key manifestations of the disease. In contrast, several studies have demonstrated that SASP factors in synovial fibroblasts trigger OA-related changes, including joint inflammation, cartilage degeneration, subchondral osteosclerosis, and ECM degradation.120,160,161 Coppé et al. reported that nutrient deficiency, hypoxia, DNA damage, reactive oxygen species (ROS), damaged organelles, or intracellular pathogens can activate various cytokines, such as IL-1, IL-6, and IL-17.101 These cytokines, typical SASP factors, can promote synovial fibroblast senescence and contribute to joint degeneration. Therefore, a deeper understanding of the relationship between SASP in synovial fluid and OA pathogenesis could elucidate the role of SASP factors in joint tissue degeneration.
The SASP of intra-articular cells plays a crucial role in the degeneration of the surrounding ECM. Xu et al. performed senescent cell transplantation and discovered that introducing aging cells into the knee joint causes leg pain, impaired mobility, and radiological as well as histological alterations characteristic of OA.144 Del Rey et al. reported that increased inflammation in rheumatoid arthritis tissue leads to the premature accumulation of senescent synovial fibroblasts.141 Senescent cells in the synovium can induce fibrous synovitis, ECM degeneration, and cartilage damage, indicating that the SASP of synovial cells can significantly alter the joint’s microenvironment.162 The degradation of the surrounding ECM is primarily mediated by: (1) SASP-released cytokines that induce the breakdown of ECM proteins, including collagen, sulfated proteoglycans, and fibronectin, by regulating the expression of IL-1, IL-6, and IL-17 in cartilage.161 (2) The action of MMPs and ADAMTS (a disintegrin and metalloproteinase with thrombospondin motifs), such as MMP13 and ADAMTS-5. The depletion of ADAMTS plays a key role in ECM degradation and is associated with chondrocyte senescence and OA progression.163 (3) Senescent chondrocytes enhance the secretion of EVs, promoting intercellular communication in bystander fibroblasts and inducing a bystander effect that drives the senescence of neighboring tissues.
Infrapatellar fat pad senescence
The patellar fat pad, the largest soft tissue structure in the knee joint, is situated between the femoral condyle, tibial plateau, and patella. Its flexible and displaceable structure helps fill the anterior gap of the knee joint, absorbing force, reducing overload, and protecting the joint.164 Additionally, the fat pad promotes uniform distribution of synovial fluid, limits excessive knee movement, and provides lubrication. Fat pads are also a source of stem cells, inflammatory factors, and neuropeptides. Inflammation of fat pads has been linked to cartilage loss and ECM degradation, indicating their potential role in driving the development and progression of OA.165,166
Studies have demonstrated that aging adipose tissue is strongly associated with several diseases, including cardiovascular and metabolic diseases.167 OA is now considered a disease of the entire “joint organ,” and substantial evidence suggests that the patellar fat pad plays a role in knee OA development.168 The infrapatellar fat pad is associated with cartilage lesions and elevated inflammatory factor production, contributing to the development of knee OA.169 Favero et al. compared the aging infrapatellar fat pad in OA patients to that in non-OA patients, confirming its crucial role in OA pathology due to its susceptibility to inflammation, vascularization, and fibrosis.165
The mechanisms by which the infrapatellar fat pad contributes to this process have been partially elucidated. This mechanism appears to be multifactorial, potentially involving a pro-inflammatory state related to aging, commonly referred to as “inflammaging”.29 Aging-related inflammation can occur both systemically and locally. Studies indicate that chronic low-grade inflammation in adipose tissue is a key mechanism driving the progression of OA.167 Researchers have investigated obesity-related changes in systemic and adipose tissue-resident immune cells, discovering that metabolic disorders in aging adipose tissue ultimately lead to an inflammatory phenotype and tissue remodeling.167,170 Aging in the patellar fat pad can trigger low-grade inflammation, disrupting the balance between acute and chronic inflammation, ultimately contributing to joint damage.171 Additionally, adipocyte hypertrophy and dysfunction in aging adipose tissue are associated with shorter telomere length, altered cell proliferation, and accelerated OA progression.11 Macroscopically, the patellar fat pad interacts with surrounding tissues, including cartilage, subchondral bone, and synovium, playing a significant role in OA pathology.165,166 Further research is required to gain a deeper understanding of this process and its underlying mechanisms. Moreover, effective strategies must be developed to prevent these degenerative processes.
Immunosenescence
Immunosenescence refers to the dysfunction of both the innate and adaptive immune systems during aging, characterized by reduced T and B cell production and the accumulation of atypical cell subsets.172 The immune system is typically activated by PRRs, which initiate inflammatory responses to infections.173 Under normal conditions, innate and adaptive immunity are tightly regulated, with damage promptly followed by repair. However, as joint cells age, various immune dysfunctions emerge within the body. This process involves both innate and adaptive immune responses, with key features including thymic degeneration and reduced T cell production; shifts in T cell populations, such as increased memory T cells and decreased naïve T cells; impaired immune surveillance; poor vaccine response and increased infection susceptibility; higher incidence of autoimmune diseases and cancer; and senescence-associated dysregulated secretion of pro-inflammatory cytokines, chemokines, and proteases. Additionally, changes in metabolic and epigenetic pathways contribute to immune system and T cell aging.172,174,175 During bone and joint aging, chronic immune responses can cause cartilage loss, ECM degradation, and subchondral sclerosis.176 This section of the review aims to outline the key effector cells and molecules while exploring the potential mechanisms of OA-related immunosenescence.
Immune cell senescence
Senescence-associated deterioration of innate and adaptive immunity in joints impairs immune defenses, leading to persistent low-grade chronic inflammation. This promotes the accumulation of senescent phenotypes and the elevated production of pro-inflammatory factors, leading to SASP in resident tissues (cartilage, synovium, subchondral bone) and increasing susceptibility to OA.177,178 A variety of cells, including macrophages, fibroblasts, and mast cells, are involved in the development of immunosenescence-related OA. Evidence indicates that monocytes/macrophages play a crucial role in OA-related inflammation and can be activated to produce excessive cytokines, MMPs, and growth factors, contributing to OA pathology.179 Senescent immune cells, such as CD28-T cells and CD14+CD16+ monocytes, are more abundant in OA patients than in healthy controls, contributing to severe chronic inflammation in OA.120,172 Studies demonstrate that depleting macrophages in cocultures with synovial cells from OA patients leads to a significant reduction in cytokines (e.g., IL-1, IL-6, TNF-α) and MMPs (MMP1, MMP3, MMP9, MMP13), modulating inflammation and OA progression.180 This process forms a closed loop, where inflammation drives immune cell senescence, and senescent immune cells, in turn, perpetuate chronic inflammation.
Chondrocyte-related immunosenescence
Chondrosenescence refers to the senescence-driven dysfunction of chondrocytes, which impairs cartilage function in OA. Although chondrocytes are not part of the immune system, they can express various innate immunity receptors and produce inflammatory effectors during OA progression. When aging chondrocytes are activated in OA, they upregulate pro-inflammatory factors such as TNF-α, IL-1, and IL-6 through the complement system.181 Additionally, senescent chondrocytes upregulate C5a receptor expression in response to the inflammatory microenvironment, further worsening articular cartilage degeneration, subchondral osteosclerosis, and synovial hyperplasia.178,182 While current studies primarily emphasize the pro-inflammatory effects of senescent chondrocytes, other joint-resident cells, including synovial cells, fibroblasts, and subchondral bone cells, also play a significant role in OA pathology.
Cytokine senescence
Immunosenescence is more pronounced in patients with OA compared to healthy controls, marked by increased pro-inflammatory cytokine production. Cytokine senescence refers to abnormal cytokine levels and activity, indicating the transformation of normal cells into a senescent, imbalanced state. Previous studies have highlighted the significant roles of cytokines, particularly TNF-α, IL-1, IL-6, and IL-17, in the initiation and progression of OA.183 A growing number of studies have shown elevated IL-17 and IL-18 expression in OA synovial fluid, identifying them as key cytokines in OA pathology.184,185 These studies suggest that cytokine senescence can trigger inflammatory reactions and tissue degeneration, driving the onset and progression of OA. Additionally, inflammation in the synovium, cartilage, and patellar fat pad leads to the secretion of inflammatory factors that infiltrate the articular cartilage, increasing the release of metabolic mediators.186 Evidence indicates that cytokines play crucial roles in promoting premature senescence in surrounding young cells. Nakajima et al. reported that IL-6 regulates senescence in multiple systems by forming IL-6/sIL-6Rα complexes with STAT3, inducing premature senescence in human fibroblasts.187 However, the underlying mechanisms remain unclear, and the role of cytokine senescence in OA is still debated, necessitating further research.
Complement system senescence
The complement system plays a vital role in the body’s defense mechanisms and is critical in protecting against diseases. An increasing number of studies show that complement activation products are elevated in the serum and synovial fluid of OA patients.188 Specifically, the complement system in aging synovial cells can be activated via the classical, alternative, and lectin pathways, forming a membrane attack complex (MAC) that induces synovial fluid inflammation in OA.189 Wang et al. conducted proteomic and transcriptomic analyses of synovial fluid and synovium in OA patients, finding that complement activation and MAC-mediated pathways play crucial roles in OA pathology.190
Chemokine senescence
In addition to cytokines, chemokines are also involved in the pathogenesis of OA. Numerous studies have shown that the levels of CC motif ligands 2 (CCL2), CCL3, CCL4, and CCL5 are elevated in the serum and synovial fluid of OA patients compared to those without OA and are positively correlated with disease severity.191 Tsuchida et al. reported that age-related stress contributed to OA development, partly due to the senescence of chemokines such as CCL2, CCL4, and GROα.192 These chemokines induced macrophage recruitment, inflammation, and pain. Zhao et al. demonstrated that multiple chemokines are involved in the inflammatory and catabolic processes of chondrocytes, potentially recruiting inflammatory cells such as neutrophils and monocytes to accelerate OA pathology.193 Acosta et al. demonstrated that the chemokine receptor CXCR2 (IL8RB) promotes senescence by binding to CXCR2 in a p53-dependent manner.194 In conclusion, chemokine-related senescence serves as an important mediator and functional pathway in joint senescence and OA pathology, either independently or in conjunction with other biological factors. This warrants further attention in understanding aging-induced OA.
New strategy for treating oa from the cellular senescence perspective
Various factors influence cellular senescence, and the development of anti-senescence drugs or strategies targeting the physiological mechanisms of senescence offers new therapeutic approaches for delaying senescence-related chronic diseases. Rapamycin, a key anti-senescence drug, induces autophagy to counteract cellular senescence caused by ROS upregulation due to increased inflammatory cytokines. Its mechanism involves promoting antioxidant protein expression by enhancing Nrf2/Keap1 signaling.195 A recent study demonstrated that coumarins, which induce mitochondrial autophagy, improve mitochondrial function and extend lifespan by inhibiting the activation of the nuclear hormone receptor DAF-12/FXR.196 L-glutamine, a common amino acid in human blood, inhibits NF-κB activity.197 This inhibition occurs via the upregulation of long non-coding RNA NKILA expression, regulated by the TGF-β1/SMAD2/3 pathway, leading to reduced expression of NO synthase, COX-2, and MMP-13. Oleanolic acid rescues mitochondrial ultrastructural abnormalities, scavenges free radicals, and regulates P450COX, thereby modulating mitochondrial integrity and autophagy in senescent cells. This modulation effectively prevents cardiac senescence by upregulating FUNDC-dependent mitochondrial autophagy, mediated by the E3 ligase MARCH5.198 Similarly, metformin, a widely known hypoglycemic agent, increases autophagy in T-cells and improves mitochondrial bioenergetics, restoring senescence-related inflammation to a normal state.199 Mitochondrial dysfunction in senescent cells increases harmful substances like ROS. Additionally, resveratrol and EVs from adipose stem cells counteract the adverse effects of ROS production.200,201 Resveratrol specifically reduces ROS levels, attenuates IL-1-induced SASP, and delays OA progression via the ROS/NF-κB axis in the ACLT rat model. Stem cell-derived EVs regulate senescence-related signaling pathways through functionally important miRNAs. As a hallmark of cellular senescence, SA-β-gal serves as a drug initiator, selectively releasing gemcitabine after activation of the prodrug SSK1, which removes senescent cells.202 Upon cellular senescence, cell-free mitochondrial DNA accumulates, enhancing immunogenicity. Additionally, the activated type I interferon response is crucial for maintaining SASP. Thus, anti-senescence can be achieved by targeting senescence-associated inflammation, using agents like the nucleoside reverse transcriptase inhibitor lamivudine, senolytics, or melatonin.46,203,204 Beyond conventional drugs, DNASE2A may enhance the clearance of excess extra-nuclear DNA in senescent cells by triggering autophagy, reducing innate immune response and SA-β-gal activity.41 Notably, these anti-senescence drugs may involve multiple mechanisms, many of which remain poorly understood, partly due to research limitations and potentially undiscovered pathways.
These anti-senescence drugs and strategies have shown efficacy, marking a breakthrough in the treatment of cartilage-damaging diseases like OA. While exercise and dietary strategies were discussed previously, this section focuses on recent advancements in anti-senescence pharmacological treatments for OA. First, several types of senescent cells and tissues within the joint cavity, including chondrocytes, stem cells, and synovial cells, can be targeted for drug intervention. However, drug specificity depends more on the cellular state than on tissue specificity, and cell state-specific drugs target senescent cell markers.205 Second, based on their effects, these drugs can be categorized into three types: maintaining normal cell phenotypes, improving the cell survival environment, and clearing senescent cells (Fig. 5). Specifically, maintaining normal cellular phenotypes involves enabling senescent cells to continue their biological functions; improving the cellular environment targets senescence-associated inflammation; and drugs for senescent cell elimination focus on enhancing autophagy and inducing apoptosis. Majority of studies focus on senescent chondrocytes in OA, but it is important to recognize that multiple cell types contribute to OA pathology. Therefore, it is crucial to investigate cellular senescence in other cell types, including adipose mesenchymal stem cells and synoviocytes.206,207 Recent studies on anti-senescence drugs for OA treatment are summarized in Table 2. Additionally, some techniques can also achieve anti-senescence effects. Pretreatment of MSCs from elderly OA patients with chondrogenic differentiation medium followed by normal growth medium rejuvenated senescent MSCs and significantly improved rabbit OA pathology. Moreover, therapeutic efficacy correlated with cell number.208

The therapeutic efficacy of representative anti-senescence drugs with different mechanisms of action in vitro and in vivo. a Gastrodin regulates phosphorylation of the PI3K-AKT pathway via SIRT3, reduces SA-β-gal positive staining in IL-1β-treated chondrocytes, and reverses cartilage destruction in the OA rat model.226 Permission of reuse obatained from copyright holder Elsevier. b Chondrocyte cultures supplemented with exosomes from umbilical cord MSCs significantly reduce SA-β-gal positive staining of OA chondrocytes and improve articular cartilage bulk structure.213 Permission of reuse obtained from copy right holder American Chemical Society. c Cerium dioxide nanoparticles reduce the percentage of SA-β-gal-positive cells in H2O2-treated synoviocytes and protect articular cartilage by scavenging ROS and inactivating the NF-κB signaling pathway.207 Permission of reuse obtained from copyright holder MDPI. d Multi-kinase inhibitor YKL-05-099 inhibit MAPK and NF-κB signaling activation by affecting kinome phosphorylation, reduce IL-1β-induced chondrocyte senescence, decrease the level of senescence markers p21Clp1 and p16INK4A in chondrocytes, and prevent subchondral bone loss effectively.212 Permission of reuse obtained from copyright holder The Author(s). e Small copper sulfide nanoparticles functionalized with anti-beta-2-microglobulin antibodies specifically induce apoptosis in senescent chondrocytes and prevent articular cartilage damage.233 Permission of reuse obtained from copyright holder The Author(s). f Rapamycin decreases the levels of senescence markers in H2O2-stimulated human chondrocytes and reduces joint P16INK4a positivity in the mouse OA model by upregulating autophagy.210 Permission of reuse obtained from copyright holder The Author(s)
In vitro experiments have demonstrated promising results with the use of anti-aging drugs alone. However, it is crucial to note that these drugs exhibit dose-dependent effects, potentially leading to high toxicity and damage to non-senescent cells.202,209 Therefore, the potential toxicological effects of anti-aging drugs in vivo are a major concern. To address this, it is necessary to achieve high local drug concentrations and prolonged duration of action, while ensuring biosafety. Various drug delivery systems for anti-aging therapies have been developed, including oral administration and intra-articular injections. Dhanabalan et al. developed a post-traumatic OA mouse model via medial meniscus destabilization and loaded rapamycin into polylactic acid-glycolic acid (PLGA) particles for slow drug release.210 The study demonstrated that intra-articular injections administered every 3 weeks effectively treated early OA in mice. Previous studies have addressed the issue of systemic toxicity resulting from frequent injections. Polymer particle-based drug delivery systems were shown to maintain a joint residence time of 19 days, which is critical for clinical translation.211 Wan et al. developed nanoliposome-based thermosensitive hydrogels that demonstrated promising results in reducing kinase inhibitor-induced cytotoxicity and enhancing protein kinase inhibitor performance.212 The therapeutic efficacy and retention time of exosomes were significantly enhanced using a two-phase system consisting of a chondrocyte-targeted polymer membrane and thiolated hyaluronic acid gel.213 This cell-free therapeutic strategy effectively restored senescent chondrocytes.
Conclusions and prospects
Our understanding of OA pathogenesis is continuously advancing, shifting focus from cartilage damage alone to the involvement of subchondral bone, synovium, infrapatellar fat, and other joint tissues. Recent studies have revealed a correlation between inflammation and cellular senescence, suggesting that cellular senescence plays a key role in the inflammatory response. The cross-talk between cellular senescence and inflammation offers a novel perspective on OA pathogenesis and the development of therapeutic strategies for cartilage-damaging diseases like OA. This review synthesizes insights from recent studies, shedding light on the role of cellular senescence in OA and its underlying mechanisms. While the role of cellular senescence in driving inflammatory responses is increasingly clear, further studies are needed to unravel the underlying molecular mechanisms linking senescence and inflammation. Additionally, several critical issues must be addressed before the clinical translation of these therapies can be realized:
-
(1)
Distinguishing normal, physiologically senescent, and pathologically senescent cells in joint tissues requires the development of novel techniques. This distinction is essential for understanding and targeting the harmful effects of pathological senescence.
-
(2)
Early Identification and Intervention: While much research has focused on mitigating the effects of established senescent cells, there is a growing need to identify and address abnormal senescence before pathological changes occur. Enhanced methods for precise identification and early intervention are crucial.
-
(3)
Numerous studies have used surgery or intra-articular drug injections to establish animal models of OA. However, it is crucial to recognize that these models do not fully replicate the natural pathological progression of OA associated with aging. Therefore, future research should prioritize investigating anti-cellular senescence therapies in aging models or in elderly OA patients.
-
(4)
The pharmacokinetics of most drugs aimed at rescuing or eliminating senescent cells in the joint cavity remain unclear. Additional high-quality in vivo data are required for further exploration.
References
Newgard, C. B. & Sharpless, N. E. Coming of age: molecular drivers of aging and therapeutic opportunities. J. Clin. Investig. 123, 946–950 (2013).
Tsuji, T., Aoshiba, K. & Nagai, A. Alveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary disease. Respiration Int. Rev. Thorac. Dis. 80, 59–70 (2010).
Cavanagh, M. M., Weyand, C. M. & Goronzy, J. J. Chronic inflammation and aging: DNA damage tips the balance. Curr. Opin. Immunol. 24, 488–493 (2012).
Del Pinto, R. & Ferri, C. J. I. Inflammation-accelerated senescence and the cardiovascular system: mechanisms and perspectives. Int. J. Mol. Sci. 19, 3701 (2018).
Ariffin, H. et al. Young adult survivors of childhood acute lymphoblastic leukemia show evidence of chronic inflammation and cellular aging. Cancer 123, 4207–4214 (2017).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961). J. E. c. r.
Childs, B. G., Durik, M., Baker, D. J. & Van Deursen, J. M. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat. Med. 21, 1424–1435 (2015).
Muñoz-Espín, D. et al. Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 (2013).
Bektas, A., Schurman, S. H., Sen, R. & Ferrucci, L. J. Aging, inflammation and the environment. Exp. Gerontol. 105, 10–18 (2018).
Aoshiba, K. & Nagai, A. J. Chronic lung inflammation in aging mice. FEBS Lett. 581, 3512–3516 (2007).
Lumeng, C. N. et al. Aging is associated with an increase in T cells and inflammatory macrophages in visceral adipose tissue. J. Immunol. 187, 6208–6216 (2011).
Sarkar, D. & Fisher, P. B. Molecular mechanisms of aging-associated inflammation. Cancer Lett. 236, 13–23 (2006).
Hy, C. et al. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Res. Rev. 8, 18–30 (2009).
Franceschi, C. et al. Inflamm‐aging: an evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 908, 244–254 (2000).
Sellam, J. & Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 6, 625–635 (2010).
Ponchel, F. et al. Changes in peripheral blood immune cell composition in osteoarthritis. Osteoarthr. Cartil. 23, 1870–1878 (2015).
Zhang, W., Ouyang, H., Dass, C. R. & Xu, Jr. Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res. 4, 15040 (2016).
Roos, E. M. & Arden, N. K. Strategies for the prevention of knee osteoarthritis. Nat. Rev. Rheumatol. 12, 92–101 (2016).
Xiong, W. et al. In situ remodeling of efferocytosis via lesion‐localized microspheres to reverse cartilage senescence. Adv. Sci. 11, 2400345 (2024).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. The hallmarks of aging. Cell 153, 1194–1217 (2013).
Krishnamurthy, J. et al. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig. 114, 1299–1307 (2004).
Scudellari, M. To stay young, kill zombies. Nature 550, 448–450 (2017).
Kuilman, T., Michaloglou, C., Mooi, W. J., Peeper, D. S. J. G. & development. The essence of senescence. Genes Dev. 24, 2463–2479 (2010).
Hou, Y. et al. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 15, 565–581 (2019).
Zhu, X. et al. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: the regulation and intervention. Signal Transduct. Target. Ther. 6, 245 (2021).
López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M. & Kroemer, G. J. C. Hallmarks of aging: an expanding universe. Cell 186, 243–278 (2023).
Krtolica, A., Parrinello, S., Lockett, S., Desprez, P.-Y. & Campisi, J. S. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc. Natl. Acad. Sci. USA 98, 12072–12077 (2001).
Herranz, N. & Gil, J. J. Mechanisms and functions of cellular senescence. J. Clin. Investig. 128, 1238–1246 (2018).
Greene, M. A. & Loeser, R. F. Aging-related inflammation in osteoarthritis. Osteoarthr. Cartil. 23, 1966–1971 (2015).
Barcena, M. L. et al. Sex and age differences in AMPK phosphorylation, mitochondrial homeostasis, and inflammation in hearts from inflammatory cardiomyopathy patients. Aging Cell 22, 13894 (2023). e13894.
Gardner, S. E., Humphry, M., Bennett, M. R., & Clarke, M. C. Senescent vascular smooth muscle cells drive inflammation through an interleukin-1α–dependent senescence-associated secretory phenotype. Arteriosclerosis Thrombosis Vasc. Biol. 35, 1963–1974 (2015).
Freund, A., Patil, C. K. & Campisi, J. J. p38MAPK is a novel DNA damage response‐independent regulator of the senescence‐associated secretory phenotype. EMBO J. 30, 1536–1548 (2011).
Loeser, R. F., Collins, J. A. & Diekman, B. O. Ageing and the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 12, 412–420 (2016).
Freund, A., Orjalo, A. V., Desprez, P.-Y. & Campisi, J. J. Inflammatory networks during cellular senescence: causes and consequences. Trends Mol. Med. 16, 238–246 (2010).
Sturmlechner, I. et al. p21 produces a bioactive secretome that places stressed cells under immunosurveillance. Science 374, eabb3420 (2021).
Sun, Y., Coppé, J.-P. & Lam, E. W.-F. Cellular senescence: the sought or the unwanted? Trends Mol. Med. 24, 871–885 (2018).
Kang, C. et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 349, aaa5612 (2015).
Vizioli, M. G. et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev. 34, 428–445 (2020).
Fulop, T. et al. Immunosenescence and inflamm-aging as two sides of the same coin: friends or foes? Front. Immunol. 8, 1960 (2017).
Zhao, B. et al. Topoisomerase 1 cleavage complex enables pattern recognition and inflammation during senescence. Nat. Commun. 11, 908 (2020).
Lan, Y. Y. et al. Extranuclear DNA accumulates in aged cells and contributes to senescence and inflammation. Aging Cell 18, 12901 (2019).
Victorelli, S. et al. Apoptotic stress causes mtDNA release during senescence and drives the. Nature 622, 627–636 (2023). SASP.
Yasuda, T. et al. Inflammation-driven senescence-associated secretory phenotype in cancer-associated fibroblasts enhances peritoneal dissemination. Cell Rep. 34, 108779 (2021).
Ong, S.-M. et al. The pro-inflammatory phenotype of the human non-classical monocyte subset is attributed to senescence. Cell Death Dis. 9, 266 (2018).
Arai, Y. et al. Inflammation, but not telomere length, predicts successful ageing at extreme old age: a longitudinal study of semi-supercentenarians. EBioMedicine 2, 1549–1558 (2015).
Iske, J. et al. Senolytics prevent mt-DNA-induced inflammation and promote the survival of aged organs following transplantation. Nat. Commun. 11, 4289 (2020).
Kling, K. M., Lopez-Rodriguez, E., Pfarrer, C., Mühlfeld, C. & Brandenberger, C. Aging exacerbates acute lung injury-induced changes of the air-blood barrier, lung function, and inflammation in the mouse. Am. J. Physiol. Lung Cell. Mol. Physiol. 312, 1 (2017).
Storci, G. et al. Genomic stability, anti-inflammatory phenotype, and up-regulation of the RNAseH2 in cells from centenarians. Cell Death Differ. 26, 1845–1858 (2019).
Huang, Y. et al. Longevity-associated transcription factor ATF7 promotes healthspan by suppressing cellular senescence and systematic inflammation. Aging Dis. 14, 1374–1389 (2023).
Glück, S. et al. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 19, 1061–1070 (2017).
Busse, P. J. & Mathur, S. K. Age-related changes in immune function: effect on airway inflammation. J. Allergy Clin. Immunol. 126, 690–699 (2010).
Ribeiro, A., et al. Uremic toxin indoxyl sulfate promotes macrophage-associated low-grade inflammation and epithelial cell senescence. Int. J. Mol. Sci. 24, 8031 (2023).
Lavandoski, P., et al. Eotaxin-1/CCL11 promotes cellular senescence in human-derived fibroblasts through pro-oxidant and pro-inflammatory pathways. Front. Immunol. 14, 1243537, (2023).
Mitchell, C. A. et al. Stromal niche inflammation mediated by IL-1 signalling is a targetable driver of haematopoietic ageing. Nat. Cell Biol. 25, 30–41 (2023).
Ortiz-Montero, P., Londoño-Vallejo, A. & Vernot, J.-P. Senescence-associated IL-6 and IL-8 cytokines induce a self-and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Communi. Signal. 15, 17 (2017).
Ortiz-Montero, P., Londoño-Vallejo, A., & Vernot, J.-P. Senescence-associated IL-6 and IL-8 cytokines induce a self-and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun. Signal. 15, 1–18 (2017).
Rodier, F. et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 11, 973–979 (2009).
Yagi, M., Endo, K., Komori, K. & Sekiya, I. Comparison of the effects of oxidative and inflammatory stresses on rat chondrocyte senescence. Sci. Rep. 13, 7697 (2023).
Ponnappan, S. & Ponnappan, U. Aging and immune function: molecular mechanisms to interventions. Antioxid. Redox Signal. 14, 1551–1585 (2011).
Jurk, D. et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2, 4172 (2014).
Wu, D. et al. Aging up-regulates expression of inflammatory mediators in mouse adipose tissue. J. Immunol. 179, 4829–4839 (2007).
Lin, L. et al. Ghrelin receptor regulates adipose tissue inflammation in aging. Aging 8, 178–191 (2016).
Mazurek, T. et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation 108, 2460–2466 (2003).
Karakasilioti, I. et al. DNA damage triggers a chronic autoinflammatory response, leading to fat depletion in NER progeria. Cell Metab. 18, 403–415 (2013).
Aulinas, A. et al. Dyslipidemia and chronic inflammation markers are correlated with telomere length shortening in Cushing’s syndrome. PLoS One 10, 0120185 (2015).
Frasca, D. & Blomberg, B. B. Adipose tissue inflammation induces B cell inflammation and decreases B cell function in aging. Front. Immunol. 8, 274580 (2017).
Shirakawa, K. et al. Obesity accelerates T cell senescence in murine visceral adipose tissue. J. Clin. Investig. 126, 4626–4639 (2016).
Matacchione, G. et al. Senescent macrophages in the human adipose tissue as a source of inflammaging. Geroscience 44, 1941–1960 (2022).
Petkevicius, K. et al. Accelerated phosphatidylcholine turnover in macrophages promotes adipose tissue inflammation in obesity. Elife 8, e47990 (2019).
Meng, F. et al. JAZF1 inhibits adipose tissue macrophages and adipose tissue inflammation in diet-induced diabetic mice. BioMed. Res. Int. 2018, 4507659 (2018).
Strong, R. et al. Nordihydroguaiaretic acid and aspirin increase lifespan of genetically heterogeneous male mice. Aging Cell 7, 641–650 (2008).
Kirkland, J. L. & Tchkonia, T. Cellular senescence: a translational perspective. EBioMedicine 21, 21–28 (2017).
Schroer, A. B. et al. Platelet factors attenuate inflammation and rescue cognition in ageing. Nature 620, 1071–1079 (2023).
Zhang, Z., et al. Increased hyaluronan by naked mole-rat HAS2 extends lifespan in mice. Nature 621, 196–205 (2023).
Lin, J.-Y. et al. Swimming exercise stimulates IGF1/PI3K/Akt and AMPK/SIRT1/PGC1α survival signaling to suppress apoptosis and inflammation in aging hippocampus. Aging 12, 6852–6864 (2020).
García-Calzón, S. et al. Dietary inflammatory index and telomere length in subjects with a high cardiovascular disease risk from the PREDIMED-NAVARRA study: cross-sectional and longitudinal analyses over 5 y. Am. J. Clin. Nutr. 102, 897–904 (2015).
Shivappa, N. et al. Association of proinflammatory diet with low-grade inflammation: results from the Moli-sani study. Nutrition 54, 182–188 (2018).
Medzhitov, R. Origin and physiological roles of inflammation. Nature 454, 428–435 (2008).
Gudkov, A. V. & Komarova, E. A. p53 and the carcinogenicity of chronic inflammation. Cold Spring Harb. Perspect. Med. 6, a026161 (2016).
Zhao, H. et al. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 6, 263 (2021).
Kong, P. et al. Inflammation and atherosclerosis: signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 7, 131 (2022).
Chen, L. et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9, 7204–7218 (2018).
Butterfield, T. A., Best, T. M. & Merrick, M. A. The dual roles of neutrophils and macrophages in inflammation: a critical balance between tissue damage and repair. J. Athl. Train. 41, 457–465 (2006).
Li, X. L., Zhao, C. L., Dong, Q. & Sun, L. R. Subchondral bone in osteoarthritis: insight into risk factors and microstructural changes. Int. Immunopharmacol. 15, 1–12 (2013).
Belluzzi, E. et al. Contribution of infrapatellar fat pad and synovial membrane to knee osteoarthritis pain. BioMed. Res. Int. 2019, 6390182 (2019).
Hsueh, M. F., Zhang, X., Wellman, S. S., Bolognesi, M. P. & Kraus, V. B. Synergistic roles of macrophages and neutrophils in osteoarthritis progression. Arthritis Rheumatol. 73, 89–99 (2021).
Griffin, T. M., Scanzello, C. R. J. C. & rheumatology, e. Innate inflammation and synovial macrophages in osteoarthritis pathophysiology. Clin. Exp. Rheumatol. 37, 57–63 (2019).
Rai, V., Dilisio, M. F., Samadi, F. & Agrawal, D. K. Counteractive effects of IL-33 and IL-37 on inflammation in osteoarthritis. Int. J. Environ. Res. Public Health 19, 5690 (2022).
Millerand, M., Berenbaum, F. & Jacques, C. Danger signals and inflammaging in osteoarthritis. Clin. Exp. Rheumatol. 37(Suppl 120), 48–56 (2019).
Yang, S., Wang, J., Brand, D. D. & Zheng, S. G. J. F. I. I. Role of TNF–TNF receptor 2 signal in regulatory T cells and its therapeutic implications. Front. Immunol. 9, 784 (2018).
Wicovsky, A. et al. Tumor necrosis factor receptor-associated factor-1 enhances proinflammatory TNF receptor-2 signaling and modifies TNFR1–TNFR2 cooperation. Oncogene 28, 1769–1781 (2009).
Rauert, H. et al. Membrane tumor necrosis factor (TNF) induces p100 processing via TNF receptor-2 (TNFR2). J. Biol. Chem. 285, 7394–7404 (2010).
Cheng, A. W. M., Stabler, T. V., Bolognesi, M. & Kraus, V. B. Selenomethionine inhibits IL-1β inducible nitric oxide synthase (iNOS) and cyclooxygenase 2 (COX2) expression in primary human chondrocytes. Osteoarthr. Cartil. 19, 118–125 (2011).
Ya Sklyarov, A., Panasyuk, N., & Fomenko, I. S. Role of nitric oxide-synthase and cyclooxygenase/lipooxygenase systems in development of experimental ulcerative colitis. J. Physiol. Pharmacol. 62, 65 (2011).
ur Rashid, H., Xu, Y., Ahmad, N., Muhammad, Y. & Wang, L. Promising anti-inflammatory effects of chalcones via inhibition of cyclooxygenase, prostaglandin E2, inducible NO synthase and nuclear factor κb activities. Bioorg. Chem. 87, 335–365 (2019).
Catheline, S. E. et al. IKKβ–NF-κB signaling in adult chondrocytes promotes the onset of age-related osteoarthritis in mice. Sci. Signal. 14, 3535 (2021).
Arra, M. et al. LDHA-mediated ROS generation in chondrocytes is a potential therapeutic target for osteoarthritis. Nat. Commun. 11, 3427 (2020).
Xie, Z. et al. Taraxasterol inhibits inflammation in osteoarthritis rat model by regulating miRNAs and NF-κB signaling pathway. Acta Biochimica Polonica 69, 811–818 (2022).
Tang, L., Sim, I., Moqbel, S., Wu, L. J. H. & Toxicology, E. Dapansutrile ameliorated chondrocyte inflammation and osteoarthritis through suppression of MAPK signaling pathway. Hum. Exp. Toxicol. 41, 09603271221145401 (2022).
Yao, M. et al. Cepharanthine ameliorates chondrocytic inflammation and osteoarthritis via regulating the MAPK/NF-κB-Autophagy pathway. Front. Pharmacol. 13, 854239 (2022).
Calabrese, G. et al. Phytochemical analysis and anti-inflammatory and anti-osteoarthritic bioactive potential of Verbascum thapsus L.(Scrophulariaceae) leaf extract evaluated in two in vitro models of inflammation and osteoarthritis. Molecules 26, 5392 (2021).
Moqbel, S. et al. Rat chondrocyte inflammation and osteoarthritis are ameliorated by madecassoside. Oxid. Med. Cell. Longev. 2020, 7540197 (2020).
Jia, Y. et al. Morusin ameliorates IL-1β-induced chondrocyte inflammation and osteoarthritis via NF-κB signal pathway. Drug Des. Dev. Ther. 14, 1227–1240 (2020).
Jia, Y. et al. Garcinol suppresses IL-1β-induced chondrocyte inflammation and osteoarthritis via inhibition of the NF-κB signaling pathway. Inflammation 42, 1754–1766 (2019).
Ran, J. et al. Schisandrin B ameliorated chondrocytes inflammation and osteoarthritis via suppression of NF-κB and MAPK signal pathways. Drug Des. Dev. Ther. 12, 1195–1204 (2018).
Takeuchi, K. et al. Downregulation of aquaporin 9 decreases catabolic factor expression through nuclear factor‑κB signaling in chondrocytes. Int. J. Mol. Med. 42, 1548–1558 (2018).
Xie, W. et al. Achyranthoside D attenuates chondrocyte loss and inflammation in osteoarthritis via targeted regulation of Wnt3a. Phytomed. Int. J. Phytother. Phytopharmacol. 111, 154663 (2023).
Wang, W., Chen, Z. & Hua, Y. J. B. Bioinformatics prediction and experimental validation identify a novel cuproptosis-related gene signature in human synovial inflammation during osteoarthritis progression. Biomolecules 13, 127 (2023).
Delmonico M. J. et al. Longitudinal study of muscle strength, quality, and adipose tissue infiltration. Am. J. Clin. Nutr. 90, 1579–1585 (2009).
Coppé, J.-P., Desprez, P.-Y., Krtolica, A. & Campisi, J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. 5, 99–118 (2010).
Izquierdo, M. C. et al. Klotho, phosphate and inflammation/ageing in chronic kidney disease. Nephrol. Dial. Transplant. 27, 6–10 (2012).
Zeng, Y., Wang, P.-H., Zhang, M., & Du, J.-R.. Aging-related renal injury and inflammation are associated with downregulation of Klotho and induction of RIG-I/NF-κB signaling pathway in senescence-accelerated mice. Aging Clin. Exp. Res. 28, 69–76 (2016).
Bailey-Downs, L. C. et al. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: a paracrine mechanism contributing to vascular redox dysregulation and inflammation. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 68, 780–792 (2013).
Ogrodnik, M. et al. Whole‐body senescent cell clearance alleviates age‐related brain inflammation and cognitive impairment in mice. Aging Cell 20, 13296 (2021).
Laberge, R.-M. et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 17, 1049–1061 (2015).
Passos, J. F. et al. Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 6, 347 (2010).
Herranz, N. et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 17, 1205–1217 (2015).
Attur, M. et al. Low‐grade inflammation in symptomatic knee osteoarthritis: prognostic value of inflammatory plasma lipids and peripheral blood leukocyte biomarkers. Arthritis Rheumatol. 67, 2905–2915 (2015).
Busse, B. et al. Decrease in the osteocyte lacunar density accompanied by hypermineralized lacunar occlusion reveals failure and delay of remodeling in aged human bone. Aging Cell 9, 1065–1075 (2010).
Coryell, P. R., Diekman, B. O. & Loeser, R. F. Mechanisms and therapeutic implications of cellular senescence in osteoarthritis. Nat. Rev. Rheumatol. 17, 47–57 (2021).
Wang, K. et al. Chondrogenic progenitor cells exhibit superiority over mesenchymal stem cells and chondrocytes in platelet-rich plasma scaffold-based cartilage regeneration. Am. J. Sports Med. 47, 2200–2215, (2019).
Lotz, M. K. et al. Cartilage cell clusters. Arthritis Rheumatism 62, 2206–2218, (2010).
Arra, M., Swarnkar, G., Alippe, Y., Mbalaviele, G. & Abu-Amer, Y. IκB-ζ signaling promotes chondrocyte inflammatory phenotype, senescence, and erosive joint pathology. Bone Res. 10, 12 (2022).
Coryell, P. R. et al. Autophagy regulates the localization and degradation of p16(INK4a). Aging Cell 19, e13171 (2020).
Martin, J. A. & Buckwalter, J. A. Telomere erosion and senescence in human articular cartilage chondrocytes. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 56, B172–179, (2001).
Loeser, R. F. Aging and osteoarthritis: the role of chondrocyte senescence and aging changes in the cartilage matrix. Osteoarthr. Cartil. 17, 971–979, (2009).
Engeland, K. Cell cycle arrest through indirect transcriptional repression by p53: I have a DREAM. Cell Death Differ. 25, 114–132, (2018).
Hernandez-Segura, A., Nehme, J. & Demaria, M. Hallmarks of cellular senescence. Trends Cell Biol. 28, 436–453, (2018).
He, S. & Sharpless, N. E. Senescence in health and disease. Cell 169, 1000–1011, (2017).
Diekman, B. O. et al. Expression of p16(INK)(4a) is a biomarker of chondrocyte aging but does not cause osteoarthritis. Aging Cell 17, e12771, (2018).
Ji, M. L.-l et al. Sirt6 attenuates chondrocyte senescence and osteoarthritis progression. Nat. Commun. 13, 7658 (2022).
Varela-Eirín, M. et al. Targeting of chondrocyte plasticity via connexin43 modulation attenuates cellular senescence and fosters a pro-regenerative environment in osteoarthritis. Cell Death Dis. 9, 1166 (2018).
Horváth, E., Sólyom, Á., Székely, J., Nagy, E. E. & Popoviciu, H. Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis. Int. J. Mol. Sci. 24, 16468 (2023).
Birch, H. L. Extracellular matrix and ageing. Subcell. Biochem. 90, 169–190, (2018).
Sun, K., Jing, X., Guo, J., Yao, X. & Guo, F. Mitophagy in degenerative joint diseases. Autophagy 17, 2082–2092, (2021).
Guo, Q. et al. STING promotes senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the NF-κB signaling pathway. Cell Death Dis. 12, 13, (2021).
Lu, H. et al. Fibroblast growth factor 21 (FGF21) alleviates senescence, apoptosis, and extracellular matrix degradation in osteoarthritis via the SIRT1-mTOR signaling pathway. Cell Death Dis. 12, 865, (2021).
Roh, K. et al. Lysosomal control of senescence and inflammation through cholesterol partitioning. Nat. Metab. 5, 398–413 (2023).
Arra, M. & Abu-Amer, Y. Cross-talk of inflammation and chondrocyte intracellular metabolism in osteoarthritis. Osteoarthr. Cartil. 31, 1012–1021 (2023).
Baker, D. J., Alimirah, F., van Deursen, J. M., Campisi, J. & Hildesheim, J. Oncogenic senescence: a multi-functional perspective. Oncotarget 8, 27661–27672, (2017).
Del Rey, M. J. et al. Senescent synovial fibroblasts accumulate prematurely in rheumatoid arthritis tissues and display an enhanced inflammatory phenotype. Immun. Ageing 16, 29, (2019).
Jeon, O. H. et al. Senescence cell-associated extracellular vesicles serve as osteoarthritis disease and therapeutic markers. JCI Insight 4, e125019 (2019).
Zhu, Y. et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14, 644–658, (2015).
Xu, M. et al. Transplanted senescent cells induce an osteoarthritis-like condition in mice. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 72, 780–785 (2017).
Liu, X. et al. Oxylipin-PPARγ-initiated adipocyte senescence propagates secondary senescence in the bone marrow. Cell Metab. 35, 667–684.e6 (2023).
Nelson, G. et al. A senescent cell bystander effect: senescence-induced senescence. Aging Cell 11, 345–349 (2012).
Waters, D. W. et al. A senescence bystander effect in human lung fibroblasts. Biomedicines 9, 1162 (2021).
Rizzo, M. G. et al. Therapeutic perspectives for inflammation and senescence in osteoarthritis using mesenchymal stem cells, mesenchymal stem cell-derived extracellular vesicles and senolytic agents. Cells 12, 1421 (2023).
Harting, M. T. et al. Inflammation-stimulated mesenchymal stromal cell-derived extracellular vesicles attenuate inflammation. Stem Cells 36, 79–90 (2018).
Bačenková, D. et al. Interaction between mesenchymal stem cells and the immune system in rheumatoid arthritis. Pharmaceuticals 15, 941 (2022).
Cavallo, C. et al. Small extracellular vesicles from adipose derived stromal cells significantly attenuate in vitro the NF-κB dependent inflammatory/catabolic environment of osteoarthritis. Sci. Rep. 11, 1053 (2021).
Yang, Y. et al. Secretive derived from hypoxia preconditioned mesenchymal stem cells promote cartilage regeneration and mitigate joint inflammation via extracellular vesicles. Bioact. Mater. 27, 98–112 (2023).
Yamanaka, S. Pluripotent stem cell-based cell therapy-promise and challenges. Cell Stem Cell 27, 523–531, (2020).
Wei, P. & Bao, R. Intra-articular mesenchymal stem cell injection for knee osteoarthritis: mechanisms and clinical evidence. Int. J. Mol. Sci. 24, 59, (2022).
Jo, C. H. et al. Intra-articular injection of mesenchymal stem cells for the treatment of osteoarthritis of the knee: a proof-of-concept clinical trial. Stem Cells 32, 1254–1266, (2014).
Ye, G. et al. ALKBH5 facilitates CYP1B1 mRNA degradation via m6A demethylation to alleviate MSC senescence and osteoarthritis progression. Exp. Mol. Med. 55, 1743–1756, (2023).
Čamernik, K. et al. Increased exhaustion of the subchondral bone-derived mesenchymal stem/ stromal cells in primary versus dysplastic osteoarthritis. Stem Cell Rev. Rep. 16, 742–754, (2020).
Cao, X. et al. Intraarticular senescent chondrocytes impair the cartilage regeneration capacity of mesenchymal stem cells. Stem Cell Res. Ther. 10, 86, (2019).
Weng, Z. et al. Mesenchymal stem/stromal cell senescence: hallmarks, mechanisms, and combating strategies. Stem Cells Transl. Med. 11, 356–371, (2022).
Zhu, J. et al. Stem cell-homing hydrogel-based miR-29b-5p delivery promotes cartilage regeneration by suppressing senescence in an osteoarthritis rat model. Sci. Adv. 8, eabk0011, (2022).
Zhang, J., Rong, Y., Luo, C. & Cui, W. Bone marrow mesenchymal stem cell-derived exosomes prevent osteoarthritis by regulating synovial macrophage polarization. Aging 12, 25138–25152, (2020).
Xie, J. et al. Cellular senescence in knee osteoarthritis: molecular mechanisms and therapeutic implications. Ageing Res. Rev. 70, 101413, (2021).
Loeser, R. F., Goldring, S. R., Scanzello, C. R., Goldring, M. B. & rheumatism. Osteoarthritis: a disease of the joint as an organ. Arthritis Rheumatism 64, 1697–1707 (2012).
Dragoo, J. L., Johnson, C. & McConnell, J. M. Evaluation and treatment of disorders of the infrapatellar fat pad. Sports Med. 42, 51–67 (2012).
Favero, M. et al. Infrapatellar fat pad features in osteoarthritis: a histopathological and molecular study. Rheumatology 56, 1784–1793 (2017).
Ioan-Facsinay, A., Kloppenburg, M. An emerging player in knee osteoarthritis: the infrapatellar fat pad. Arthritis. Res. Ther. 15, 1–9 (2013).
Reyes-Farias, M., Fos-Domenech, J., Serra, D., Herrero, L. & Sánchez-Infantes, D. P. White adipose tissue dysfunction in obesity and aging. Biochemical Pharmacol. 192, 114723 (2021).
Xie, C. & Chen, Q. J. C. r. r. Adipokines: new therapeutic target for osteoarthritis? Curr. Rheumatol. Rep. 21, 71 (2019).
Belluzzi, E. et al. Systemic and local adipose tissue in knee osteoarthritis. J. Cell. Physiol. 232, 1971–1978 (2017).
Zeng, N., Yan, Z.-P., Chen, X.-Y. & Ni, G. X. Infrapatellar fat pad and knee osteoarthritis. Aging Dis. 11, 1317–1328 (2020). & disease.
Eymard, F. & Chevalier, X. Inflammation of the infrapatellar fat pad. Jt. Bone Spine 83, 389–393 (2016).
Liu, Z. et al. Immunosenescence: molecular mechanisms and diseases. Signal Transduct. Target. Ther. 8, 200 (2023).
Brubaker, S. W., Bonham, K. S., Zanoni, I. & Kagan, J. C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 33, 257–290 (2015).
Lian, J., Yue, Y., Yu, W. & Zhang, Y. Immunosenescence: a key player in cancer development. J. Hematol. Oncol. 13, 151 (2020).
Shive, C. & Pandiyan, P. Inflammation, immune senescence, and dysregulated immune regulation in the elderly. Front. Aging 3, 840827 (2022).
Motta, F., Barone, E., Sica, A. & Selmi, C. J. Inflammaging and osteoarthritis. Clin. Rev. Allergy Immunol. 64, 222–238 (2023).
Huang, K., Cai, H.-l, Bao, J.-p & Wu, L. D. Dehydroepiandrosterone and age-related musculoskeletal diseases: Connections and therapeutic implications. Ageing Res. Rev. 62, 101132 (2020).
Mobasheri, A., Matta, C., Zákány, R. & Musumeci, G. Chondrosenescence: definition, hallmarks and potential role in the pathogenesis of osteoarthritis. Maturitas 80, 237–244 (2015).
Bondeson, J. et al. The role of synovial macrophages and macrophage-produced mediators in driving inflammatory and destructive responses in osteoarthritis (INTECH Open Access Publisher, 2012).
Bondeson, J. et al. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis. Res. Ther. 8, 1–12 (2006).
Bradley, K. et al. Synthesis of classical pathway complement components by chondrocytes. Immunology 88, 648–656 (1996).
Onuma, H. et al. Expression of the anaphylatoxin receptor C5aR (CD88) by human articular chondrocytes. Rheumatol. Int. 22, 52–55 (2002).
Van den Bosch, M., van Lent, P., Van der Kraan, P. J. O. & Cartilage. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthr. Cartil. 28, 532–543 (2020).
Melchiorri, C. et al. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis. Rheumatism 41, 2165–2174 (1998).
Scanzello, C. R. et al. Local cytokine profiles in knee osteoarthritis: elevated synovial fluid interleukin-15 differentiates early from end-stage disease. Osteoarthr. Cartil. 17, 1040–1048 (2009).
Jotanovic, Z., Mihelic, R., Sestan, B. & Dembic, Z. Emerging pathways and promising agents with possible disease modifying effect in osteoarthritis treatment. Curr. Drug Targets 15, 635–661 (2014).
Kojima, H., Inoue, T., Kunimoto, H. & Nakajima, K. IL-6-STAT3 signaling and premature senescence. JAK-STAT 2, e25763 (2013).
Struglics, A. et al. The complement system is activated in synovial fluid from subjects with knee injury and from patients with osteoarthritis. Arthritis. Res. Ther. 18, 223 (2016).
Corvetta, A. et al. Terminal complement complex in synovial tissue from patients affected by rheumatoid arthritis, osteoarthritis and acute joint trauma. Clin. Exp. Rheumatol. 10, 433–438 (1992).
Wang, Q. et al. Identification of a central role for complement in osteoarthritis. Nat. Med. 17, 1674–1679 (2011).
Beekhuizen, M. et al. An explorative study comparing levels of soluble mediators in control and osteoarthritic synovial fluid. Osteoarthr. Cartil. 21, 918–922 (2013).
Tsuchida, A. I. et al. Cytokine profiles in the joint depend on pathology, but are different between synovial fluid, cartilage tissue and cultured chondrocytes. Arthritis Res. Ther. 16, 441 (2014).
Zhao, X. Y. et al. CCL3 serves as a potential plasma biomarker in knee degeneration (osteoarthritis). Osteoarthr. Cartil. 23, 1405–1411 (2015).
Acosta, J. C. et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133, 1006–1018 (2008).
Zuo, R. et al. Rapamycin induced autophagy inhibits inflammation-mediated endplate degeneration by enhancing Nrf2/Keap1 signaling of cartilage endplate stem cells. Stem Cells 37, 828–840 (2019).
Chamoli, M. et al. A drug-like molecule engages nuclear hormone receptor DAF-12/FXR to regulate mitophagy and extend lifespan. Nat Aging 3, 1529–1543 (2023).
Ma, X. et al. L-Glutamine alleviates osteoarthritis by regulating lncRNA-NKILA expression through the TGF-β1/SMAD2/3 signalling pathway. Clin. Sci. 136, 1053–1069 (2022).
Gong, Y. et al. Pentacyclic triterpene oleanolic acid protects against cardiac aging through regulation of mitophagy and mitochondrial integrity. Biochim. Biophys. Acta Mol. Basis Dis. 1868, 166402 (2022).
Bharath, L. P. et al. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 32, 44–55 (2020).
Chen, X. et al. Alcohol induces cellular senescence and impairs osteogenic potential in bone marrow-derived mesenchymal stem cells. Alcohol Alcohol. 52, 289–297 (2017).
Xu, P. et al. Extracellular vesicles from adipose-derived stem cells ameliorate ultraviolet B-induced skin photoaging by attenuating reactive oxygen species production and inflammation. Stem Cell Res. Ther. 11, 1–14 (2020).
Cai, Y. et al. Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res. 30, 574–589 (2020).
De Cecco, M. et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78 (2019).
Rodríguez, M. I. et al. Chronic melatonin treatment reduces the age‐dependent inflammatory process in senescence‐accelerated mice. J. Pineal Res. 42, 272–279 (2007).
Yang, H. et al. Navitoclax (ABT263) reduces inflammation and promotes chondrogenic phenotype by clearing senescent osteoarthritic chondrocytes in osteoarthritis. Aging 12, 12750–12770 (2020).
Li, Z. et al. Metformin ameliorates senescence of adipose-derived mesenchymal stem cells and attenuates osteoarthritis progression via the ampk-dependent autophagy pathway. Oxid. Med. Cell Longev. 2022, 4620254 (2022).
Ren, X., Zhuang, H., Jiang, F., Zhang, Y. & Zhou Ceria Nanoparticles alleviated osteoarthritis through attenuating senescence and senescence-associated secretory phenotype in synoviocytes. Int. J. Mol. Sci. 24, 5056 (2023).
Zong, Z. et al. Rejuvenated ageing mesenchymal stem cells by stepwise preconditioning ameliorates surgery-induced osteoarthritis in rabbits. Bone Jt. Res. 10, 10–21 (2021).
Schafer, M. J. et al. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun. 8, 14532 (2017).
Dhanabalan, K. M. et al. Intra‐articular injection of rapamycin microparticles prevent senescence and effectively treat osteoarthritis. Bioeng. Transl. Med. 8, 10298 (2023).
Dhanabalan, K. M., Gupta, V. K. & Agarwal, R. Rapamycin–PLGA microparticles prevent senescence, sustain cartilage matrix production under stress and exhibit prolonged retention in mouse joints. Biomater. Sci. 8, 4308–4321 (2020).
Wan, J. et al. Novel strategy of senescence elimination via toxicity-exempted kinome perturbations by nanoliposome-based thermosensitive hydrogel for osteoarthritis therapy. Adv. Composites Hybrid Mater. 6, 104 (2023).
Cao, H. et al. Cell-free osteoarthritis treatment with sustained-release of chondrocyte-targeting exosomes from umbilical cord-derived mesenchymal stem cells to rejuvenate aging chondrocytes. ACS Nano 17, 13358–13376 (2023).
Zhang, R., Chen, W. & Adams, P. D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 27, 2343–2358 (2007).
Contrepois, K. et al. Histone variant H2A. J accumulates in senescent cells and promotes inflammatory gene expression. Nat. Commun. 8, 14995 (2017).
Salama, R., Sadaie, M., Hoare, M. & Narita, M. Cellular senescence and its effector programs. Genes Dev. 28, 99–114 (2014).
Davalos, A. R. et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J. Cell Biol. 201, 613–629 (2013).
Olivieri, F. et al. MiR-146a as marker of senescence-associated pro-inflammatory status in cells involved in vascular remodelling. Age 35, 1157–1172 (2013).
Bhaumik, D. et al. MicroRNAs miR-146a/b negatively modulate the senescence-associated inflammatory mediators IL-6 and IL-8. Aging 1, 402–411 (2009).
Collins, C. J. & Sedivy, J. M. J. Involvement of the INK4a/Arf gene locus in senescence. Aging Cell 2, 145–150 (2003). A. c.
Simboeck, E. & Di Croce, L. p16INK4a in cellular senescence. Aging 5, 590–591 (2013).
Wan, M., Gray-Gaillard, E. F. & Elisseeff, J. H. Cellular senescence in musculoskeletal homeostasis, diseases, and regeneration. Bone Res. 9, 41 (2021).
Collado, M. & Serrano, M. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer 10, 51–57 (2010).
Lei, Q. et al. Microvesicles as potential biomarkers for the identification of senescence in human mesenchymal stem cells. Theranostics 7, 2673–2689 (2017).
Xia, G. et al. β-Hydroxybutyrate alleviates cartilage senescence through hnRNP A1-mediated up-regulation of PTEN. Exp. Gerontol. 175, 112140 (2023).
Zhang, Y. et al. Gastrodin alleviates rat chondrocyte senescence and mitochondrial dysfunction through Sirt3. Int. Immunopharmacol. 118, 110022 (2023).
Bi, J. et al. Protective effect of vildagliptin on TNF‐α‐induced chondrocyte senescence. IUBMB life 71, 978–985 (2019).
Platas, J. et al. Anti-senescence and anti-inflammatory effects of the c-terminal moiety of PTHrP peptides in OA osteoblasts. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 72, 624–631 (2017).
Zhang, C., Jiang, S., Lu, Y. & Yuan, F. Butorphanol tartrate mitigates cellular senescence against tumor necrosis factor–α (TNF-α) in human HC-A chondrocytes. Bioengineered 13, 5434–5442 (2022).
Clérigues, V. et al. Heme oxygenase-1 mediates protective effects on inflammatory, catabolic and senescence responses induced by interleukin-1β in osteoarthritic osteoblasts. Biochemical Pharmacol. 83, 395–405 (2012).
Dai, H. et al. Eliminating senescent chondrogenic progenitor cells enhances chondrogenesis under intermittent hydrostatic pressure for the treatment of OA. Stem Cell Res. Ther. 11, 1–18 (2020).
Miura, Y., Endo, K., Komori, K., Sekiya, I. & Therapy. Clearance of senescent cells with ABT-263 improves biological functions of synovial mesenchymal stem cells from osteoarthritis patients. Stem Cell Res. Ther. 13, 222 (2022).
Wang, X. et al. Conversion of senescent cartilage into a pro-chondrogenic microenvironment with antibody-functionalized copper sulfate nanoparticles for efficient osteoarthritis therapy. J. Nanobiotechnol. 21, 258 (2023).
Sacitharan, P. K., Lwin, S., Gharios, G. B. & Edwards, J. R. Spermidine restores dysregulated autophagy and polyamine synthesis in aged and osteoarthritic chondrocytes via EP300. Exp. Mol. Med. 50, 1–10 (2018).
Liao, B., Ding, M., Wang, Y., Xu, H. & Shangguan, L. Strontium ion attenuates osteoarthritis through inhibiting senescence and enhancing autophagy in fibroblast-like synoviocytes. Mol. Biol. Rep. 50, 1437–1446 (2023).
Acknowledgements
This review work was supported by the Beijing Hospitals Authority’s Ascent Plan, the National Natural Science Foundation of China (82272473, 82402805), the Talent Development Plan for High-level Public Health Technical Personnel Project (Subject leader -02-23), the Beijing Natural Science Foundation Haidian Original Innovation Joint Fund (L212010), the Priming Scientific Research Foundation for the Junior Researcher in Beijing Tongren Hospital (2023-YJJ-ZZL-016).
Author information
Authors and Affiliations
Contributions
M.Z. designed the concept of the study. Z.H. and K.W. wrote the manuscript. S.D and M.Z. revised the paper. All the authors read the paper, and M.Z. approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, Z., Wang, K., Ding, S. et al. Cross-talk of inflammation and cellular senescence: a new insight into the occurrence and progression of osteoarthritis. Bone Res 12, 69 (2024). https://doi.org/10.1038/s41413-024-00375-z
Received:
Revised:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41413-024-00375-z
This article is cited by
-
Epigenetic silencing of SPHK1-IRF7 axis drives inflammaging in age-related meniscus degeneration via sphingolipid-immune dysregulation
Journal of Orthopaedic Surgery and Research (2025)
-
Senescent bone marrow mesenchymal stem cells exacerbate subchondral bone sclerosis and osteoarthritis via the senescence-associated secretory phenotype
Stem Cell Research & Therapy (2025)
-
Arthritis and increased risk of cardiovascular disease: is there an interaction with other chronic diseases? A nationwide cohort study
BMC Cardiovascular Disorders (2025)
-
The role of the immune system in osteoarthritis: mechanisms, challenges and future directions
Nature Reviews Rheumatology (2025)
-
Single-cell sequencing reveals a senescent immune landscape in bone marrow lesions inducing articular cartilage damage in osteoarthritis
Bone Research (2025)
